
ATF3 Promotes Arsenic-Induced Apoptosis and Oppositely Regulates DR5 and Bcl-xL Expression in Human Bronchial Epithelial Cells


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
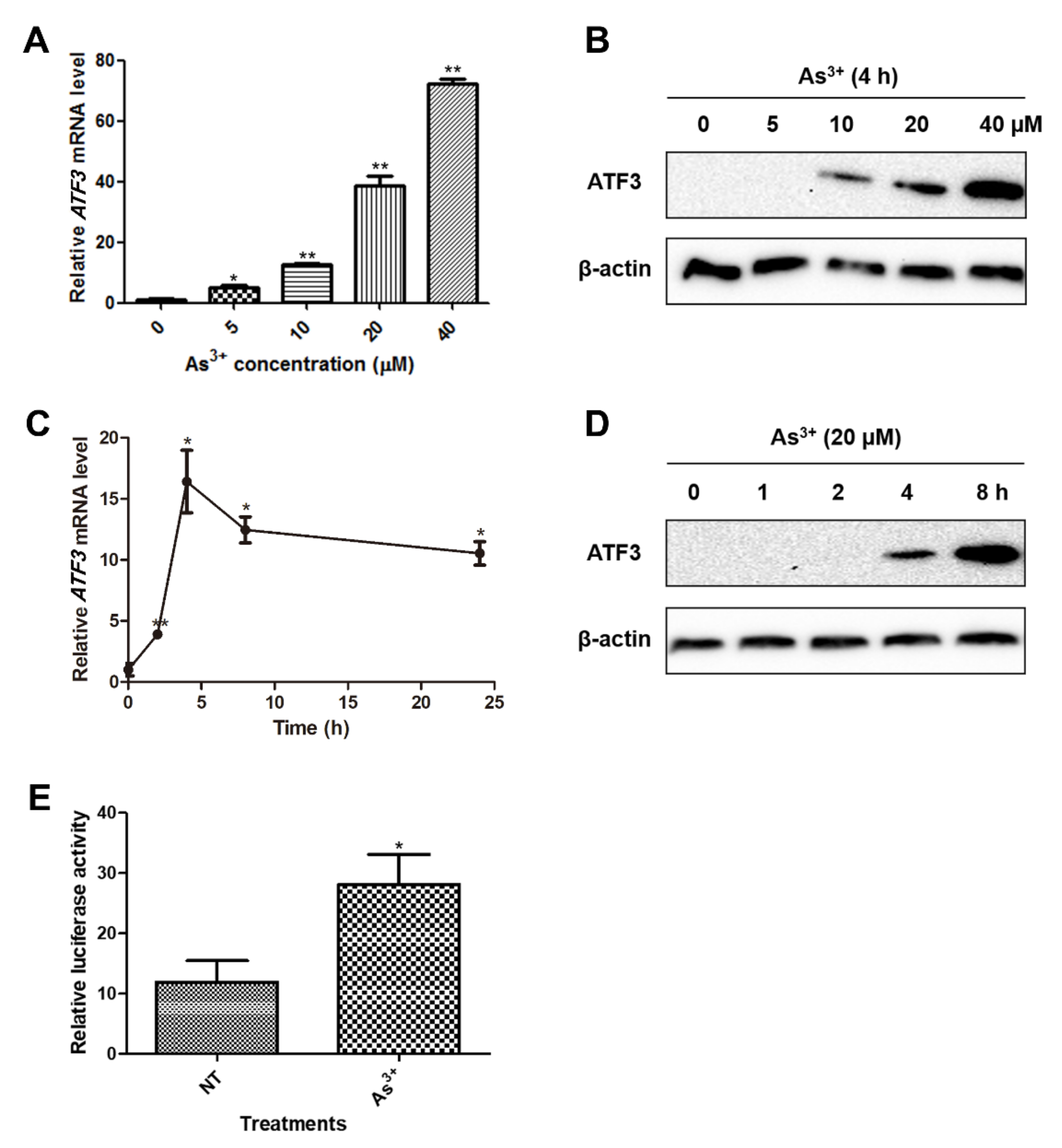
2.1. ATF3 Is Induced in BEAS-2B Cells upon Arsenic Exposure
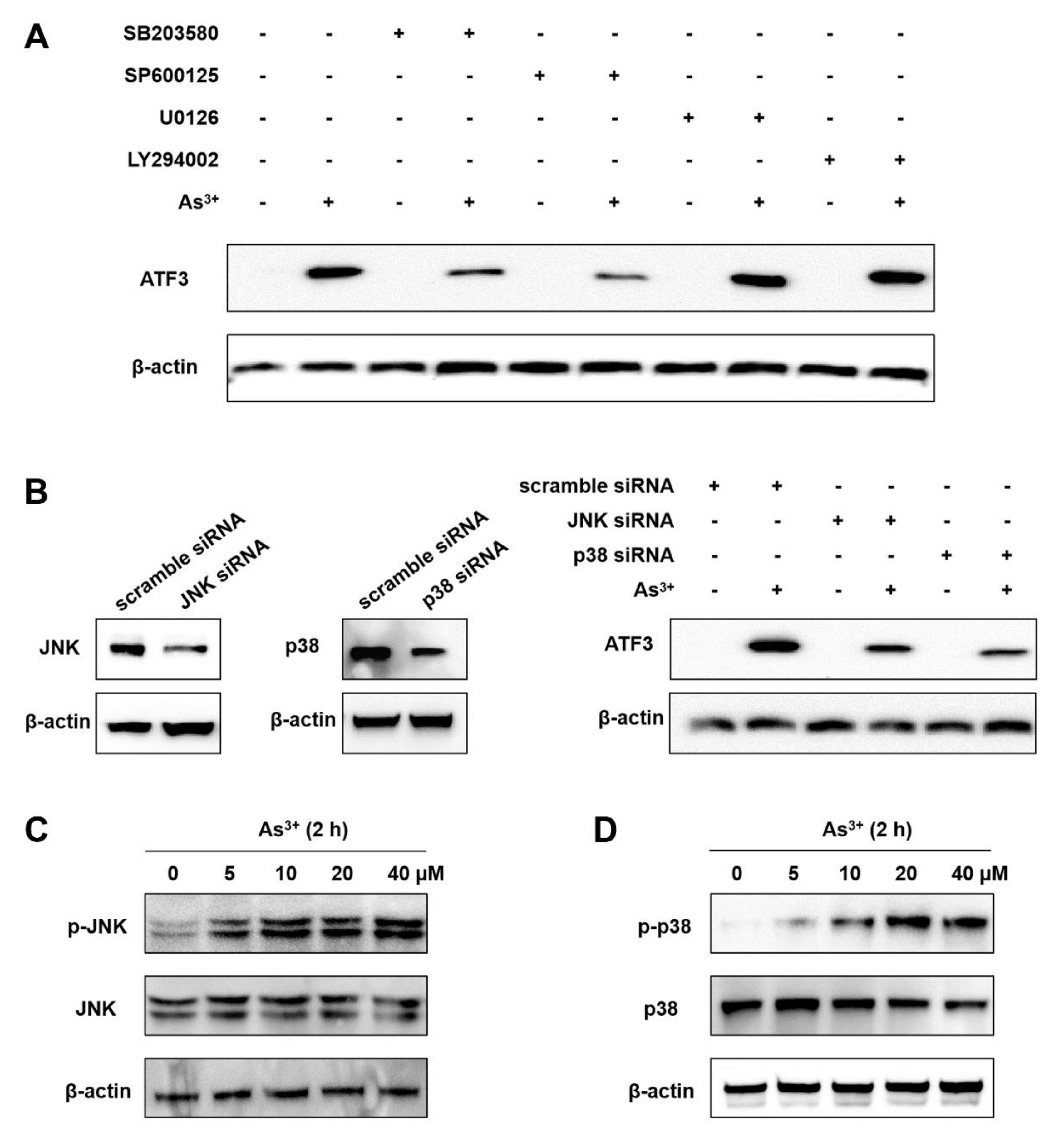
2.2. The Arsenic-Induced ATF3 Expression Relies on JNK and p38
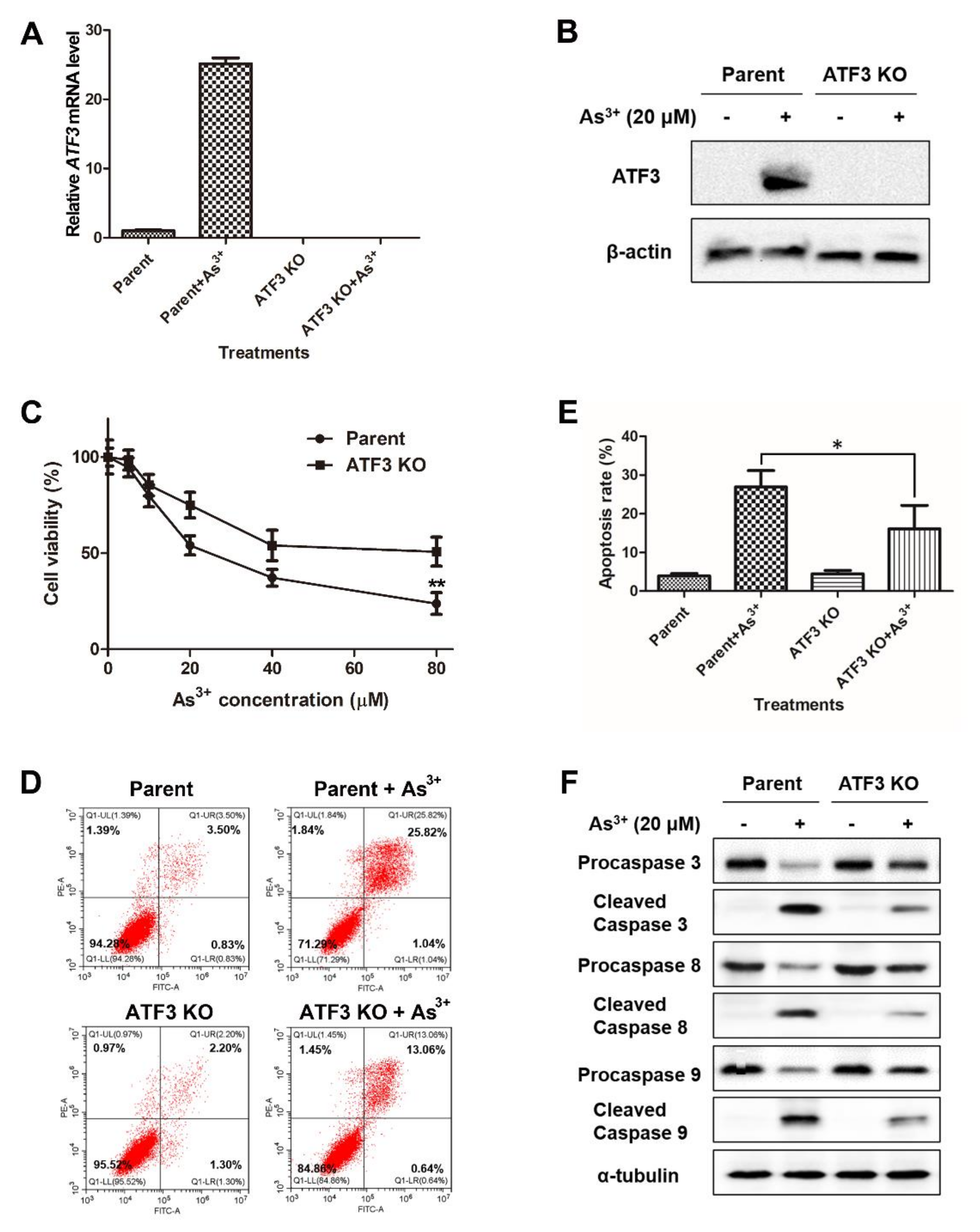
2.3. ATF3 Mediates the Arsenic-Induced Apoptosis
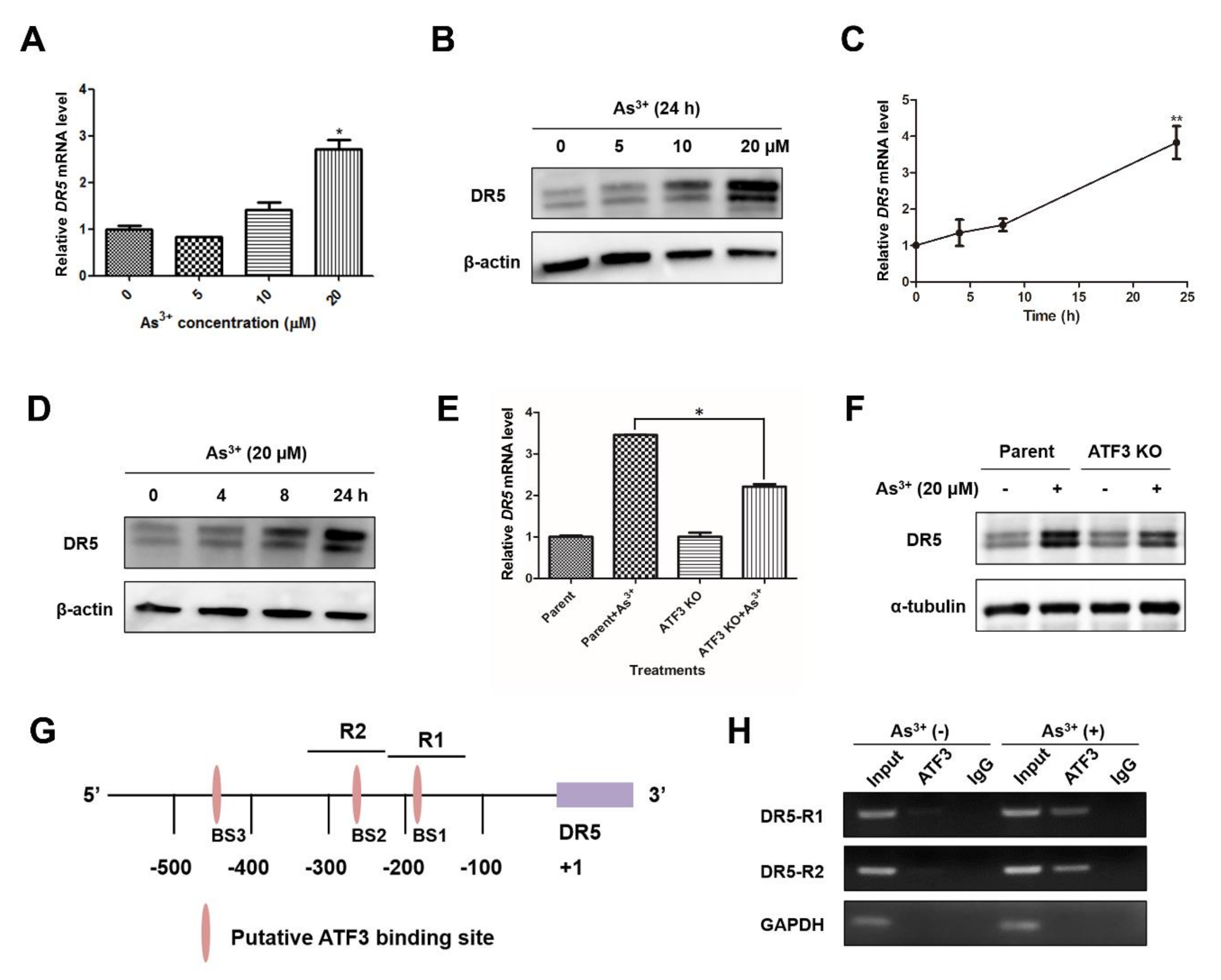
2.4. Arsenic-Induced ATF3 Activates DR5 Transcription by Directly Binding to the DR5 Promoter
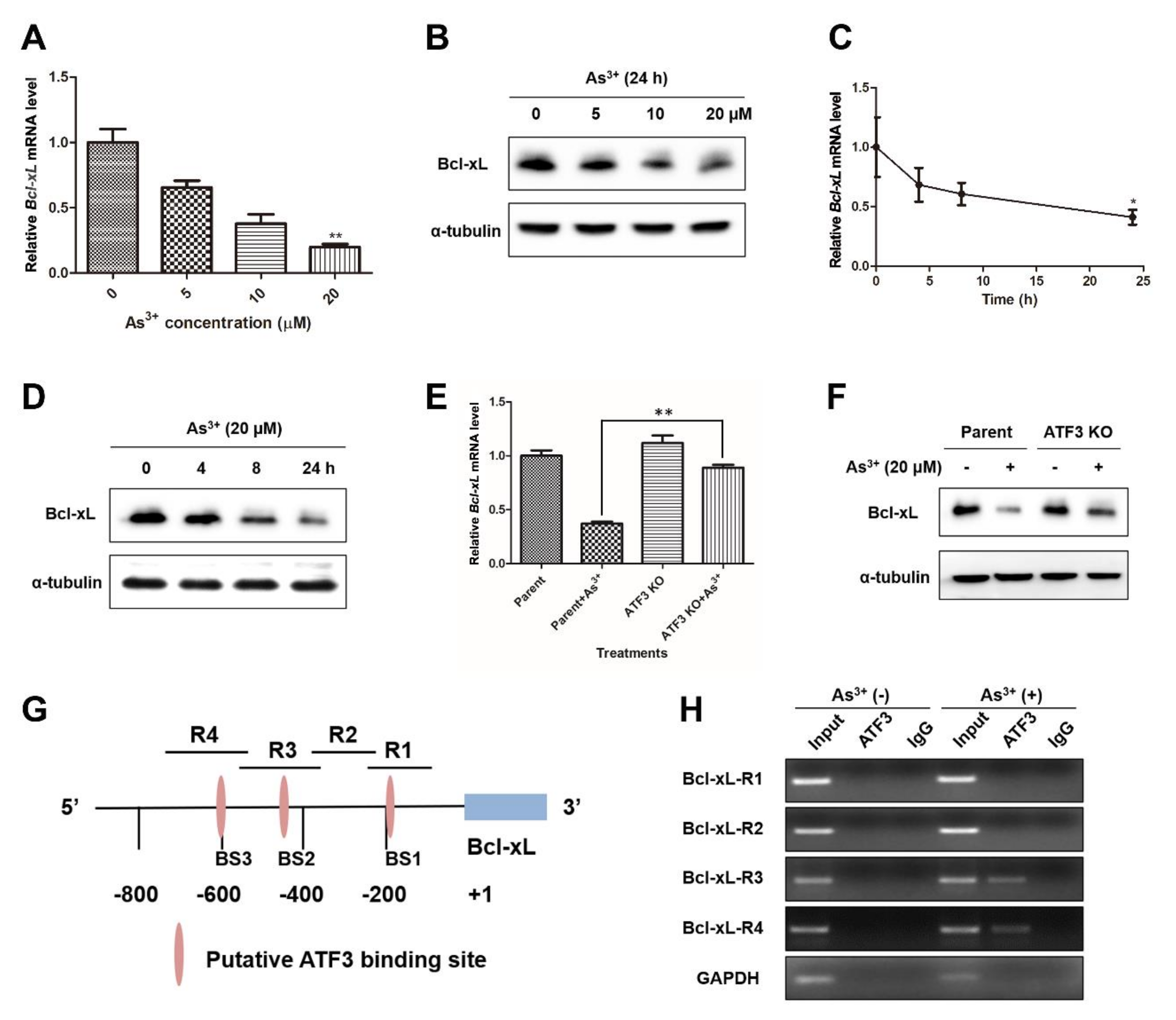
2.5. ATF3 Is Recruited onto the Bcl-xL Promoter and Represses Bcl-xL Expression upon Arsenic Exposure
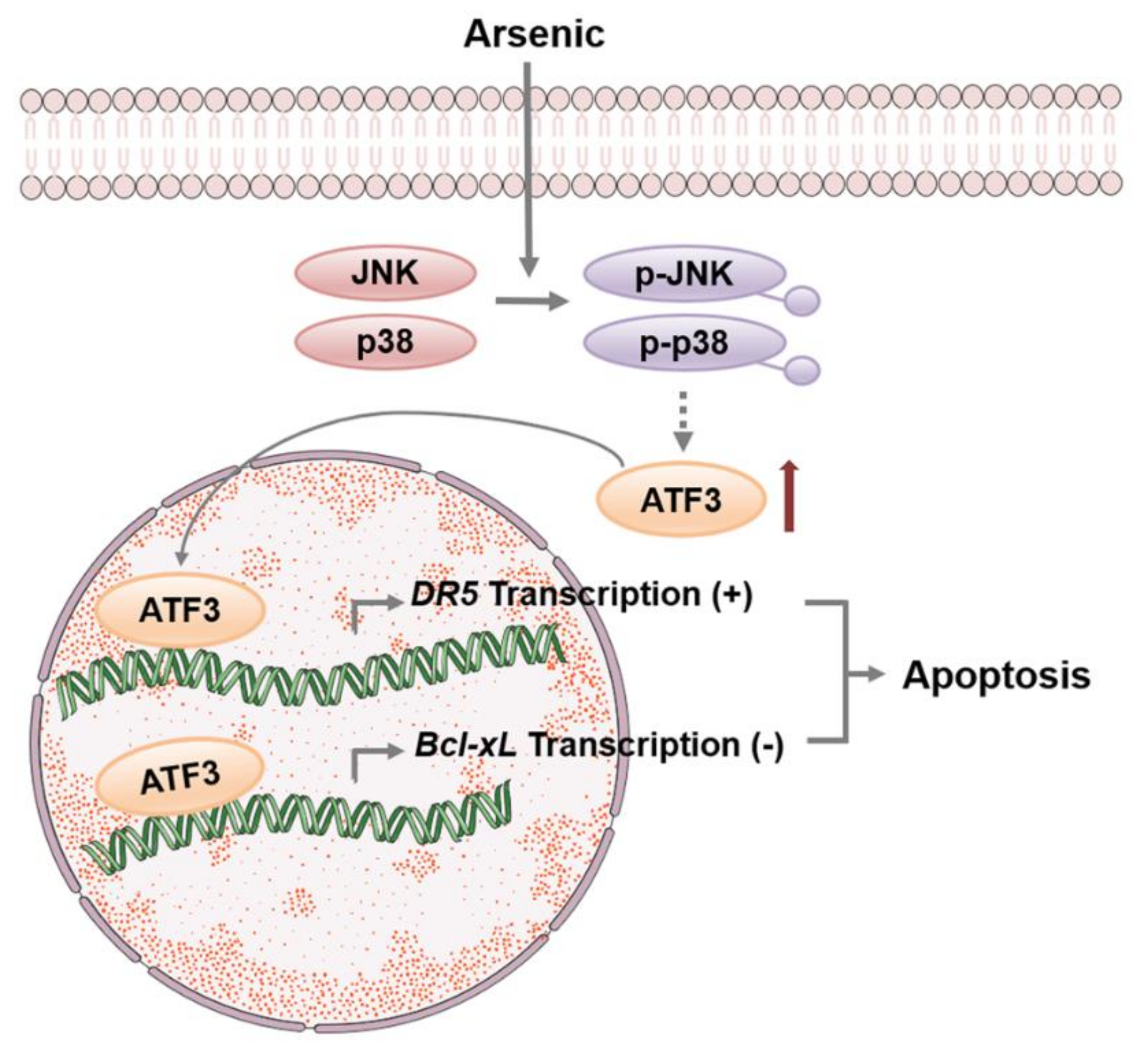
3. Discussion
4. Materials and Methods
4.1. Cell Culture and Reagents
4.2. Plasmid Construct and Dual Luciferase Assay
4.3. RNA Isolation and Quantitative RT-PCR
4.4. Protein Isolation and Western Blot Analysis
4.5. Apoptosis Analysis by Flow Cytometry
4.6. Cell Viability Assay
4.7. Chromatin Immunoprecipitation (ChIP) Assay
4.8. Transfection of siRNA and Plasmids
4.9. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Abdul, K.S.; Jayasinghe, S.S.; Chandana, E.P.; Jayasumana, C.; De Silva, P.M. Arsenic and human health effects: A review. Environ. Toxicol. Pharmacol. 2015, 40, 828–846. [Google Scholar] [CrossRef]
