
Anti-Inflammatory Therapies for Treatment of Inflammation-Related Preterm Brain Injury

 , and
, and 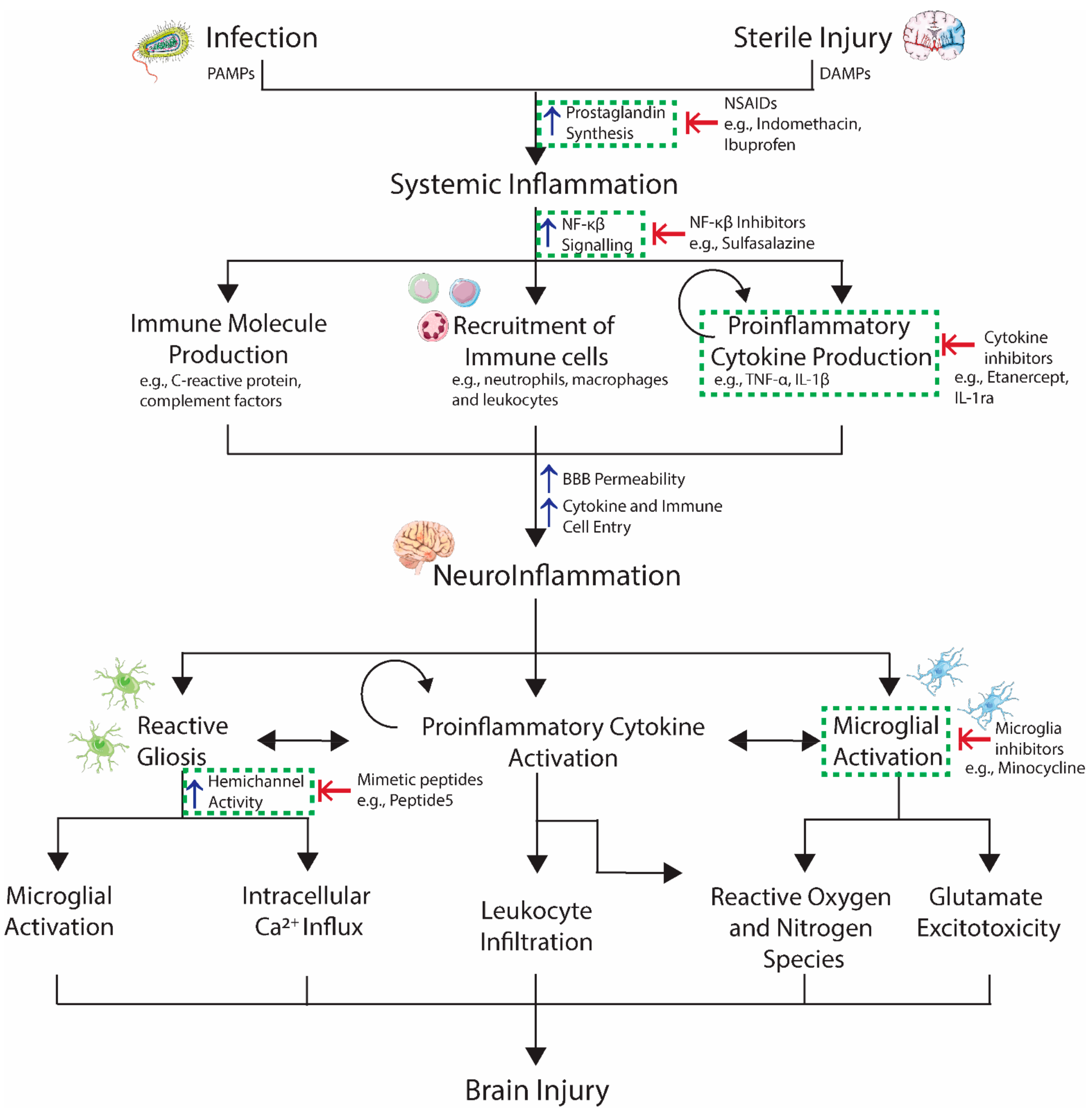{kind=link}
{kind=link}
Abstract
:1. Introduction
2. The Neonatal Immune System
Protection from Pathogens
3. From Systemic Inflammation to Neuroinflammation
4. Causes of Perinatal Neuroinflammation
4.1. Intrauterine Infection
4.2. Postnatal Infection
4.3. Nonspecific/Sterile Inflammation
5. Preterm Brain Injury
6. Molecular Mechanisms of Inflammation-Related Brain Injury
6.1. An Imbalance of Cytokines
6.2. Microglia
6.3. Astrocytes
7. Anti-Inflammatory Agents in the Treatment of Inflammation-Related Brain Injury
7.1. Inhibition of the Immune Response
7.1.1. Indomethacin
7.1.2. Ibuprofen
7.2. Inhibition of Cytokine Production
7.3. Inhibition of Specific Cytokines
7.3.1. TNF-α
7.3.2. IL-1
7.4. Inhibition of Microglial Activation
7.5. Inhibition of Astrocyte Activation
8. Concluding Remarks
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Blencowe, H.; Lee, A.C.; Cousens, S.; Bahalim, A.; Narwal, R.; Zhong, N.; Chou, D.; Say, L.; Modi, N.; Katz, J.; et al. Preterm birth-associated neurodevelopmental impairment estimates at regional and global levels for 2010. Pediatr. Res. 2013, 74 (Suppl. 1), 17–34. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, L.; Oza, S.; Hogan, D.; Chu, Y.; Perin, J.; Zhu, J.; Lawn, J.E.; Cousens, S.; Mathers, C.; Black, R.E. Global, regional, and national causes of under-5 mortality in 2000-15: An updated systematic analysis with implications for the Sustainable Development Goals. Lancet 2016, 388, 3027–3035. [Google Scholar] [CrossRef] [Green Version]
- Helenius, K.; Sjors, G.; Shah, P.S.; Modi, N.; Reichman, B.; Morisaki, N.; Kusuda, S.; Lui, K.; Darlow, B.A.; Bassler, D.; et al. Survival in Very Preterm Infants: An International Comparison of 10 National Neonatal Networks. Pediatrics 2017, 140, e20171264. [Google Scholar] [CrossRef] [Green Version]
- Kerstjens, J.M.; De Winter, A.F.; Bocca-Tjeertes, I.F.; Bos, A.F.; Reijneveld, S.A. Risk of developmental delay increases exponentially as gestational age of preterm infants decreases: A cohort study at age 4 years. Dev. Med. Child. Neurol. 2012, 54, 1096–1101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tronnes, H.; Wilcox, A.J.; Lie, R.T.; Markestad, T.; Moster, D. Risk of cerebral palsy in relation to pregnancy disorders and preterm birth: A national cohort study. Dev. Med. Child. Neurol. 2014, 56, 779–785. [Google Scholar] [CrossRef] [Green Version]
- Stavsky, M.; Mor, O.; Mastrolia, S.A.; Greenbaum, S.; Than, N.G.; Erez, O. Cerebral Palsy-Trends in Epidemiology and Recent Development in Prenatal Mechanisms of Disease, Treatment, and Prevention. Front. Pediatr. 2017, 5, 21. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Inder, T.E.; Warfield, S.K.; Wang, H.; Huppi, P.S.; Volpe, J.J. Abnormal cerebral structure is present at term in premature infants. Pediatrics 2005, 115, 286–294. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheong, J.L.; Doyle, L.W.; Burnett, A.C.; Lee, K.J.; Walsh, J.M.; Potter, C.R.; Treyvaud, K.; Thompson, D.K.; Olsen, J.E.; Anderson, P.J.; et al. Association Between Moderate and Late Preterm Birth and Neurodevelopment and Social-Emotional Development at Age 2 Years. JAMA Pediatr. 2017, 171, e164805. [Google Scholar] [CrossRef]
- Butler, A.S.; Behrman, R.E. (Eds.) Preterm Birth: Causes, Consequences, and Prevention; National Academy of Sciences: Washington, DC, USA, 2007. [Google Scholar] [CrossRef]
- Goldenberg, R.L.; Culhane, J.F.; Iams, J.D.; Romero, R. Epidemiology and causes of preterm birth. Lancet 2008, 371, 75–84. [Google Scholar] [CrossRef]
- Kemp, M.W. Preterm birth, intrauterine infection, and fetal inflammation. Front. Immunol. 2014, 5, 574. [Google Scholar] [CrossRef] [Green Version]
- Yates, N.; Gunn, A.J.; Bennet, L.; Dhillon, S.K.; Davidson, J.O. Preventing Brain Injury in the Preterm Infant-Current Controversies and Potential Therapies. Int. J. Mol. Sci. 2021, 7, 1671. [Google Scholar] [CrossRef]
- Stoll, B.J.; Hansen, N.I.; Adams-Chapman, I.; Fanaroff, A.A.; Hintz, S.R.; Vohr, B.; Higgins, R.D.; National Institute of Child Health; Human Development Neonatal Research Network. Neurodevelopmental and growth impairment among extremely low-birth-weight infants with neonatal infection. JAMA 2004, 292, 2357–2365. [Google Scholar] [CrossRef] [Green Version]
- Yanni, D.; Korzeniewski, S.J.; Allred, E.N.; Fichorova, R.N.; O’Shea, T.M.; Kuban, K.; Dammann, O.; Leviton, A. Both antenatal and postnatal inflammation contribute information about the risk of brain damage in extremely preterm newborns. Pediatr. Res. 2017, 82, 691–696. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klein, R.S.; Hunter, C.A. Protective and Pathological Immunity during Central Nervous System Infections. Immunity 2017, 46, 891–909. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Comi, C.; Tondo, G. Insights into the protective role of immunity in neurodegenerative disease. Neural Regen. Res. 2017, 12, 64–65. [Google Scholar] [CrossRef] [PubMed]
- Thornton, C.; Rousset, C.I.; Kichev, A.; Miyakuni, Y.; Vontell, R.; Baburamani, A.A.; Fleiss, B.; Gressens, P.; Hagberg, H. Molecular mechanisms of neonatal brain injury. Neurol. Res. Int. 2012, 2012, 506320. [Google Scholar] [CrossRef] [PubMed]
- Fleiss, B.; Gressens, P. Tertiary mechanisms of brain damage: A new hope for treatment of cerebral palsy? Lancet Neurol. 2012, 11, 556–566. [Google Scholar] [CrossRef]
- Lear, C.A.; Davidson, J.O.; Booth, L.C.; Wassink, G.; Galinsky, R.; Drury, P.P.; Fraser, M.; Bennet, L.; Gunn, A.J. Biphasic changes in fetal heart rate variability in preterm fetal sheep developing hypotension after acute on chronic lipopolysaccharide exposure. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2014, 307, R387–R395. [Google Scholar] [CrossRef] [Green Version]
- Mathai, S.; Booth, L.C.; Davidson, J.O.; Drury, P.P.; Fraser, M.; Jensen, E.C.; George, S.; Naylor, A.; Gunn, A.J.; Bennet, L. Acute on chronic exposure to endotoxin in preterm fetal sheep. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2013, 304, R189–R197. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Stridh, L.; Li, W.; Dean, J.; Elmgren, A.; Gan, L.; Eriksson, K.; Hagberg, H.; Mallard, C. Lipopolysaccharide sensitizes neonatal hypoxic-ischemic brain injury in a MyD88-dependent manner. J. Immunol. 2009, 183, 7471–7477. [Google Scholar] [CrossRef] [Green Version]
- Eklind, S.; Mallard, C.; Arvidsson, P.; Hagberg, H. Lipopolysaccharide induces both a primary and a secondary phase of sensitization in the developing rat brain. Pediatr. Res. 2005, 58, 112–116. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- van den Heuij, L.G.; Mathai, S.; Davidson, J.O.; Lear, C.A.; Booth, L.C.; Fraser, M.; Gunn, A.J.; Bennet, L. Synergistic white matter protection with acute-on-chronic endotoxin and subsequent asphyxia in preterm fetal sheep. J. Neuroinflamm. 2014, 11, 89. [Google Scholar] [CrossRef] [Green Version]
- Dhillon, S.K.; Gunn, A.J.; Jung, Y.; Mathai, S.; Bennet, L.; Fraser, M. Lipopolysaccharide-Induced Preconditioning Attenuates Apoptosis and Differentially Regulates TLR4 and TLR7 Gene Expression after Ischemia in the Preterm Ovine Fetal Brain. Dev. Neurosci. 2015, 37, 497–514. [Google Scholar] [CrossRef] [PubMed]
- Kallapur, S.G.; Jobe, A.H.; Ball, M.K.; Nitsos, I.; Moss, T.J.; Hillman, N.H.; Newnham, J.P.; Kramer, B.W. Pulmonary and systemic endotoxin tolerance in preterm fetal sheep exposed to chorioamnionitis. J. Immunol. 2007, 179, 8491–8499. [Google Scholar] [CrossRef] [PubMed]
- Melville, J.M.; Moss, T.J. The immune consequences of preterm birth. Front. Neurosci. 2013, 7, 79. [Google Scholar] [CrossRef] [Green Version]
- Strunk, T.; Currie, A.; Richmond, P.; Simmer, K.; Burgner, D. Innate immunity in human newborn infants: Prematurity means more than immaturity. J. Matern. Fetal Neonatal Med. 2011, 24, 25–31. [Google Scholar] [CrossRef]
- Marchant, E.A.; Kan, B.; Sharma, A.A.; van Zanten, A.; Kollmann, T.R.; Brant, R.; Lavoie, P.M. Attenuated innate immune defenses in very premature neonates during the neonatal period. Pediatr. Res. 2015, 78, 492–497. [Google Scholar] [CrossRef]
- Levy, O. Innate immunity of the newborn: Basic mechanisms and clinical correlates. Nat. Rev. Immunol. 2007, 7, 379–390. [Google Scholar] [CrossRef]
- O’Driscoll, D.N.; Greene, C.M.; Molloy, E.J. Immune function? A missing link in the gender disparity in preterm neonatal outcomes. Expert Rev. Clin. Immunol. 2017, 13, 1061–1071. [Google Scholar] [CrossRef]
- Joyner, J.L.; Augustine, N.H.; Taylor, K.A.; La Pine, T.R.; Hill, H.R. Effects of group B streptococci on cord and adult mononuclear cell interleukin-12 and interferon-gamma mRNA accumulation and protein secretion. J. Infect. Dis. 2000, 182, 974–977. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chelvarajan, R.L.; Collins, S.M.; Doubinskaia, I.E.; Goes, S.; Van Willigen, J.; Flanagan, D.; De Villiers, W.J.; Bryson, J.S.; Bondada, S. Defective macrophage function in neonates and its impact on unresponsiveness of neonates to polysaccharide antigens. J. Leukoc. Biol. 2004, 75, 982–994. [Google Scholar] [CrossRef]
- Carr, R.; Modi, N. Haemopoietic colony stimulating factors for preterm neonates. Arch. Dis. Child. Fetal Neonatal Ed. 1997, 76, F128–F133. [Google Scholar] [CrossRef] [Green Version]
- Strunk, T.; Temming, P.; Gembruch, U.; Reiss, I.; Bucsky, P.; Schultz, C. Differential maturation of the innate immune response in human fetuses. Pediatr. Res. 2004, 56, 219–226. [Google Scholar] [CrossRef] [Green Version]
- Bjorkqvist, M.; Jurstrand, M.; Bodin, L.; Fredlund, H.; Schollin, J. Defective neutrophil oxidative burst in preterm newborns on exposure to coagulase-negative staphylococci. Pediatr. Res. 2004, 55, 966–971. [Google Scholar] [CrossRef] [Green Version]
