
Disease-Induced Modulation of Drug Transporters at the Blood–Brain Barrier Level

 , , ,
, , ,  and
and 
Abstract
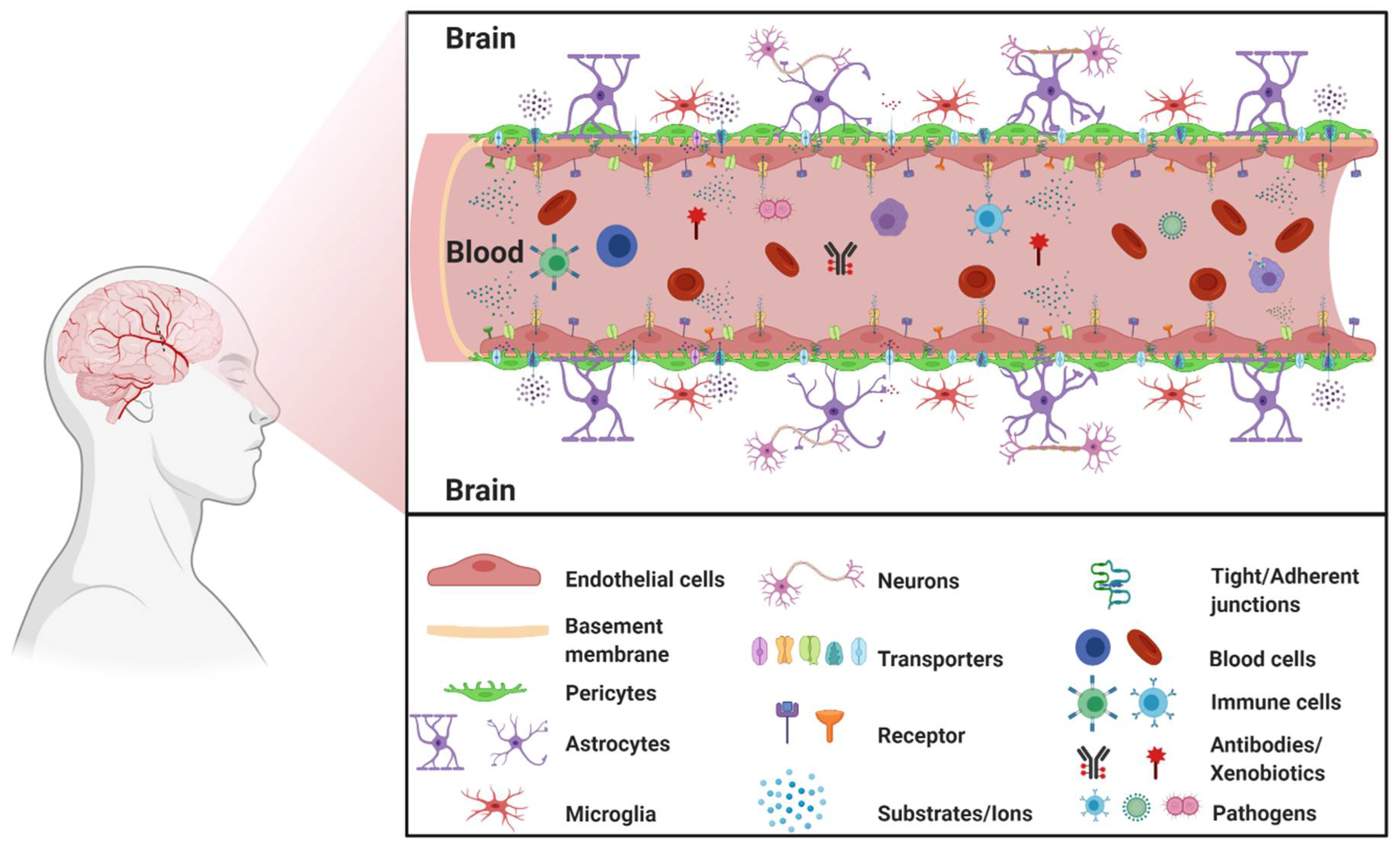
:1. The Blood–Brain Barrier
1.1. BBB Facts and Figures
1.2. The Neurovascular Unit
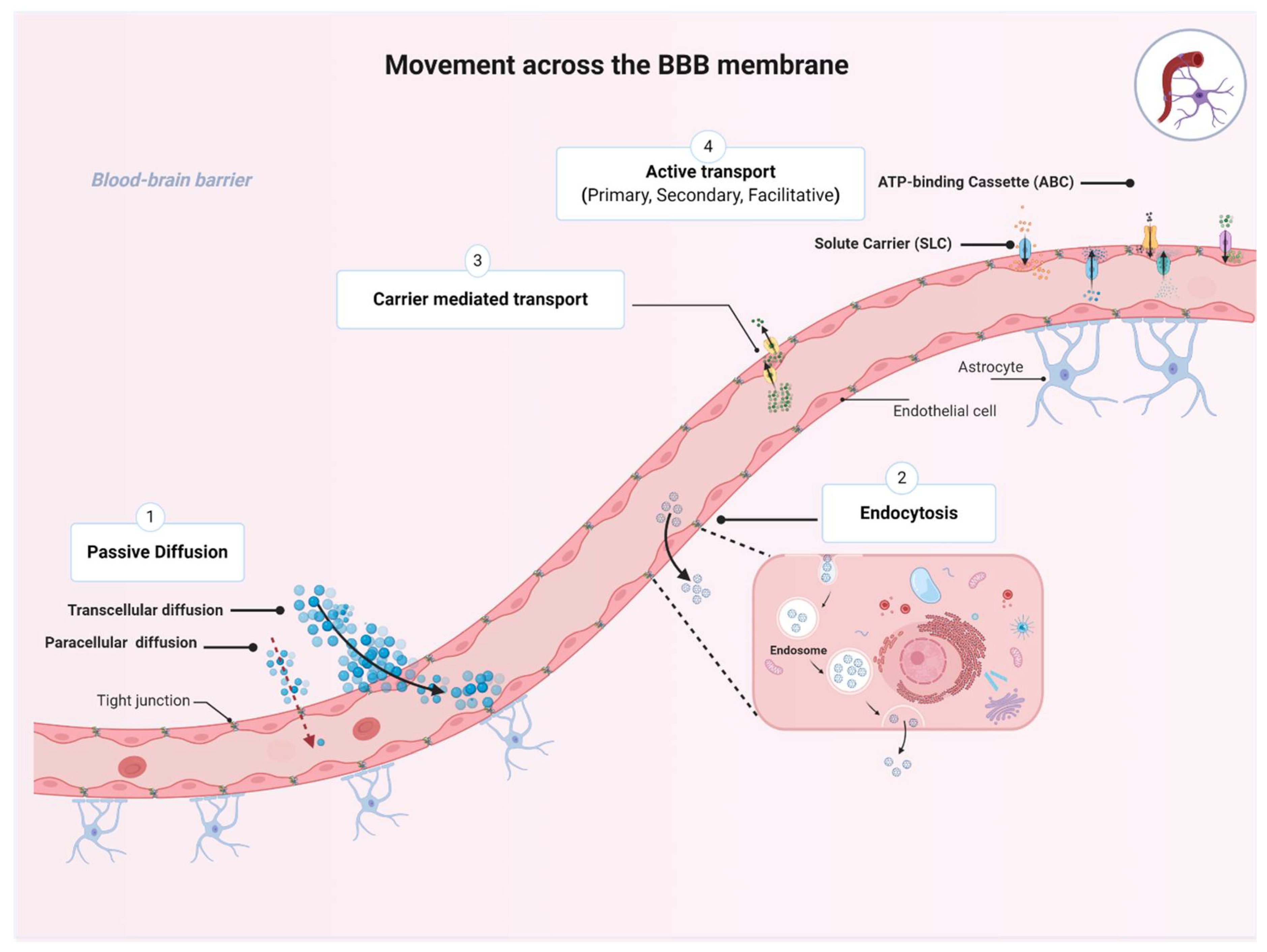
1.3. Movement across the BBB
2. Transporters Expressed at the BBB
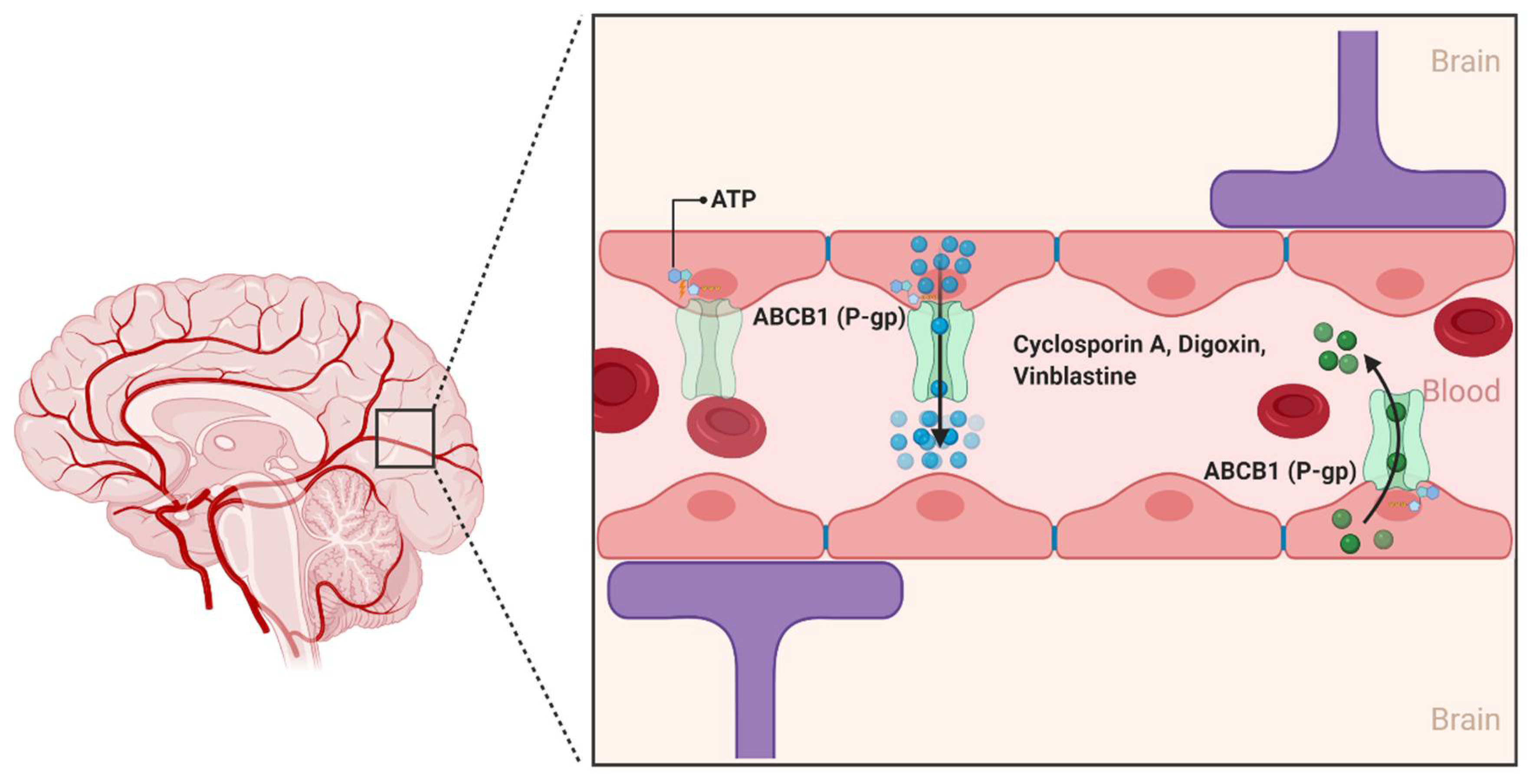
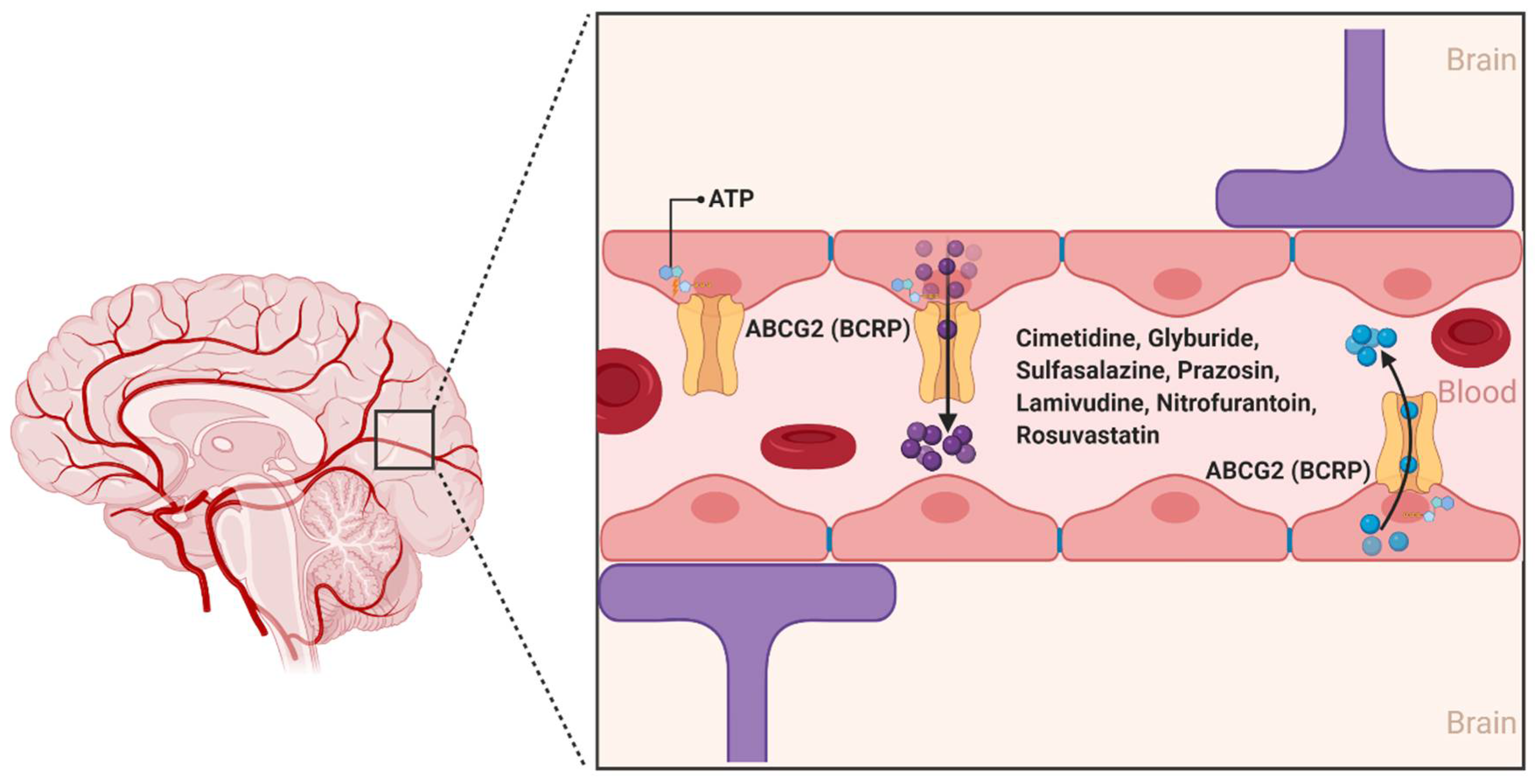
2.1. ATP-Binding Cassette (ABC) Transporters
2.1.1. ABCB1
2.1.2. ABCG2
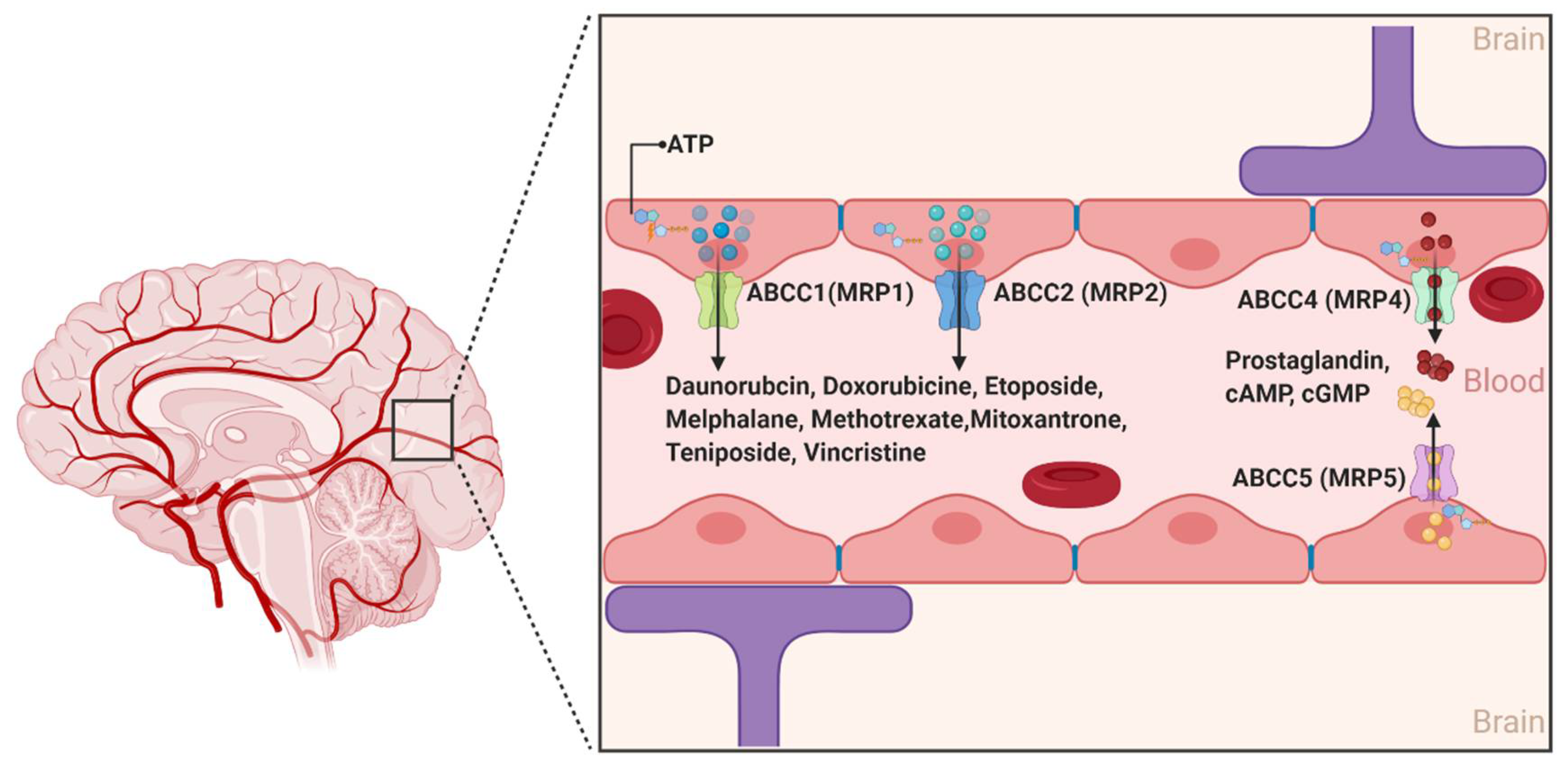
2.1.3. ABCCs
2.2. The Solute Carrier (SLCs) Superfamily
2.2.1. Organic Anion Transporting Polypeptides (SLCOs and Formally OATPs)
2.2.2. Organic Anion Transporters (SLC22s Formally OATs)
2.2.3. Organic Cation Transporters (SLC22s Formally OCTs)
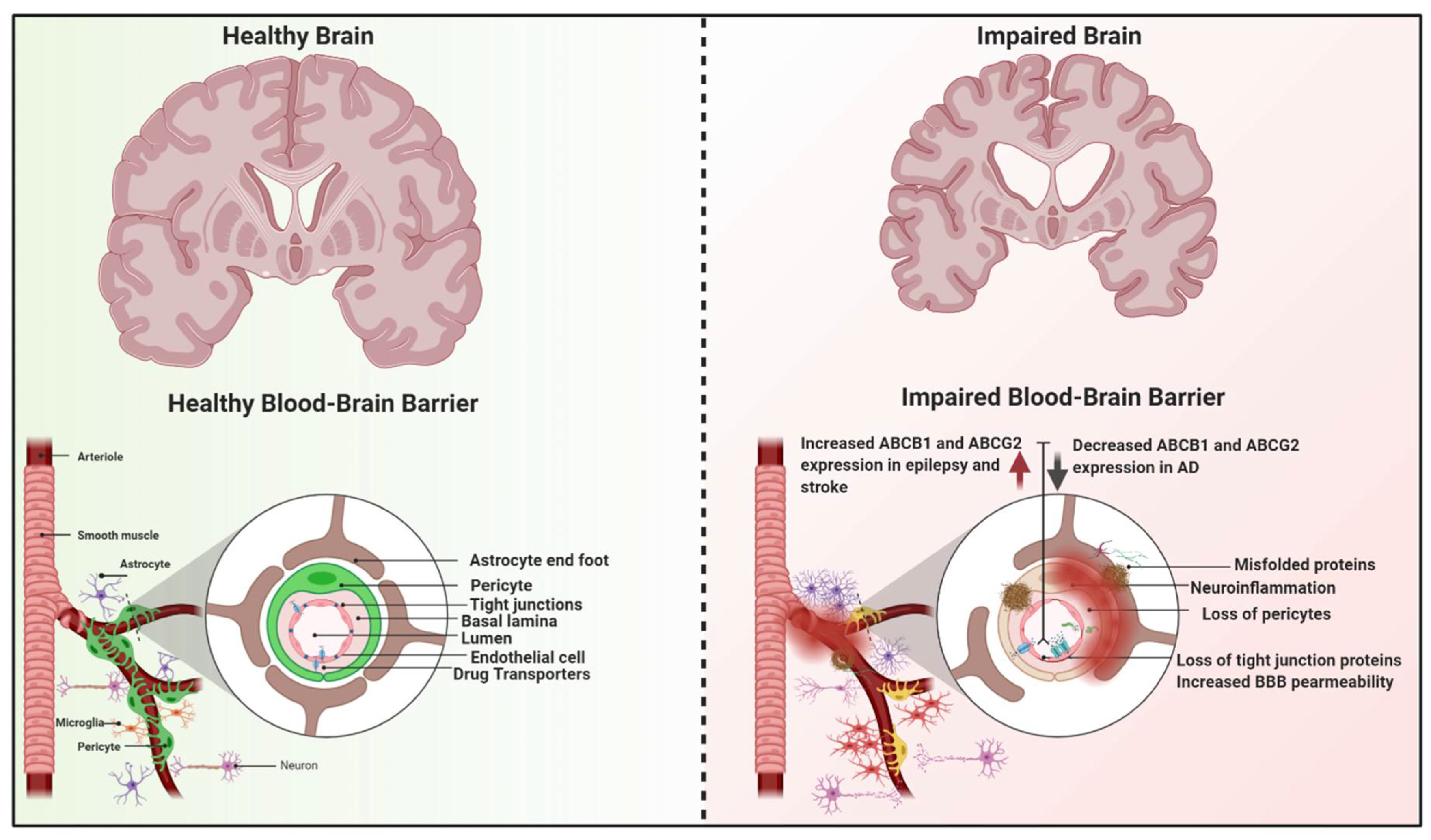
3. Dysfunction of the BBB Transporters in Neurological Diseases
3.1. Neuroinflammation and BBB
3.2. Alzheimer’s Disease (AD)
3.3. Epilepsy
3.4. Stroke
3.5. Amyotrophic Lateral Sclerosis (ALS)
3.6. Multiple Sclerosis
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Azevedo, F.A.; Carvalho, L.R.; Grinberg, L.T.; Farfel, J.M.; Ferretti, R.E.; Leite, R.E.; Jacob Filho, W.; Lent, R.; Herculano-Houzel, S. Equal numbers of neuronal and nonneuronal cells make the human brain an isometrically scaled-up primate brain. J. Comp. Neurol. 2009, 513, 532–541. [Google Scholar] [CrossRef]
- Hossmann, K.A. Pathophysiology and therapy of experimental stroke. Cell Mol. Neurobiol. 2006, 26, 1057–1083. [Google Scholar] [CrossRef]
- Chandra, A.; Li, W.A.; Stone, C.R.; Geng, X.; Ding, Y. The cerebral circulation and cerebrovascular disease I: Anatomy. Brain Circ. 2017, 3, 45–56. [Google Scholar] [CrossRef] [PubMed]
- Raichle, M.E.; Gusnard, D.A. Appraising the brain’s energy budget. Proc. Natl. Acad. Sci. USA 2002, 99, 10237–10239. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cipolla, M.J. The Cerebral Circulation; Morgan & Claypool Life Sciences: San Rafael, CA, USA, 2009.