- Chen, Q.Y.; Li, J.; Sun, H.; Wu, F.; Zhu, Y.; Kluz, T.; Jordan, A.; DesMarais, T.; Zhang, X.; Murphy, A.; et al. Role of miR-31 and SATB2 in arsenic-induced malignant BEAS-2B cell transformation. Mol. Carcinog. 2018, 57, 968–977. [Google Scholar] [CrossRef]
- Chen, Q.Y.; Costa, M. PI3K/Akt/mTOR Signaling Pathway and the Biphasic Effect of Arsenic in Carcinogenesis. Mol. Pharmacol. 2018, 94, 784–792. [Google Scholar] [CrossRef]
- Zhu, H.H.; Hu, J.; Lo-Coco, F.; Jin, J. The simpler, the better: Oral arsenic for acute promyelocytic leukemia. Blood 2019, 134, 597–605. [Google Scholar] [CrossRef]
- Mahalanobish, S.; Saha, S.; Dutta, S.; Sil, P.C. Mangiferin alleviates arsenic induced oxidative lung injury via upregulation of the Nrf2-HO1 axis. Food Chem. Toxicol. 2019, 126, 41–55. [Google Scholar] [CrossRef]
- Dodson, M.; de la Vega, M.R.; Harder, B.; Castro-Portuguez, R.; Rodrigues, S.D.; Wong, P.K.; Chapman, E.; Zhang, D.D. Low-level arsenic causes proteotoxic stress and not oxidative stress. Toxicol. Appl. Pharmacol. 2018, 341, 106–113. [Google Scholar] [CrossRef] [PubMed]
- Qi, Y.; Li, H.; Zhang, M.; Zhang, T.; Frank, J.; Chen, G. Autophagy in arsenic carcinogenesis. Exp. Toxicol. Pathol. 2014, 66, 163–168. [Google Scholar] [CrossRef]
- Zhao, F.; Severson, P.; Pacheco, S.; Futscher, B.W.; Klimecki, W.T. Arsenic exposure induces the Warburg effect in cultured human cells. Toxicol. Appl. Pharmacol. 2013, 271, 72–77. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lau, A.; Zheng, Y.; Tao, S.; Wang, H.; Whitman, S.A.; White, E.; Zhang, D.D. Arsenic inhibits autophagic flux, activating the Nrf2-Keap1 pathway in a p62-dependent manner. Mol. Cell. Biol. 2013, 33, 2436–2446. