
The Interplay between Dysregulated Ion Transport and Mitochondrial Architecture as a Dangerous Liaison in Cancer


{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
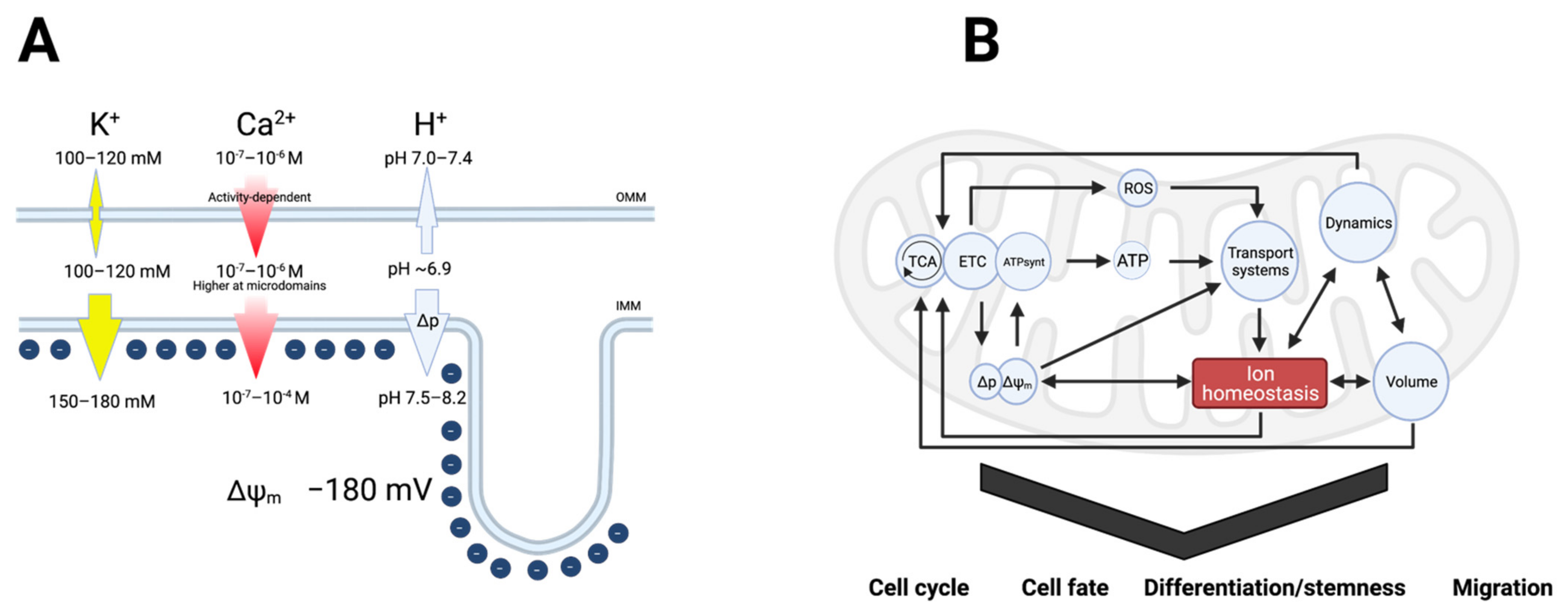
2. Interdependent Ion Fluxes Drive Mitochondrial Function
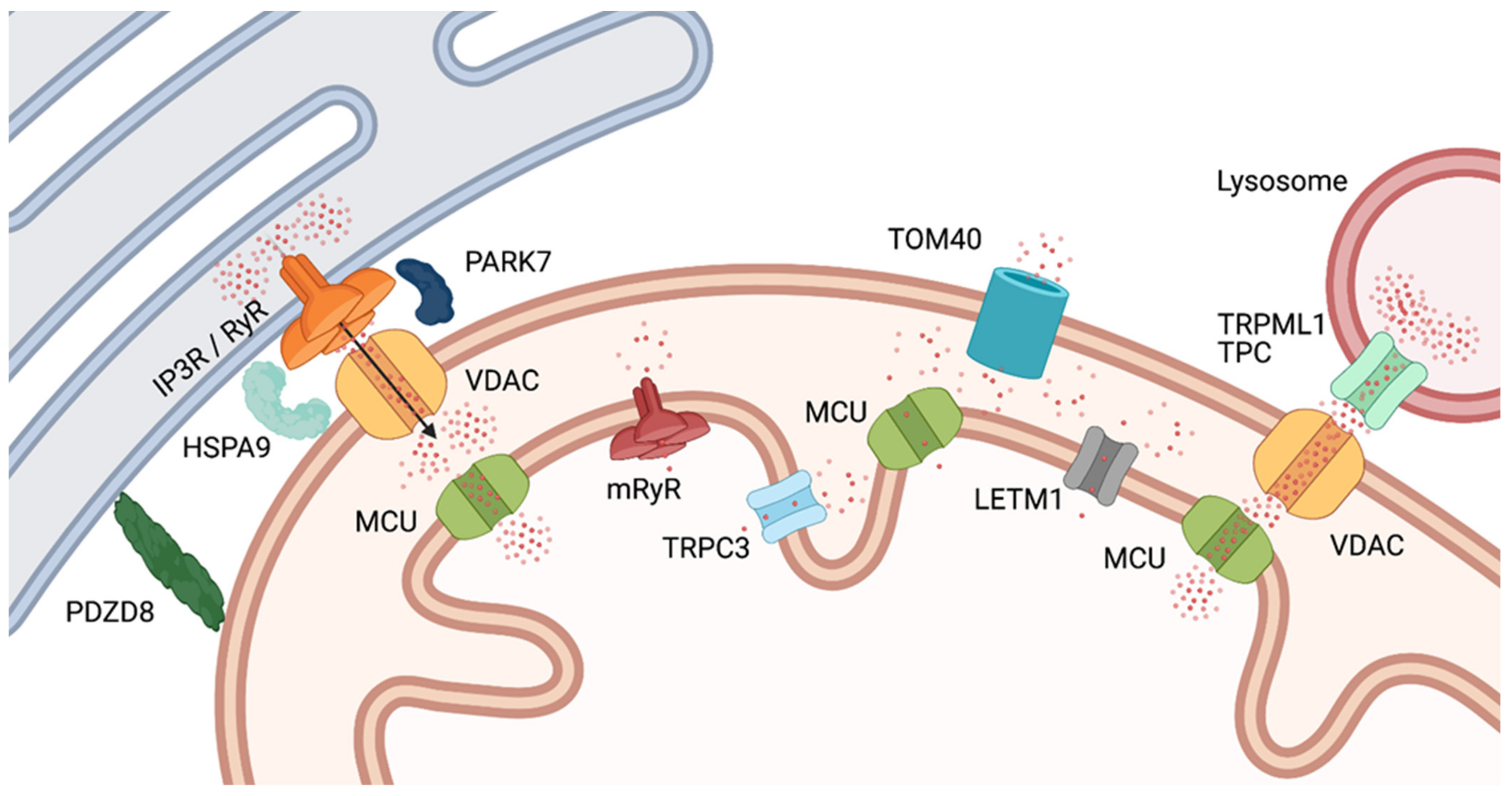
2.1. Ca2+ in Mitochondrial Function
2.2. Protons in Mitochondrial Function
2.3. Potassium in Mitochondrial Function
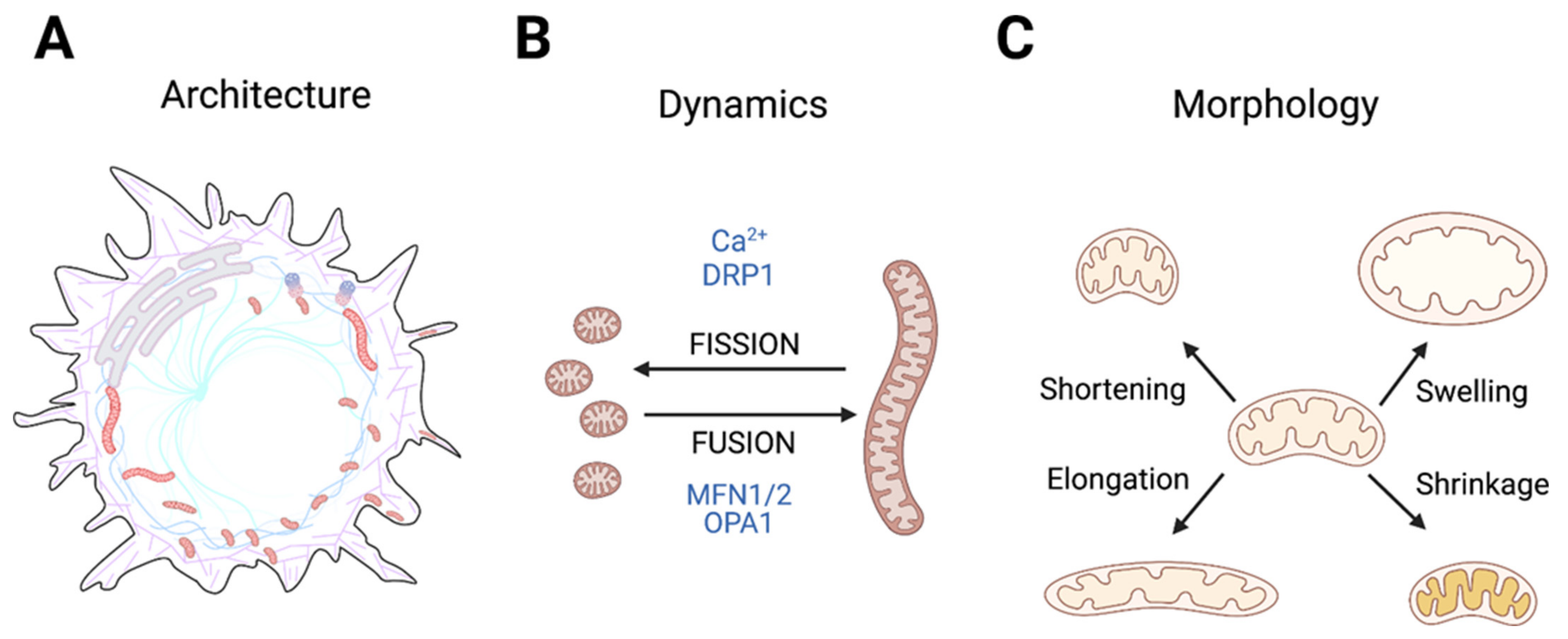
3. Mitochondrial Architecture, Dynamics, and Morphology in Normal Cells
3.1. Physiological Processes Dependent on Changes in Mitochondrial Architecture
3.2. Regulation of Mitochondrial Architecture: A Highly Ca2+-Dependent Process
3.3. Impact of H+ and K+ Homeostasis on Mitochondrial Architecture—and Vice Versa
4. General Features of Mitochondrial Architecture in Cancer Cells
5. Cancer Cell Functions Driven by Changes in Mitochondrial Architecture
5.1. Survival in the Harsh Tumor Microenvironment
5.2. Cell Proliferation, Stemness, and Senescence
5.3. Cell Migration and Invasion
5.4. ROS Production and Signaling
5.5. Resisting Cell Death
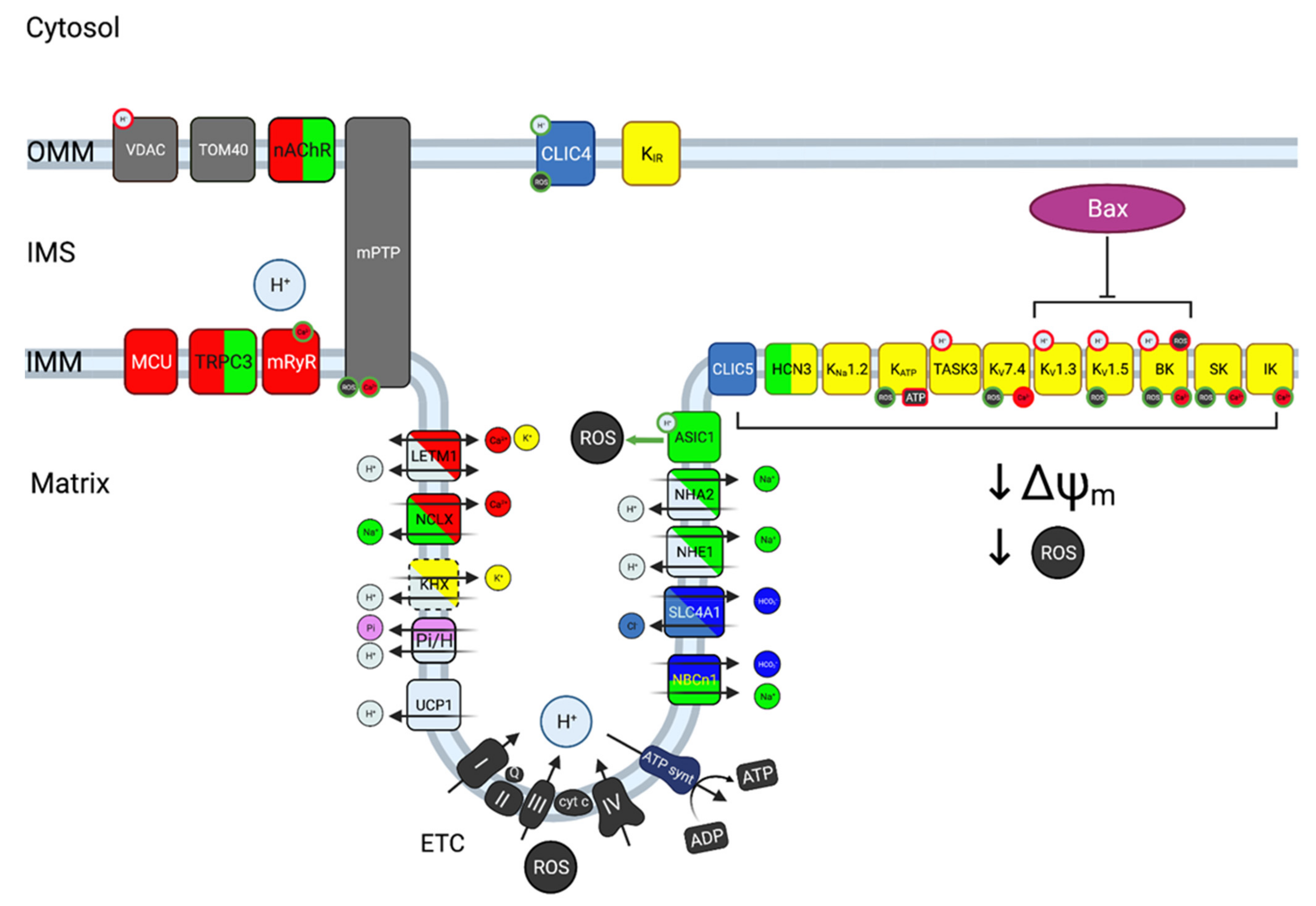
6. Dysregulation of Mitochondrial Ion Channels and Transporters in Cancer
7. Conclusions and Perspectives
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Ernster, L.; Schatz, G. Mitochondria: A historical review. J. Cell Biol. 1981, 91, 227s–255s. [Google Scholar] [CrossRef] [PubMed]
- Audano, M.; Pedretti, S.; Ligorio, S.; Crestani, M.; Caruso, D.; De Fabiani, E.; Mitro, N. “The Loss of Golden Touch”: Mitochondria-Organelle Interactions, Metabolism, and Cancer. Cells 2020, 9, 2519. [Google Scholar] [CrossRef] [PubMed]
- Fenton, A.R.; Jongens, T.A.; Holzbaur, E.L.F. Mitochondrial dynamics: Shaping and remodeling an organelle network. Curr. Opin. Cell Biol. 2020, 68, 28–36. [Google Scholar] [CrossRef] [PubMed]
- Vyas, S.; Zaganjor, E.; Haigis, M.C. Mitochondria and Cancer. Cell 2016, 166, 555–566. [Google Scholar] [CrossRef]
- Wallace, D.C. Mitochondria and cancer. Nat. Rev. Cancer 2012, 12, 685–698. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gillies, R.J.; Verduzco, D.; Gatenby, R.A. Evolutionary dynamics of carcinogenesis and why targeted therapy does not work. Nat. Rev. Cancer 2012, 12, 487–493. [Google Scholar] [CrossRef] [PubMed]
- Corbet, C.; Feron, O. Tumour acidosis: From the passenger to the driver’s seat. Nat. Rev. Cancer 2017, 17, 577–593. [Google Scholar] [CrossRef] [PubMed]
- White, K.A.; Grillo-Hill, B.K.; Barber, D.L. Cancer cell behaviors mediated by dysregulated pH dynamics at a glance. J. Cell Sci. 2017, 130, 663–669. [Google Scholar] [CrossRef] [Green Version]
- Boedtkjer, E.; Pedersen, S.F. The Acidic Tumor Microenvironment as a Driver of Cancer. Annu. Rev. Physiol. 2020, 82, 103–126. [Google Scholar] [CrossRef] [Green Version]
- Pardo, L.A.; Stühmer, W. The roles of K+ channels in cancer. Nat. Rev. Cancer 2014, 14, 39–48. [Google Scholar] [CrossRef]
- Prevarskaya, N.; Skryma, R.; Shuba, Y. Ion Channels in Cancer: Are Cancer Hallmarks Oncochannelopathies? Physiol. Rev. 2018, 98, 559–621. [Google Scholar] [CrossRef] [Green Version]
- Porporato, P.E.; Filigheddu, N.; Pedro, J.M.B.; Kroemer, G.; Galluzzi, L. Mitochondrial metabolism and cancer. Cell Res. 2018, 28, 265–280. [Google Scholar] [CrossRef]
- Kozoriz, M.G.; Church, J.; Ozog, M.A.; Naus, C.C.; Krebs, C. Temporary sequestration of potassium by mitochondria in astrocytes. J. Biol. Chem. 2010, 285, 31107–31119. [Google Scholar] [CrossRef] [Green Version]
- Garlid, K.D. Cation transport in mitochondria—The potassium cycle. Biochim. Biophys. Acta 1996, 1275, 123–126. [Google Scholar] [CrossRef] [Green Version]
- Kowaltowski, A.J.; Cosso, R.G.; Campos, C.B.; Fiskum, G. Effect of Bcl-2 overexpression on mitochondrial structure and function. J. Biol. Chem. 2002, 277, 42802–42807. [Google Scholar] [CrossRef] [Green Version]
- Kaasik, A.; Safiulina, D.; Zharkovsky, A.; Veksler, V. Regulation of mitochondrial matrix volume. Am. J. Physiol. Cell Physiol. 2007, 292, C157–C163. [Google Scholar] [CrossRef]
- Sorgato, M.C.; Keller, B.U.; Stuhmer, W. Patch-clamping of the inner mitochondrial membrane reveals a voltage-dependent ion channel. Nature 1987, 330, 498–500. [Google Scholar] [CrossRef]
- Fahanik-Babaei, J.; Rezaee, B.; Nazari, M.; Torabi, N.; Saghiri, R.; Sauve, R.; Eliassi, A. A new brain mitochondrial sodium-sensitive potassium channel: Effect of sodium ions on respiratory chain activity. J. Cell Sci. 2020, 133, jcs242446. [Google Scholar] [CrossRef]
- Parrasia, S.; Mattarei, A.; Furlan, A.; Zoratti, M.; Biasutto, L. Small-Molecule Modulators of Mitochondrial Channels as Chemotherapeutic Agents. Cell Physiol. Biochem. 2019, 53, 11–43. [Google Scholar] [CrossRef] [Green Version]
- Bednarczyk, P.; Wieckowski, M.R.; Broszkiewicz, M.; Skowronek, K.; Siemen, D.; Szewczyk, A. Putative Structural and Functional Coupling of the Mitochondrial BKCa Channel to the Respiratory Chain. PLoS ONE 2013, 8, e68125. [Google Scholar] [CrossRef]
- Szabo, I.; Bock, J.; Grassme, H.; Soddemann, M.; Wilker, B.; Lang, F.; Zoratti, M.; Gulbins, E. Mitochondrial potassium channel Kv1.3 mediates Bax-induced apoptosis in lymphocytes. Proc. Natl. Acad. Sci. USA 2008, 105, 14861–14866. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Skalska, J.; Piwonska, M.; Wyroba, E.; Surmacz, L.; Wieczorek, R.; Koszela-Piotrowska, I.; Zielinska, J.; Bednarczyk, P.; Dolowy, K.; Wilczynski, G.M.; et al. A novel potassium channel in skeletal muscle mitochondria. Biochim. Biophys. Acta 2008, 1777, 651–659. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Savic Azoulay, I.; Liu, F.; Hu, Q.; Rozenfeld, M.; Ben Kasus Nissim, T.; Zhu, M.X.; Sekler, I.; Xu, T.L. ASIC1a channels regulate mitochondrial ion signaling and energy homeostasis in neurons. J. Neurochem. 2020, 153, 203–215. [Google Scholar] [CrossRef] [PubMed]
- Malinska, D.; Mirandola, S.R.; Kunz, W.S. Mitochondrial potassium channels and reactive oxygen species. FEBS Lett. 2010, 584, 2043–2048. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fornazari, M.; de Paula, J.G.; Castilho, R.F.; Kowaltowski, A.J. Redox properties of the adenoside triphosphate-sensitive K+ channel in brain mitochondria. J. Neurosci. Res. 2008, 86, 1548–1556. [Google Scholar] [CrossRef] [PubMed]
- Raffaello, A.; Mammucari, C.; Gherardi, G.; Rizzuto, R. Calcium at the Center of Cell Signaling: Interplay between Endoplasmic Reticulum, Mitochondria, and Lysosomes. Trends Biochem. Sci. 2016, 41, 1035–1049. [Google Scholar] [CrossRef] [Green Version]
- Trombetta-Lima, M.; Krabbendam, I.E.; Dolga, A.M. Calcium-activated potassium channels: Implications for aging and age-related neurodegeneration. Int. J. Biochem. Cell Biol. 2020, 123, 105748. [Google Scholar] [CrossRef]
- Szabo, I.; Zoratti, M.; Biasutto, L. Targeting mitochondrial ion channels for cancer therapy. Redox Biol. 2020, 42, 101846. [Google Scholar] [CrossRef]