- Wong, A.D.; Ye, M.; Levy, A.F.; Rothstein, J.D.; Bergles, D.E.; Searson, P.C. The blood-brain barrier: An engineering perspective. Front. Neuroeng. 2013, 6, 7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cohen, Z.; Bonvento, G.; Lacombe, P.; Hamel, E. Serotonin in the regulation of brain microcirculation. Prog. Neurobiol. 1996, 50, 335–362. [Google Scholar] [CrossRef]
- Hawkins, B.T.; Davis, T.P. The blood-brain barrier/neurovascular unit in health and disease. Pharmacol. Rev. 2005, 57, 173–185. [Google Scholar] [CrossRef] [PubMed]
- Grotta, J.C. Stroke Progress Review Group. Stroke 2013, 44, S111–S113. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- National Institute of Neurological Disorders and Stroke. Stroke Progress Review Group. Available online: https://www.ninds.nih.gov/About-NINDS/Strategic-Plans-Evaluations/Strategic-Plans/Stroke-Progress-Review-Group (accessed on 29 January 2021).
- Muoio, V.; Persson, P.B.; Sendeski, M.M. The neurovascular unit—Concept review. Acta Physiol. 2014, 210, 790–798. [Google Scholar] [CrossRef]
- Jullienne, A.; Badaut, J. Molecular contributions to neurovascular unit dysfunctions after brain injuries: Lessons for target-specific drug development. Future Neurol. 2013, 8, 677–689. [Google Scholar] [CrossRef] [Green Version]
- Pardridge, W.M. Drug transport across the blood-brain barrier. J. Cereb. Blood Flow Metab. 2012, 32, 1959–1972. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Covarrubias, L.; Slosky, L.M.; Thompson, B.J.; Davis, T.P.; Ronaldson, P.T. Transporters at CNS barrier sites: Obstacles or opportunities for drug delivery? Curr. Pharm. Des. 2014, 20, 1422–1449. [Google Scholar] [CrossRef] [Green Version]
- Banks, W.A. From blood-brain barrier to blood-brain interface: New opportunities for CNS drug delivery. Nat. Rev. Drug Discov. 2016, 15, 275–292. [Google Scholar] [CrossRef] [PubMed]
- Laterra, J.; Keep, R.; Betz, L.A.; Goldstein, G. Blood-Brain Barrier. In Basic Neurochemistry: Molecular, Cellular and Medical Aspects, 6th ed.; Siegel, G.J., Agranoff, B.W., Albers, R.W., Fisher, S.K., Uhler, M.D., Eds.; Lippincott-Raven: Philadelphia, PA, USA, 1999. [Google Scholar]
- Wang, Y.; Tajkhorshid, E. Nitric oxide conduction by the brain aquaporin AQP4. Proteins 2010, 78, 661–670. [Google Scholar] [CrossRef] [Green Version]
- Herve, F.; Ghinea, N.; Scherrmann, J.M. CNS delivery via adsorptive transcytosis. AAPS J. 2008, 10, 455–472. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- King, G.L.; Johnson, S.M. Receptor-mediated transport of insulin across endothelial cells. Science 1985, 227, 1583–1586. [Google Scholar] [CrossRef]
- Fishman, J.B.; Rubin, J.B.; Handrahan, J.V.; Connor, J.R.; Fine, R.E. Receptor-mediated transcytosis of transferrin across the blood-brain barrier. J. Neurosci. Res. 1987, 18, 299–304. [Google Scholar] [CrossRef] [PubMed]
- Dickens, D.; Radisch, S.; Pirmohamed, M. Chapter 5 Drug Transporters at the Blood–Brain Barrier. In Drug Transporters: Volume 1: Role and Importance in ADME and Drug Development; The Royal Society of Chemistry: London, UK, 2016; Volume 1, pp. 151–183. [Google Scholar]
- Abbott, N.J.; Ronnback, L.; Hansson, E. Astrocyte-endothelial interactions at the blood-brain barrier. Nat. Rev. Neurosci. 2006, 7, 41–53. [Google Scholar] [CrossRef]
- Pan, Y.; Nicolazzo, J.A. Impact of aging, Alzheimer’s disease and Parkinson’s disease on the blood-brain barrier transport of therapeutics. Adv. Drug Deliv. Rev. 2018, 135, 62–74. [Google Scholar] [CrossRef] [PubMed]
- Harilal, S.; Jose, J.; Parambi, D.G.T.; Kumar, R.; Unnikrishnan, M.K.; Uddin, M.S.; Mathew, G.E.; Pratap, R.; Marathakam, A.; Mathew, B. Revisiting the blood-brain barrier: A hard nut to crack in the transportation of drug molecules. Brain Res. Bull. 2020, 160, 121–140. [Google Scholar] [CrossRef] [PubMed]
- Ye, Z.; Lu, Y.; Wu, T. The impact of ATP-binding cassette transporters on metabolic diseases. Nutr. Metab. 2020, 17, 61. [Google Scholar] [CrossRef]
- Dean, M.; Rzhetsky, A.; Allikmets, R. The human ATP-binding cassette (ABC) transporter superfamily. Genome Res. 2001, 11, 1156–1166. [Google Scholar] [CrossRef]
- Dalvi, S.; On, N.; Nguyen, H.; Pogorzelec, M.; Miller, D.W.; Hatch, G.M. The blood brain barrier—Regulation of fatty acid and drug transport. In Neurochemistry; IntechOpen: London, UK, 2014; pp. 1–33. [Google Scholar]
- Mahringer, A.; Fricker, G. ABC transporters at the blood-brain barrier. Expert Opin. Drug Metab. Toxicol. 2016, 12, 499–508. [Google Scholar] [CrossRef]
- Gil-Martins, E.; Barbosa, D.J.; Silva, V.; Remiao, F.; Silva, R. Dysfunction of ABC transporters at the blood-brain barrier: Role in neurological disorders. Pharmacol. Ther. 2020, 213, 107554. [Google Scholar] [CrossRef]
- Ling, V.; Thompson, L.H. Reduced permeability in CHO cells as a mechanism of resistance to colchicine. J. Cell Physiol. 1974, 83, 103–116. [Google Scholar] [CrossRef]
- Abdullahi, W.; Davis, T.P.; Ronaldson, P.T. Functional Expression of P-glycoprotein and Organic Anion Transporting Polypeptides at the Blood-Brain Barrier: Understanding Transport Mechanisms for Improved CNS Drug Delivery? AAPS J. 2017, 19, 931–939. [Google Scholar] [CrossRef] [Green Version]
- Schinkel, A.H. P-Glycoprotein, a gatekeeper in the blood-brain barrier. Adv. Drug Deliv. Rev. 1999, 36, 179–194. [Google Scholar] [CrossRef]
- Demeule, M.; Regina, A.; Jodoin, J.; Laplante, A.; Dagenais, C.; Berthelet, F.; Moghrabi, A.; Beliveau, R. Drug transport to the brain: Key roles for the efflux pump P-glycoprotein in the blood-brain barrier. Vascul. Pharmacol. 2002, 38, 339–348. [Google Scholar] [CrossRef]
- Robberecht, W.; Philips, T. The changing scene of amyotrophic lateral sclerosis. Nat. Rev. Neurosci. 2013, 14, 248–264. [Google Scholar] [CrossRef] [PubMed]
- Mohamed, L.A.; Markandaiah, S.; Bonanno, S.; Pasinelli, P.; Trotti, D. Blood-Brain Barrier Driven Pharmacoresistance in Amyotrophic Lateral Sclerosis and Challenges for Effective Drug Therapies. AAPS J. 2017, 19, 1600–1614. [Google Scholar] [CrossRef] [PubMed]
- Jablonski, M.R.; Markandaiah, S.S.; Jacob, D.; Meng, N.J.; Li, K.; Gennaro, V.; Lepore, A.C.; Trotti, D.; Pasinelli, P. Inhibiting drug efflux transporters improves efficacy of ALS therapeutics. Ann. Clin. Transl. Neurol. 2014, 1, 996–1005. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chiu, C.; Miller, M.C.; Monahan, R.; Osgood, D.P.; Stopa, E.G.; Silverberg, G.D. P-glycoprotein expression and amyloid accumulation in human aging and Alzheimer’s disease: Preliminary observations. Neurobiol. Aging 2015, 36, 2475–2482. [Google Scholar] [CrossRef]
- Van Assema, D.M.; Lubberink, M.; Rizzu, P.; van Swieten, J.C.; Schuit, R.C.; Eriksson, J.; Scheltens, P.; Koepp, M.; Lammertsma, A.A.; van Berckel, B.N. Blood-brain barrier P-glycoprotein function in healthy subjects and Alzheimer’s disease patients: Effect of polymorphisms in the ABCB1 gene. EJNMMI Res. 2012, 2, 57. [Google Scholar] [CrossRef] [PubMed]
- Zhong, X.; Liu, M.Y.; Sun, X.H.; Wei, M.J. Association between ABCB1 polymorphisms and haplotypes and Alzheimer’s disease: A meta-analysis. Sci. Rep. 2016, 6, 32708. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.N.; Mickley, L.A.; Schwartz, A.M.; Acton, E.M.; Hwang, J.L.; Fojo, A.T. Characterization of adriamycin-resistant human breast cancer cells which display overexpression of a novel resistance-related membrane protein. J. Biol. Chem. 1990, 265, 10073–10080. [Google Scholar] [CrossRef]
- Doyle, L.A.; Yang, W.; Abruzzo, L.V.; Krogmann, T.; Gao, Y.; Rishi, A.K.; Ross, D.D. A multidrug resistance transporter from human MCF-7 breast cancer cells. Proc. Natl. Acad. Sci. USA 1998, 95, 15665–15670. [Google Scholar] [CrossRef] [Green Version]