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rohini, M.; Haritha Menon, A.; Selvamurugan, N. Role of activating transcription factor 3 and its interacting proteins under physiological and pathological conditions. Int. J. Biol. Macromol. 2018, 120, 310–317. [Google Scholar] [CrossRef]
- Wang, L.; Liu, Y.; Du, T.; Yang, H.; Lei, L.; Guo, M.; Ding, H.F.; Zhang, J.; Wang, H.; Chen, X.; et al. ATF3 promotes erastin-induced ferroptosis by suppressing system Xc(). Cell Death Differ. 2020, 27, 662–675. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Z.; He, Y.; Deng, W.; Lang, L.; Yang, H.; Jin, B.; Kolhe, R.; Ding, H.F.; Zhang, J.; Hai, T.; et al. Atf3 deficiency promotes genome instability and spontaneous tumorigenesis in mice. Oncogene 2018, 37, 18–27. [Google Scholar] [CrossRef]
- Sood, V.; Sharma, K.B.; Gupta, V.; Saha, D.; Dhapola, P.; Sharma, M.; Sen, U.; Kitajima, S.; Chowdhury, S.; Kalia, M.; et al. ATF3 negatively regulates cellular antiviral signaling and autophagy in the absence of type I interferons. Sci. Rep. 2017, 7, 8789. [Google Scholar] [CrossRef] [Green Version]
- Cui, H.; Guo, M.; Xu, D.; Ding, Z.C.; Zhou, G.; Ding, H.F.; Zhang, J.; Tang, Y.; Yan, C. The stress-responsive gene ATF3 regulates the histone acetyltransferase Tip60. Nat. Commun. 2015, 6, 6752. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cui, H.; Li, X.; Han, C.; Wang, Q.E.; Wang, H.; Ding, H.F.; Zhang, J.; Yan, C. The Stress-responsive Gene ATF3 Mediates Dichotomous UV Responses by Regulating the Tip60 and p53 Proteins. J. Biol. Chem. 2016, 291, 10847–10857. [Google Scholar] [CrossRef] [Green Version]