- Nowikovsky, K.; Schweyen, R.J.; Bernardi, P. Pathophysiology of mitochondrial volume homeostasis: Potassium transport and permeability transition. Biochim. Biophys. Acta 2009, 1787, 345–350. [Google Scholar] [CrossRef]
- Peng, K.; Hu, J.; Xiao, J.; Dan, G.; Yang, L.; Ye, F.; Zou, Z.; Cao, J.; Sai, Y. Mitochondrial ATP-sensitive potassium channel regulates mitochondrial dynamics to participate in neurodegeneration of Parkinson’s disease. Biochim. Biophys. Acta Mol. Basis Dis. 2018, 1864, 1086–1103. [Google Scholar] [CrossRef]
- Bauer, T.M.; Murphy, E. Role of Mitochondrial Calcium and the Permeability Transition Pore in Regulating Cell Death. Circ. Res. 2020, 126, 280–293. [Google Scholar] [CrossRef] [PubMed]
- Chapman, C.G.; Meadows, H.J.; Godden, R.J.; Campbell, D.A.; Duckworth, M.; Kelsell, R.E.; Murdock, P.R.; Randall, A.D.; Rennie, G.I.; Gloger, I.S. Cloning, localisation and functional expression of a novel human, cerebellum specific, two pore domain potassium channel. Brain Res. Mol. Brain Res. 2000, 82, 74–83. [Google Scholar] [CrossRef]
- Rajan, S.; Wischmeyer, E.; Liu, X.G.; Preisig-Muller, R.; Daut, J.; Karschin, A.; Derst, C. TASK-3, a novel tandem pore domain acid-sensitive K+ channel. An extracellular histiding as pH sensor. J. Biol. Chem. 2000, 275, 16650–16657. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, Y.; Xia, X.M.; Lingle, C.J. BK channel inhibition by strong extracellular acidification. Elife 2018, 7, e38060. [Google Scholar] [CrossRef] [PubMed]
- Avdonin, V.; Tang, X.D.; Hoshi, T. Stimulatory action of internal protons on Slo1 BK channels. Biophys. J. 2003, 84, 2969–2980. [Google Scholar] [CrossRef] [Green Version]
- Kellenberger, S.; Rash, L.D. Acid-sensing (proton-gated) ion channels (ASICs) (version 2020.5) in the IUPHAR/BPS Guide to Pharmacology Database. IUPHAR BPS Guide Pharmacol. CITE 2020, 2020. [Google Scholar] [CrossRef]
- Poburko, D.; Santo-Domingo, J.; Demaurex, N. Dynamic regulation of the mitochondrial proton gradient during cytosolic calcium elevations. J. Biol. Chem. 2011, 286, 11672–11684. [Google Scholar] [CrossRef] [Green Version]
- Berridge, M.J.; Lipp, P.; Bootman, M.D. The versatility and universality of calcium signalling. Nat. Rev. Mol. Cell Biol. 2000, 1, 11–21. [Google Scholar] [CrossRef]
- Clapham, D.E. Calcium signaling. Cell 2007, 131, 1047–1058. [Google Scholar] [CrossRef] [Green Version]
- Contreras, L.; Drago, I.; Zampese, E.; Pozzan, T. Mitochondria: The calcium connection. Biochim. Biophys. Acta 2010, 1797, 607–618. [Google Scholar] [CrossRef] [Green Version]
- Peruzzo, R.; Costa, R.; Bachmann, M.; Leanza, L.; Szabo, I. Mitochondrial Metabolism, Contact Sites and Cellular Calcium Signaling: Implications for Tumorigenesis. Cancers 2020, 12, 2574. [Google Scholar] [CrossRef] [PubMed]
- Denton, R.M. Regulation of mitochondrial dehydrogenases by calcium ions. Biochim. Biophys. Acta 2009, 1787, 1309–1316. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peng, T.I.; Jou, M.J. Oxidative stress caused by mitochondrial calcium overload. Ann. N. Y. Acad. Sci. 2010, 1201, 183–188. [Google Scholar] [CrossRef] [PubMed]
- Gorlach, A.; Bertram, K.; Hudecova, S.; Krizanova, O. Calcium and ROS: A mutual interplay. Redox Biol. 2015, 6, 260–271. [Google Scholar] [CrossRef] [Green Version]
- Delierneux, C.; Kouba, S.; Shanmughapriya, S.; Potier-Cartereau, M.; Trebak, M.; Hempel, N. Mitochondrial Calcium Regulation of Redox Signaling in Cancer. Cells 2020, 9, 432. [Google Scholar] [CrossRef] [Green Version]
- Wolf, S.G.; Mutsafi, Y.; Dadosh, T.; Ilani, T.; Lansky, Z.; Horowitz, B.; Rubin, S.; Elbaum, M.; Fass, D. 3D visualization of mitochondrial solid-phase calcium stores in whole cells. Elife 2017, 6, e29929. [Google Scholar] [CrossRef]
- Montero, M.; Alonso, M.T.; Carnicero, E.; Cuchillo-Ibanez, I.; Albillos, A.; Garcia, A.G.; Garcia-Sancho, J.; Alvarez, J. Chromaffin-cell stimulation triggers fast millimolar mitochondrial Ca2+ transients that modulate secretion. Nat. Cell Biol. 2000, 2, 57–61. [Google Scholar] [CrossRef]
- Sander, P.; Gudermann, T.; Schredelseker, J. A Calcium Guard in the Outer Membrane: Is VDAC a Regulated Gatekeeper of Mitochondrial Calcium Uptake? Int. J. Mol. Sci. 2021, 22, 946. [Google Scholar] [CrossRef]
- Kruger, V.; Becker, T.; Becker, L.; Montilla-Martinez, M.; Ellenrieder, L.; Vogtle, F.N.; Meyer, H.E.; Ryan, M.T.; Wiedemann, N.; Warscheid, B.; et al. Identification of new channels by systematic analysis of the mitochondrial outer membrane. J. Cell Biol. 2017, 216, 3485–3495. [Google Scholar] [CrossRef]
- Fieni, F.; Parkar, A.; Misgeld, T.; Kerschensteiner, M.; Lichtman, J.W.; Pasinelli, P.; Trotti, D. Voltage-dependent inwardly rectifying potassium conductance in the outer membrane of neuronal mitochondria. J. Biol. Chem. 2010, 285, 27411–27417. [Google Scholar] [CrossRef] [Green Version]
- Gergalova, G.; Lykhmus, O.; Kalashnyk, O.; Koval, L.; Chernyshov, V.; Kryukova, E.; Tsetlin, V.; Komisarenko, S.; Skok, M. Mitochondria express alpha7 nicotinic acetylcholine receptors to regulate Ca2+ accumulation and cytochrome c release: Study on isolated mitochondria. PLoS ONE 2012, 7, e31361. [Google Scholar] [CrossRef]
- Ponnalagu, D.; Gururaja Rao, S.; Farber, J.; Xin, W.; Hussain, A.T.; Shah, K.; Tanda, S.; Berryman, M.; Edwards, J.C.; Singh, H. Molecular identity of cardiac mitochondrial chloride intracellular channel proteins. Mitochondrion 2016, 27, 6–14. [Google Scholar] [CrossRef]
- Tan, W.; Colombini, M. VDAC closure increases calcium ion flux. Biochim. Biophys. Acta 2007, 1768, 2510–2515. [Google Scholar] [CrossRef] [Green Version]
- Preto, J.; Krimm, I. The intrinsically disordered N-terminus of the voltage-dependent anion channel. PLoS Comput. Biol. 2021, 17, e1008750. [Google Scholar] [CrossRef]
- Szabadkai, G.; Bianchi, K.; Varnai, P.; De Stefani, D.; Wieckowski, M.R.; Cavagna, D.; Nagy, A.I.; Balla, T.; Rizzuto, R. Chaperone-mediated coupling of endoplasmic reticulum and mitochondrial Ca2+ channels. J. Cell Biol. 2006, 175, 901–911. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Ma, X.; Fujioka, H.; Liu, J.; Chen, S.; Zhu, X. DJ-1 regulates the integrity and function of ER-mitochondria association through interaction with IP3R3-Grp75-VDAC1. Proc. Natl. Acad. Sci. USA 2019, 116, 25322–25328. [Google Scholar] [CrossRef]
- Rosencrans, W.M.; Rajendran, M.; Bezrukov, S.M.; Rostovtseva, T.K. VDAC regulation of mitochondrial calcium flux: From channel biophysics to disease. Cell Calcium. 2021, 94, 102356. [Google Scholar] [CrossRef]
- Hirabayashi, Y.; Kwon, S.K.; Paek, H.; Pernice, W.M.; Paul, M.A.; Lee, J.; Erfani, P.; Raczkowski, A.; Petrey, D.S.; Pon, L.A.; et al. ER-mitochondria tethering by PDZD8 regulates Ca(2+) dynamics in mammalian neurons. Science 2017, 358, 623–630. [Google Scholar] [CrossRef] [Green Version]
- Kirichok, Y.; Krapivinsky, G.; Clapham, D.E. The mitochondrial calcium uniporter is a highly selective ion channel. Nature 2004, 427, 360–364. [Google Scholar] [CrossRef]