- Nicolazzo, J.A.; Katneni, K. Drug transport across the blood-brain barrier and the impact of breast cancer resistance protein (ABCG2). Curr. Top. Med. Chem. 2009, 9, 130–147. [Google Scholar] [CrossRef]
- Mao, Q.; Unadkat, J.D. Role of the breast cancer resistance protein (ABCG2) in drug transport. AAPS J. 2005, 7, E118–E133. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mao, Q.; Unadkat, J.D. Role of the breast cancer resistance protein (BCRP/ABCG2) in drug transport—An update. AAPS J. 2015, 17, 65–82. [Google Scholar] [CrossRef] [Green Version]
- Robey, R.W.; Steadman, K.; Polgar, O.; Bates, S.E. ABCG2-mediated transport of photosensitizers: Potential impact on photodynamic therapy. Cancer Biol. Ther. 2005, 4, 187–194. [Google Scholar] [CrossRef] [Green Version]
- Kruh, G.D.; Belinsky, M.G. The MRP family of drug efflux pumps. Oncogene 2003, 22, 7537–7552. [Google Scholar] [CrossRef] [PubMed]
- Miller, D.S. Regulation of ABC transporters at the blood-brain barrier. Clin. Pharmacol. Ther. 2015, 97, 395–403. [Google Scholar] [CrossRef] [PubMed]
- Cole, S.P.; Sparks, K.E.; Fraser, K.; Loe, D.W.; Grant, C.E.; Wilson, G.M.; Deeley, R.G. Pharmacological characterization of multidrug resistant MRP-transfected human tumor cells. Cancer Res. 1994, 54, 5902–5910. [Google Scholar] [PubMed]
- Dallas, S.; Miller, D.S.; Bendayan, R. Multidrug resistance-associated proteins: Expression and function in the central nervous system. Pharmacol. Rev. 2006, 58, 140–161. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Su, W.; Pasternak, G.W. The role of multidrug resistance-associated protein in the blood-brain barrier and opioid analgesia. Synapse 2013, 67, 609–619. [Google Scholar] [CrossRef] [Green Version]
- Graff, C.L.; Pollack, G.M. Drug transport at the blood-brain barrier and the choroid plexus. Curr. Drug Metab. 2004, 5, 95–108. [Google Scholar] [CrossRef]
- Lingineni, K.; Belekar, V.; Tangadpalliwar, S.R.; Garg, P. The role of multidrug resistance protein (MRP-1) as an active efflux transporter on blood-brain barrier (BBB) permeability. Mol. Divers. 2017, 21, 355–365. [Google Scholar] [CrossRef]
- Choudhuri, S.; Klaassen, C.D. Structure, function, expression, genomic organization, and single nucleotide polymorphisms of human ABCB1 (MDR1), ABCC (MRP), and ABCG2 (BCRP) efflux transporters. Int. J. Toxicol. 2006, 25, 231–259. [Google Scholar] [CrossRef]
- Cole, S.P.; Deeley, R.G. Multidrug resistance mediated by the ATP-binding cassette transporter protein MRP. Bioessays 1998, 20, 931–940. [Google Scholar] [CrossRef]
- Reid, G.; Wielinga, P.; Zelcer, N.; van der Heijden, I.; Kuil, A.; de Haas, M.; Wijnholds, J.; Borst, P. The human multidrug resistance protein MRP4 functions as a prostaglandin efflux transporter and is inhibited by nonsteroidal antiinflammatory drugs. Proc. Natl. Acad. Sci. USA 2003, 100, 9244–9249. [Google Scholar] [CrossRef] [Green Version]
- Jedlitschky, G.; Burchell, B.; Keppler, D. The multidrug resistance protein 5 functions as an ATP-dependent export pump for cyclic nucleotides. J. Biol. Chem. 2000, 275, 30069–30074. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stieger, B.; Gao, B. Drug transporters in the central nervous system. Clin. Pharmacokinet. 2015, 54, 225–242. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- BioParadigms. SLC Tables. Available online: http://slc.bioparadigms.org/ (accessed on 29 January 2021).
- Kusuhara, H.; Sugiyama, Y. Active efflux across the blood-brain barrier: Role of the solute carrier family. NeuroRx 2005, 2, 73–85. [Google Scholar] [CrossRef] [PubMed]
- Morris, M.E.; Rodriguez-Cruz, V.; Felmlee, M.A. SLC and ABC Transporters: Expression, Localization, and Species Differences at the Blood-Brain and the Blood-Cerebrospinal Fluid Barriers. AAPS J. 2017, 19, 1317–1331. [Google Scholar] [CrossRef]
- Hu, C.; Tao, L.; Cao, X.; Chen, L. The solute carrier transporters and the brain: Physiological and pharmacological implications. Asian J. Pharm. Sci. 2020, 15, 131–144. [Google Scholar] [CrossRef]
- Ferreira, G.C.; McKenna, M.C. L-Carnitine and Acetyl-L-carnitine Roles and Neuroprotection in Developing Brain. NeuroChem. Res. 2017, 42, 1661–1675. [Google Scholar] [CrossRef]
- Roth, M.; Obaidat, A.; Hagenbuch, B. OATPs, OATs and OCTs: The organic anion and cation transporters of the SLCO and SLC22A gene superfamilies. Br. J. Pharmacol. 2012, 165, 1260–1287. [Google Scholar] [CrossRef] [Green Version]
- Hagenbuch, B.; Stieger, B. The SLCO (former SLC21) superfamily of transporters. Mol. Aspects Med. 2013, 34, 396–412. [Google Scholar] [CrossRef] [Green Version]
- Meier-Abt, F.; Mokrab, Y.; Mizuguchi, K. Organic anion transporting polypeptides of the OATP/SLCO superfamily: Identification of new members in nonmammalian species, comparative modeling and a potential transport mode. J. Membr. Biol. 2005, 208, 213–227. [Google Scholar] [CrossRef]
- Thompson, B.J.; Sanchez-Covarrubias, L.; Slosky, L.M.; Zhang, Y.; Laracuente, M.L.; Ronaldson, P.T. Hypoxia/reoxygenation stress signals an increase in organic anion transporting polypeptide 1a4 (Oatp1a4) at the blood-brain barrier: Relevance to CNS drug delivery. J. Cereb. Blood Flow Metab. 2014, 34, 699–707. [Google Scholar] [CrossRef]
- Ronaldson, P.T.; Davis, T.P. Targeted drug delivery to treat pain and cerebral hypoxia. Pharmacol. Rev. 2013, 65, 291–314. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brzica, H.; Abdullahi, W.; Reilly, B.G.; Ronaldson, P.T. Sex-specific differences in organic anion transporting polypeptide 1a4 (Oatp1a4) functional expression at the blood-brain barrier in Sprague-Dawley rats. Fluids Barriers CNS 2018, 15, 25. [Google Scholar] [CrossRef] [PubMed]
- Gao, B.; Vavricka, S.R.; Meier, P.J.; Stieger, B. Differential cellular expression of organic anion transporting peptides OATP1A2 and OATP2B1 in the human retina and brain: Implications for carrier-mediated transport of neuropeptides and neurosteriods in the CNS. Pflugers Arch. 2015, 467, 1481–1493. [Google Scholar] [CrossRef] [PubMed]
- Ose, A.; Kusuhara, H.; Endo, C.; Tohyama, K.; Miyajima, M.; Kitamura, S.; Sugiyama, Y. Functional characterization of mouse organic anion transporting peptide 1a4 in the uptake and efflux of drugs across the blood-brain barrier. Drug Metab. Dispos. 2010, 38, 168–176. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burckhardt, G.; Burckhardt, B.C. In vitro and in vivo evidence of the importance of organic anion transporters (OATs) in drug therapy. Handb. Exp. Pharmacol. 2011, 29–104. [Google Scholar] [CrossRef]
- Koepsell, H.; Endou, H. The SLC22 drug transporter family. Pflugers Arch. 2004, 447, 666–676. [Google Scholar] [CrossRef]
- Koepsell, H. The SLC22 family with transporters of organic cations, anions and zwitterions. Mol. Aspects Med. 2013, 34, 413–435. [Google Scholar] [CrossRef]
- Bush, K.T.; Nagle, M.; Truong, D.M.; Bhatnagar, V.; Kaler, G.; Eraly, S.A.; Wu, W.; Nigam, S.K. Chapter 3 Organic Anion Transporters. In Drug Transporters: Molecular Characterization and Role in Drug Disposition, 2nd ed.; You, G., Morris, M.E., Wang, B., Eds.; John Wiley & Sons: Hoboken, NJ, USA, 2017. [Google Scholar]
- Uchida, Y.; Ohtsuki, S.; Katsukura, Y.; Ikeda, C.; Suzuki, T.; Kamiie, J.; Terasaki, T. Quantitative targeted absolute proteomics of human blood-brain barrier transporters and receptors. J. Neurochem. 2011, 117, 333–345. [Google Scholar] [CrossRef]
- Kikuchi, R.; Kusuhara, H.; Sugiyama, D.; Sugiyama, Y. Contribution of Organic Anion Transporter 3 (Slc22a8) to the Elimination of Aminohippuric Acid and Benzylpenicillin across the Blood-Brain Barrier. J. Pharmacol. Exp. Ther. 2003, 306, 51–58. [Google Scholar] [CrossRef] [Green Version]