- You, Z.; Xu, J.; Li, B.; Ye, H.; Chen, L.; Liu, Y.; Xiong, X. The mechanism of ATF3 repression of epithelial-mesenchymal transition and suppression of cell viability in cholangiocarcinoma via p53 signal pathway. J. Cell. Mol. Med. 2019, 23, 2184–2193. [Google Scholar] [CrossRef] [PubMed]
- Wei, S.; Li, T.; Xie, R.; Ye, B.; Xiang, J.; Liu, K.; Chen, Z.; Gao, X. The role of ATF3 in ZnO nanoparticle-induced genotoxicity and cytotoxicity in bronchial epithelial cells. Int. J. Biochem. Cell Biol. 2019, 113, 95–102. [Google Scholar] [CrossRef]
- Park, Y.H.; Kim, D.; Dai, J.; Zhang, Z. Human bronchial epithelial BEAS-2B cells, an appropriate in vitro model to study heavy metals induced carcinogenesis. Toxicol. Appl. Pharmacol. 2015, 287, 240–245. [Google Scholar] [CrossRef] [Green Version]
- Alamolhodaei, N.S.; Shirani, K.; Karimi, G. Arsenic cardiotoxicity: An overview. Environ. Toxicol. Pharmacol. 2015, 40, 1005–1014. [Google Scholar] [CrossRef]
- Taketani, K.; Kawauchi, J.; Tanaka-Okamoto, M.; Ishizaki, H.; Tanaka, Y.; Sakai, T.; Miyoshi, J.; Maehara, Y.; Kitajima, S. Key role of ATF3 in p53-dependent DR5 induction upon DNA damage of human colon cancer cells. Oncogene 2012, 31, 2210–2221. [Google Scholar] [CrossRef] [Green Version]