- Nussbaum, C.; Gloning, A.; Pruenster, M.; Frommhold, D.; Bierschenk, S.; Genzel-Boroviczeny, O.; von Andrian, U.H.; Quackenbush, E.; Sperandio, M. Neutrophil and endothelial adhesive function during human fetal ontogeny. J. Leukoc. Biol. 2013, 93, 175–184. [Google Scholar] [CrossRef] [Green Version]
- Kallman, J.; Schollin, J.; Schalen, C.; Erlandsson, A.; Kihlstrom, E. Impaired phagocytosis and opsonisation towards group B streptococci in preterm neonates. Arch. Dis. Child. Fetal Neonatal Ed. 1998, 78, F46–F50. [Google Scholar] [CrossRef]
- Sadeghi, K.; Berger, A.; Langgartner, M.; Prusa, A.R.; Hayde, M.; Herkner, K.; Pollak, A.; Spittler, A.; Forster-Waldl, E. Immaturity of infection control in preterm and term newborns is associated with impaired toll-like receptor signaling. J. Infect. Dis. 2007, 195, 296–302. [Google Scholar] [CrossRef]
- Currie, A.J.; Curtis, S.; Strunk, T.; Riley, K.; Liyanage, K.; Prescott, S.; Doherty, D.; Simmer, K.; Richmond, P.; Burgner, D. Preterm infants have deficient monocyte and lymphocyte cytokine responses to group B streptococcus. Infect. Immun. 2011, 79, 1588–1596. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- van den Berg, J.P.; Westerbeek, E.A.; van der Klis, F.R.; Berbers, G.A.; van Elburg, R.M. Transplacental transport of IgG antibodies to preterm infants: A review of the literature. Early Hum. Dev. 2011, 87, 67–72. [Google Scholar] [CrossRef] [PubMed]
- Walker, J.C.; Smolders, M.A.; Gemen, E.F.; Antonius, T.A.; Leuvenink, J.; de Vries, E. Development of lymphocyte subpopulations in preterm infants. Scand. J. Immunol. 2011, 73, 53–58. [Google Scholar] [CrossRef] [PubMed]
- Duggan, P.J.; Maalouf, E.F.; Watts, T.L.; Sullivan, M.H.; Counsell, S.J.; Allsop, J.; Al-Nakib, L.; Rutherford, M.A.; Battin, M.; Roberts, I.; et al. Intrauterine T-cell activation and increased proinflammatory cytokine concentrations in preterm infants with cerebral lesions. Lancet 2001, 358, 1699–1700. [Google Scholar] [CrossRef]
- Mallard, C. Innate immune regulation by toll-like receptors in the brain. ISRN Neurol. 2012, 2012, 701950. [Google Scholar] [CrossRef] [Green Version]
- Hagberg, H.; Gressens, P.; Mallard, C. Inflammation during fetal and neonatal life: Implications for neurologic and neuropsychiatric disease in children and adults. Ann. Neurol. 2012, 71, 444–457. [Google Scholar] [CrossRef]
- Carr, R. Neutrophil production and function in newborn infants. Br. J. Haematol. 2000, 110, 18–28. [Google Scholar] [CrossRef] [PubMed]
- Zeni, P.; Doepker, E.; Schulze-Topphoff, U.; Huewel, S.; Tenenbaum, T.; Galla, H.J. MMPs contribute to TNF-alpha-induced alteration of the blood-cerebrospinal fluid barrier in vitro. Am. J. Physiol. Cell Physiol. 2007, 293, C855–C864. [Google Scholar] [CrossRef] [PubMed]
- Tilling, T.; Korte, D.; Hoheisel, D.; Galla, H.J. Basement membrane proteins influence brain capillary endothelial barrier function in vitro. J. Neurochem. 1998, 71, 1151–1157. [Google Scholar] [CrossRef] [PubMed]
- Malaeb, S.; Dammann, O. Fetal inflammatory response and brain injury in the preterm newborn. J. Child. Neurol. 2009, 24, 1119–1126. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vitkovic, L.; Konsman, J.P.; Bockaert, J.; Dantzer, R.; Homburger, V.; Jacque, C. Cytokine signals propagate through the brain. Mol. Psychiatry 2000, 5, 604–615. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Osburg, B.; Peiser, C.; Dömling, D.; Schomburg, L.; Ko, Y.T.; Voigt, K.; Bickel, U. Effect of endotoxin on expression of TNF receptors and transport of TNF-α at the blood-brain barrier of the rat. Am. J. Physiol. Endocrinol. Metab. 2002, 283, E899–E908. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Banks, W.A.; Ortiz, L.; Plotkin, S.R.; Kastin, A.J. Human interleukin (IL) 1 alpha, murine IL-1 alpha and murine IL-1 beta are transported from blood to brain in the mouse by a shared saturable mechanism. J. Pharm. Exp. 1991, 259, 988–996. [Google Scholar]
- Galinsky, R.; Lear, C.A.; Dean, J.M.; Wassink, G.; Dhillon, S.K.; Fraser, M.; Davidson, J.O.; Bennet, L.; Gunn, A.J. Complex interactions between hypoxia-ischemia and inflammation in preterm brain injury. Dev. Med. Child. Neurol. 2018, 60, 126–133. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Becroft, D.M.O.; Thompson, J.M.D.; Mitchell, E.A. Placental Chorioamnionitis at Term: Epidemiology and Follow-Up in Childhood. Pediatric Dev. Pathol. 2010, 13, 282–290. [Google Scholar] [CrossRef] [PubMed]
- Strunk, T.; Inder, T.; Wang, X.; Burgner, D.; Mallard, C.; Levy, O. Infection-induced inflammation and cerebral injury in preterm infants. Lancet Infect. Dis. 2014, 14, 751–762. [Google Scholar] [CrossRef] [Green Version]
- Bastek, J.A.; Weber, A.L.; McShea, M.A.; Ryan, M.E.; Elovitz, M.A. Prenatal inflammation is associated with adverse neonatal outcomes. Am. J. Obstet. Gynecol. 2014, 210, 450.e1–450.e10. [Google Scholar] [CrossRef] [PubMed]
- Bierstone, D.; Wagenaar, N.; Gano, D.L.; Guo, T.; Georgio, G.; Groenendaal, F.; de Vries, L.S.; Varghese, J.; Glass, H.C.; Chung, C.; et al. Association of Histologic Chorioamnionitis With Perinatal Brain Injury and Early Childhood Neurodevelopmental Outcomes Among Preterm Neonates. JAMA Pediatr. 2018, 172, 534–541. [Google Scholar] [CrossRef] [PubMed]
- Chau, V.; Poskitt, K.J.; McFadden, D.E.; Bowen-Roberts, T.; Synnes, A.; Brant, R.; Sargent, M.A.; Soulikias, W.; Miller, S.P. Effect of chorioamnionitis on brain development and injury in premature newborns. Ann. Neurol. 2009, 66, 155–164. [Google Scholar] [CrossRef] [Green Version]
- Quinn, P.A.; Butany, J.; Taylor, J.; Hannah, W. Chorioamnionitis: Its association with pregnancy outcome and microbial infection. Am. J. Obs. Gynecol. 1987, 156, 379–387. [Google Scholar] [CrossRef]
- Gomez, R.; Romero, R.; Ghezzi, F.; Yoon, B.H.; Mazor, M.; Berry, S.M. The fetal inflammatory response syndrome. Am. J. Obs. Gynecol. 1998, 179, 194–202. [Google Scholar] [CrossRef]
- Romero, R.; Gotsch, F.; Pineles, B.; Kusanovic, J.P. Inflammation in pregnancy: Its roles in reproductive physiology, obstetrical complications, and fetal injury. Nutr. Rev. 2007, 65, S194–S202. [Google Scholar] [CrossRef]
- Rand, K.M.; Austin, N.C.; Inder, T.E.; Bora, S.; Woodward, L.J. Neonatal Infection and Later Neurodevelopmental Risk in the Very Preterm Infant. J. Pediatr. 2016, 170, 97–104. [Google Scholar] [CrossRef] [Green Version]
- O’Shea, T.M.; Allred, E.N.; Kuban, K.C.K.; Dammann, O.; Paneth, N.; Fichorova, R.; Hirtz, D.; Leviton, A.; Age, E.L.G. Elevated Concentrations of Inflammation-Related Proteins in Postnatal Blood Predict Severe Developmental Delay at 2 Years of Age in Extremely Preterm Infants. J. Pediatr. 2012, 160, 395–401. [Google Scholar] [CrossRef] [Green Version]
- O’Shea, T.M.; Joseph, R.M.; Kuban, K.C.; Allred, E.N.; Ware, J.; Coster, T.; Fichorova, R.N.; Dammann, O.; Leviton, A.; Investigators, E.S. Elevated blood levels of inflammation-related proteins are associated with an attention problem at age 24 mo in extremely preterm infants. Pediatr. Res. 2014, 75, 781–787. [Google Scholar] [CrossRef]
- Chau, V.; Brant, R.; Poskitt, K.J.; Tam, E.W.; Synnes, A.; Miller, S.P. Postnatal infection is associated with widespread abnormalities of brain development in premature newborns. Pediatr. Res. 2012, 71, 274–279. [Google Scholar] [CrossRef] [Green Version]
- Glass, H.C.; Bonifacio, S.L.; Chau, V.; Glidden, D.; Poskitt, K.; Barkovich, A.J.; Ferriero, D.M.; Miller, S.P. Recurrent postnatal infections are associated with progressive white matter injury in premature infants. Pediatrics 2008, 122, 299–305. [Google Scholar] [CrossRef]
- Glass, T.J.A.; Chau, V.; Grunau, R.E.; Synnes, A.; Guo, T.; Duerden, E.G.; Foong, J.; Poskitt, K.J.; Miller, S.P. Multiple Postnatal Infections in Newborns Born Preterm Predict Delayed Maturation of Motor Pathways at Term-Equivalent Age with Poorer Motor Outcomes at 3 Years. J. Pediatr. 2018, 196, 91–97.e91. [Google Scholar] [CrossRef]
- Rocha-Ferreira, E.; Hristova, M. Antimicrobial peptides and complement in neonatal hypoxia-ischemia induced brain damage. Front. Immunol 2015, 6, 56. [Google Scholar] [CrossRef] [Green Version]
- Bartha, A.I.; Foster-Barber, A.; Miller, S.P.; Vigneron, D.B.; Glidden, D.V.; Barkovich, A.J.; Ferriero, D.M. Neonatal encephalopathy: Association of cytokines with MR spectroscopy and outcome. Pediatr. Res. 2004, 56, 960–966. [Google Scholar] [CrossRef] [Green Version]
- Buser, J.R.; Maire, J.; Riddle, A.; Gong, X.; Nguyen, T.; Nelson, K.; Luo, N.L.; Ren, J.; Struve, J.; Sherman, L.S.; et al. Arrested preoligodendrocyte maturation contributes to myelination failure in premature infants. Ann. Neurol. 2012, 71, 93–109. [Google Scholar] [CrossRef] [PubMed]
- McElrath, T.F.; Allred, E.N.; Van Marter, L.; Fichorova, R.N.; Leviton, A.; Investigators, E.S. Perinatal systemic inflammatory responses of growth-restricted preterm newborns. Acta Paediatr. 2013, 102, e439–e442. [Google Scholar] [CrossRef] [PubMed]
- Leviton, A.; Fichorova, R.N.; O’Shea, T.M.; Kuban, K.; Paneth, N.; Dammann, O.; Allred, E.N.; Investigators, E.S. Two-hit model of brain damage in the very preterm newborn: Small for gestational age and postnatal systemic inflammation. Pediatr. Res. 2013, 73, 362–370. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bohrer, B.; Silveira, R.C.; Neto, E.C.; Procianoy, R.S. Mechanical ventilation of newborns infant changes in plasma pro- and anti-inflammatory cytokines. J. Pediatr. 2010, 156, 16–19. [Google Scholar] [CrossRef] [PubMed]
- Bose, C.L.; Laughon, M.M.; Allred, E.N.; O’Shea, T.M.; Van Marter, L.J.; Ehrenkranz, R.A.; Fichorova, R.N.; Leviton, A.; Investigators, E.S. Systemic inflammation associated with mechanical ventilation among extremely preterm infants. Cytokine 2013, 61, 315–322. [Google Scholar] [CrossRef] [Green Version]