- Cha, S.H.; Sekine, T.; Kusuhara, H.; Yu, E.; Kim, J.Y.; Kim, D.K.; Sugiyama, Y.; Kanai, Y.; Endou, H. Molecular cloning and characterization of multispecific organic anion transporter 4 expressed in the placenta. J. Biol. Chem. 2000, 275, 4507–4512. [Google Scholar] [CrossRef] [Green Version]
- Kimura, H.; Takeda, M.; Narikawa, S.; Enomoto, A.; Ichida, K.; Endou, H. Human organic anion transporters and human organic cation transporters mediate renal transport of prostaglandins. J. Pharmacol. Exp. Ther. 2002, 301, 293–298. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koepsell, H.; Schmitt, B.M.; Gorboulev, V. Organic cation transporters. Rev. Physiol. Biochem. Pharmacol. 2003, 150, 36–90. [Google Scholar] [CrossRef]
- Apparsundaram, S.; Ferguson, S.M.; George, A.L., Jr.; Blakely, R.D. Molecular cloning of a human, hemicholinium-3-sensitive choline transporter. Biochem. Biophys Res. Commun. 2000, 276, 862–867. [Google Scholar] [CrossRef] [PubMed]
- Blakely, R.D.; De Felice, L.J.; Hartzell, H.C. Molecular physiology of norepinephrine and serotonin transporters. J. Exp. Biol. 1994, 196, 263–281. [Google Scholar] [PubMed]
- Nigam, S.K. The SLC22 Transporter Family: A Paradigm for the Impact of Drug Transporters on Metabolic Pathways, Signaling, and Disease. Annu. Rev. Pharmacol. Toxicol. 2018, 58, 663–687. [Google Scholar] [CrossRef] [PubMed]
- Rindi, G.; Laforenza, U. Thiamine intestinal transport and related issues: Recent aspects. Proc. Soc. Exp. Biol. Med. 2000, 224, 246–255. [Google Scholar] [CrossRef]
- Nies, A.T.; Koepsell, H.; Damme, K.; Schwab, M. Organic cation transporters (OCTs, MATEs), in vitro and in vivo evidence for the importance in drug therapy. Handb. Exp. Pharmacol. 2011, 105–167. [Google Scholar] [CrossRef]
- Dickens, D.; Owen, A.; Alfirevic, A.; Giannoudis, A.; Davies, A.; Weksler, B.; Romero, I.A.; Couraud, P.O.; Pirmohamed, M. Lamotrigine is a substrate for OCT1 in brain endothelial cells. Biochem. Pharmacol. 2012, 83, 805–814. [Google Scholar] [CrossRef]
- Dos Santos Pereira, J.N.; Tadjerpisheh, S.; Abu Abed, M.; Saadatmand, A.R.; Weksler, B.; Romero, I.A.; Couraud, P.O.; Brockmoller, J.; Tzvetkov, M.V. The poorly membrane permeable antipsychotic drugs amisulpride and sulpiride are substrates of the organic cation transporters from the SLC22 family. AAPS J. 2014, 16, 1247–1258. [Google Scholar] [CrossRef] [Green Version]
- Okura, T.; Hattori, A.; Takano, Y.; Sato, T.; Hammarlund-Udenaes, M.; Terasaki, T.; Deguchi, Y. Involvement of the pyrilamine transporter, a putative organic cation transporter, in blood-brain barrier transport of oxycodone. Drug Metab. Dispos. 2008, 36, 2005–2013. [Google Scholar] [CrossRef]
- Kido, Y.; Tamai, I.; Ohnari, A.; Sai, Y.; Kagami, T.; Nezu, J.; Nikaido, H.; Hashimoto, N.; Asano, M.; Tsuji, A. Functional relevance of carnitine transporter OCTN2 to brain distribution of L-carnitine and acetyl-L-carnitine across the blood-brain barrier. J. Neurochem. 2001, 79, 959–969. [Google Scholar] [CrossRef] [PubMed]
- Tamai, I.; Yabuuchi, H.; Nezu, J.; Sai, Y.; Oku, A.; Shimane, M.; Tsuji, A. Cloning and characterization of a novel human pH-dependent organic cation transporter, OCTN1. FEBS Lett. 1997, 419, 107–111. [Google Scholar] [CrossRef] [Green Version]
- Abdullahi, W.; Tripathi, D.; Ronaldson, P.T. Blood-brain barrier dysfunction in ischemic stroke: Targeting tight junctions and transporters for vascular protection. Am. J. Physiol. Cell Physiol. 2018, 315, C343–C356. [Google Scholar] [CrossRef]
- Rosenberg, G.A. Neurological diseases in relation to the blood-brain barrier. J. Cereb. Blood Flow Metab. 2012, 32, 1139–1151. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takeshita, Y.; Ransohoff, R.M. Blood–brain barrier and neurological diseases. Clin. Exp. Neuroimmunol. 2015, 6, 351–361. [Google Scholar] [CrossRef]
- Qosa, H.; Mohamed, L.A.; Alqahtani, S.; Abuasal, B.S.; Hill, R.A.; Kaddoumi, A. Transporters as Drug Targets in Neurological Diseases. Clin. Pharmacol. Ther. 2016, 100, 441–453. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Erickson, M.A.; Dohi, K.; Banks, W.A. Neuroinflammation: A common pathway in CNS diseases as mediated at the blood-brain barrier. Neuroimmunomodulation 2012, 19, 121–130. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shi, L.; Zeng, M.; Fu, B. Temporal Effects of Vascular Endothelial Growth Factor and 3,5-Cyclic Monophosphate on Blood-Brain Barrier Solute Permeability In Vivo. J. Neurosci. Res. 2014, 92. [Google Scholar] [CrossRef] [Green Version]
- Geiseler, S.J.; Morland, C. The Janus Face of VEGF in Stroke. Int. J. Mol. Sci. 2018, 19, 1362. [Google Scholar] [CrossRef] [Green Version]
- Van Tellingen, O.; Yetkin-Arik, B.; de Gooijer, M.C.; Wesseling, P.; Wurdinger, T.; de Vries, H.E. Overcoming the blood-brain tumor barrier for effective glioblastoma treatment. Drug Resist. Updates 2015, 19, 1–12. [Google Scholar] [CrossRef]
- Von Wedel-Parlow, M.; Wolte, P.; Galla, H.J. Regulation of major efflux transporters under inflammatory conditions at the blood-brain barrier in vitro. J. Neurochem. 2009, 111, 111–118. [Google Scholar] [CrossRef]
- Sharma, P.L.; Crumpacker, C.S. Decreased processivity of human immunodeficiency virus type 1 reverse transcriptase (RT) containing didanosine-selected mutation Leu74Val: A comparative analysis of RT variants Leu74Val and lamivudine-selected Met184Val. J. Virol. 1999, 73, 8448–8456. [Google Scholar] [CrossRef] [Green Version]
- Sonar, S.A.; Lal, G. Blood-brain barrier and its function during inflammation and autoimmunity. J. Leukoc. Biol. 2018, 103, 839–853. [Google Scholar] [CrossRef]
- DiSabato, D.J.; Quan, N.; Godbout, J.P. Neuroinflammation: The devil is in the details. J. Neurochem. 2016, 139 (Suppl. S2), 136–153. [Google Scholar] [CrossRef] [Green Version]
- Skrobik, Y.; Leger, C.; Cossette, M.; Michaud, V.; Turgeon, J. Factors predisposing to coma and delirium: Fentanyl and midazolam exposure; CYP3A5, ABCB1, and ABCG2 genetic polymorphisms; and inflammatory factors. Crit. Care Med. 2013, 41, 999–1008. [Google Scholar] [CrossRef]
- Begley, D.J. ABC transporters and the blood-brain barrier. Curr. Pharm. Des. 2004, 10, 1295–1312. [Google Scholar] [CrossRef]
- Bauer, B.; Hartz, A.M.; Miller, D.S. Tumor necrosis factor alpha and endothelin-1 increase P-glycoprotein expression and transport activity at the blood-brain barrier. Mol. Pharmacol. 2007, 71, 667–675. [Google Scholar] [CrossRef] [Green Version]
- Goralski, K.B.; Hartmann, G.; Piquette-Miller, M.; Renton, K.W. Downregulation of mdr1a expression in the brain and liver during CNS inflammation alters the in vivo disposition of digoxin. Br. J. Pharmacol. 2003, 139, 35–48. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Poller, B.; Drewe, J.; Krähenbühl, S.; Huwyler, J.; Gutmann, H. Regulation of BCRP (ABCG2) and P-glycoprotein (ABCB1) by cytokines in a model of the human blood-brain barrier. Cell Mol. Neurobiol. 2010, 30, 63–70. [Google Scholar] [CrossRef] [PubMed]
- Eisenblatter, T.; Galla, H.J. A new multidrug resistance protein at the blood-brain barrier. Biochem. Biophys. Res. Commun. 2002, 293, 1273–1278. [Google Scholar] [CrossRef]
- Makrides, V.; Dolgodilina, E.; Virgintino, D. Blood–Brain Barrier Transporters and Neuroinflammation: Partners in Neuroprotection and in Pathology. In The Blood Brain Barrier and Inflammation; Springer: Berlin/Heidelberg, Germany, 2017; pp. 103–151. [Google Scholar]
- Alzheimer’s Association. What Is Alzheimer’s Disease? Available online: https://www.alz.org/alzheimers-dementia/what-is-alzheimers (accessed on 29 January 2021).