- Edagawa, M.; Kawauchi, J.; Hirata, M.; Goshima, H.; Inoue, M.; Okamoto, T.; Murakami, A.; Maehara, Y.; Kitajima, S. Role of activating transcription factor 3 (ATF3) in endoplasmic reticulum (ER) stress-induced sensitization of p53-deficient human colon cancer cells to tumor necrosis factor (TNF)-related apoptosis-inducing ligand (TRAIL)-mediated apoptosis through up-regulation of death receptor 5 (DR5) by zerumbone and celecoxib. J. Biol. Chem. 2014, 289, 21544–21561. [Google Scholar] [CrossRef] [Green Version]
- Liu, Z.; Shi, Q.; Song, X.; Wang, Y.; Song, E.; Song, Y. Activating Transcription Factor 4 (ATF4)-ATF3-C/EBP Homologous Protein (CHOP) Cascade Shows an Essential Role in the ER Stress-Induced Sensitization of Tetrachlorobenzoquinone-Challenged PC12 Cells to ROS-Mediated Apoptosis via Death Receptor 5 (DR5) Signaling. Chem. Res. Toxicol. 2016, 29, 1510–1518. [Google Scholar] [CrossRef]
- Chen, N.; Chen, X.; Huang, R.; Zeng, H.; Gong, J.; Meng, W.; Lu, Y.; Zhao, F.; Wang, L.; Zhou, Q. BCL-xL is a target gene regulated by hypoxia-inducible factor-1{alpha}. J. Biol. Chem. 2009, 284, 10004–10012. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chueh, A.C.; Tse, J.W.T.; Dickinson, M.; Ioannidis, P.; Jenkins, L.; Togel, L.; Tan, B.; Luk, I.; Davalos-Salas, M.; Nightingale, R.; et al. ATF3 Repression of BCL-XL Determines Apoptotic Sensitivity to HDAC Inhibitors across Tumor Types. Clin. Cancer Res. 2017, 23, 5573–5584. [Google Scholar] [CrossRef] [Green Version]