- Beutner, G.; Sharma, V.K.; Giovannucci, D.R.; Yule, D.I.; Sheu, S.S. Identification of a ryanodine receptor in rat heart mitochondria. J. Biol. Chem. 2001, 276, 21482–21488. [Google Scholar] [CrossRef] [Green Version]
- Ryu, S.Y.; Beutner, G.; Dirksen, R.T.; Kinnally, K.W.; Sheu, S.S. Mitochondrial ryanodine receptors and other mitochondrial Ca2+ permeable channels. FEBS Lett. 2010, 584, 1948–1955. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feng, S.; Li, H.; Tai, Y.; Huang, J.; Su, Y.; Abramowitz, J.; Zhu, M.X.; Birnbaumer, L.; Wang, Y. Canonical transient receptor potential 3 channels regulate mitochondrial calcium uptake. Proc. Natl. Acad. Sci. USA 2013, 110, 11011–11016. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Naumova, N.; Sachl, R. Regulation of Cell Death by Mitochondrial Transport Systems of Calcium and Bcl-2 Proteins. Membranes 2020, 10, 299. [Google Scholar] [CrossRef] [PubMed]
- Belosludtsev, K.N.; Dubinin, M.V.; Belosludtseva, N.V.; Mironova, G.D. Mitochondrial Ca2+ Transport: Mechanisms, Molecular Structures, and Role in Cells. Biochemistry 2019, 84, 593–607. [Google Scholar] [CrossRef]
- Fieni, F.; Lee, S.B.; Jan, Y.N.; Kirichok, Y. Activity of the mitochondrial calcium uniporter varies greatly between tissues. Nat. Commun. 2012, 3, 1317. [Google Scholar] [CrossRef] [Green Version]
- Wong, Y.C.; Ysselstein, D.; Krainc, D. Mitochondria-lysosome contacts regulate mitochondrial fission via RAB7 GTP hydrolysis. Nature 2018, 554, 382–386. [Google Scholar] [CrossRef]
- Peng, W.; Wong, Y.C.; Krainc, D. Mitochondria-lysosome contacts regulate mitochondrial Ca(2+) dynamics via lysosomal TRPML1. Proc. Natl. Acad. Sci. USA 2020, 117, 19266–19275. [Google Scholar] [CrossRef]
- Patel, S.; Muallem, S. Acidic Ca(2+) stores come to the fore. Cell Calcium. 2011, 50, 109–112. [Google Scholar] [CrossRef]
- Briston, T.; Selwood, D.L.; Szabadkai, G.; Duchen, M.R. Mitochondrial Permeability Transition: A Molecular Lesion with Multiple Drug Targets. Trends Pharmacol. Sci. 2019, 40, 50–70. [Google Scholar] [CrossRef]
- Beretta, M.; Santos, C.X.; Molenaar, C.; Hafstad, A.D.; Miller, C.C.; Revazian, A.; Betteridge, K.; Schroder, K.; Streckfuss-Bomeke, K.; Doroshow, J.H.; et al. Nox4 regulates InsP3 receptor-dependent Ca(2+) release into mitochondria to promote cell survival. EMBO J. 2020, 39, e103530. [Google Scholar] [CrossRef]
- Mitchell, P.; Moyle, J. Estimation of membrane potential and pH difference across the cristae membrane of rat liver mitochondria. Eur. J. Biochem. 1969, 7, 471–484. [Google Scholar] [CrossRef] [PubMed]
- Nicholls, D.G. The influence of respiration and ATP hydrolysis on the proton-electrochemical gradient across the inner membrane of rat-liver mitochondria as determined by ion distribution. Eur. J. Biochem. 1974, 50, 305–315. [Google Scholar] [CrossRef] [PubMed]
- Rottenberg, H. The measurement of transmembrane electrochemical proton gradients. J. Bioenerg. 1975, 7, 61–74. [Google Scholar] [CrossRef] [PubMed]
- Lemasters, J.J.; Chacon, E.; Ohata, H.; Harper, I.S.; Nieminen, A.L.; Tesfai, S.A.; Herman, B. Measurement of electrical potential, pH, and free calcium ion concentration in mitochondria of living cells by laser scanning confocal microscopy. Methods Enzymol. 1995, 260, 428–444. [Google Scholar] [CrossRef]
- Llopis, J.; McCaffery, J.M.; Miyawaki, A.; Farquhar, M.G.; Tsien, R.Y. Measurement of cytosolic, mitochondrial, and Golgi pH in single living cells with green fluorescent proteins. Proc. Natl. Acad. Sci. USA 1998, 95, 6803–6808. [Google Scholar] [CrossRef] [Green Version]
- Abad, M.F.; Di Benedetto, G.; Magalhaes, P.J.; Filippin, L.; Pozzan, T. Mitochondrial pH monitored by a new engineered green fluorescent protein mutant. J. Biol. Chem. 2004, 279, 11521–11529. [Google Scholar] [CrossRef] [Green Version]
- Porcelli, A.M.; Ghelli, A.; Zanna, C.; Pinton, P.; Rizzuto, R.; Rugolo, M. pH difference across the outer mitochondrial membrane measured with a green fluorescent protein mutant. Biochem. Biophys. Res. Commun. 2005, 326, 799–804. [Google Scholar] [CrossRef]
- Saraste, M. Oxidative phosphorylation at the fin de siecle. Science 1999, 283, 1488–1493. [Google Scholar] [CrossRef]
- Santo-Domingo, J.; Demaurex, N. Perspectives on: SGP symposium on mitochondrial physiology and medicine: The renaissance of mitochondrial pH. J. Gen. Physiol. 2012, 139, 415–423. [Google Scholar] [CrossRef] [Green Version]
- Jiang, D.; Zhao, L.; Clapham, D.E. Genome-wide RNAi screen identifies Letm1 as a mitochondrial Ca2+/H+ antiporter. Science 2009, 326, 144–147. [Google Scholar] [CrossRef] [Green Version]
- Natarajan, G.K.; Mishra, J.; Camara, A.K.S.; Kwok, W.M. LETM1: A Single Entity With Diverse Impact on Mitochondrial Metabolism and Cellular Signaling. Front. Physiol. 2021, 12, 637852. [Google Scholar] [CrossRef]
- Lin, Q.T.; Lee, R.; Feng, A.L.; Kim, M.S.; Stathopulos, P.B. The leucine zipper EF-hand containing transmembrane protein-1 EF-hand is a tripartite calcium, temperature, and pH sensor. Protein. Sci. 2021, 30, 855–872. [Google Scholar] [CrossRef]
- Piao, L.; Li, Y.; Kim, S.J.; Byun, H.S.; Huang, S.M.; Hwang, S.K.; Yang, K.J.; Park, K.A.; Won, M.; Hong, J.; et al. Association of LETM1 and MRPL36 contributes to the regulation of mitochondrial ATP production and necrotic cell death. Cancer Res. 2009, 69, 3397–3404. [Google Scholar] [CrossRef] [Green Version]
- Brett, C.L.; Wei, Y.; Donowitz, M.; Rao, R. Human Na(+)/H(+) exchanger isoform 6 is found in recycling endosomes of cells, not in mitochondria. Am. J. Physiol. Cell Physiol. 2002, 282, C1031–C1041. [Google Scholar] [CrossRef] [Green Version]
- Javadov, S.; Rajapurohitam, V.; Kilic, A.; Hunter, J.C.; Zeidan, A.; Said Faruq, N.; Escobales, N.; Karmazyn, M. Expression of mitochondrial fusion-fission proteins during post-infarction remodeling: The effect of NHE-1 inhibition. Basic Res. Cardiol. 2011, 106, 99–109. [Google Scholar] [CrossRef]
- Fuster, D.G.; Zhang, J.; Shi, M.; Bobulescu, I.A.; Andersson, S.; Moe, O.W. Characterization of the sodium/hydrogen exchanger NHA2. J. Am. Soc. Nephrol. 2008, 19, 1547–1556. [Google Scholar] [CrossRef] [Green Version]
- Pedersen, S.F.; Counillon, L. The SLC9A-C Mammalian Na(+)/H(+) Exchanger Family: Molecules, Mechanisms, and Physiology. Physiol. Rev. 2019, 99, 2015–2113. [Google Scholar] [CrossRef]
- Austin, S.; Nowikovsky, K. LETM1: Essential for Mitochondrial Biology and Cation Homeostasis? Trends Biochem. Sci. 2019, 44, 648–658. [Google Scholar] [CrossRef] [Green Version]
- Palmieri, F. The mitochondrial transporter family (SLC25): Physiological and pathological implications. Pflug. Arch. 2004, 447, 689–709. [Google Scholar] [CrossRef]
- Dodgson, S.J.; Forster, R.E., 2nd; Storey, B.T.; Mela, L. Mitochondrial carbonic anhydrase. Proc. Natl. Acad. Sci. USA 1980, 77, 5562–5566. [Google Scholar] [CrossRef] [Green Version]