- Kempuraj, D.; Thangavel, R.; Natteru, P.A.; Selvakumar, G.P.; Saeed, D.; Zahoor, H.; Zaheer, S.; Iyer, S.S.; Zaheer, A. Neuroinflammation Induces Neurodegeneration. J. Neurol. Neurosurg. Spine 2016, 1, 1003. [Google Scholar]
- Sweeney, M.D.; Zhao, Z.; Montagne, A.; Nelson, A.R.; Zlokovic, B.V. Blood-Brain Barrier: From Physiology to Disease and Back. Physiol. Rev. 2019, 99, 21–78. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Z.; Nelson, A.R.; Betsholtz, C.; Zlokovic, B.V. Establishment and Dysfunction of the Blood-Brain Barrier. Cell 2015, 163, 1064–1078. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Montagne, A.; Barnes, S.R.; Sweeney, M.D.; Halliday, M.R.; Sagare, A.P.; Zhao, Z.; Toga, A.W.; Jacobs, R.E.; Liu, C.Y.; Amezcua, L.; et al. Blood-brain barrier breakdown in the aging human hippocampus. Neuron 2015, 85, 296–302. [Google Scholar] [CrossRef] [Green Version]
- Ashraf, T.; Kis, O.; Banerjee, N.; Bendayan, R. Drug transporters at brain barriers: Expression and regulation by neurological disorders. Adv. Exp. Med. Biol. 2012, 763, 20–69. [Google Scholar]
- Shibata, M.; Yamada, S.; Kumar, S.R.; Calero, M.; Bading, J.; Frangione, B.; Holtzman, D.M.; Miller, C.A.; Strickland, D.K.; Ghiso, J.; et al. Clearance of Alzheimer’s amyloid-ss(1-40) peptide from brain by LDL receptor-related protein-1 at the blood-brain barrier. J. Clin. Invest. 2000, 106, 1489–1499. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brenn, A.; Grube, M.; Peters, M.; Fischer, A.; Jedlitschky, G.; Kroemer, H.K.; Warzok, R.W.; Vogelgesang, S. Beta-Amyloid Downregulates MDR1-P-Glycoprotein (Abcb1) Expression at the Blood-Brain Barrier in Mice. Int. J. Alzheimers Dis. 2011, 2011, 690121. [Google Scholar] [CrossRef] [Green Version]
- Hendrikse, N.H.; de Vries, E.G.; Eriks-Fluks, L.; van der Graaf, W.T.; Hospers, G.A.; Willemsen, A.T.; Vaalburg, W.; Franssen, E.J. A new in vivo method to study P-glycoprotein transport in tumors and the blood-brain barrier. Cancer Res. 1999, 59, 2411–2416. [Google Scholar]
- Bart, J.; Willemsen, A.T.; Groen, H.J.; van der Graaf, W.T.; Wegman, T.D.; Vaalburg, W.; de Vries, E.G.; Hendrikse, N.H. Quantitative assessment of P-glycoprotein function in the rat blood-brain barrier by distribution volume of [11C]verapamil measured with PET. Neuroimage 2003, 20, 1775–1782. [Google Scholar] [CrossRef]
- Hendrikse, N.H.; Bart, J.; de Vries, E.G.; Groen, H.J.; van der Graaf, W.T.; Vaalburg, W. P-glycoprotein at the blood-brain barrier and analysis of drug transport with positron-emission tomography. J. Clin. Pharmacol. 2001, 41, 48s–54s. [Google Scholar] [CrossRef]
- Storelli, F.; Billington, S.; Kumar, A.R.; Unadkat, J.D. Abundance of P-Glycoprotein and Other Drug Transporters at the Human Blood-Brain Barrier in Alzheimer’s Disease: A Quantitative Targeted Proteomic Study. Clin. Pharmacol. Ther. 2020, 109, 667–675. [Google Scholar] [CrossRef] [PubMed]
- Toornvliet, R.; van Berckel, B.N.; Luurtsema, G.; Lubberink, M.; Geldof, A.A.; Bosch, T.M.; Oerlemans, R.; Lammertsma, A.A.; Franssen, E.J. Effect of age on functional P-glycoprotein in the blood-brain barrier measured by use of (R)-[(11)C]verapamil and positron emission tomography. Clin. Pharmacol. Ther. 2006, 79, 540–548. [Google Scholar] [CrossRef] [PubMed]
- Cirrito, J.R.; Deane, R.; Fagan, A.M.; Spinner, M.L.; Parsadanian, M.; Finn, M.B.; Jiang, H.; Prior, J.L.; Sagare, A.; Bales, K.R.; et al. P-glycoprotein deficiency at the blood-brain barrier increases amyloid-beta deposition in an Alzheimer disease mouse model. J. Clin. Investig. 2005, 115, 3285–3290. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Al Rihani, S.B.; Lan, R.S.; Kaddoumi, A. Granisetron Alleviates Alzheimer’s Disease Pathology in TgSwDI Mice Through Calmodulin-Dependent Protein Kinase II/cAMP-Response Element Binding Protein Pathway. J. Alzheimers Dis. 2019, 72, 1097–1117. [Google Scholar] [CrossRef]
- Mohamed, L.A.; Keller, J.N.; Kaddoumi, A. Role of P-glycoprotein in mediating rivastigmine effect on amyloid-beta brain load and related pathology in Alzheimer’s disease mouse model. Biochim. Biophys. Acta 2016, 1862, 778–787. [Google Scholar] [CrossRef] [PubMed]
- Behl, T.; Kaur, I.; Sehgal, A.; Kumar, A.; Uddin, M.S.; Bungau, S. The Interplay of ABC Transporters in Abeta Translocation and Cholesterol Metabolism: Implicating Their Roles in Alzheimer’s Disease. Mol. Neurobiol. 2020, 58, 1564–1582. [Google Scholar] [CrossRef]
- Tai, L.M.; Loughlin, A.J.; Male, D.K.; Romero, I.A. P-glycoprotein and breast cancer resistance protein restrict apical-to-basolateral permeability of human brain endothelium to amyloid-beta. J. Cereb. Blood Flow Metab. 2009, 29, 1079–1083. [Google Scholar] [CrossRef]
- Krohn, M.; Lange, C.; Hofrichter, J.; Scheffler, K.; Stenzel, J.; Steffen, J.; Schumacher, T.; Bruning, T.; Plath, A.S.; Alfen, F.; et al. Cerebral amyloid-beta proteostasis is regulated by the membrane transport protein ABCC1 in mice. J. Clin. Investig. 2011, 121, 3924–3931. [Google Scholar] [CrossRef]
- Xiong, H.; Callaghan, D.; Jones, A.; Bai, J.; Rasquinha, I.; Smith, C.; Pei, K.; Walker, D.; Lue, L.F.; Stanimirovic, D.; et al. ABCG2 is upregulated in Alzheimer’s brain with cerebral amyloid angiopathy and may act as a gatekeeper at the blood-brain barrier for Abeta(1-40) peptides. J. Neurosci. 2009, 29, 5463–5475. [Google Scholar] [CrossRef] [Green Version]
- Steinberg, S.; Stefansson, H.; Jonsson, T.; Johannsdottir, H.; Ingason, A.; Helgason, H.; Sulem, P.; Magnusson, O.T.; Gudjonsson, S.A.; Unnsteinsdottir, U.; et al. Loss-of-function variants in ABCA7 confer risk of Alzheimer’s disease. Nat. Genet. 2015, 47, 445–447. [Google Scholar] [CrossRef] [Green Version]
- Abe-Dohmae, S.; Ueda, K.; Yokoyama, S. ABCA7, a molecule with unknown function. FEBS Lett. 2006, 580, 1178–1182. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, Q.F.; Wan, Y.; Wang, H.F.; Sun, F.R.; Hao, X.K.; Tan, M.S.; Tan, C.C.; Zhang, D.Q.; Tan, L.; Yu, J.T.; et al. ABCA7 Genotypes Confer Alzheimer’s Disease Risk by Modulating Amyloid-beta Pathology. J. Alzheimers Dis. 2016, 52, 693–703. [Google Scholar] [CrossRef] [PubMed]
- De Roeck, A.; Van Broeckhoven, C.; Sleegers, K. The role of ABCA7 in Alzheimer’s disease: Evidence from genomics, transcriptomics and methylomics. Acta Neuropathol. 2019, 138, 201–220. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kalilani, L.; Sun, X.; Pelgrims, B.; Noack-Rink, M.; Villanueva, V. The epidemiology of drug-resistant epilepsy: A systematic review and meta-analysis. Epilepsia 2018, 59, 2179–2193. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aronica, E.; Sisodiya, S.M.; Gorter, J.A. Cerebral expression of drug transporters in epilepsy. Adv. Drug Deliv. Rev. 2012, 64, 919–929. [Google Scholar] [CrossRef]
- Marchi, N.; Gonzalez-Martinez, J.; Nguyen, M.T.; Granata, T.; Janigro, D. Transporters in drug-refractory epilepsy: Clinical significance. Clin. Pharmacol. Ther. 2010, 87, 13–15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- van Vliet, E.A.; van Schaik, R.; Edelbroek, P.M.; Redeker, S.; Aronica, E.; Wadman, W.J.; Marchi, N.; Vezzani, A.; Gorter, J.A. Inhibition of the multidrug transporter P-glycoprotein improves seizure control in phenytoin-treated chronic epileptic rats. Epilepsia 2006, 47, 672–680. [Google Scholar] [CrossRef]
- Dombrowski, S.M.; Desai, S.Y.; Marroni, M.; Cucullo, L.; Goodrich, K.; Bingaman, W.; Mayberg, M.R.; Bengez, L.; Janigro, D. Overexpression of multiple drug resistance genes in endothelial cells from patients with refractory epilepsy. Epilepsia 2001, 42, 1501–1506. [Google Scholar] [CrossRef] [PubMed]
- Loscher, W. Drug transporters in the epileptic brain. Epilepsia 2007, 48 (Suppl. S1), 8–13. [Google Scholar] [CrossRef] [PubMed]
- Feldmann, M.; Asselin, M.C.; Liu, J.; Wang, S.; McMahon, A.; Anton-Rodriguez, J.; Walker, M.; Symms, M.; Brown, G.; Hinz, R.; et al. P-glycoprotein expression and function in patients with temporal lobe epilepsy: A case-control study. Lancet Neurol. 2013, 12, 777–785. [Google Scholar] [CrossRef]
- Banerjee Dixit, A.; Sharma, D.; Srivastava, A.; Banerjee, J.; Tripathi, M.; Prakash, D.; Sarat Chandra, P. Upregulation of breast cancer resistance protein and major vault protein in drug resistant epilepsy. Seizure 2017, 47, 9–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cornford, E.M.; Hyman, S.; Cornford, M.E.; Landaw, E.M.; Delgado-Escueta, A.V. Interictal seizure resections show two configurations of endothelial Glut1 glucose transporter in the human blood-brain barrier. J. Cereb. Blood Flow Metab. 1998, 18, 26–42. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lauritzen, F.; de Lanerolle, N.C.; Lee, T.S.; Spencer, D.D.; Kim, J.H.; Bergersen, L.H.; Eid, T. Monocarboxylate transporter 1 is deficient on microvessels in the human epileptogenic hippocampus. Neurobiol. Dis. 2011, 41, 577–584. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rambeck, B.; Jurgens, U.H.; May, T.W.; Pannek, H.W.; Behne, F.; Ebner, A.; Gorji, A.; Straub, H.; Speckmann, E.J.; Pohlmann-Eden, B.; et al. Comparison of brain extracellular fluid, brain tissue, cerebrospinal fluid, and serum concentrations of antiepileptic drugs measured intraoperatively in patients with intractable epilepsy. Epilepsia 2006, 47, 681–694. [Google Scholar] [CrossRef]