- Koksal, N.; Kayik, B.; Cetinkaya, M.; Ozkan, H.; Budak, F.; Kilic, S.; Canitez, Y.; Oral, B. Value of serum and bronchoalveolar fluid lavage pro- and anti-inflammatory cytokine levels for predicting bronchopulmonary dysplasia in premature infants. Eur. Cytokine Netw. 2012, 23, 29–35. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Laptook, A.R.; O’Shea, T.M.; Shankaran, S.; Bhaskar, B.; Network, N.N. Adverse neurodevelopmental outcomes among extremely low birth weight infants with a normal head ultrasound: Prevalence and antecedents. Pediatrics 2005, 115, 673–680. [Google Scholar] [CrossRef] [PubMed]
- Nott, F.; Jane Pillow, J.; Dahl, M.; Kelly, S.B.; Melville, J.; McDonald, C.; Nitsos, I.; Lim, R.; Wallace, E.M.; Jenkin, G.; et al. Brain inflammation and injury at 48 h is not altered by human amnion epithelial cells in ventilated preterm lambs. Pediatr. Res. 2020, 88, 27–37. [Google Scholar] [CrossRef]
- Ballabh, P.; de Vries, L.S. White matter injury in infants with intraventricular haemorrhage: Mechanisms and therapies. Nat. Rev. Neurol. 2021. [Google Scholar] [CrossRef]
- Shooman, D.; Portess, H.; Sparrow, O. A review of the current treatment methods for posthaemorrhagic hydrocephalus of infants. Cereb. Fluid Res. 2009, 6, 1. [Google Scholar] [CrossRef] [Green Version]
- Atienza-Navarro, I.; Alves-Martinez, P.; Lubian-Lopez, S.; Garcia-Alloza, M. Germinal Matrix-Intraventricular Hemorrhage of the Preterm Newborn and Preclinical Models: Inflammatory Considerations. Int. J. Mol. Sci. 2020, 21, 8343. [Google Scholar] [CrossRef]
- Savman, K.; Blennow, M.; Hagberg, H.; Tarkowski, E.; Thoresen, M.; Whitelaw, A. Cytokine response in cerebrospinal fluid from preterm infants with posthaemorrhagic ventricular dilatation. Acta Paediatr. 2002, 91, 1357–1363. [Google Scholar] [CrossRef]
- Supramaniam, V.; Vontell, R.; Srinivasan, L.; Wyatt-Ashmead, J.; Hagberg, H.; Rutherford, M. Microglia activation in the extremely preterm human brain. Pediatr. Res. 2013, 73, 301–309. [Google Scholar] [CrossRef]
- Khwaja, O.; Volpe, J.J. Pathogenesis of cerebral white matter injury of prematurity. Arch. Dis. Child. Fetal Neonatal Ed. 2008, 93, F153–F161. [Google Scholar] [CrossRef]
- Hamrick, S.E.; Miller, S.P.; Leonard, C.; Glidden, D.V.; Goldstein, R.; Ramaswamy, V.; Piecuch, R.; Ferriero, D.M. Trends in severe brain injury and neurodevelopmental outcome in premature newborn infants: The role of cystic periventricular leukomalacia. J. Pediatr. 2004, 145, 593–599. [Google Scholar] [CrossRef]
- Back, S.A. Perinatal white matter injury: The changing spectrum of pathology and emerging insights into pathogenetic mechanisms. Ment. Retard Dev. Disabil. Res. Rev. 2006, 12, 129–140. [Google Scholar] [CrossRef]
- van Tilborg, E.; Heijnen, C.J.; Benders, M.J.; van Bel, F.; Fleiss, B.; Gressens, P.; Nijboer, C.H. Impaired oligodendrocyte maturation in preterm infants: Potential therapeutic targets. Prog. Neurobiol. 2016, 136, 28–49. [Google Scholar] [CrossRef] [PubMed]
- Resch, B.; Muhlanger, A.; Maurer-Fellbaum, U.; Pichler-Stachl, E.; Resch, E.; Urlesberger, B. Quality of Life of Children with Cystic Periventricular Leukomalacia—A Prospective Analysis with the Child Health Questionnaire-Parent Form 50. Front. Pediatr. 2016, 4, 50. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sellier, E.; Platt, M.J.; Andersen, G.L.; Krageloh-Mann, I.; De La Cruz, J.; Cans, C.; Surveillance of Cerebral Palsy Network. Decreasing prevalence in cerebral palsy: A multi-site European population-based study, 1980 to 2003. Dev. Med. Child. Neurol. 2016, 58, 85–92. [Google Scholar] [CrossRef]
- Back, S.A. White matter injury in the preterm infant: Pathology and mechanisms. Acta Neuropathol. 2017, 134, 331–349. [Google Scholar] [CrossRef] [PubMed]
- Counsell, S.J.; Allsop, J.M.; Harrison, M.C.; Larkman, D.J.; Kennea, N.L.; Kapellou, O.; Cowan, F.M.; Hajnal, J.V.; Edwards, A.D.; Rutherford, M.A. Diffusion-weighted imaging of the brain in preterm infants with focal and diffuse white matter abnormality. Pediatrics 2003, 112, 1–7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Back, S.A.; Riddle, A.; McClure, M.M. Maturation-dependent vulnerability of perinatal white matter in premature birth. Stroke 2007, 38, 724–730. [Google Scholar] [CrossRef] [Green Version]
- Riddle, A.; Maire, J.; Gong, X.; Chen, K.X.; Kroenke, C.D.; Hohimer, A.R.; Back, S.A. Differential susceptibility to axonopathy in necrotic and non-necrotic perinatal white matter injury. Stroke 2012, 43, 178–184. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Counsell, S.J.; Edwards, A.D.; Chew, A.T.; Anjari, M.; Dyet, L.E.; Srinivasan, L.; Boardman, J.P.; Allsop, J.M.; Hajnal, J.V.; Rutherford, M.A.; et al. Specific relations between neurodevelopmental abilities and white matter microstructure in children born preterm. Brain 2008, 131, 3201–3208. [Google Scholar] [CrossRef] [Green Version]
- Woodward, L.J.; Anderson, P.J.; Austin, N.C.; Howard, K.; Inder, T.E. Neonatal MRI to predict neurodevelopmental outcomes in preterm infants. N. Engl. J. Med. 2006, 355, 685–694. [Google Scholar] [CrossRef] [Green Version]
- Woodward, L.J.; Edgin, J.O.; Thompson, D.; Inder, T.E. Object working memory deficits predicted by early brain injury and development in the preterm infant. Brain 2005, 128, 2578–2587. [Google Scholar] [CrossRef] [Green Version]
- Ajayi-Obe, M.; Saeed, N.; Cowan, F.M.; Rutherford, M.A.; Edwards, A.D. Reduced development of cerebral cortex in extremely preterm infants. Lancet 2000, 356, 1162–1163. [Google Scholar] [CrossRef]
- Nagy, Z.; Ashburner, J.; Andersson, J.; Jbabdi, S.; Draganski, B.; Skare, S.; Bohm, B.; Smedler, A.C.; Forssberg, H.; Lagercrantz, H. Structural correlates of preterm birth in the adolescent brain. Pediatrics 2009, 124, e964–e972. [Google Scholar] [CrossRef] [PubMed]
- Nosarti, C.; Mechelli, A.; Herrera, A.; Walshe, M.; Shergill, S.S.; Murray, R.M.; Rifkin, L.; Allin, M.P. Structural covariance in the cortex of very preterm adolescents: A voxel-based morphometry study. Hum. Brain Mapp. 2011, 32, 1615–1625. [Google Scholar] [CrossRef] [PubMed]
- Peterson, B.S. Brain imaging studies of the anatomical and functional consequences of preterm birth for human brain development. Ann. N. Y. Acad. Sci. 2003, 1008, 219–237. [Google Scholar] [CrossRef]
- Rathbone, R.; Counsell, S.J.; Kapellou, O.; Dyet, L.; Kennea, N.; Hajnal, J.; Allsop, J.M.; Cowan, F.; Edwards, A.D. Perinatal cortical growth and childhood neurocognitive abilities. Neurology 2011, 77, 1510–1517. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nosarti, C.; Nam, K.W.; Walshe, M.; Murray, R.M.; Cuddy, M.; Rifkin, L.; Allin, M.P. Preterm birth and structural brain alterations in early adulthood. Neuroimage Clin. 2014, 6, 180–191. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nam, K.W.; Castellanos, N.; Simmons, A.; Froudist-Walsh, S.; Allin, M.P.; Walshe, M.; Murray, R.M.; Evans, A.; Muehlboeck, J.S.; Nosarti, C. Alterations in cortical thickness development in preterm-born individuals: Implications for high-order cognitive functions. Neuroimage 2015, 115, 64–75. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kalpakidou, A.K.; Allin, M.P.; Walshe, M.; Giampietro, V.; McGuire, P.K.; Rifkin, L.; Murray, R.M.; Nosarti, C. Functional neuroanatomy of executive function after neonatal brain injury in adults who were born very preterm. PLoS ONE 2014, 9, e113975. [Google Scholar] [CrossRef] [Green Version]
- Marín-Padilla, M. Developmental neuropathology and impact of perinatal brain damage. II: White matter lesions of the neocortex. J. Neuropathol. Exp. Neurol. 1997, 56, 219–235. [Google Scholar] [CrossRef] [Green Version]
- Inder, T.E.; Huppi, P.S.; Warfield, S.; Kikinis, R.; Zientara, G.P.; Barnes, P.D.; Jolesz, F.; Volpe, J.J. Periventricular white matter injury in the premature infant is followed by reduced cerebral cortical gray matter volume at term. Ann. Neurol. 1999, 46, 755–760. [Google Scholar] [CrossRef]
- Andiman, S.E.; Haynes, R.L.; Trachtenberg, F.L.; Billiards, S.S.; Folkerth, R.D.; Volpe, J.J.; Kinney, H.C. The cerebral cortex overlying periventricular leukomalacia: Analysis of pyramidal neurons. Brain Pathol. 2010, 20, 803–814. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pierson, C.R.; Folkerth, R.D.; Billiards, S.S.; Trachtenberg, F.L.; Drinkwater, M.E.; Volpe, J.J.; Kinney, H.C. Gray matter injury associated with periventricular leukomalacia in the premature infant. Acta Neuropathol. 2007, 114, 619–631. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Back, S.A.; Luo, N.L.; Mallinson, R.A.; O’Malley, J.P.; Wallen, L.D.; Frei, B.; Morrow, J.D.; Petito, C.K.; Roberts, C.T., Jr.; Murdoch, G.H.; et al. Selective vulnerability of preterm white matter to oxidative damage defined by F2-isoprostanes. Ann. Neurol. 2005, 58, 108–120. [Google Scholar] [CrossRef]
- McClendon, E.; Chen, K.; Gong, X.; Sharifnia, E.; Hagen, M.; Cai, V.; Shaver, D.C.; Riddle, A.; Dean, J.M.; Gunn, A.J.; et al. Prenatal cerebral ischemia triggers dysmaturation of caudate projection neurons. Ann. Neurol 2014, 75, 508–524. [Google Scholar] [CrossRef]
- Dean, J.M.; McClendon, E.; Hansen, K.; Azimi-Zonooz, A.; Chen, K.; Riddle, A.; Gong, X.; Sharifnia, E.; Hagen, M.; Ahmad, T.; et al. Prenatal cerebral ischemia disrupts MRI-defined cortical microstructure through disturbances in neuronal arborization. Sci. Transl. Med. 2013, 5, 168ra7. [Google Scholar] [CrossRef] [Green Version]
- Prasad, J.D.; van de Looij, Y.; Gunn, K.C.; Ranchhod, S.M.; White, P.B.; Berry, M.J.; Bennet, L.; Sizonenko, S.V.; Gunn, A.J.; Dean, J.M. Long-term coordinated microstructural disruptions of the developing neocortex and subcortical white matter after early postnatal systemic inflammation. Brain Behav. Immun. 2020. [Google Scholar] [CrossRef] [PubMed]