- Ostedgaard, L.S.; Jennings, M.L.; Karniski, L.P.; Schuster, V.L. A 45-kDa protein antigenically related to band 3 is selectively expressed in kidney mitochondria. Proc. Natl. Acad. Sci. USA 1991, 88, 981–985. [Google Scholar] [CrossRef] [Green Version]
- Kumar, S.; Flacke, J.P.; Kostin, S.; Appukuttan, A.; Reusch, H.P.; Ladilov, Y. SLC4A7 sodium bicarbonate co-transporter controls mitochondrial apoptosis in ischaemic coronary endothelial cells. Cardiovasc. Res. 2011, 89, 392–400. [Google Scholar] [CrossRef] [Green Version]
- Acin-Perez, R.; Salazar, E.; Kamenetsky, M.; Buck, J.; Levin, L.R.; Manfredi, G. Cyclic AMP produced inside mitochondria regulates oxidative phosphorylation. Cell Metab. 2009, 9, 265–276. [Google Scholar] [CrossRef] [Green Version]
- Litvin, T.N.; Kamenetsky, M.; Zarifyan, A.; Buck, J.; Levin, L.R. Kinetic properties of “soluble” adenylyl cyclase. Synergism between calcium and bicarbonate. J. Biol. Chem. 2003, 278, 15922–15926. [Google Scholar] [CrossRef] [Green Version]
- Korge, P.; Honda, H.M.; Weiss, J.N. K+-dependent regulation of matrix volume improves mitochondrial function under conditions mimicking ischemia-reperfusion. Am. J. Physiol. Heart Circ. Physiol. 2005, 289, H66–H77. [Google Scholar] [CrossRef] [Green Version]
- Paggio, A.; Checchetto, V.; Campo, A.; Menabo, R.; Di Marco, G.; Di Lisa, F.; Szabo, I.; Rizzuto, R.; De Stefani, D. Identification of an ATP-sensitive potassium channel in mitochondria. Nature 2019, 572, 609–613. [Google Scholar] [CrossRef]
- Foster, D.B.; Ho, A.S.; Rucker, J.; Garlid, A.O.; Chen, L.; Sidor, A.; Garlid, K.D.; O’Rourke, B. Mitochondrial ROMK channel is a molecular component of mitoK(ATP). Circ. Res. 2012, 111, 446–454. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ye, B.; Kroboth, S.L.; Pu, J.L.; Sims, J.J.; Aggarwal, N.T.; McNally, E.M.; Makielski, J.C.; Shi, N.Q. Molecular identification and functional characterization of a mitochondrial sulfonylurea receptor 2 splice variant generated by intraexonic splicing. Circ. Res. 2009, 105, 1083–1093. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stowe, D.F.; Gadicherla, A.K.; Zhou, Y.; Aldakkak, M.; Cheng, Q.; Kwok, W.M.; Jiang, M.T.; Heisner, J.S.; Yang, M.; Camara, A.K. Protection against cardiac injury by small Ca(2+)-sensitive K(+) channels identified in guinea pig cardiac inner mitochondrial membrane. Biochim. Biophys. Acta 2013, 1828, 427–442. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Marchi, U.; Sassi, N.; Fioretti, B.; Catacuzzeno, L.; Cereghetti, G.M.; Szabo, I.; Zoratti, M. Intermediate conductance Ca2+-activated potassium channel (KCa3.1) in the inner mitochondrial membrane of human colon cancer cells. Cell Calcium. 2009, 45, 509–516. [Google Scholar] [CrossRef]
- Sassi, N.; De Marchi, U.; Fioretti, B.; Biasutto, L.; Gulbins, E.; Franciolini, F.; Szabo, I.; Zoratti, M. An investigation of the occurrence and properties of the mitochondrial intermediate-conductance Ca2+-activated K+ channel mtKCa3.1. Biochim. Biophys. Acta 2010, 1797, 1260–1267. [Google Scholar] [CrossRef] [Green Version]
- Balderas, E.; Zhang, J.; Stefani, E.; Toro, L. Mitochondrial BKCa channel. Front. Physiol. 2015, 6, 104. [Google Scholar] [CrossRef] [Green Version]
- Aldrich, R.; Chandy, K.G.; Grissmer, S.; Gutman, G.A.; Kaczmarek, L.K.; Wei, A.D.; Wulff, H. Calcium- and sodium-activated potassium channels (version 2019.4) in the IUPHAR/BPS Guide to Pharmacology Database. IUPHAR BPS Guide Pharmacol. CITE 2019, 2019. [Google Scholar] [CrossRef] [Green Version]
- Sesti, F.; Liu, S.; Cai, S.Q. Oxidation of potassium channels by ROS: A general mechanism of aging and neurodegeneration? Trends Cell Biol. 2010, 20, 45–51. [Google Scholar] [CrossRef]
- Smith, C.O.; Wang, Y.T.; Nadtochiy, S.M.; Miller, J.H.; Jonas, E.A.; Dirksen, R.T.; Nehrke, K.; Brookes, P.S. Cardiac metabolic effects of KNa1.2 channel deletion and evidence for its mitochondrial localization. FASEB J. 2018, 32, fj201800139R. [Google Scholar] [CrossRef]
- Attali, B.; Chandy, K.G.; Giese, M.H.; Grissmer, S.; Gutman, G.A.; Jan, L.Y.; Lazdunski, M.; Mckinnon, D.; Nerbonne, J.; Pardo, L.A.; et al. Voltage-gated potassium channels (version 2019.4) in the IUPHAR/BPS Guide to Pharmacology Database. IUPHAR/BPS Guide Pharmacol. CITE 2019. [Google Scholar] [CrossRef] [Green Version]
- Bonnet, S.; Archer, S.L.; Allalunis-Turner, J.; Haromy, A.; Beaulieu, C.; Thompson, R.; Lee, C.T.; Lopaschuk, G.D.; Puttagunta, L.; Bonnet, S.; et al. A mitochondria-K+ channel axis is suppressed in cancer and its normalization promotes apoptosis and inhibits cancer growth. Cancer Cell 2007, 11, 37–51. [Google Scholar] [CrossRef] [Green Version]
- Testai, L.; Barrese, V.; Soldovieri, M.V.; Ambrosino, P.; Martelli, A.; Vinciguerra, I.; Miceli, F.; Greenwood, I.A.; Curtis, M.J.; Breschi, M.C.; et al. Expression and function of Kv7.4 channels in rat cardiac mitochondria: Possible targets for cardioprotection. Cardiovasc. Res. 2016, 110, 40–50. [Google Scholar] [CrossRef] [Green Version]
- Rusznak, Z.; Bakondi, G.; Kosztka, L.; Pocsai, K.; Dienes, B.; Fodor, J.; Telek, A.; Gonczi, M.; Szucs, G.; Csernoch, L. Mitochondrial expression of the two-pore domain TASK-3 channels in malignantly transformed and non-malignant human cells. Virchows Arch. 2008, 452, 415–426. [Google Scholar] [CrossRef]
- Mishra, P.; Chan, D.C. Mitochondrial dynamics and inheritance during cell division, development and disease. Nat. Rev. Mol. Cell Biol. 2014, 15, 634–646. [Google Scholar] [CrossRef] [Green Version]
- Tondera, D.; Grandemange, S.; Jourdain, A.; Karbowski, M.; Mattenberger, Y.; Herzig, S.; da Cruz, S.; Clerc, P.; Raschke, I.; Merkwirth, C.; et al. SLP-2 is required for stress-induced mitochondrial hyperfusion. EMBO J. 2009, 28, 1589–1600. [Google Scholar] [CrossRef] [Green Version]
- Rambold, A.S.; Kostelecky, B.; Elia, N.; Lippincott-Schwartz, J. Tubular network formation protects mitochondria from autophagosomal degradation during nutrient starvation. Proc. Natl. Acad. Sci. USA 2011, 108, 10190–10195. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Palikaras, K.; Lionaki, E.; Tavernarakis, N. Mechanisms of mitophagy in cellular homeostasis, physiology and pathology. Nat. Cell Biol. 2018, 20, 1013–1022. [Google Scholar] [CrossRef]
- Poole, L.P.; Macleod, K.F. Mitophagy in tumorigenesis and metastasis. Cell. Mol. Life Sci. 2021, 78, 3817–3851. [Google Scholar] [CrossRef] [PubMed]
- Gomes, L.C.; di Benedetto, G.; Scorrano, L. During autophagy mitochondria elongate, are spared from degradation and sustain cell viability. Nat. Cell Biol. 2011, 13, 589–598. [Google Scholar] [CrossRef] [Green Version]
- Brahimi-Horn, M.C.; Lacas-Gervais, S.; Adaixo, R.; Ilc, K.; Rouleau, M.; Notte, A.; Dieu, M.; Michiels, C.; Voeltzel, T.; Maguer-Satta, V.; et al. Local mitochondrial-endolysosomal microfusion cleaves voltage-dependent anion channel 1 to promote survival in hypoxia. Mol. Cell Biol. 2015, 35, 1491–1505. [Google Scholar] [CrossRef] [Green Version]