- Dickens, D.; Yusof, S.R.; Abbott, N.J.; Weksler, B.; Romero, I.A.; Couraud, P.-O.; Alfirevic, A.; Pirmohamed, M.; Owen, A. A Multi-System Approach Assessing the Interaction of Anticonvulsants with P-gp. PLoS ONE 2013, 8, e64854. [Google Scholar] [CrossRef] [PubMed]
- Luna-Tortos, C.; Rambeck, B.; Jurgens, U.H.; Loscher, W. The antiepileptic drug topiramate is a substrate for human P-glycoprotein but not multidrug resistance proteins. Pharm. Res. 2009, 26, 2464–2470. [Google Scholar] [CrossRef] [PubMed]
- Siddiqui, A.; Kerb, R.; Weale, M.E.; Brinkmann, U.; Smith, A.; Goldstein, D.B.; Wood, N.W.; Sisodiya, S.M. Association of Multidrug Resistance in Epilepsy with a Polymorphism in the Drug-Transporter Gene ABCB1. N. Engl. J. Med. 2003, 348, 1442–1448. [Google Scholar] [CrossRef] [Green Version]
- Sills, G.J.; Mohanraj, R.; Butler, E.; McCrindle, S.; Collier, L.; Wilson, E.A.; Brodie, M.J. Lack of association between the C3435T polymorphism in the human multidrug resistance (MDR1) gene and response to antiepileptic drug treatment. Epilepsia 2005, 46, 643–647. [Google Scholar] [CrossRef]
- Liu, J.Y.; Thom, M.; Catarino, C.B.; Martinian, L.; Figarella-Branger, D.; Bartolomei, F.; Koepp, M.; Sisodiya, S.M. Neuropathology of the blood-brain barrier and pharmaco-resistance in human epilepsy. Brain 2012, 135, 3115–3133. [Google Scholar] [CrossRef] [Green Version]
- Löscher, W.; Potschka, H. Role of multidrug transporters in pharmacoresistance to antiepileptic drugs. J. Pharmacol. Exp. Ther. 2002, 301, 7–14. [Google Scholar] [CrossRef] [Green Version]
- Maschio, M. Brain tumor-related epilepsy. Curr. Neuropharmacol. 2012, 10, 124–133. [Google Scholar] [CrossRef] [PubMed]
- Calatozzolo, C.; Pollo, B.; Botturi, A.; Dinapoli, L.; Carosi, M.; Salmaggi, A.; Maschio, M. Multidrug resistance proteins expression in glioma patients with epilepsy. J. Neurooncol 2012, 110, 129–135. [Google Scholar] [CrossRef]
- Munoz, J.L.; Walker, N.D.; Scotto, K.W.; Rameshwar, P. Temozolomide competes for P-glycoprotein and contributes to chemoresistance in glioblastoma cells. Cancer Lett. 2015, 367, 69–75. [Google Scholar] [CrossRef] [PubMed]
- Gallagher, P.; Leach, J.P.; Grant, R. Time to focus on brain tumor-related epilepsy trials. Neurooncol. Pract. 2014, 1, 123–133. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Katan, M.; Luft, A. Global Burden of Stroke. Semin. Neurol. 2018, 38, 208–211. [Google Scholar] [CrossRef] [Green Version]
- Brzica, H.; Abdullahi, W.; Ibbotson, K.; Ronaldson, P.T. Role of Transporters in Central Nervous System Drug Delivery and Blood-Brain Barrier Protection: Relevance to Treatment of Stroke. J. Cent. Nerv. Syst. Dis. 2017, 9. [Google Scholar] [CrossRef]
- Virani, S.S.; Alonso, A.; Benjamin, E.J.; Bittencourt, M.S.; Callaway, C.W.; Carson, A.P.; Chamberlain, A.M.; Chang, A.R.; Cheng, S.; Delling, F.N.; et al. Heart Disease and Stroke Statistics-2020 Update: A Report From the American Heart Association. Circulation 2020, 141, e139–e596. [Google Scholar] [CrossRef]
- Hoffmann, A.; Dege, T.; Kunze, R.; Ernst, A.-S.; Lorenz, H.; Böhler, L.-I.; Korff, T.; Marti, H.H.; Heiland, S.; Bendszus, M.; et al. Early Blood–Brain Barrier Disruption in Ischemic Stroke Initiates Multifocally Around Capillaries/Venules. Stroke 2018, 49, 1479–1487. [Google Scholar] [CrossRef]
- Nadareishvili, Z.; Simpkins, A.N.; Hitomi, E.; Reyes, D.; Leigh, R. Post-Stroke Blood-Brain Barrier Disruption and Poor Functional Outcome in Patients Receiving Thrombolytic Therapy. Cerebrovasc. Dis. 2019, 47, 135–142. [Google Scholar] [CrossRef] [PubMed]
- Dirnagl, U.; Iadecola, C.; Moskowitz, M.A. Pathobiology of ischaemic stroke: An integrated view. Trends Neurosci. 1999, 22, 391–397. [Google Scholar] [CrossRef]
- DeMars, K.M.; Yang, C.; Hawkins, K.E.; McCrea, A.O.; Siwarski, D.M.; Candelario-Jalil, E. Spatiotemporal Changes in P-glycoprotein Levels in Brain and Peripheral Tissues Following Ischemic Stroke in Rats. J. Exp. Neurosci. 2017, 11, 1179069517701741. [Google Scholar] [CrossRef] [PubMed]
- Neuhaus, W.; Burek, M.; Djuzenova, C.S.; Thal, S.C.; Koepsell, H.; Roewer, N.; Forster, C.Y. Addition of NMDA-receptor antagonist MK801 during oxygen/glucose deprivation moderately attenuates the upregulation of glucose uptake after subsequent reoxygenation in brain endothelial cells. Neurosci. Lett. 2012, 506, 44–49. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, F.; Vannucci, S.J.; Philp, N.J.; Simpson, I.A. Monocarboxylate transporter expression in the spontaneous hypertensive rat: Effect of stroke. J. Neurosci. Res. 2005, 79, 139–145. [Google Scholar] [CrossRef] [PubMed]
- Comerford, K.M.; Wallace, T.J.; Karhausen, J.; Louis, N.A.; Montalto, M.C.; Colgan, S.P. Hypoxia-inducible factor-1-dependent regulation of the multidrug resistance (MDR1) gene. Cancer Res. 2002, 62, 3387–3394. [Google Scholar] [PubMed]
- Spudich, A.; Kilic, E.; Xing, H.; Kilic, U.; Rentsch, K.M.; Wunderli-Allenspach, H.; Bassetti, C.L.; Hermann, D.M. Inhibition of multidrug resistance transporter-1 facilitates neuroprotective therapies after focal cerebral ischemia. Nat. Neurosci. 2006, 9, 487–488. [Google Scholar] [CrossRef] [PubMed]
- Vaughan, C.J.; Delanty, N. Neuroprotective properties of statins in cerebral ischemia and stroke. Stroke 1999, 30, 1969–1973. [Google Scholar] [CrossRef]
- Sierra, S.; Ramos, M.C.; Molina, P.; Esteo, C.; Vazquez, J.A.; Burgos, J.S. Statins as neuroprotectants: A comparative in vitro study of lipophilicity, blood-brain-barrier penetration, lowering of brain cholesterol, and decrease of neuron cell death. J. Alzheimers Dis. 2011, 23, 307–318. [Google Scholar] [CrossRef]
- Butterfield, D.A.; Barone, E.; Di Domenico, F.; Cenini, G.; Sultana, R.; Murphy, M.P.; Mancuso, C.; Head, E. Atorvastatin treatment in a dog preclinical model of Alzheimer’s disease leads to up-regulation of haem oxygenase-1 and is associated with reduced oxidative stress in brain. Int. J. Neuropsychopharmacol. 2012, 15, 981–987. [Google Scholar] [CrossRef] [Green Version]
- Brown, R.H.; Al-Chalabi, A. Amyotrophic Lateral Sclerosis. N. Engl. J. Med. 2017, 377, 162–172. [Google Scholar] [CrossRef] [Green Version]
- Pfohl, S.R.; Kim, R.B.; Coan, G.S.; Mitchell, C.S. Unraveling the Complexity of Amyotrophic Lateral Sclerosis Survival Prediction. Front. Neuroinf. 2018, 12. [Google Scholar] [CrossRef] [Green Version]
- Brotman, R.G.; Moreno-Escobar, M.C.; Joseph, J.; Pawar, G. Amyotrophic Lateral Sclerosis. In StatPearls; StatPearls Publishing LLC.: Treasure Island, FL, USA, 2021. [Google Scholar]
- Rosen, D.R.; Siddique, T.; Patterson, D.; Figlewicz, D.A.; Sapp, P.; Hentati, A.; Donaldson, D.; Goto, J.; O’Regan, J.P.; Deng, H.X.; et al. Mutations in Cu/Zn superoxide dismutase gene are associated with familial amyotrophic lateral sclerosis. Nature 1993, 362, 59–62. [Google Scholar] [CrossRef]
- Blair, I.P.; Williams, K.L.; Warraich, S.T.; Durnall, J.C.; Thoeng, A.D.; Manavis, J.; Blumbergs, P.C.; Vucic, S.; Kiernan, M.C.; Nicholson, G.A. FUS mutations in amyotrophic lateral sclerosis: Clinical, pathological, neurophysiological and genetic analysis. J. Neurol. Neurosurg. Psychiatry 2010, 81, 639–645. [Google Scholar] [CrossRef] [Green Version]
- Gitcho, M.A.; Baloh, R.H.; Chakraverty, S.; Mayo, K.; Norton, J.B.; Levitch, D.; Hatanpaa, K.J.; White, C.L., 3rd; Bigio, E.H.; Caselli, R.; et al. TDP-43 A315T mutation in familial motor neuron disease. Ann. Neurol. 2008, 63, 535–538. [Google Scholar] [CrossRef] [Green Version]
- Sreedharan, J.; Blair, I.P.; Tripathi, V.B.; Hu, X.; Vance, C.; Rogelj, B.; Ackerley, S.; Durnall, J.C.; Williams, K.L.; Buratti, E.; et al. TDP-43 mutations in familial and sporadic amyotrophic lateral sclerosis. Science 2008, 319, 1668–1672. [Google Scholar] [CrossRef] [PubMed]
- Lagier-Tourenne, C.; Cleveland, D.W. Rethinking ALS: The FUS about TDP-43. Cell 2009, 136, 1001–1004. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vance, C.; Rogelj, B.; Hortobágyi, T.; De Vos, K.J.; Nishimura, A.L.; Sreedharan, J.; Hu, X.; Smith, B.; Ruddy, D.; Wright, P.; et al. Mutations in FUS, an RNA processing protein, cause familial amyotrophic lateral sclerosis type 6. Science 2009, 323, 1208–1211. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kwiatkowski, T.J., Jr.; Bosco, D.A.; Leclerc, A.L.; Tamrazian, E.; Vanderburg, C.R.; Russ, C.; Davis, A.; Gilchrist, J.; Kasarskis, E.J.; Munsat, T.; et al. Mutations in the FUS/TLS gene on chromosome 16 cause familial amyotrophic lateral sclerosis. Science 2009, 323, 1205–1208. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Al-Chalabi, A.; van den Berg, L.H.; Veldink, J. Gene discovery in amyotrophic lateral sclerosis: Implications for clinical management. Nat. Rev. Neurol. 2017, 13, 96–104. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garbuzova-Davis, S.; Sanberg, P.R. Blood-CNS Barrier Impairment in ALS patients versus an animal model. Front. Cell Neurosci. 2014, 8, 21. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leonardi, A.; Abbruzzese, G.; Arata, L.; Cocito, L.; Vische, M. Cerebrospinal fluid (CSF) findings in amyotrophic lateral sclerosis. J. Neurol. 1984, 231, 75–78. [Google Scholar] [CrossRef]