- Dodmane, P.R.; Arnold, L.L.; Kakiuchi-Kiyota, S.; Qiu, F.; Liu, X.; Rennard, S.I.; Cohen, S.M. Cytotoxicity and gene expression changes induced by inorganic and organic trivalent arsenicals in human cells. Toxicology 2013, 312, 18–29. [Google Scholar] [CrossRef]
- Sanchez, T.R.; Powers, M.; Perzanowski, M.; George, C.M.; Graziano, J.H.; Navas-Acien, A. A Meta-analysis of Arsenic Exposure and Lung Function: Is There Evidence of Restrictive or Obstructive Lung Disease? Curr. Environ. Health Rep. 2018, 5, 244–254. [Google Scholar] [CrossRef]
- Shih, Y.H.; Argos, M.; Turyk, M.E. Urinary arsenic concentration, airway inflammation, and lung function in the U.S. adult population. Environ. Res. 2019, 175, 308–315. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Q.; Xi, S. A review on arsenic carcinogenesis: Epidemiology, metabolism, genotoxicity and epigenetic changes. Regul. Toxicol. Pharmacol. 2018, 99, 78–88. [Google Scholar] [CrossRef]
- Li, J.; Zhao, L.; Zhang, Y.; Li, W.; Duan, X.; Chen, J.; Guo, Y.; Yang, S.; Sun, G.; Li, B. Imbalanced immune responses involving inflammatory molecules and immune-related pathways in the lung of acute and subchronic arsenic-exposed mice. Environ. Res. 2017, 159, 381–393. [Google Scholar] [CrossRef]
- Henderson, M.W.; Madenspacher, J.H.; Whitehead, G.S.; Thomas, S.Y.; Aloor, J.J.; Gowdy, K.M.; Fessler, M.B. Effects of Orally Ingested Arsenic on Respiratory Epithelial Permeability to Bacteria and Small Molecules in Mice. Environ. Health Perspect. 2017, 125, 097024. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nguyen, C.T.; Kim, E.H.; Luong, T.T.; Pyo, S.; Rhee, D.K. TLR4 mediates pneumolysin-induced ATF3 expression through the JNK/p38 pathway in Streptococcus pneumoniae-infected RAW 264.7 cells. Mol. Cells 2015, 38, 58–64. [Google Scholar] [CrossRef] [Green Version]
- Lu, D.; Chen, J.; Hai, T. The regulation of ATF3 gene expression by mitogen-activated protein kinases. Biochem. J. 2007, 401, 559–567. [Google Scholar] [CrossRef] [Green Version]
- Tang, Q.; Ren, L.; Liu, J.; Li, W.; Zheng, X.; Wang, J.; Du, G. Withaferin A triggers G2/M arrest and intrinsic apoptosis in glioblastoma cells via ATF4-ATF3-CHOP axis. Cell Prolif. 2020, 53, e12706. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yan, J.; Zhong, N.; Liu, G.; Chen, K.; Liu, X.; Su, L.; Singhal, S. Usp9x- and Noxa-mediated Mcl-1 downregulation contributes to pemetrexed-induced apoptosis in human non-small-cell lung cancer cells. Cell Death Dis. 2014, 5, e1316. [Google Scholar] [CrossRef] [Green Version]
- Bueno, M.; Brands, J.; Voltz, L.; Fiedler, K.; Mays, B.; St Croix, C.; Sembrat, J.; Mallampalli, R.K.; Rojas, M.; Mora, A.L. ATF3 represses PINK1 gene transcription in lung epithelial cells to control mitochondrial homeostasis. Aging Cell 2018, 17. [Google Scholar] [CrossRef] [PubMed]
- Hsu, Y.J.; Hsu, S.C.; Chang, Y.L.; Huang, S.M.; Shih, C.C.; Tsai, C.S.; Lin, C.Y. Indoxyl sulfate upregulates the cannabinoid type 1 receptor gene via an ATF3/c-Jun complex-mediated signaling pathway in the model of uremic cardiomyopathy. Int. J. Cardiol. 2018, 252, 128–135. [Google Scholar] [CrossRef] [PubMed]