- Pahima, H.; Reina, S.; Tadmor, N.; Dadon-Klein, D.; Shteinfer-Kuzmine, A.; Mazure, N.M.; de Pinto, V.; Shoshan-Barmatz, V. Hypoxic-induced truncation of voltage-dependent anion channel 1 is mediated by both asparagine endopeptidase and calpain 1 activities. Oncotarget 2018, 9, 12825–12841. [Google Scholar] [CrossRef] [Green Version]
- Cogliati, S.; Enriquez, J.A.; Scorrano, L. Mitochondrial Cristae: Where Beauty Meets Functionality. Trends Biochem. Sci. 2016, 41, 261–273. [Google Scholar] [CrossRef] [Green Version]
- Horbay, R.; Bilyy, R. Mitochondrial dynamics during cell cycling. Apoptosis Int. J. Program. Cell Death 2016, 21, 1327–1335. [Google Scholar] [CrossRef]
- Mitra, K.; Wunder, C.; Roysam, B.; Lin, G.; Lippincott-Schwartz, J. A hyperfused mitochondrial state achieved at G1-S regulates cyclin E buildup and entry into S phase. Proc. Natl. Acad. Sci. USA 2009, 106, 11960–11965. [Google Scholar] [CrossRef] [Green Version]
- Abrisch, R.G.; Gumbin, S.C.; Wisniewski, B.T.; Lackner, L.L.; Voeltz, G.K. Fission and fusion machineries converge at ER contact sites to regulate mitochondrial morphology. J. Cell Biol. 2020, 219, e201911122. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harper, C.S.; White, A.J.; Lackner, L.L. The multifunctional nature of mitochondrial contact site proteins. Curr. Opin. Cell Biol. 2020, 65, 58–65. [Google Scholar] [CrossRef]
- Lawrence, E.J.; Mandato, C.A. Mitochondria localize to the cleavage furrow in mammalian cytokinesis. PLoS ONE 2013, 8, e72886. [Google Scholar] [CrossRef]
- Kasahara, A.; Cipolat, S.; Chen, Y.; Dorn, G.W., 2nd; Scorrano, L. Mitochondrial fusion directs cardiomyocyte differentiation via calcineurin and Notch signaling. Science 2013, 342, 734–737. [Google Scholar] [CrossRef] [Green Version]
- Khacho, M.; Clark, A.; Svoboda, D.S.; Azzi, J.; MacLaurin, J.G.; Meghaizel, C.; Sesaki, H.; Lagace, D.C.; Germain, M.; Harper, M.E.; et al. Mitochondrial Dynamics Impacts Stem Cell Identity and Fate Decisions by Regulating a Nuclear Transcriptional Program. Cell Stem. Cell 2016, 19, 232–247. [Google Scholar] [CrossRef] [Green Version]
- Wu, M.J.; Chen, Y.S.; Kim, M.R.; Chang, C.C.; Gampala, S.; Zhang, Y.; Wang, Y.; Chang, C.Y.; Yang, J.Y.; Chang, C.J. Epithelial-Mesenchymal Transition Directs Stem Cell Polarity via Regulation of Mitofusin. Cell Metab. 2019, 29, 993–1002.e1006. [Google Scholar] [CrossRef] [Green Version]
- Ponte, S.; Carvalho, L.; Gagliardi, M.; Campos, I.; Oliveira, P.J.; Jacinto, A. Drp1-mediated mitochondrial fission regulates calcium and F-actin dynamics during wound healing. Biol. Open 2020, 9, bio048629. [Google Scholar] [CrossRef] [Green Version]
- Kashatus, D.F. The regulation of tumor cell physiology by mitochondrial dynamics. Biochem. Biophys. Res. Commun. 2018, 500, 9–16. [Google Scholar] [CrossRef]
- Senft, D.; Ronai, Z.A. Regulators of mitochondrial dynamics in cancer. Curr. Opin. Cell Biol. 2016, 39, 43–52. [Google Scholar] [CrossRef] [Green Version]
- Desai, S.P.; Bhatia, S.N.; Toner, M.; Irimia, D. Mitochondrial localization and the persistent migration of epithelial cancer cells. Biophys. J. 2013, 104, 2077–2088. [Google Scholar] [CrossRef] [Green Version]
- Arismendi-Morillo, G.; Hoa, N.T.; Ge, L.; Jadus, M.R. Mitochondrial network in glioma’s invadopodia displays an activated state both in situ and in vitro: Potential functional implications. Ultrastruct. Pathol. 2012, 36, 409–414. [Google Scholar] [CrossRef] [PubMed]
- Bock, F.J.; Tait, S.W.G. Mitochondria as multifaceted regulators of cell death. Nat. Rev. Mol. Cell Biol. 2020, 21, 85–100. [Google Scholar] [CrossRef] [PubMed]
- Izzo, V.; Bravo-San Pedro, J.M.; Sica, V.; Kroemer, G.; Galluzzi, L. Mitochondrial Permeability Transition: New Findings and Persisting Uncertainties. Trends Cell Biol. 2016, 26, 655–667. [Google Scholar] [CrossRef] [PubMed]
- Alirol, E.; Martinou, J.C. Mitochondria and cancer: Is there a morphological connection? Oncogene 2006, 25, 4706–4716. [Google Scholar] [CrossRef] [Green Version]
- Szabadkai, G.; Simoni, A.M.; Chami, M.; Wieckowski, M.R.; Youle, R.J.; Rizzuto, R. Drp-1-dependent division of the mitochondrial network blocks intraorganellar Ca2+ waves and protects against Ca2+-mediated apoptosis. Mol. Cell 2004, 16, 59–68. [Google Scholar] [CrossRef]
- Tan, J.X.; Finkel, T. Mitochondria as intracellular signaling platforms in health and disease. J. Cell Biol. 2020, 219, e202002179. [Google Scholar] [CrossRef] [Green Version]
- Moloney, J.N.; Cotter, T.G. ROS signalling in the biology of cancer. Semin. Cell Dev. Biol. 2018, 80, 50–64. [Google Scholar] [CrossRef]
- Uchi, J.; Ryu, S.Y.; Jhun, B.S.; Hurst, S.; Sheu, S.S. Mitochondrial ion channels/transporters as sensors and regulators of cellular redox signaling. Antioxid. Redox Signal 2014, 21, 987–1006. [Google Scholar] [CrossRef] [Green Version]
- Kolossov, V.L.; Sivaguru, M.; Huff, J.; Luby, K.; Kanakaraju, K.; Gaskins, H.R. Airyscan super-resolution microscopy of mitochondrial morphology and dynamics in living tumor cells. Microsc. Res. Tech. 2018, 81, 115–128. [Google Scholar] [CrossRef]
- Giacomello, M.; Pyakurel, A.; Glytsou, C.; Scorrano, L. The cell biology of mitochondrial membrane dynamics. Nat. Rev. Mol. Cell Biol. 2020, 21, 204–224. [Google Scholar] [CrossRef]
- Cribbs, J.T.; Strack, S. Reversible phosphorylation of Drp1 by cyclic AMP-dependent protein kinase and calcineurin regulates mitochondrial fission and cell death. EMBO Rep. 2007, 8, 939–944. [Google Scholar] [CrossRef] [Green Version]
- Favaro, G.; Romanello, V.; Varanita, T.; Andrea Desbats, M.; Morbidoni, V.; Tezze, C.; Albiero, M.; Canato, M.; Gherardi, G.; De Stefani, D.; et al. DRP1-mediated mitochondrial shape controls calcium homeostasis and muscle mass. Nat. Commun. 2019, 10, 2576. [Google Scholar] [CrossRef]
- Kowaltowski, A.J.; Menezes-Filho, S.L.; Assali, E.A.; Goncalves, I.G.; Cabral-Costa, J.V.; Abreu, P.; Miller, N.; Nolasco, P.; Laurindo, F.R.M.; Bruni-Cardoso, A.; et al. Mitochondrial morphology regulates organellar Ca(2+) uptake and changes cellular Ca(2+) homeostasis. FASEB J. 2019, 33, 13176–13188. [Google Scholar] [CrossRef] [Green Version]
- MacAskill, A.F.; Kittler, J.T. Control of mitochondrial transport and localization in neurons. Trends Cell Biol. 2010, 20, 102–112. [Google Scholar] [CrossRef]
- Chen, Y.; Sheng, Z.H. Kinesin-1-syntaphilin coupling mediates activity-dependent regulation of axonal mitochondrial transport. J. Cell Biol. 2013, 202, 351–364. [Google Scholar] [CrossRef]
- Schonichen, A.; Webb, B.A.; Jacobson, M.P.; Barber, D.L. Considering protonation as a posttranslational modification regulating protein structure and function. Annu. Rev. Biophys. 2013, 42, 289–314. [Google Scholar] [CrossRef] [Green Version]
- Khacho, M.; Tarabay, M.; Patten, D.; Khacho, P.; MacLaurin, J.G.; Guadagno, J.; Bergeron, R.; Cregan, S.P.; Harper, M.E.; Park, D.S.; et al. Acidosis overrides oxygen deprivation to maintain mitochondrial function and cell survival. Nat. Commun. 2014, 5, 3550. [Google Scholar] [CrossRef] [Green Version]