- Bilbo, S.D.; Schwarz, J.M. The immune system and developmental programming of brain and behavior. Front. Neuroendocr. 2012, 33, 267–286. [Google Scholar] [CrossRef] [Green Version]
- Mehler, M.F.; Marmur, R.; Gross, R.; Mabie, P.C.; Zang, Z.; Papavasiliou, A.; Kessler, J.A. Cytokines regulate the cellular phenotype of developing neural lineage species. Int. J. Dev. Neurosci. Off. J. Int. Soc. Dev. Neurosci. 1995, 13, 213–240. [Google Scholar] [CrossRef]
- Dammann, O.; O’Shea, T.M. Cytokines and perinatal brain damage. Clin. Perinatol. 2008, 35, 643–663. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rees, S.; Harding, R.; Walker, D. The biological basis of injury and neuroprotection in the fetal and neonatal brain. Int. J. Dev. Neurosci. Off. J. Int. Soc. Dev. Neurosci. 2011, 29, 551–563. [Google Scholar] [CrossRef] [Green Version]
- Dinarello, C.A. Proinflammatory cytokines. Chest 2000, 118, 503–508. [Google Scholar] [CrossRef]
- Yoon, B.H.; Jun, J.K.; Romero, R.; Park, K.H.; Gomez, R.; Choi, J.-H.; Kim, I.-O. Amniotic fluid inflammatory cytokines (interleukin-6, interleukin-1β, and tumor necrosis factor-α), neonatal brain white matter lesions, and cerebral palsy. Am. J. Obstet. Gynecol. 1997, 177, 19–26. [Google Scholar] [CrossRef]
- Miller, L.C.; Isa, S.; LoPreste, G.; Schaller, J.G.; Dinarello, C.A. Neonatal interleukin-1 beta, interleukin-6, and tumor necrosis factor: Cord blood levels and cellular production. J. Pediatr. 1990, 117, 961–965. [Google Scholar] [CrossRef]
- Leviton, A.; Kuban, K.; O’Shea, T.M.; Paneth, N.; Fichorova, R.; Allred, E.N.; Dammann, O. The relationship between early concentrations of 25 blood proteins and cerebral white matter injury in preterm newborns: The ELGAN study. J. Pediatr. 2011, 158, 897–903. [Google Scholar] [CrossRef] [PubMed]
- Mustafa, M.M.; Lebel, M.H.; Ramilo, O.; Olsen, K.D.; Reisch, J.S.; Beutler, B.; McCracken, G.H., Jr. Correlation of interleukin-1 beta and cachectin concentrations in cerebrospinal fluid and outcome from bacterial meningitis. J. Pediatr. 1989, 115, 208–213. [Google Scholar] [CrossRef]
- Kadhim, H.; Tabarki, B.; Verellen, G.; De Prez, C.; Rona, A.M.; Sebire, G. Inflammatory cytokines in the pathogenesis of periventricular leukomalacia. Neurology 2001, 56, 1278–1284. [Google Scholar] [CrossRef]
- Leviton, A.; Dammann, O.; Allred, E.N.; Joseph, R.M.; Fichorova, R.N.; O’Shea, T.M.; Kuban, K.C.K. Neonatal systemic inflammation and the risk of low scores on measures of reading and mathematics achievement at age 10 years among children born extremely preterm. Int. J. Dev. Neurosci. Off. J. Int. Soc. Dev. Neurosci. 2018, 66, 45–53. [Google Scholar] [CrossRef]
- McAdams, R.M.; Juul, S.E. The role of cytokines and inflammatory cells in perinatal brain injury. Neurol. Res. Int. 2012, 2012, 561494. [Google Scholar] [CrossRef]
- Ådén, U.; Favrais, G.; Plaisant, F.; Winerdal, M.; Felderhoff-Mueser, U.; Lampa, J.; Lelièvre, V.; Gressens, P. Systemic inflammation sensitizes the neonatal brain to excitotoxicity through a pro-/anti-inflammatory imbalance: Key role of TNFα pathway and protection by etanercept. Brain Behav. Immun. 2010, 24, 747–758. [Google Scholar] [CrossRef]
- Sebire, G.; Emilie, D.; Wallon, C.; Hery, C.; Devergne, O.; Delfraissy, J.F.; Galanaud, P.; Tardieu, M. In vitro production of IL-6, IL-1 beta, and tumor necrosis factor-alpha by human embryonic microglial and neural cells. J. Immunol. 1993, 150, 1517–1523. [Google Scholar]
- Lee, S.C.; Liu, W.; Dickson, D.W.; Brosnan, C.F.; Berman, J.W. Cytokine production by human fetal microglia and astrocytes. Differential induction by lipopolysaccharide and IL-1 beta. J. Immunol. 1993, 150, 2659–2667. [Google Scholar] [PubMed]
- Andrews, T.; Zhang, P.; Bhat, N.R. TNFα potentiates IFNγ-induced cell death in oligodendrocyte progenitors. J. Neurosci. Res. 1998, 54, 574–583. [Google Scholar] [CrossRef]
- Kadhim, H.; Tabarki, B.; De Prez, C.; Rona, A.M.; Sebire, G. Interleukin-2 in the pathogenesis of perinatal white matter damage. Neurology 2002, 58, 1125–1128. [Google Scholar] [CrossRef] [PubMed]
- Feldhaus, B.; Dietzel, I.D.; Heumann, R.; Berger, R. Effects of interferon-gamma and tumor necrosis factor-alpha on survival and differentiation of oligodendrocyte progenitors. J. Soc. Gynecol. Investig. 2004, 11, 89–96. [Google Scholar] [CrossRef] [PubMed]
- Kuzumaki, N.; Ikegami, D.; Imai, S.; Narita, M.; Tamura, R.; Yajima, M.; Suzuki, A.; Miyashita, K.; Niikura, K.; Takeshima, H.; et al. Enhanced IL-1beta production in response to the activation of hippocampal glial cells impairs neurogenesis in aged mice. Synapse 2010, 64, 721–728. [Google Scholar] [CrossRef]
- Bonora, M.; De Marchi, E.; Patergnani, S.; Suski, J.M.; Celsi, F.; Bononi, A.; Giorgi, C.; Marchi, S.; Rimessi, A.; Duszynski, J.; et al. Tumor necrosis factor-alpha impairs oligodendroglial differentiation through a mitochondria-dependent process. Cell Death Differ. 2014, 21, 1198–1208. [Google Scholar] [CrossRef] [Green Version]
- Yanowitz, T.D.; Jordan, J.A.; Gilmour, C.H.; Towbin, R.; Bowen, A.; Roberts, J.M.; Brozanski, B.S. Hemodynamic disturbances in premature infants born after chorioamnionitis: Association with cord blood cytokine concentrations. Pediatr. Res. 2002, 51, 310–316. [Google Scholar] [CrossRef] [Green Version]
- van der Poll, T.; Buller, H.R.; ten Cate, H.; Wortel, C.H.; Bauer, K.A.; van Deventer, S.J.; Hack, C.E.; Sauerwein, H.P.; Rosenberg, R.D.; ten Cate, J.W. Activation of coagulation after administration of tumor necrosis factor to normal subjects. N. Engl. J. Med. 1990, 322, 1622–1627. [Google Scholar] [CrossRef] [PubMed]
- Broussard, S.R.; McCusker, R.H.; Novakofski, J.E.; Strle, K.; Shen, W.H.; Johnson, R.W.; Freund, G.G.; Dantzer, R.; Kelley, K.W. Cytokine-hormone interactions: Tumor necrosis factor alpha impairs biologic activity and downstream activation signals of the insulin-like growth factor I receptor in myoblasts. Endocrinology 2003, 144, 2988–2996. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Venters, H.D.; Tang, Q.; Liu, Q.; VanHoy, R.W.; Dantzer, R.; Kelley, K.W. A new mechanism of neurodegeneration: A proinflammatory cytokine inhibits receptor signaling by a survival peptide. Proc. Natl. Acad. Sci. USA 1999, 96, 9879–9884. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barrientos, R.M.; Sprunger, D.B.; Campeau, S.; Higgins, E.A.; Watkins, L.R.; Rudy, J.W.; Maier, S.F. Brain-derived neurotrophic factor mRNA downregulation produced by social isolation is blocked by intrahippocampal interleukin-1 receptor antagonist. Neuroscience 2003, 121, 847–853. [Google Scholar] [CrossRef]
- Lapchak, P.A.; Araujo, D.M.; Hefti, F. Systemic interleukin-1 beta decreases brain-derived neurotrophic factor messenger RNA expression in the rat hippocampal formation. Neuroscience 1993, 53, 297–301. [Google Scholar] [CrossRef]
- Banks, W.A. The blood-brain barrier in neuroimmunology: Tales of separation and assimilation. Brain Behav. Immun. 2015, 44, 1–8. [Google Scholar] [CrossRef] [Green Version]
- Biernacki, K.; Prat, A.; Blain, M.; Antel, J.P. Regulation of cellular and molecular trafficking across human brain endothelial cells by Th1- and Th2-polarized lymphocytes. J. Neuropathol. Exp. Neurol. 2004, 63, 223–232. [Google Scholar] [CrossRef] [Green Version]
- Ramesh, G.; MacLean, A.G.; Philipp, M.T. Cytokines and Chemokines at the Crossroads of Neuroinflammation, Neurodegeneration, and Neuropathic Pain. Mediat. Inflamm. 2013, 2013, 480739. [Google Scholar] [CrossRef] [Green Version]
- Ueno, M.; Fujita, Y.; Tanaka, T.; Nakamura, Y.; Kikuta, J.; Ishii, M.; Yamashita, T. Layer V cortical neurons require microglial support for survival during postnatal development. Nat. Neurosci. 2013, 16, 543–551. [Google Scholar] [CrossRef]
- Wlodarczyk, A.; Holtman, I.R.; Krueger, M.; Yogev, N.; Bruttger, J.; Khorooshi, R.; Benmamar-Badel, A.; de Boer-Bergsma, J.J.; Martin, N.A.; Karram, K.; et al. A novel microglial subset plays a key role in myelinogenesis in developing brain. EMBO J. 2017, 36, 3292–3308. [Google Scholar] [CrossRef]
- Pogledic, I.; Kostovic, I.; Fallet-Bianco, C.; Adle-Biassette, H.; Gressens, P.; Verney, C. Involvement of the subplate zone in preterm infants with periventricular white matter injury. Brain Pathol. 2014, 24, 128–141. [Google Scholar] [CrossRef]
- Paolicelli, R.C.; Bolasco, G.; Pagani, F.; Maggi, L.; Scianni, M.; Panzanelli, P.; Giustetto, M.; Ferreira, T.A.; Guiducci, E.; Dumas, L.; et al. Synaptic Pruning by Microglia Is Necessary for Normal Brain Development. Science 2011, 333, 1456–1458. [Google Scholar] [CrossRef] [Green Version]
- Eyo, U.B.; Dailey, M.E. Microglia: Key elements in neural development, plasticity, and pathology. J. Neuroimmune Pharm. 2013, 8, 494–509. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schwarz, J.M.; Sholar, P.W.; Bilbo, S.D. Sex differences in microglial colonization of the developing rat brain. J. Neurochem. 2012, 120, 948–963. [Google Scholar] [CrossRef] [PubMed]
- Murabe, Y.; Sano, Y. Morphological studies on neuroglia. Cell Tissue Res. 1982, 225, 469–485. [Google Scholar] [CrossRef]
- Monier, A.; Adle-Biassette, H.; Delezoide, A.L.; Evrard, P.; Gressens, P.; Verney, C. Entry and distribution of microglial cells in human embryonic and fetal cerebral cortex. J. Neuropathol. Exp. Neurol. 2007, 66, 372–382. [Google Scholar] [CrossRef] [PubMed]
- Verney, C.; Pogledic, I.; Biran, V.; Adle-Biassette, H.; Fallet-Bianco, C.; Gressens, P. Microglial reaction in axonal crossroads is a hallmark of noncystic periventricular white matter injury in very preterm infants. J. Neuropathol. Exp. Neurol. 2012, 71, 251–264. [Google Scholar] [CrossRef] [Green Version]