- Shi, Q.; Zhao, L.; Xu, C.; Zhang, L.; Zhao, H. High Molecular Weight Hyaluronan Suppresses Macrophage M1 Polarization and Enhances IL-10 Production in PM2.5-Induced Lung Inflammation. Molecules 2019, 24. [Google Scholar] [CrossRef] [Green Version]
- Xu, C.; Shi, Q.; Zhang, L.; Zhao, H. High molecular weight hyaluronan attenuates fine particulate matter-induced acute lung injury through inhibition of ROS-ASK1-p38/JNK-mediated epithelial apoptosis. Environ. Toxicol. Pharmacol. 2018, 59, 190–198. [Google Scholar] [CrossRef] [PubMed]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shi, Q.; Hu, B.; Yang, C.; Zhao, L.; Wu, J.; Qi, N. ATF3 Promotes Arsenic-Induced Apoptosis and Oppositely Regulates DR5 and Bcl-xL Expression in Human Bronchial Epithelial Cells. Int. J. Mol. Sci. 2021, 22, 4223. https://doi.org/10.3390/ijms22084223
Shi Q, Hu B, Yang C, Zhao L, Wu J, Qi N. ATF3 Promotes Arsenic-Induced Apoptosis and Oppositely Regulates DR5 and Bcl-xL Expression in Human Bronchial Epithelial Cells. International Journal of Molecular Sciences. 2021; 22(8):4223. https://doi.org/10.3390/ijms22084223
Chicago/Turabian StyleShi, Qiwen, Bei Hu, Chen Yang, Lan Zhao, Jing Wu, and Nan Qi. 2021. "ATF3 Promotes Arsenic-Induced Apoptosis and Oppositely Regulates DR5 and Bcl-xL Expression in Human Bronchial Epithelial Cells" International Journal of Molecular Sciences 22, no. 8: 4223. https://doi.org/10.3390/ijms22084223