- Matsuyama, S.; Reed, J.C. Mitochondria-dependent apoptosis and cellular pH regulation. Cell Death Differ. 2000, 7, 1155–1165. [Google Scholar] [CrossRef] [Green Version]
- Khalifat, N.; Puff, N.; Bonneau, S.; Fournier, J.B.; Angelova, M.I. Membrane deformation under local pH gradient: Mimicking mitochondrial cristae dynamics. Biophys. J. 2008, 95, 4924–4933. [Google Scholar] [CrossRef] [Green Version]
- Santo-Domingo, J.; Giacomello, M.; Poburko, D.; Scorrano, L.; Demaurex, N. OPA1 promotes pH flashes that spread between contiguous mitochondria without matrix protein exchange. EMBO J. 2013, 32, 1927–1940. [Google Scholar] [CrossRef] [Green Version]
- Liu, X.; Hajnoczky, G. Altered fusion dynamics underlie unique morphological changes in mitochondria during hypoxia-reoxygenation stress. Cell Death Differ. 2011, 18, 1561–1572. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hernandez-Resendiz, I.; Pacheu-Grau, D.; Sanchez, A.; Pardo, L.A. Inhibition of Kv10.1 Channels Sensitizes Mitochondria of Cancer Cells to Antimetabolic Agents. Cancers 2020, 12, 920. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arismendi-Morillo, G. Electron microscopy morphology of the mitochondrial network in human cancer. Int. J. Biochem. Cell Biol. 2009, 41, 2062–2068. [Google Scholar] [CrossRef] [PubMed]
- Serasinghe, M.N.; Wieder, S.Y.; Renault, T.T.; Elkholi, R.; Asciolla, J.J.; Yao, J.L.; Jabado, O.; Hoehn, K.; Kageyama, Y.; Sesaki, H.; et al. Mitochondrial division is requisite to RAS-induced transformation and targeted by oncogenic MAPK pathway inhibitors. Mol. Cell 2015, 57, 521–536. [Google Scholar] [CrossRef] [Green Version]
- Von Eyss, B.; Jaenicke, L.A.; Kortlever, R.M.; Royla, N.; Wiese, K.E.; Letschert, S.; McDuffus, L.A.; Sauer, M.; Rosenwald, A.; Evan, G.I.; et al. A MYC-Driven Change in Mitochondrial Dynamics Limits YAP/TAZ Function in Mammary Epithelial Cells and Breast Cancer. Cancer Cell 2015, 28, 743–757. [Google Scholar] [CrossRef] [Green Version]
- Grieco, J.P.; Allen, M.E.; Perry, J.B.; Wang, Y.; Song, Y.; Rohani, A.; Compton, S.L.E.; Smyth, J.W.; Swami, N.S.; Brown, D.A.; et al. Progression-Mediated Changes in Mitochondrial Morphology Promotes Adaptation to Hypoxic Peritoneal Conditions in Serous Ovarian Cancer. Front. Oncol. 2020, 10, 600113. [Google Scholar] [CrossRef]
- Zhao, J.; Zhang, J.; Yu, M.; Xie, Y.; Huang, Y.; Wolff, D.W.; Abel, P.W.; Tu, Y. Mitochondrial dynamics regulates migration and invasion of breast cancer cells. Oncogene 2013, 32, 4814–4824. [Google Scholar] [CrossRef]
- Castello-Cros, R.; Bonuccelli, G.; Molchansky, A.; Capozza, F.; Witkiewicz, A.K.; Birbe, R.C.; Howell, A.; Pestell, R.G.; Whitaker-Menezes, D.; Sotgia, F.; et al. Matrix remodeling stimulates stromal autophagy, “fueling” cancer cell mitochondrial metabolism and metastasis. Cell Cycle 2011, 10, 2021–2034. [Google Scholar] [CrossRef] [Green Version]
- Martinez-Outschoorn, U.E.; Lisanti, M.P.; Sotgia, F. Catabolic cancer-associated fibroblasts transfer energy and biomass to anabolic cancer cells, fueling tumor growth. Semin. Cancer Biol. 2014, 25, 47–60. [Google Scholar] [CrossRef]
- Elingaard-Larsen, L.O.; Rolver, M.G.; Sorensen, E.E.; Pedersen, S.F. How reciprocal interactions between the tumor microenvironment and ion transport proteins drive cancer progression. Rev. Physiol. Biochem. Pharmacol. 2020, in press. [Google Scholar]
- Rossignol, R.; Gilkerson, R.; Aggeler, R.; Yamagata, K.; Remington, S.J.; Capaldi, R.A. Energy substrate modulates mitochondrial structure and oxidative capacity in cancer cells. Cancer Res. 2004, 64, 985–993. [Google Scholar] [CrossRef] [Green Version]
- Qian, W.; Choi, S.; Gibson, G.A.; Watkins, S.C.; Bakkenist, C.J.; Van Houten, B. Mitochondrial hyperfusion induced by loss of the fission protein Drp1 causes ATM-dependent G2/M arrest and aneuploidy through DNA replication stress. J. Cell Sci. 2012, 125, 5745–5757. [Google Scholar] [CrossRef] [Green Version]
- Rehman, J.; Zhang, H.J.; Toth, P.T.; Zhang, Y.; Marsboom, G.; Hong, Z.; Salgia, R.; Husain, A.N.; Wietholt, C.; Archer, S.L. Inhibition of mitochondrial fission prevents cell cycle progression in lung cancer. FASEB J. 2012, 26, 2175–2186. [Google Scholar] [CrossRef] [Green Version]
- Yu, M.; Nguyen, N.D.; Huang, Y.; Lin, D.; Fujimoto, T.N.; Molkentine, J.M.; Deorukhkar, A.; Kang, Y.; San Lucas, F.A.; Fernandes, C.J.; et al. Mitochondrial fusion exploits a therapeutic vulnerability of pancreatic cancer. JCI Insight 2019, 5, e126915. [Google Scholar] [CrossRef]
- Sessions, D.T.; Kashatus, D.F. Mitochondrial dynamics in cancer stem cells. Cell Mol. Life Sci. 2021, 78, 3803–3816. [Google Scholar] [CrossRef]
- Peiris-Pages, M.; Bonuccelli, G.; Sotgia, F.; Lisanti, M.P. Mitochondrial fission as a driver of stemness in tumor cells: mDIVI1 inhibits mitochondrial function, cell migration and cancer stem cell (CSC) signalling. Oncotarget 2018, 9, 13254–13275. [Google Scholar] [CrossRef] [Green Version]
- Hu, P.; Li, S.; Tian, N.; Wu, F.; Hu, Y.; Li, D.; Qi, Y.; Wei, Z.; Wei, Q.; Li, Y.; et al. Acidosis enhances the self-renewal and mitochondrial respiration of stem cell-like glioma cells through CYP24A1-mediated reduction of vitamin D. Cell Death Dis. 2019, 10, 25. [Google Scholar] [CrossRef]
- Chapman, J.; Fielder, E.; Passos, J.F. Mitochondrial dysfunction and cell senescence: Deciphering a complex relationship. FEBS Lett. 2019, 593, 1566–1579. [Google Scholar] [CrossRef] [Green Version]
- Coppe, J.P.; Desprez, P.Y.; Krtolica, A.; Campisi, J. The senescence-associated secretory phenotype: The dark side of tumor suppression. Annu. Rev. Pathol. 2010, 5, 99–118. [Google Scholar] [CrossRef] [Green Version]
- Sun, X.; Cao, H.; Zhan, L.; Yin, C.; Wang, G.; Liang, P.; Li, J.; Wang, Z.; Liu, B.; Huang, Q.; et al. Mitochondrial fission promotes cell migration by Ca2+/CaMKII/ERK/FAK pathway in hepatocellular carcinoma. Liver Int. 2018, 38, 1263–1272. [Google Scholar] [CrossRef]
- Jung, J.U.; Ravi, S.; Lee, D.W.; McFadden, K.; Kamradt, M.L.; Toussaint, L.G.; Sitcheran, R. NIK/MAP3K14 Regulates Mitochondrial Dynamics and Trafficking to Promote Cell Invasion. Curr. Biol. 2016, 26, 3288–3302. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Caino, M.C.; Ghosh, J.C.; Chae, Y.C.; Vaira, V.; Rivadeneira, D.B.; Faversani, A.; Rampini, P.; Kossenkov, A.V.; Aird, K.M.; Zhang, R.; et al. PI3K therapy reprograms mitochondrial trafficking to fuel tumor cell invasion. Proc. Natl. Acad. Sci. USA 2015, 112, 8638–8643. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Caino, M.C.; Seo, J.H.; Aguinaldo, A.; Wait, E.; Bryant, K.G.; Kossenkov, A.V.; Hayden, J.E.; Vaira, V.; Morotti, A.; Ferrero, S.; et al. A neuronal network of mitochondrial dynamics regulates metastasis. Nat. Commun. 2016, 7, 13730. [Google Scholar] [CrossRef] [PubMed]
- Caino, M.C.; Seo, J.H.; Wang, Y.; Rivadeneira, D.B.; Gabrilovich, D.I.; Kim, E.T.; Weeraratna, A.T.; Languino, L.R.; Altieri, D.C. Syntaphilin controls a mitochondrial rheostat for proliferation-motility decisions in cancer. J. Clin. Invest. 2017, 127, 3755–3769. [Google Scholar] [CrossRef] [Green Version]