- Merrill, J.E. Effects of interleukin-1 and tumor necrosis factor-alpha on astrocytes, microglia, oligodendrocytes, and glial precursors in vitro. Dev. Neurosci. 1991, 13, 130–137. [Google Scholar] [CrossRef]
- Lehnardt, S.; Lachance, C.; Patrizi, S.; Lefebvre, S.; Follett, P.L.; Jensen, F.E.; Rosenberg, P.A.; Volpe, J.J.; Vartanian, T. The toll-like receptor TLR4 is necessary for lipopolysaccharide-induced oligodendrocyte injury in the CNS. J. Neurosci. 2002, 22, 2478–2486. [Google Scholar] [CrossRef]
- Merrill, J.E.; Ignarro, L.J.; Sherman, M.P.; Melinek, J.; Lane, T.E. Microglial cell cytotoxicity of oligodendrocytes is mediated through nitric oxide. J. Immunol. 1993, 151, 2132–2141. [Google Scholar]
- Burd, I.; Balakrishnan, B.; Kannan, S. Models of fetal brain injury, intrauterine inflammation, and preterm birth. Am. J. Reprod. Immunol. 2012, 67, 287–294. [Google Scholar] [CrossRef]
- Domercq, M.; Sanchez-Gomez, M.V.; Sherwin, C.; Etxebarria, E.; Fern, R.; Matute, C. System xc- and glutamate transporter inhibition mediates microglial toxicity to oligodendrocytes. J. Immunol. 2007, 178, 6549–6556. [Google Scholar] [CrossRef] [Green Version]
- Azevedo, E.P.; Ledo, J.H.; Barbosa, G.; Sobrinho, M.; Diniz, L.; Fonseca, A.C.; Gomes, F.; Romao, L.; Lima, F.R.; Palhano, F.L.; et al. Activated microglia mediate synapse loss and short-term memory deficits in a mouse model of transthyretin-related oculoleptomeningeal amyloidosis. Cell Death Dis. 2013, 4, e789. [Google Scholar] [CrossRef] [Green Version]
- Dean, J.M.; Wang, X.; Kaindl, A.M.; Gressens, P.; Fleiss, B.; Hagberg, H.; Mallard, C. Microglial MyD88 signaling regulates acute neuronal toxicity of LPS-stimulated microglia in vitro. Brain Behav. Immun. 2010, 24, 776–783. [Google Scholar] [CrossRef]
- Pang, Y.; Campbell, L.; Zheng, B.; Fan, L.; Cai, Z.; Rhodes, P. Lipopolysaccharide-activated microglia induce death of oligodendrocyte progenitor cells and impede their development. Neuroscience 2010, 166, 464–475. [Google Scholar] [CrossRef]
- Smith, K.L.; Kassem, M.S.; Clarke, D.J.; Kuligowski, M.P.; Bedoya-Perez, M.A.; Todd, S.M.; Lagopoulos, J.; Bennett, M.R.; Arnold, J.C. Microglial cell hyper-ramification and neuronal dendritic spine loss in the hippocampus and medial prefrontal cortex in a mouse model of PTSD. Brain Behav. Immun. 2019, 80, 889–899. [Google Scholar] [CrossRef]
- Chhor, V.; Le Charpentier, T.; Lebon, S.; Ore, M.V.; Celador, I.L.; Josserand, J.; Degos, V.; Jacotot, E.; Hagberg, H.; Savman, K.; et al. Characterization of phenotype markers and neuronotoxic potential of polarised primary microglia in vitro. Brain Behav. Immun. 2013, 32, 70–85. [Google Scholar] [CrossRef]
- Boche, D.; Perry, V.H.; Nicoll, J.A. Review: Activation patterns of microglia and their identification in the human brain. Neuropathol. Appl. Neurobiol. 2013, 39, 3–18. [Google Scholar] [CrossRef] [PubMed]
- Fleiss, B.; Gressens, P. Chapter 15—Neuroprotection of the preterm brain. In Handbook of Clinical Neurology; de Vries, L.S., Glass, H.C., Eds.; Elsevier: Amsterdam, The Netherlands, 2019; Volume 162, pp. 315–328. [Google Scholar]
- Smith, G.M.; Rutishauser, U.; Silver, J.; Miller, R.H. Maturation of astrocytes in vitro alters the extent and molecular basis of neurite outgrowth. Dev. Biol. 1990, 138, 377–390. [Google Scholar] [CrossRef]
- Raff, M.C.; Abney, E.R.; Fok-Seang, J. Reconstitution of a developmental clock in vitro: A critical role for astrocytes in the timing of oligodendrocyte differentiation. Cell 1985, 42, 61–69. [Google Scholar] [CrossRef]
- Ullian, E.M.; Sapperstein, S.K.; Christopherson, K.S.; Barres, B.A. Control of synapse number by glia. Science 2001, 291, 657–661. [Google Scholar] [CrossRef]
- Machler, P.; Wyss, M.T.; Elsayed, M.; Stobart, J.; Gutierrez, R.; von Faber-Castell, A.; Kaelin, V.; Zuend, M.; San Martin, A.; Romero-Gomez, I.; et al. In Vivo Evidence for a Lactate Gradient from Astrocytes to Neurons. Cell Metab. 2016, 23, 94–102. [Google Scholar] [CrossRef] [Green Version]
- Wiese, S.; Karus, M.; Faissner, A. Astrocytes as a source for extracellular matrix molecules and cytokines. Front. Pharm. 2012, 3, 120. [Google Scholar] [CrossRef] [Green Version]
- Rudge, J.S.; Alderson, R.F.; Pasnikowski, E.; McClain, J.; Ip, N.Y.; Lindsay, R.M. Expression of Ciliary Neurotrophic Factor and the Neurotrophins-Nerve Growth Factor, Brain-Derived Neurotrophic Factor and Neurotrophin 3-in Cultured Rat Hippocampal Astrocytes. Eur. J. Neurosci. 1992, 4, 459–471. [Google Scholar] [CrossRef]
- Prat, A.; Biernacki, K.; Wosik, K.; Antel, J.P. Glial cell influence on the human blood-brain barrier. Glia 2001, 36, 145–155. [Google Scholar] [CrossRef] [PubMed]
- Bokobza, C.; Van Steenwinckel, J.; Mani, S.; Mezger, V.; Fleiss, B.; Gressens, P. Neuroinflammation in preterm babies and autism spectrum disorders. Pediatr. Res. 2019, 85, 155–165. [Google Scholar] [CrossRef] [PubMed]
- Benveniste, E.N. Astrocyte-microglia interactions. Astrocytes Pharmacol. Funct. 1993, 15, 355–382. [Google Scholar]
- Mallard, C.; Davidson, J.O.; Tan, S.; Green, C.R.; Bennet, L.; Robertson, N.J.; Gunn, A.J. Astrocytes and microglia in acute cerebral injury underlying cerebral palsy associated with preterm birth. Pediatr. Res. 2014, 75, 234–240. [Google Scholar] [CrossRef] [PubMed]
- Elovitz, M.A.; Mrinalini, C.; Sammel, M.D. Elucidating the early signal transduction pathways leading to fetal brain injury in preterm birth. Pediatr. Res. 2006, 59, 50–55. [Google Scholar] [CrossRef] [Green Version]
- Dean, J.M.; Riddle, A.; Maire, J.; Hansen, K.D.; Preston, M.; Barnes, A.P.; Sherman, L.S.; Back, S.A. An organotypic slice culture model of chronic white matter injury with maturation arrest of oligodendrocyte progenitors. Mol. Neurodegener. 2011, 6, 46. [Google Scholar] [CrossRef] [Green Version]
- See, J.; Zhang, X.; Eraydin, N.; Mun, S.B.; Mamontov, P.; Golden, J.A.; Grinspan, J.B. Oligodendrocyte maturation is inhibited by bone morphogenetic protein. Mol. Cell Neurosci. 2004, 26, 481–492. [Google Scholar] [CrossRef]
- Wang, Y.; Cheng, X.; He, Q.; Zheng, Y.; Kim, D.H.; Whittemore, S.R.; Cao, Q.L. Astrocytes from the contused spinal cord inhibit oligodendrocyte differentiation of adult oligodendrocyte precursor cells by increasing the expression of bone morphogenetic proteins. J. Neurosci. 2011, 31, 6053–6058. [Google Scholar] [CrossRef] [Green Version]
- Gard, A.L.; Burrell, M.R.; Pfeiffer, S.E.; Rudge, J.S.; Williams, W.C., 2nd. Astroglial control of oligodendrocyte survival mediated by PDGF and leukemia inhibitory factor-like protein. Development 1995, 121, 2187–2197. [Google Scholar]
- Hammond, T.R.; Gadea, A.; Dupree, J.; Kerninon, C.; Nait-Oumesmar, B.; Aguirre, A.; Gallo, V. Astrocyte-derived endothelin-1 inhibits remyelination through notch activation. Neuron 2014, 81, 588–602. [Google Scholar] [CrossRef] [Green Version]
- Feigenson, K.; Reid, M.; See, J.; Crenshaw, E.B., 3rd; Grinspan, J.B. Wnt signaling is sufficient to perturb oligodendrocyte maturation. Mol. Cell Neurosci. 2009, 42, 255–265. [Google Scholar] [CrossRef] [PubMed]
- Shiow, L.R.; Favrais, G.; Schirmer, L.; Schang, A.L.; Cipriani, S.; Andres, C.; Wright, J.N.; Nobuta, H.; Fleiss, B.; Gressens, P.; et al. Reactive astrocyte COX2-PGE2 production inhibits oligodendrocyte maturation in neonatal white matter injury. Glia 2017, 65, 2024–2037. [Google Scholar] [CrossRef] [PubMed]
- Ibi, D.; Nagai, T.; Nakajima, A.; Mizoguchi, H.; Kawase, T.; Tsuboi, D.; Kano, S.; Sato, Y.; Hayakawa, M.; Lange, U.C.; et al. Astroglial IFITM3 mediates neuronal impairments following neonatal immune challenge in mice. Glia 2013, 61, 679–693. [Google Scholar] [CrossRef] [PubMed]
- Yamada, S.; Itoh, N.; Nagai, T.; Nakai, T.; Ibi, D.; Nakajima, A.; Nabeshima, T.; Yamada, K. Innate immune activation of astrocytes impairs neurodevelopment via upregulation of follistatin-like 1 and interferon-induced transmembrane protein 3. J. Neuroinflamm. 2018, 15, 295. [Google Scholar] [CrossRef]
- Bezzi, P.; Domercq, M.; Brambilla, L.; Galli, R.; Schols, D.; De Clercq, E.; Vescovi, A.; Bagetta, G.; Kollias, G.; Meldolesi, J.; et al. CXCR4-activated astrocyte glutamate release via TNFalpha: Amplification by microglia triggers neurotoxicity. Nat. Neurosci. 2001, 4, 702–710. [Google Scholar] [CrossRef] [PubMed]
- Farina, C.; Aloisi, F.; Meinl, E. Astrocytes are active players in cerebral innate immunity. Trends Immunol. 2007, 28, 138–145. [Google Scholar] [CrossRef] [PubMed]
- Meme, W.; Calvo, C.F.; Froger, N.; Ezan, P.; Amigou, E.; Koulakoff, A.; Giaume, C. Proinflammatory cytokines released from microglia inhibit gap junctions in astrocytes: Potentiation by beta-amyloid. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2006, 20, 494–496. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Retamal, M.A.; Froger, N.; Palacios-Prado, N.; Ezan, P.; Saez, P.J.; Saez, J.C.; Giaume, C. Cx43 hemichannels and gap junction channels in astrocytes are regulated oppositely by proinflammatory cytokines released from activated microglia. J. Neurosci. 2007, 27, 13781–13792. [Google Scholar] [CrossRef] [PubMed]
- Faustmann, P.M.; Haase, C.G.; Romberg, S.; Hinkerohe, D.; Szlachta, D.; Smikalla, D.; Krause, D.; Dermietzel, R. Microglia activation influences dye coupling and Cx43 expression of the astrocytic network. Glia 2003, 42, 101–108. [Google Scholar] [CrossRef] [PubMed]
- Peng, W.; Cotrina, M.L.; Han, X.; Yu, H.; Bekar, L.; Blum, L.; Takano, T.; Tian, G.F.; Goldman, S.A.; Nedergaard, M. Systemic administration of an antagonist of the ATP-sensitive receptor P2X7 improves recovery after spinal cord injury. Proc. Natl. Acad. Sci. USA 2009, 106, 12489–12493. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Froger, N.; Orellana, J.A.; Calvo, C.F.; Amigou, E.; Kozoriz, M.G.; Naus, C.C.; Saez, J.C.; Giaume, C. Inhibition of cytokine-induced connexin43 hemichannel activity in astrocytes is neuroprotective. Mol. Cell Neurosci. 2010, 45, 37–46. [Google Scholar] [CrossRef] [PubMed]