- Stock, C.; Schwab, A. Ion channels and transporters in metastasis. Biochim. Biophys. Acta Biomembr. 2015, 1848, 2638–2646. [Google Scholar] [CrossRef] [Green Version]
- Tsai, F.C.; Meyer, T. Ca2+ pulses control local cycles of lamellipodia retraction and adhesion along the front of migrating cells. Curr. Biol. 2012, 22, 837–842. [Google Scholar] [CrossRef] [Green Version]
- Demaurex, N.; Poburko, D.; Frieden, M. Regulation of plasma membrane calcium fluxes by mitochondria. Biochim. Biophys. Acta 2009, 1787, 1383–1394. [Google Scholar] [CrossRef]
- Panieri, E.; Santoro, M.M. ROS homeostasis and metabolism: A dangerous liason in cancer cells. Cell Death Dis. 2016, 7, e2253. [Google Scholar] [CrossRef]
- Chio, I.I.C.; Tuveson, D.A. ROS in Cancer: The Burning Question. Trends Mol. Med. 2017, 23, 411–429. [Google Scholar] [CrossRef] [Green Version]
- Nazarewicz, R.R.; Dikalova, A.; Bikineyeva, A.; Ivanov, S.; Kirilyuk, I.A.; Grigor’ev, I.A.; Dikalov, S.I. Does scavenging of mitochondrial superoxide attenuate cancer prosurvival signaling pathways? Antioxid. Redox Signal 2013, 19, 344–349. [Google Scholar] [CrossRef] [Green Version]
- Schwarzlander, M.; Murphy, M.P.; Duchen, M.R.; Logan, D.C.; Fricker, M.D.; Halestrap, A.P.; Muller, F.L.; Rizzuto, R.; Dick, T.P.; Meyer, A.J.; et al. Mitochondrial ‘flashes’: A radical concept repHined. Trends Cell Biol. 2012, 22, 503–508. [Google Scholar] [CrossRef]
- Johnson, D.; Allman, E.; Nehrke, K. Regulation of acid-base transporters by reactive oxygen species following mitochondrial fragmentation. Am. J. Physiol. Cell Physiol. 2012, 302, C1045. [Google Scholar] [CrossRef] [Green Version]
- LaMonte, G.; Tang, X.; Chen, J.L.; Wu, J.; Ding, C.K.; Keenan, M.M.; Sangokoya, C.; Kung, H.N.; Ilkayeva, O.; Boros, L.G.; et al. Acidosis induces reprogramming of cellular metabolism to mitigate oxidative stress. Cancer Metab. 2013, 1, 23. [Google Scholar] [CrossRef] [Green Version]
- Anderson, G.R.; Wardell, S.E.; Cakir, M.; Yip, C.; Ahn, Y.-R.; Ali, M.; Yllanes, A.P.; Chao, C.A.; McDonnell, D.P.; Wood, K.C. Dysregulation of mitochondrial dynamics proteins are a targetable feature of human tumors. Nat. Commun. 2018, 9, 1677. [Google Scholar] [CrossRef] [Green Version]
- Han, Y.; Kim, B.; Cho, U.; Park, I.S.; Kim, S.I.; Dhanasekaran, D.N.; Tsang, B.K.; Song, Y.S. Mitochondrial fission causes cisplatin resistance under hypoxic conditions via ROS in ovarian cancer cells. Oncogene 2019, 38, 7089–7105. [Google Scholar] [CrossRef]
- Shen, L.; Sun, B.; Sheng, J.; Yu, S.; Li, Y.; Xu, H.; Su, J.; Sun, L. PGC1alpha promotes cisplatin resistance in human ovarian carcinoma cells through upregulation of mitochondrial biogenesis. Int. J. Oncol. 2018, 53, 404–416. [Google Scholar] [CrossRef] [Green Version]
- Sperandio, S.; de Belle, I.; Bredesen, D.E. An alternative, nonapoptotic form of programmed cell death. Proc. Natl. Acad. Sci. USA 2000, 97, 14376–14381. [Google Scholar] [CrossRef] [Green Version]
- Fontana, F.; Raimondi, M.; Marzagalli, M.; Di Domizio, A.; Limonta, P. The emerging role of paraptosis in tumor cell biology: Perspectives for cancer prevention and therapy with natural compounds. Biochim. Biophys. Acta Rev. Cancer 2020, 1873, 188338. [Google Scholar] [CrossRef]
- Rolver, M.G.; Elingaard-Larsen, L.O.; Andersen, A.P.; Counillon, L.; Pedersen, S.F. Pyrazine ring-based Na+/H+ exchanger (NHE) inhibitors potently inhibit cancer cell growth in 3D culture, independent of NHE1. Sci. Rep. 2020, 10, 5800. [Google Scholar] [CrossRef]
- Leanza, L.; Zoratti, M.; Gulbins, E.; Szabo, I. Induction of apoptosis in macrophages via Kv1.3 and Kv1.5 potassium channels. Curr. Med. Chem. 2012, 19, 5394–5404. [Google Scholar] [CrossRef]
- Leanza, L.; Zoratti, M.; Gulbins, E.; Szabo, I. Mitochondrial ion channels as oncological targets. Oncogene 2014, 33, 5569–5581. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shoshan-Barmatz, V.; Ben-Hail, D.; Admoni, L.; Krelin, Y.; Tripathi, S.S. The mitochondrial voltage-dependent anion channel 1 in tumor cells. Biochim. Biophys. Acta 2015, 1848, 2547–2575. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mazure, N.M. VDAC in cancer. Biochim. Biophys. Acta Bioenerg. 2017, 1858, 665–673. [Google Scholar] [CrossRef] [PubMed]
- Shoshan-Barmatz, V.; Krelin, Y.; Shteinfer-Kuzmine, A.; Arif, T. Voltage-Dependent Anion Channel 1 As an Emerging Drug Target for Novel Anti-Cancer Therapeutics. Front. Oncol. 2017, 7, 154. [Google Scholar] [CrossRef] [PubMed]
- Tosatto, A.; Sommaggio, R.; Kummerow, C.; Bentham, R.B.; Blacker, T.S.; Berecz, T.; Duchen, M.R.; Rosato, A.; Bogeski, I.; Szabadkai, G.; et al. The mitochondrial calcium uniporter regulates breast cancer progression via HIF-1alpha. EMBO Mol. Med. 2016, 8, 569–585. [Google Scholar] [CrossRef] [PubMed]
- Jin, M.; Wang, J.; Ji, X.; Cao, H.; Zhu, J.; Chen, Y.; Yang, J.; Zhao, Z.; Ren, T.; Xing, J. MCUR1 facilitates epithelial-mesenchymal transition and metastasis via the mitochondrial calcium dependent ROS/Nrf2/Notch pathway in hepatocellular carcinoma. J. Exp. Clin. Cancer Res. 2019, 38, 136. [Google Scholar] [CrossRef] [Green Version]
- Ren, T.; Wang, J.; Zhang, H.; Yuan, P.; Zhu, J.; Wu, Y.; Huang, Q.; Guo, X.; Zhang, J.; Ji, L.; et al. MCUR1-Mediated Mitochondrial Calcium Signaling Facilitates Cell Survival of Hepatocellular Carcinoma via Reactive Oxygen Species-Dependent P53 Degradation. Antioxid. Redox Signal 2018, 28, 1120–1136. [Google Scholar] [CrossRef]
- Ren, T.; Zhang, H.; Wang, J.; Zhu, J.; Jin, M.; Wu, Y.; Guo, X.; Ji, L.; Huang, Q.; Zhang, H.; et al. MCU-dependent mitochondrial Ca(2+) inhibits NAD(+)/SIRT3/SOD2 pathway to promote ROS production and metastasis of HCC cells. Oncogene 2017, 36, 5897–5909. [Google Scholar] [CrossRef]
- Marchi, S.; Lupini, L.; Patergnani, S.; Rimessi, A.; Missiroli, S.; Bonora, M.; Bononi, A.; Corra, F.; Giorgi, C.; De Marchi, E.; et al. Downregulation of the mitochondrial calcium uniporter by cancer-related miR-25. Curr. Biol. 2013, 23, 58–63. [Google Scholar] [CrossRef] [Green Version]
- Chen, L.; Sun, Q.; Zhou, D.; Song, W.; Yang, Q.; Ju, B.; Zhang, L.; Xie, H.; Zhou, L.; Hu, Z.; et al. HINT2 triggers mitochondrial Ca(2+) influx by regulating the mitochondrial Ca(2+) uniporter (MCU) complex and enhances gemcitabine apoptotic effect in pancreatic cancer. Cancer Lett. 2017, 411, 106–116. [Google Scholar] [CrossRef]
- Tiapko, O.; Groschner, K. TRPC3 as a Target of Novel Therapeutic Interventions. Cells 2018, 7, 83. [Google Scholar] [CrossRef] [Green Version]
- Li, S.; Jiang, K.; Li, J.; Hao, X.; Chu, W.; Luo, C.; Zhu, Y.; Xie, R.; Chen, B. Estrogen enhances the proliferation and migration of ovarian cancer cells by activating transient receptor potential channel C3. J. Ovarian Res. 2020, 13, 20. [Google Scholar] [CrossRef] [Green Version]