- Song, L.; Pei, L.; Yao, S.; Wu, Y.; Shang, Y. NLRP3 Inflammasome in Neurological Diseases, from Functions to Therapies. Front. Cell Neurosci. 2017, 11, 63. [Google Scholar] [CrossRef] [Green Version]
- Kim, Y.; Davidson, J.O.; Gunn, K.C.; Phillips, A.R.; Green, C.R.; Gunn, A.J. Role of Hemichannels in CNS Inflammation and the Inflammasome Pathway. Adv. Protein Chem. Struct. Biol. 2016, 104, 1–37. [Google Scholar]
- Galinsky, R.; Davidson, J.O.; Dean, J.M.; Green, C.R.; Bennet, L.; Gunn, A.J. Glia and hemichannels: Key mediators of perinatal encephalopathy. Neural Regen. Res. 2018, 13, 181–189. [Google Scholar] [CrossRef]
- Hauck, W.; Samlalsingh-Parker, J.; Glibetic, M.; Ricard, G.; Beaudoin, M.C.; Noya, F.J.; Aranda, J.V. Deregulation of cyclooxygenase and nitric oxide synthase gene expression in the inflammatory cascade triggered by experimental group B streptococcal meningitis in the newborn brain and cerebral microvessels. Semin. Perinatol. 1999, 23, 250–260. [Google Scholar] [CrossRef]
- Cao, C.; Matsumura, K.; Watanabe, Y. Induction of cyclooxygenase-2 in the brain by cytokines. Ann. N. Y. Acad. Sci. 1997, 813, 307–309. [Google Scholar] [CrossRef]
- Dubois, R.N.; Abramson, S.B.; Crofford, L.; Gupta, R.A.; Simon, L.S.; Van De Putte, L.B.; Lipsky, P.E. Cyclooxygenase in biology and disease. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 1998, 12, 1063–1073. [Google Scholar] [CrossRef] [Green Version]
- Rocca, B.; FitzGerald, G.A. Cyclooxygenases and prostaglandins: Shaping up the immune response. Int. Immunopharmacol. 2002, 2, 603–630. [Google Scholar] [CrossRef]
- Weaver-Mikaere, L.; Gunn, A.J.; Mitchell, M.D.; Bennet, L.; Fraser, M. LPS and TNF alpha modulate AMPA/NMDA receptor subunit expression and induce PGE2 and glutamate release in preterm fetal ovine mixed glial cultures. J. Neuroinflamm. 2013, 10, 153. [Google Scholar] [CrossRef] [Green Version]
- Takemiya, T.; Matsumura, K.; Sugiura, H.; Yasuda, S.; Uematsu, S.; Akira, S.; Yamagata, K. Endothelial microsomal prostaglandin E synthase-1 facilitates neurotoxicity by elevating astrocytic Ca2+ levels. Neurochem. Int. 2011, 58, 489–496. [Google Scholar] [CrossRef] [PubMed]
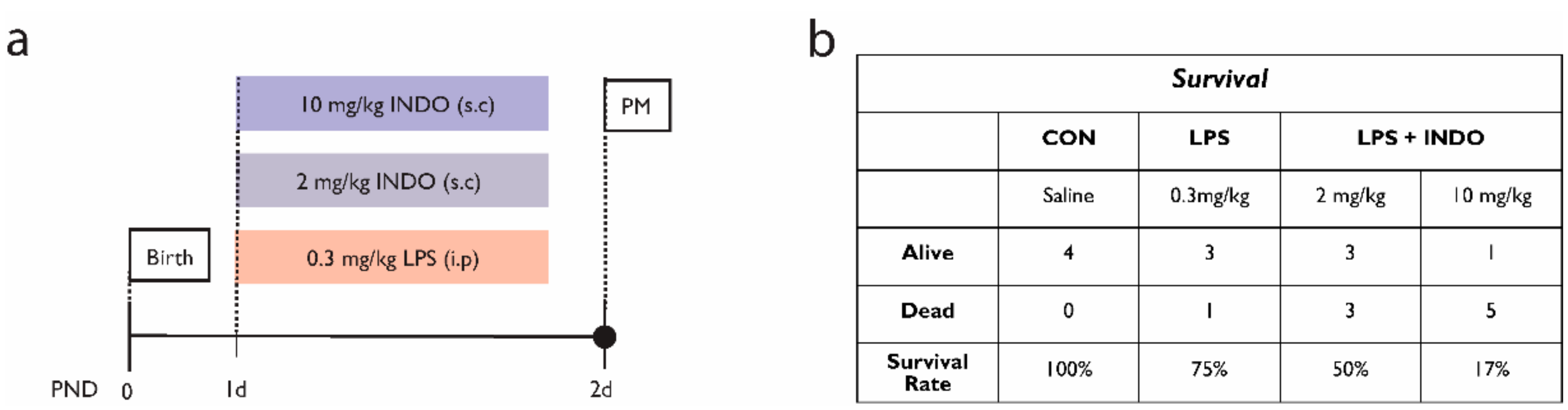
- El-Mashad, A.E.; El-Mahdy, H.; El Amrousy, D.; Elgendy, M. Comparative study of the efficacy and safety of paracetamol, ibuprofen, and indomethacin in closure of patent ductus arteriosus in preterm neonates. Eur. J. Pediatr. 2017, 176, 233–240. [Google Scholar] [CrossRef]
- Antonucci, R.; Zaffanello, M.; Puxeddu, E.; Porcella, A.; Cuzzolin, L.; Pilloni, M.D.; Fanos, V. Use of Non-steroidal Anti-inflammatory Drugs in Pregnancy: Impact on the Fetus and Newborn. Curr. Drug Metab. 2012, 13, 474–490. [Google Scholar] [CrossRef]
- Fowlie, P.W.; Davis, P.G.; McGuire, W. Prophylactic intravenous indomethacin for preventing mortality and morbidity in preterm infants. Cochrane Database Syst. Rev. 2010. [Google Scholar] [CrossRef]
- Miller, S.P.; Mayer, E.E.; Clyman, R.I.; Glidden, D.V.; Hamrick, S.E.G.; Barkovich, A.J. Prolonged indomethacin exposure is associated with decreased white matter injury detected with magnetic resonance imaging in premature newborns at 24 to 28 weeks’ gestation at birth. Pediatrics 2006, 117, 1626–1631. [Google Scholar] [CrossRef] [PubMed]
- Gano, D.; Andersen, S.K.; Partridge, J.C.; Bonifacio, S.L.; Xu, D.; Glidden, D.V.; Ferriero, D.M.; Barkovich, A.J.; Glass, H.C. Diminished white matter injury over time in a cohort of premature newborns. J. Pediatr. 2015, 166, 39–43. [Google Scholar] [CrossRef] [Green Version]
- Schmidt, B.; Davis, P.; Moddemann, D.; Ohlsson, A.; Roberts, R.S.; Saigal, S.; Solimano, A.; Vincer, M.; Wright, L.L.; Trial of Indomethacin Prophylaxis in Preterms Investigators. Long-term effects of indomethacin prophylaxis in extremely-low-birth-weight infants. N. Engl. J. Med. 2001, 344, 1966–1972. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pacifici, G.M. Differential renal adverse effects of ibuprofen and indomethacin in preterm infants: A review. Clin. Pharm. 2014, 6, 111–116. [Google Scholar] [CrossRef] [Green Version]
- Patel, J.; Roberts, I.; Azzopardi, D.; Hamilton, P.; Edwards, A.D. Randomized double-blind controlled trial comparing the effects of ibuprofen with indomethacin on cerebral hemodynamics in preterm infants with patent ductus arteriosus. Pediatr. Res. 2000, 47, 36–42. [Google Scholar] [CrossRef] [Green Version]
- Norton, M.E.; Merrill, J.; Cooper, B.A.; Kuller, J.A.; Clyman, R.I. Neonatal complications after the administration of indomethacin for preterm labor. N. Engl. J. Med. 1993, 329, 1602–1607. [Google Scholar] [CrossRef]
- Favrais, G.; Schwendimann, L.; Gressens, P.; Lelievre, V. Cyclooxygenase-2 mediates the sensitizing effects of systemic IL-1-beta on excitotoxic brain lesions in newborn mice. Neurobiol. Dis. 2007, 25, 496–505. [Google Scholar] [CrossRef]
- Taskin, E.; Ozcan, K.; Canacankatan, N.; Satar, M.; Yapicioglu, H.Y.; Erdogan, S. The effects of indomethacin on caspases, glutathione level and lipid peroxidation in the newborn rats with hypoxic-ischemic cerebral injury. Brain Res. 2009, 1289, 118–123. [Google Scholar] [CrossRef] [PubMed]
- Herson, V.C.; Krause, P.J.; Eisenfeld, L.I.; Pontius, L.; Maderazo, E.G. Indomethacin-associated sepsis in very-low-birth-weight infants. Am. J. Dis. Child. 1988, 142, 555–558. [Google Scholar] [CrossRef] [PubMed]
- Aranda, J.; Salomone, F.; Valencia, G.; Beharry, K. Non-steroidal Anti-inflammatory Drugs in Newborns and Infants. Pediatric Clin. North. Am. 2017, 64, 1327–1340. [Google Scholar] [CrossRef]
- Ricciotti, E.; FitzGerald, G.A. Prostaglandins and inflammation. Arterioscler. Thromb. Vasc. Biol. 2011, 31, 986–1000. [Google Scholar] [CrossRef] [PubMed]
- Gournay, V.; Savagner, C.; Thiriez, G.; Kuster, A.; Roze, J.C. Pulmonary hypertension after ibuprofen prophylaxis in very preterm infants. Lancet 2002, 359, 1486–1488. [Google Scholar] [CrossRef]
- Zecca, E.; Romagnoli, C.; De Carolis, M.P.; Costa, S.; Marra, R.; De Luca, D. Does Ibuprofen increase neonatal hyperbilirubinemia? Pediatrics 2009, 124, 480–484. [Google Scholar] [CrossRef]
- Erdeve, O.; Sarici, S.U.; Sari, E.; Gok, F. Oral-ibuprofen-induced acute renal failure in a preterm infant. Pediatr. Nephrol. 2008, 23, 1565–1567. [Google Scholar] [CrossRef]
- Aranda, J.V.; Thomas, R. Systematic review: Intravenous Ibuprofen in preterm newborns. Semin. Perinatol. 2006, 30, 114–120. [Google Scholar] [CrossRef] [Green Version]
- Dani, C.; Bertini, G.; Pezzati, M.; Poggi, C.; Guerrini, P.; Martano, C.; Rubaltelli, F.F. Prophylactic ibuprofen for the prevention of intraventricular hemorrhage among preterm infants: A multicenter, randomized study. Pediatrics 2005, 115, 1529–1535. [Google Scholar] [CrossRef]
- Ohlsson, A.; Walia, R.; Shah, S.S. Ibuprofen for the treatment of patent ductus arteriosus in preterm or low birth weight (or both) infants. Cochrane Database Syst. Rev. 2018. [Google Scholar] [CrossRef]
- Gournay, V.; Roze, J.C.; Kuster, A.; Daoud, P.; Cambonie, G.; Hascoet, J.M.; Chamboux, C.; Blanc, T.; Fichtner, C.; Savagner, C.; et al. Prophylactic ibuprofen versus placebo in very premature infants: A randomised, double-blind, placebo-controlled trial. Lancet 2004, 364, 1939–1944. [Google Scholar] [CrossRef]
- Carty, M.L.; Wixey, J.A.; Reinebrant, H.E.; Gobe, G.; Colditz, P.B.; Buller, K.M. Ibuprofen inhibits neuroinflammation and attenuates white matter damage following hypoxia–ischemia in the immature rodent brain. Brain Res. 2011, 1402, 9–19. [Google Scholar] [CrossRef]
- Wixey, J.A.; Reinebrant, H.E.; Buller, K.M. Post-insult ibuprofen treatment attenuates damage to the serotonergic system after hypoxia-ischemia in the immature rat brain. J. Neuropathol. Exp. Neurol. 2012, 71, 1137–1148. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wixey, J.A.; Sukumar, K.R.; Pretorius, R.; Lee, K.M.; Colditz, P.B.; Bjorkman, S.T.; Chand, K.K. Ibuprofen Treatment Reduces the Neuroinflammatory Response and Associated Neuronal and White Matter Impairment in the Growth Restricted Newborn. Front. Physiol. 2019, 10, 541. [Google Scholar] [CrossRef] [PubMed]
- Lawrence, T. The nuclear factor NF-kappaB pathway in inflammation. Cold Spring Harb. Perspect. Biol. 2009, 1, a001651. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wahl, C.; Liptay, S.; Adler, G.; Schmid, R.M. Sulfasalazine: A potent and specific inhibitor of nuclear factor kappa B. J. Clin. Invest. 1998, 101, 1163–1174. [Google Scholar] [CrossRef] [Green Version]
- Zenlea, T.; Peppercorn, M.A. Immunosuppressive therapies for inflammatory bowel disease. World J. Gastroenterol. WJG 2014, 20, 3146–3152. [Google Scholar] [CrossRef] [PubMed]
- Norgard, B.; Czeizel, A.E.; Rockenbauer, M.; Olsen, J.; Sorensen, H.T. Population-based case control study of the safety of sulfasalazine use during pregnancy. Aliment. Pharm. 2001, 15, 483–486. [Google Scholar] [CrossRef] [PubMed]
- Esbjorner, E.; Jarnerot, G.; Wranne, L. Sulphasalazine and sulphapyridine serum levels in children to mothers treated with sulphasalazine during pregnancy and lactation. Acta Paediatr. Scand. 1987, 76, 137–142. [Google Scholar] [CrossRef]
- Rahimi, R.; Nikfar, S.; Rezaie, A.; Abdollahi, M. Pregnancy outcome in women with inflammatory bowel disease following exposure to 5-aminosalicylic acid drugs: A meta-analysis. Reprod. Toxicol. 2008, 25, 271–275. [Google Scholar] [CrossRef]
- Keelan, J.A.; Khan, S.; Yosaatmadja, F.; Mitchell, M.D. Prevention of inflammatory activation of human gestational membranes in an ex vivo model using a pharmacological NF-kappaB inhibitor. J. Immunol. 2009, 183, 5270–5278. [Google Scholar] [CrossRef] [Green Version]