- Yang, S.L.; Cao, Q.; Zhou, K.C.; Feng, Y.J.; Wang, Y.Z. Transient receptor potential channel C3 contributes to the progression of human ovarian cancer. Oncogene 2009, 28, 1320–1328. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Qi, Y.X.; Qi, Z.; Tsang, S.Y. TRPC3 Regulates the Proliferation and Apoptosis Resistance of Triple Negative Breast Cancer Cells through the TRPC3/RASA4/MAPK Pathway. Cancers 2019, 11, 558. [Google Scholar] [CrossRef] [Green Version]
- Wang, C.A.; Liu, Q.; Chen, Y.; Liu, S.; Xu, J.; Cui, X.; Zhang, Y.; Piao, L. Clinical implication of leucine zipper/EF hand-containing transmembrane-1 overexpression in the prognosis of triple-negative breast cancer. Exp. Mol. Pathol. 2015, 98, 254–259. [Google Scholar] [CrossRef]
- Zhang, Y.; Chen, L.; Cao, Y.; Chen, S.; Xu, C.; Xing, J.; Zhang, K. LETM1 Promotes Gastric Cancer Cell Proliferation, Migration, and Invasion via the PI3K/Akt Signaling Pathway. J. Gastric. Cancer 2020, 20, 139–151. [Google Scholar] [CrossRef]
- Zhou, B.; Yang, C.; Yan, X.; Shi, Z.; Xiao, H.; Wei, X.; Jiang, N.; Wu, Z. LETM1 Knockdown Promotes Autophagy and Apoptosis Through AMP-Activated Protein Kinase Phosphorylation-Mediated Beclin-1/Bcl-2 Complex Dissociation in Hepatocellular Carcinoma. Front. Oncol. 2020, 10, 606790. [Google Scholar] [CrossRef]
- Andersen, A.P.; Samsoe-Petersen, J.; Oernbo, E.K.; Boedtkjer, E.; Moreira, J.M.A.; Kveiborg, M.; Pedersen, S.F. The net acid extruders NHE1, NBCn1 and MCT4 promote mammary tumor growth through distinct but overlapping mechanisms. Int. J. Cancer 2018, 142, 2529–2542. [Google Scholar] [CrossRef] [Green Version]
- Lee, S.; Axelsen, T.V.; Andersen, A.P.; Vahl, P.; Pedersen, S.F.; Boedtkjer, E. Disrupting Na+, HCO3−-cotransporter NBCn1 (Slc4a7) delays murine breast cancer development. Oncogene 2016, 35, 2112–2122. [Google Scholar] [CrossRef]
- Cardone, R.A.; Greco, M.R.; Zeeberg, K.; Zaccagnino, A.; Saccomano, M.; Bellizzi, A.; Bruns, P.; Menga, M.; Pilarsky, C.; Schwab, A.; et al. A novel NHE1-centered signaling cassette drives epidermal growth factor receptor-dependent pancreatic tumor metastasis and is a target for combination therapy. Neoplasia 2015, 17, 155–166. [Google Scholar] [CrossRef] [Green Version]
- Intlekofer, A.M.; Wang, B.; Liu, H.; Shah, H.; Carmona-Fontaine, C.; Rustenburg, A.S.; Salah, S.; Gunner, M.R.; Chodera, J.D.; Cross, J.R.; et al. L-2-Hydroxyglutarate production arises from noncanonical enzyme function at acidic pH. Nat. Chem. Biol. 2017, 13, 494–500. [Google Scholar] [CrossRef] [PubMed]
- Quast, S.-A.; Berger, A.; Buttstädt, N.; Friebel, K.; Schönherr, R.; Eberle, J. General Sensitization of melanoma cells for TRAIL-induced apoptosis by the potassium channel inhibitor TRAM-34 depends on release of SMAC. PLoS ONE 2012, 7, e39290. [Google Scholar] [CrossRef] [Green Version]
- Meuth, S.G.; Herrmann, A.M.; Ip, C.W.; Kanyshkova, T.; Bittner, S.; Weishaupt, A.; Budde, T.; Wiendl, H. The two-pore domain potassium channel TASK3 functionally impacts glioma cell death. J. Neurooncol. 2008, 87, 263–270. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Y.; Gulbins, E.; Siemen, D. Activation of the permeability transition pore by Bax via inhibition of the mitochondrial BK channel. Cell Physiol. Biochem. 2011, 27, 191–200. [Google Scholar] [CrossRef]
- McFerrin, M.B.; Turner, K.L.; Cuddapah, V.A.; Sontheimer, H. Differential role of IK and BK potassium channels as mediators of intrinsic and extrinsic apoptotic cell death. Am. J. Physiol. Cell Physiol. 2012, 303, C1070–C1078. [Google Scholar] [CrossRef] [Green Version]
- Ma, Y.G.; Liu, W.C.; Dong, S.; Du, C.; Wang, X.J.; Li, J.S.; Xie, X.P.; Wu, L.; Ma, D.C.; Yu, Z.B.; et al. Activation of BK(Ca) channels in zoledronic acid-induced apoptosis of MDA-MB-231 breast cancer cells. PLoS ONE 2012, 7, e37451. [Google Scholar] [CrossRef] [Green Version]
- Ma, Y.G.; Dong, L.; Ye, X.L.; Deng, C.L.; Cheng, J.H.; Liu, W.C.; Ma, J.; Chang, Y.M.; Xie, M.J. Activation of cloned BK(Ca) channels in nitric oxide-induced apoptosis of HEK293 cells. Apoptosis Int. J. Program. Cell Death 2010, 15, 426–438. [Google Scholar] [CrossRef]
- Leanza, L.; Romio, M.; Becker, K.A.; Azzolini, M.; Trentin, L.; Manago, A.; Venturini, E.; Zaccagnino, A.; Mattarei, A.; Carraretto, L.; et al. Direct Pharmacological Targeting of a Mitochondrial Ion Channel Selectively Kills Tumor Cells In Vivo. Cancer Cell 2017, 31, 516–531. [Google Scholar] [CrossRef] [Green Version]
- Zaccagnino, A.; Manago, A.; Leanza, L.; Gontarewitz, A.; Linder, B.; Azzolini, M.; Biasutto, L.; Zoratti, M.; Peruzzo, R.; Legler, K.; et al. Tumor-reducing effect of the clinically used drug clofazimine in a SCID mouse model of pancreatic ductal adenocarcinoma. Oncotarget 2017, 8, 38276–38293. [Google Scholar] [CrossRef]
- Ward, D.M.; Cloonan, S.M. Mitochondrial Iron in Human Health and Disease. Annu. Rev. Physiol. 2019, 81, 453–482. [Google Scholar] [CrossRef]
- Paul, B.T.; Manz, D.H.; Torti, F.M.; Torti, S.V. Mitochondria and Iron: Current questions. Expert Rev. Hematol. 2017, 10, 65–79. [Google Scholar] [CrossRef] [Green Version]
- Torti, S.V.; Torti, F.M. Iron and cancer: More ore to be mined. Nat. Rev. Cancer 2013, 13, 342–355. [Google Scholar] [CrossRef] [Green Version]
- Yamanaka, R.; Tabata, S.; Shindo, Y.; Hotta, K.; Suzuki, K.; Soga, T.; Oka, K. Mitochondrial Mg(2+) homeostasis decides cellular energy metabolism and vulnerability to stress. Sci. Rep. 2016, 6, 30027. [Google Scholar] [CrossRef]
- Ackerman, C.M.; Chang, C.J. Copper signaling in the brain and beyond. J. Biol. Chem. 2018, 293, 4628–4635. [Google Scholar] [CrossRef] [Green Version]
- Zhu, X.; Boulet, A.; Buckley, K.M.; Phillips, C.B.; Gammon, M.G.; Oldfather, L.E.; Moore, S.A.; Leary, S.C.; Cobine, P.A. Mitochondrial copper and phosphate transporter specificity was defined early in the evolution of eukaryotes. Elife 2021, 10, e64690. [Google Scholar] [CrossRef]
- Cho, H.M.; Ryu, J.R.; Jo, Y.; Seo, T.W.; Choi, Y.N.; Kim, J.H.; Chung, J.M.; Cho, B.; Kang, H.C.; Yu, S.W.; et al. Drp1-Zip1 Interaction Regulates Mitochondrial Quality Surveillance System. Mol. Cell 2019, 73, 364–376.e368. [Google Scholar] [CrossRef] [Green Version]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pedersen, S.F.; Flinck, M.; Pardo, L.A. The Interplay between Dysregulated Ion Transport and Mitochondrial Architecture as a Dangerous Liaison in Cancer. Int. J. Mol. Sci. 2021, 22, 5209. https://doi.org/10.3390/ijms22105209
Pedersen SF, Flinck M, Pardo LA. The Interplay between Dysregulated Ion Transport and Mitochondrial Architecture as a Dangerous Liaison in Cancer. International Journal of Molecular Sciences. 2021; 22(10):5209. https://doi.org/10.3390/ijms22105209
Chicago/Turabian StylePedersen, Stine F., Mette Flinck, and Luis A. Pardo. 2021. "The Interplay between Dysregulated Ion Transport and Mitochondrial Architecture as a Dangerous Liaison in Cancer" International Journal of Molecular Sciences 22, no. 10: 5209. https://doi.org/10.3390/ijms22105209