- Nath, C.A.; Ananth, C.V.; Smulian, J.C.; Peltier, M.R. Can sulfasalazine prevent infection-mediated pre-term birth in a murine model? Am. J. Reprod. Immunol. 2010, 63, 144–149. [Google Scholar] [CrossRef] [PubMed]
- Bertucci, M.C.; Loose, J.M.; Wallace, E.M.; Jenkin, G.; Miller, S.L. Anti-inflammatory therapy in an ovine model of fetal hypoxia induced by single umbilical artery ligation. Reprod. Fertil. Dev. 2011, 23, 346–352. [Google Scholar] [CrossRef]
- O’Neill, L.A.J.; Kaltschmidt, C. NF-kB: A crucial transcription factor for glial and neuronal cell function. Trends Neurosci. 1997, 20, 252–258. [Google Scholar] [CrossRef]
- Philippe, O.; Rio, M.; Malan, V.; Van Esch, H.; Baujat, G.; Bahi-Buisson, N.; Valayannopoulos, V.; Gesny, R.; Bonnefont, J.P.; Munnich, A.; et al. NF-kappaB signalling requirement for brain myelin formation is shown by genotype/MRI phenotype correlations in patients with Xq28 duplications. Eur. J. Hum. Genet. 2013, 21, 195–199. [Google Scholar] [CrossRef] [Green Version]
- Leviton, A.; Kuban, K.C.; Allred, E.N.; Fichorova, R.N.; O’Shea, T.M.; Paneth, N.; Investigators, E.S. Early postnatal blood concentrations of inflammation-related proteins and microcephaly two years later in infants born before the 28th post-menstrual week. Early Hum. Dev. 2011, 87, 325–330. [Google Scholar] [CrossRef]
- Kogo, J.; Takeba, Y.; Kumai, T.; Kitaoka, Y.; Matsumoto, N.; Ueno, S.; Kobayashi, S. Involvement of TNF-alpha in glutamate-induced apoptosis in a differentiated neuronal cell line. Brain Res. 2006, 1122, 201–208. [Google Scholar] [CrossRef]
- Gilmore, J.H.; Fredrik Jarskog, L.; Vadlamudi, S.; Lauder, J.M. Prenatal infection and risk for schizophrenia: IL-1beta, IL-6, and TNFalpha inhibit cortical neuron dendrite development. Neuropsychopharmacology 2004, 29, 1221–1229. [Google Scholar] [CrossRef] [PubMed]
- Favrais, G.; van de Looij, Y.; Fleiss, B.; Ramanantsoa, N.; Bonnin, P.; Stoltenburg-Didinger, G.; Lacaud, A.; Saliba, E.; Dammann, O.; Gallego, J.; et al. Systemic inflammation disrupts the developmental program of white matter. Ann. Neurol. 2011, 70, 550–565. [Google Scholar] [CrossRef] [PubMed]
- Cai, Z.; Lin, S.; Pang, Y.; Rhodes, P.G. Brain injury induced by intracerebral injection of interleukin-1beta and tumor necrosis factor-alpha in the neonatal rat. Pediatr. Res. 2004, 56, 377–384. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tohmi, M.; Tsuda, N.; Zheng, Y.; Mizuno, M.; Sotoyama, H.; Shibuya, M.; Kawamura, M.; Kakita, A.; Takahashi, H.; Nawa, H. The cellular and behavioral consequences of interleukin-1 alpha penetration through the blood-brain barrier of neonatal rats: A critical period for efficacy. Neuroscience 2007, 150, 234–250. [Google Scholar] [CrossRef] [PubMed]
- Deverman, B.E.; Patterson, P.H. Cytokines and CNS development. Neuron 2009, 64, 61–78. [Google Scholar] [CrossRef] [Green Version]
- Leviton, A.; Hecht, J.L.; Allred, E.N.; Yamamoto, H.; Fichorova, R.N.; Dammann, O.; Investigators, E.S. Persistence after birth of systemic inflammation associated with umbilical cord inflammation. J. Reprod. Immunol. 2011, 90, 235–243. [Google Scholar] [CrossRef]
- O’Shea, T.M.; Shah, B.; Allred, E.N.; Fichorova, R.N.; Kuban, K.C.K.; Dammann, O.; Leviton, A.; Investigators, E.S. Inflammation-initiating illnesses, inflammation-related proteins, and cognitive impairment in extremely preterm infants. Brain Behav. Immun. 2013, 29, 104–112. [Google Scholar] [CrossRef] [Green Version]
- Kuban, K.C.K.; Joseph, R.M.; O’Shea, T.M.; Heeren, T.; Fichorova, R.N.; Douglass, L.; Jara, H.; Frazier, J.A.; Hirtz, D.; Rollins, J.V.; et al. Circulating Inflammatory-Associated Proteins in the First Month of Life and Cognitive Impairment at Age 10 Years in Children Born Extremely Preterm. J. Pediatr. 2017, 180, 116–123. [Google Scholar] [CrossRef] [Green Version]
- Sinha, A.; Patient, C. Rheumatoid arthritis in pregnancy: Successful outcome with anti-TNF agent (Etanercept). J. Obs. Gynaecol. 2006, 26, 689–691. [Google Scholar] [CrossRef]
- Otermin, I.; Elizondo, G.; Zabaleta, J.; Amigot, A. Etanercept y embarazo. An. Del Sist. Sanit. De Navar. 2007, 30, 491–493. [Google Scholar] [CrossRef]
- Berthelsen, B.G.; Fjeldsoe-Nielsen, H.; Nielsen, C.T.; Hellmuth, E. Etanercept concentrations in maternal serum, umbilical cord serum, breast milk and child serum during breastfeeding. Rheumatology 2010, 49, 2225–2227. [Google Scholar] [CrossRef] [Green Version]
- Diav-Citrin, O.; Otcheretianski-Volodarsky, A.; Shechtman, S.; Ornoy, A. Pregnancy outcome following gestational exposure to TNF-alpha-inhibitors: A prospective, comparative, observational study. Reprod. Toxicol. 2014, 43, 78–84. [Google Scholar] [CrossRef]
- Roux, C.H.; Brocq, O.; Breuil, V.; Albert, C.; Euller-Ziegler, L. Pregnancy in rheumatology patients exposed to anti-tumour necrosis factor (TNF)-alpha therapy. Rheumatology 2007, 46, 695–698. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chambers, C.; Johnson, D.; Jones, K. the OTIS Collaborative Research Group: Pregnancy outcome in women exposed to anti-TNF-alpha medications: The OTIS Rheumatoid Arthritis in Pregnancy Study. Dermatology 2005, 152, 205. [Google Scholar]
- Verstappen, S.M.; King, Y.; Watson, K.D.; Symmons, D.P.; Hyrich, K.L.; BSRBR Control Centre Consortium; BSR Biologics Register. Anti-TNF therapies and pregnancy: Outcome of 130 pregnancies in the British Society for Rheumatology Biologics Register. Ann. Rheum Dis. 2011, 70, 823–826. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berthelot, J.M.; De Bandt, M.; Goupille, P.; Solau-Gervais, E.; Liote, F.; Goeb, V.; Azais, I.; Martin, A.; Pallot-Prades, B.; Maugars, Y.; et al. Exposition to anti-TNF drugs during pregnancy: Outcome of 15 cases and review of the literature. Jt. Bone Spine 2009, 76, 28–34. [Google Scholar] [CrossRef] [PubMed]
- Johansen, C.B.; Jimenez-Solem, E.; Haerskjold, A.; Sand, F.L.; Thomsen, S.F. The Use and Safety of TNF Inhibitors during Pregnancy in Women with Psoriasis: A Review. Int. J. Mol. Sci. 2018, 19, 1349. [Google Scholar] [CrossRef] [Green Version]
- Fidel, P.L., Jr.; Romero, R.; Cutright, J.; Wolf, N.; Gomez, R.; Araneda, H.; Ramirez, M.; Yoon, B.H. Treatment with the interleukin-I receptor antagonist and soluble tumor necrosis factor receptor Fc fusion protein does not prevent endotoxin-induced preterm parturition in mice. J. Soc. Gynecol. Investig. 1997, 4, 22–26. [Google Scholar] [CrossRef]
- Holmgren, C.; Esplin, M.S.; Hamblin, S.; Molenda, M.; Simonsen, S.; Silver, R. Evaluation of the use of anti-TNF-alpha in an LPS-induced murine model. J. Reprod. Immunol. 2008, 78, 134–139. [Google Scholar] [CrossRef] [PubMed]
- Cunningham, M.W.; Jayaram, A.; Deer, E.; Amaral, L.M.; Vaka, V.R.; Ibrahim, T.; Cornelius, D.C.; LaMarca, B. Abstract P1110: Tumor Necrosis Factor Alpha Blockade Improves Natural Killer Cell Activation, Hypertension, and Mitochondrial Oxidative Stress in a Preclinical Rat Model of Preeclampsia. Hypertension 2019, 74, AP1110. [Google Scholar] [CrossRef]
- Chen, B.; Deng, X.; Wang, B.; Liu, H. Etanercept, an inhibitor of TNF-a, prevents propofol-induced neurotoxicity in the developing brain. Int. J. Dev. Neurosci. Off. J. Int. Soc. Dev. Neurosci. 2016, 55, 91–100. [Google Scholar] [CrossRef] [PubMed]
- Shin, S.H.; Kim, E.K.; Lee, K.Y.; Kim, H.S. TNF-alpha antagonist attenuates systemic lipopolysaccharide-induced brain white matter injury in neonatal rats. BMC Neurosci. 2019, 20, 45. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Galinsky, R.; Dhillon, S.K.; Dean, J.M.; Davidson, J.O.; Lear, C.A.; Wassink, G.; Nott, F.; Kelly, S.B.; Fraser, M.; Yuill, C.; et al. Tumor necrosis factor inhibition attenuates white matter gliosis after systemic inflammation in preterm fetal sheep. J. Neuroinflamm. 2020, 17, 92. [Google Scholar] [CrossRef] [PubMed]
- Cai, Z.; Pang, Y.; Lin, S.; Rhodes, P.G. Differential roles of tumor necrosis factor-alpha and interleukin-1 beta in lipopolysaccharide-induced brain injury in the neonatal rat. Brain Res. 2003, 975, 37–47. [Google Scholar] [CrossRef]
- Goshen, I.; Kreisel, T.; Ounallah-Saad, H.; Renbaum, P.; Zalzstein, Y.; Ben-Hur, T.; Levy-Lahad, E.; Yirmiya, R. A dual role for interleukin-1 in hippocampal-dependent memory processes. Psychoneuroendocrinology 2007, 32, 1106–1115. [Google Scholar] [CrossRef]
- Matsuki, T.; Horai, R.; Sudo, K.; Iwakura, Y. IL-1 plays an important role in lipid metabolism by regulating insulin levels under physiological conditions. J. Exp. Med. 2003, 198, 877–888. [Google Scholar] [CrossRef] [Green Version]
- Romero, R.; Mazor, M.; Tartakovsky, B. Systemic administration of interleukin-1 induces preterm parturition in mice. Am. J. Obs. Gynecol. 1991, 165, 969–971. [Google Scholar] [CrossRef]
- Lee, J.Y.; Shin, N.E.; Na, Q.; Dong, J.; Chudnovets, A.; Li, S.; Novak, C.M.; McLane, M.W.; Lei, J.; Burd, I. Exposure to systemic and intrauterine inflammation leads to decreased pup survival via different placental mechanisms. J. Reprod. Immunol. 2019, 133, 52–62. [Google Scholar] [CrossRef]
- Novak, C.M.; Lee, J.Y.; Ozen, M.; Tsimis, M.E.; Kucirka, L.M.; McLane, M.W.; Xie, L.; Kelleher, M.; Xie, H.; Jia, B.; et al. Increased placental T cell trafficking results in adverse neurobehavioral outcomes in offspring exposed to sub-chronic maternal inflammation. Brain Behav. Immun. 2019, 75, 129–136. [Google Scholar] [CrossRef]
- Mertens, M.; Singh, J.A. Anakinra for rheumatoid arthritis. Cochrane Database Syst. Rev. 2009. [Google Scholar] [CrossRef] [PubMed]
- Goldbach-Mansky, R.; Dailey, N.J.; Canna, S.W.; Gelabert, A.; Jones, J.; Rubin, B.I.; Kim, H.J.; Brewer, C.; Zalewski, C.; Wiggs, E.; et al. Neonatal-onset multisystem inflammatory disease responsive to interleukin-1beta inhibition. N. Engl. J. Med. 2006, 355, 581–592. [Google Scholar] [CrossRef] [Green Version]
- Opal, S.M.; Fisher, C.J., Jr.; Dhainaut, J.F.; Vincent, J.L.; Brase, R.; Lowry, S.F.; Sadoff, J.C.; Slotman, G.J.; Levy, H.; Balk, R.A.; et al. Confirmatory interleukin-1 receptor antagonist trial in severe sepsis: A phase III, randomized, double-blind, placebo-controlled, multicenter trial. The Interleukin-1 Receptor Antagonist Sepsis Investigator Group. Crit. Care Med. 1997, 25, 1115–1124. [Google Scholar] [CrossRef]
- Romero, R.; Sepulveda, W.; Mazor, M.; Brandt, F.; Cotton, D.; Dinarello, C.; Mitchell, M. The natural interleukin-1 receptor antagonist in tem and preterm parturition. Am. J. Obstet. Gynecol. 1992, 167, 863–872. [Google Scholar] [CrossRef]
- Romero, R.; Tartakovsky, B. The natural interleukin-1 receptor antagonist prevents interleukin-l-induced preterm delivery in mice. Am. J. Obstet. Gynecol. 1992, 167, 1041–1045. [Google Scholar] [CrossRef]
- Nadeau-Vallee, M.; Chin, P.Y.; Belarbi, L.; Brien, M.E.; Pundir, S.; Berryer, M.H.; Beaudry-Richard, A.; Madaan, A.; Sharkey, D.J.; Lupien-Meilleur, A.; et al. Antenatal Suppression of IL-1 Protects against Inflammation-Induced Fetal Injury and Improves Neonatal and Developmental Outcomes in Mice. J. Immunol. 2017, 198, 2047–2062. [Google Scholar] [CrossRef]
- Girard, S.; Tremblay, L.; Lepage, M.; Sebire, G. IL-1 receptor antagonist protects against placental and neurodevelopmental defects induced by maternal inflammation. J. Immunol. 2010, 184, 3997–4005. [Google Scholar] [CrossRef]
- Girard, S.; Sebire, H.; Brochu, M.E.; Briota, S.; Sarret, P.; Sebire, G. Postnatal administration of IL-1Ra exerts neuroprotective effects following perinatal inflammation and/or hypoxic-ischemic injuries. Brain Behav. Immun. 2012, 26, 1331–1339. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mattucci, K.F.; Levin, W.J.; Habib, M.A. Acute bacterial sinusitis. Minocycline vs amoxicillin. Arch. Otolaryngol. Head Neck Surg. 1986, 112, 73–76. [Google Scholar] [CrossRef] [PubMed]
- Esterly, N.B.; Koransky, J.S.; Furey, N.L.; Trevisan, M. Neutrophil chemotaxis in patients with acne receiving oral tetracycline therapy. Arch. Derm. 1984, 120, 1308–1313. [Google Scholar] [CrossRef]
- Stone, M.; Fortin, P.R.; Pacheco-Tena, C.; Inman, R.D. Should tetracycline treatment be used more extensively for rheumatoid arthritis? Metaanalysis demonstrates clinical benefit with reduction in disease activity. J. Rheumatol. 2003, 30, 2112. [Google Scholar] [PubMed]
- Zabad, R.K.; Metz, L.M.; Todoruk, T.R.; Zhang, Y.; Mitchell, J.R.; Yeung, M.; Patry, D.G.; Bell, R.B.; Yong, V.W. The clinical response to minocycline in multiple sclerosis is accompanied by beneficial immune changes: A pilot study. Mult. Scler. 2007, 13, 517–526. [Google Scholar] [CrossRef] [PubMed]
- Elewa, H.F.; Hilali, H.; Hess, D.C.; Machado, L.S.; Fagan, S.C. Minocycline for short-term neuroprotection. Pharmacotherapy 2006, 26, 515–521. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arvin, K.L.; Han, B.H.; Du, Y.; Lin, S.Z.; Paul, S.M.; Holtzman, D.M. Minocycline markedly protects the neonatal brain against hypoxic-ischemic injury. Ann. Neurol. 2002, 52, 54–61. [Google Scholar] [CrossRef] [PubMed]
- Carty, M.L.; Wixey, J.A.; Colditz, P.B.; Buller, K.M. Post-insult minocycline treatment attenuates hypoxia-ischemia-induced neuroinflammation and white matter injury in the neonatal rat: A comparison of two different dose regimens. Int. J. Dev. Neurosci. Off. J. Int. Soc. Dev. Neurosci. 2008, 26, 477–485. [Google Scholar] [CrossRef] [PubMed]
- Wixey, J.A.; Reinebrant, H.E.; Spencer, S.J.; Buller, K.M. Efficacy of post-insult minocycline administration to alter long-term hypoxia-ischemia-induced damage to the serotonergic system in the immature rat brain. Neuroscience 2011, 182, 184–192. [Google Scholar] [CrossRef] [PubMed]
- Fan, L.W.; Lin, S.; Pang, Y.; Rhodes, P.G.; Cai, Z. Minocycline attenuates hypoxia-ischemia-induced neurological dysfunction and brain injury in the juvenile rat. Eur. J. Neurosci. 2006, 24, 341–350. [Google Scholar] [CrossRef]
- Suk, K. Minocycline suppresses hypoxic activation of rodent microglia in culture. Neurosci. Lett. 2004, 366, 167–171. [Google Scholar] [CrossRef]
- Cai, Z.; Lin, S.; Fan, L.W.; Pang, Y.; Rhodes, P.G. Minocycline alleviates hypoxic–ischemic injury to developing oligodendrocytes in the neonatal rat brain. Neuroscience 2006, 137, 425–435. [Google Scholar] [CrossRef]
- Fan, L.W.; Pang, Y.; Lin, S.; Rhodes, P.G.; Cai, Z. Minocycline attenuates lipopolysaccharide-induced white matter injury in the neonatal rat brain. Neuroscience 2005, 133, 159–168. [Google Scholar] [CrossRef]
- Fan, L.W.; Pang, Y.; Lin, S.; Tien, L.T.; Ma, T.; Rhodes, P.G.; Cai, Z. Minocycline reduces lipopolysaccharide-induced neurological dysfunction and brain injury in the neonatal rat. J. Neurosci. Res. 2005, 82, 71–82. [Google Scholar] [CrossRef] [PubMed]
- Zhu, F.; Zheng, Y.; Liu, Y.; Zhang, X.; Zhao, J. Minocycline alleviates behavioral deficits and inhibits microglial activation in the offspring of pregnant mice after administration of polyriboinosinic-polyribocytidilic acid. Psychiatry Res. 2014, 219, 680–686. [Google Scholar] [CrossRef] [PubMed]
- Zhu, F.; Zheng, Y.; Ding, Y.Q.; Liu, Y.; Zhang, X.; Wu, R.; Guo, X.; Zhao, J. Minocycline and risperidone prevent microglia activation and rescue behavioral deficits induced by neonatal intrahippocampal injection of lipopolysaccharide in rats. PLoS ONE 2014, 9, e93966. [Google Scholar] [CrossRef] [PubMed]
- Tsuji, M.; Wilson, M.A.; Lange, M.S.; Johnston, M.V. Minocycline worsens hypoxic-ischemic brain injury in a neonatal mouse model. Exp. Neurol. 2004, 189, 58–65. [Google Scholar] [CrossRef] [PubMed]
- Strahan, J.A.; Walker, W.H., 2nd; Montgomery, T.R.; Forger, N.G. Minocycline causes widespread cell death and increases microglial labeling in the neonatal mouse brain. Dev. Neurobiol. 2017, 77, 753–766. [Google Scholar] [CrossRef]
- Arnoux, I.; Hoshiko, M.; Sanz Diez, A.; Audinat, E. Paradoxical effects of minocycline in the developing mouse somatosensory cortex. Glia 2014, 62, 399–410. [Google Scholar] [CrossRef]
- Shigemoto-Mogami, Y.; Hoshikawa, K.; Goldman, J.E.; Sekino, Y.; Sato, K. Microglia enhance neurogenesis and oligodendrogenesis in the early postnatal subventricular zone. J. Neurosci. 2014, 34, 2231–2243. [Google Scholar] [CrossRef] [Green Version]
- Karpuk, N.; Burkovetskaya, M.; Fritz, T.; Angle, A.; Kielian, T. Neuroinflammation leads to region-dependent alterations in astrocyte gap junction communication and hemichannel activity. J. Neurosci. 2011, 31, 414–425. [Google Scholar] [CrossRef] [Green Version]
- O’Carroll, S.J.; Alkadhi, M.; Nicholson, L.F.; Green, C.R. Connexin 43 mimetic peptides reduce swelling, astrogliosis, and neuronal cell death after spinal cord injury. Cell Commun. Adhes. 2008, 15, 27–42. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, Y.; Griffin, J.M.; Harris, P.W.; Chan, S.H.; Nicholson, L.F.; Brimble, M.A.; O’Carroll, S.J.; Green, C.R. Characterizing the mode of action of extracellular Connexin43 channel blocking mimetic peptides in an in vitro ischemia injury model. Biochim. Biophys. Acta Gen. Subj. 2017, 1861, 68–78. [Google Scholar] [CrossRef]
- Galinsky, R.; Davidson, J.O.; Lear, C.A.; Bennet, L.; Green, C.R.; Gunn, A.J. Connexin hemichannel blockade improves survival of striatal GABA-ergic neurons after global cerebral ischaemia in term-equivalent fetal sheep. Sci. Rep. 2017, 7, 6304. [Google Scholar] [CrossRef] [PubMed]
- Davidson, J.O.; Green, C.R.; Nicholson, L.F.B.; Bennet, L.; Gunn, A.J. Connexin hemichannel blockade is neuroprotective after, but not during, global cerebral ischemia in near-term fetal sheep. Exp. Neurol. 2013, 248, 301–308. [Google Scholar] [CrossRef] [PubMed]
- Davidson, J.O.; Green, C.R.; Nicholson, L.F.; O’Carroll, S.J.; Fraser, M.; Bennet, L.; Gunn, A.J. Connexin hemichannel blockade improves outcomes in a model of fetal ischemia. Ann. Neurol. 2012, 71, 121–132. [Google Scholar] [CrossRef] [PubMed]
- Yang, P.; Davidson, J.O.; Fowke, T.M.; Galinsky, R.; Wassink, G.; Karunasinghe, R.N.; Prasad, J.D.; Ranasinghe, S.; Green, C.R.; Bennet, L.; et al. Connexin Hemichannel Mimetic Peptide Attenuates Cortical Interneuron Loss and Perineuronal Net Disruption Following Cerebral Ischemia in Near-Term Fetal Sheep. Int. J. Mol. Sci. 2020, 21, 6475. [Google Scholar] [CrossRef]
- Davidson, J.O.; Drury, P.P.; Green, C.R.; Nicholson, L.F.; Bennet, L.; Gunn, A.J. Connexin hemichannel blockade is neuroprotective after asphyxia in preterm fetal sheep. PLoS ONE 2014, 9, e96558. [Google Scholar] [CrossRef]
- Davidson, J.O.; Green, C.R.; Nicholson, L.F.; Bennet, L.; Gunn, A.J. Deleterious effects of high dose connexin 43 mimetic peptide infusion after cerebral ischaemia in near-term fetal sheep. Int. J. Mol. Sci. 2012, 13, 6303–6319. [Google Scholar] [CrossRef] [Green Version]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Prasad, J.D.; Gunn, K.C.; Davidson, J.O.; Galinsky, R.; Graham, S.E.; Berry, M.J.; Bennet, L.; Gunn, A.J.; Dean, J.M. Anti-Inflammatory Therapies for Treatment of Inflammation-Related Preterm Brain Injury. Int. J. Mol. Sci. 2021, 22, 4008. https://doi.org/10.3390/ijms22084008
Prasad JD, Gunn KC, Davidson JO, Galinsky R, Graham SE, Berry MJ, Bennet L, Gunn AJ, Dean JM. Anti-Inflammatory Therapies for Treatment of Inflammation-Related Preterm Brain Injury. International Journal of Molecular Sciences. 2021; 22(8):4008. https://doi.org/10.3390/ijms22084008
Chicago/Turabian StylePrasad, Jaya D., Katherine C. Gunn, Joanne O. Davidson, Robert Galinsky, Scott E. Graham, Mary J. Berry, Laura Bennet, Alistair J. Gunn, and Justin M. Dean. 2021. "Anti-Inflammatory Therapies for Treatment of Inflammation-Related Preterm Brain Injury" International Journal of Molecular Sciences 22, no. 8: 4008. https://doi.org/10.3390/ijms22084008