- Annunziata, P.; Volpi, N. High levels of C3c in the cerebrospinal fluid from amyotrophic lateral sclerosis patients. Acta Neurol. Scand. 1985, 72, 61–64. [Google Scholar] [CrossRef] [PubMed]
- Mineyeva, M.F.; Kudrin, V.S.; Rayevsky, K.S. Brain tyrosine hydroxylase: Kinetic properties and regulation of the activity. Ann. Ist. Super. Sanita. 1978, 14, 83–88. [Google Scholar]
- Garbuzova-Davis, S.; Hernandez-Ontiveros, D.G.; Rodrigues, M.C.; Haller, E.; Frisina-Deyo, A.; Mirtyl, S.; Sallot, S.; Saporta, S.; Borlongan, C.V.; Sanberg, P.R. Impaired blood-brain/spinal cord barrier in ALS patients. Brain Res. 2012, 1469, 114–128. [Google Scholar] [CrossRef] [PubMed]
- Garbuzova-Davis, S.; Haller, E.; Saporta, S.; Kolomey, I.; Nicosia, S.V.; Sanberg, P.R. Ultrastructure of blood-brain barrier and blood-spinal cord barrier in SOD1 mice modeling ALS. Brain Res. 2007, 1157, 126–137. [Google Scholar] [CrossRef] [PubMed]
- Jablonski, M.R.; Jacob, D.A.; Campos, C.; Miller, D.S.; Maragakis, N.J.; Pasinelli, P.; Trotti, D. Selective increase of two ABC drug efflux transporters at the blood-spinal cord barrier suggests induced pharmacoresistance in ALS. Neurobiol. Dis. 2012, 47, 194–200. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qosa, H.; Lichter, J.; Sarlo, M.; Markandaiah, S.S.; McAvoy, K.; Richard, J.P.; Jablonski, M.R.; Maragakis, N.J.; Pasinelli, P.; Trotti, D. Astrocytes drive upregulation of the multidrug resistance transporter ABCB1 (P-Glycoprotein) in endothelial cells of the blood-brain barrier in mutant superoxide dismutase 1-linked amyotrophic lateral sclerosis. Glia 2016, 64, 1298–1313. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Banks, D.B.; Chan, G.N.; Evans, R.A.; Miller, D.S.; Cannon, R.E. Lysophosphatidic acid and amitriptyline signal through LPA1R to reduce P-glycoprotein transport at the blood-brain barrier. J. Cereb. Blood Flow Metab. 2018, 38, 857–868. [Google Scholar] [CrossRef]
- Milane, A.; Fernandez, C.; Dupuis, L.; Buyse, M.; Loeffler, J.P.; Farinotti, R.; Meininger, V.; Bensimon, G. P-glycoprotein expression and function are increased in an animal model of amyotrophic lateral sclerosis. Neurosci. Lett. 2010, 472, 166–170. [Google Scholar] [CrossRef]
- Chan, G.N.; Evans, R.A.; Banks, D.B.; Mesev, E.V.; Miller, D.S.; Cannon, R.E. Selective induction of P-glycoprotein at the CNS barriers during symptomatic stage of an ALS animal model. Neurosci. Lett. 2017, 639, 103–113. [Google Scholar] [CrossRef] [Green Version]
- Kirkinezos, I.G.; Hernandez, D.; Bradley, W.G.; Moraes, C.T. An ALS mouse model with a permeable blood–brain barrier benefits from systemic cyclosporine—A treatment. J. Neurochem. 2004, 88, 821–826. [Google Scholar] [CrossRef]
- Boston-Howes, W.; Williams, E.O.; Bogush, A.; Scolere, M.; Pasinelli, P.; Trotti, D. Nordihydroguaiaretic acid increases glutamate uptake in vitro and in vivo: Therapeutic implications for amyotrophic lateral sclerosis. Exp. Neurol. 2008, 213, 229–237. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, T.; Ferrill, L.; Gallant, L.; McGillicuddy, S.; Fernandes, T.; Schields, N.; Bai, S. Verapamil and riluzole cocktail liposomes overcome pharmacoresistance by inhibiting P-glycoprotein in brain endothelial and astrocyte cells: A potent approach to treat amyotrophic lateral sclerosis. Eur. J. Pharm. Sci. 2018, 120, 30–39. [Google Scholar] [CrossRef] [PubMed]
- Milane, A.; Vautier, S.; Chacun, H.; Meininger, V.; Bensimon, G.; Farinotti, R.; Fernandez, C. Interactions between riluzole and ABCG2/BCRP transporter. Neurosci. Lett. 2009, 452, 12–16. [Google Scholar] [CrossRef] [PubMed]
- Friese, M.A.; Schattling, B.; Fugger, L. Mechanisms of neurodegeneration and axonal dysfunction in multiple sclerosis. Nat. Rev. Neurol. 2014, 10, 225–238. [Google Scholar] [CrossRef]
- Nelson, L.M.; Wallin, M.T.; Marrie, R.A.; Culpepper, W.J.; Langer-Gould, A.; Campbell, J.; Buka, S.; Tremlett, H.; Cutter, G.; Kaye, W.; et al. A new way to estimate neurologic disease prevalence in the United States: Illustrated with MS. Neurology 2019, 92, 469–480. [Google Scholar] [CrossRef]
- Multiple Sclerosis News Today. MS Statistics. Available online: https://multiplesclerosisnewstoday.com/multiple-sclerosis-overview/statistics/ (accessed on 30 March 2021).
- Tafti, D.; Ehsan, M.; Xixis, K.L. Multiple Sclerosis. Available online: https://www.ncbi.nlm.nih.gov/books/NBK499849 (accessed on 30 March 2021).
- Brownlee, W.J.; Hardy, T.A.; Fazekas, F.; Miller, D.H. Diagnosis of multiple sclerosis: Progress and challenges. Lancet 2017, 389, 1336–1346. [Google Scholar] [CrossRef]
- Kooij, G.; Mizee, M.R.; van Horssen, J.; Reijerkerk, A.; Witte, M.E.; Drexhage, J.A.R.; van der Pol, S.M.A.; van Het Hof, B.; Scheffer, G.; Scheper, R.; et al. Adenosine triphosphate-binding cassette transporters mediate chemokine (C-C motif) ligand 2 secretion from reactive astrocytes: Relevance to multiple sclerosis pathogenesis. Brain 2011, 134, 555–570. [Google Scholar] [CrossRef] [Green Version]
- Kooij, G.; van Horssen, J.; de Lange, E.C.M.; Reijerkerk, A.; van der Pol, S.M.A.; van Het Hof, B.; Drexhage, J.; Vennegoor, A.; Killestein, J.; Scheffer, G.; et al. T lymphocytes impair P-glycoprotein function during neuroinflammation. J. Autoimmun. 2010, 34, 416–425. [Google Scholar] [CrossRef]
- Kooij, G.; van Horssen, J.; Bandaru, V.V.R.; Haughey, N.; De Vries, H. The Role of ATP-Binding Cassette Transporters in Neuro-Inflammation: Relevance for Bioactive Lipids. Front. Pharmacol. 2012, 3, 74. [Google Scholar] [CrossRef] [Green Version]
- Kooij, G.; Backer, R.; Koning, J.J.; Reijerkerk, A.; van Horssen, J.; van der Pol, S.M.A.; Drexhage, J.; Schinkel, A.; Dijkstra, C.D.; den Haan, J.M.M.; et al. P-Glycoprotein Acts as an Immunomodulator during Neuroinflammation. PLoS ONE 2009, 4, e8212. [Google Scholar] [CrossRef]
- Vogelgesang, S.; Cascorbi, I.; Schroeder, E.; Pahnke, J.; Kroemer, H.K.; Siegmund, W.; Kunert-Keil, C.; Walker, L.C.; Warzok, R.W. Deposition of Alzheimer’s beta-amyloid is inversely correlated with P-glycoprotein expression in the brains of elderly non-demented humans. Pharmacogenetics 2002, 12, 535–541. [Google Scholar] [CrossRef] [PubMed]
- Lam, F.C.; Liu, R.; Lu, P.; Shapiro, A.B.; Renoir, J.M.; Sharom, F.J.; Reiner, P.B. beta-Amyloid efflux mediated by p-glycoprotein. J. Neurochem. 2001, 76, 1121–1128. [Google Scholar] [CrossRef] [PubMed]
- Kuhnke, D.; Jedlitschky, G.; Grube, M.; Krohn, M.; Jucker, M.; Mosyagin, I.; Cascorbi, I.; Walker, L.C.; Kroemer, H.K.; Warzok, R.W.; et al. MDR1-P-Glycoprotein (ABCB1) Mediates Transport of Alzheimer’s amyloid-beta peptides--implications for the mechanisms of Abeta clearance at the blood-brain barrier. Brain Pathol. 2007, 17, 347–353. [Google Scholar] [CrossRef] [PubMed]
- Hartz, A.M.S.; Zhong, Y.; Wolf, A.; LeVine, H.; Miller, D.S.; Bauer, B. Aβ40 Reduces P-Glycoprotein at the Blood–Brain Barrier through the Ubiquitin–Proteasome Pathway. J. Neurosci. 2016, 36, 1930–1941. [Google Scholar] [CrossRef] [PubMed]
- Wijesuriya, H.C.; Bullock, J.Y.; Faull, R.L.; Hladky, S.B.; Barrand, M.A. ABC efflux transporters in brain vasculature of Alzheimer’s subjects. Brain Res. 2010, 1358, 228–238. [Google Scholar] [CrossRef]
- Abuznait, A.H.; Cain, C.; Ingram, D.; Burk, D.; Kaddoumi, A. Up-regulation of P-glycoprotein reduces intracellular accumulation of beta amyloid: Investigation of P-glycoprotein as a novel therapeutic target for Alzheimer’s disease. J. Pharm. Pharmacol. 2011, 63, 1111–1118. [Google Scholar] [CrossRef] [Green Version]
- Wang, W.; Bodles-Brakhop, A.M.; Barger, S.W. A Role for P-Glycoprotein in Clearance of Alzheimer Amyloid β -Peptide from the Brain. Curr. Alzheimer Res. 2016, 13, 615–620. [Google Scholar] [CrossRef]
- Kooij, G.; Kroon, J.; Paul, D.; Reijerkerk, A.; Geerts, D.; van der Pol, S.M.; van Het Hof, B.; Drexhage, J.A.; van Vliet, S.J.; Hekking, L.H.; et al. P-glycoprotein regulates trafficking of CD8(+) T cells to the brain parenchyma. Acta Neuropathol. 2014, 127, 699–711. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Liu, M.; Yang, L.; Huang, F.; Lan, Y.; Li, H.; Wu, H.; Zhang, B.; Shi, H.; Wu, X. P-glycoprotein Inhibitor Tariquidar Potentiates Efficacy of Astragaloside IV in Experimental Autoimmune Encephalomyelitis Mice. Molecules 2019, 24, 561. [Google Scholar] [CrossRef] [Green Version]
- Tishler, D.M.; Weinberg, K.I.; Hinton, D.R.; Barbaro, N.; Annett, G.M.; Raffel, C. MDR1 gene expression in brain of patients with medically intractable epilepsy. Epilepsia 1995, 36, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Sisodiya, S.M.; Heffernan, J.; Squier, M.V. Over-expression of P-glycoprotein in malformations of cortical development. Neuroreport 1999, 10, 3437–3441. [Google Scholar] [CrossRef]
- Milane, A.; Fernandez, C.; Vautier, S.; Bensimon, G.; Meininger, V.; Farinotti, R. Minocycline and riluzole brain disposition: Interactions with p-glycoprotein at the blood-brain barrier. J. Neurochem. 2007, 103, 164–173. [Google Scholar] [CrossRef]
- Samoto, K.; Ikezaki, K.; Yokoyama, N.; Fukui, M. P-glycoprotein expression in brain capillary endothelial cells after focal ischaemia in the rat. Neurol. Res. 1994, 16, 217–223. [Google Scholar] [CrossRef]
- Dazert, P.; Suofu, Y.; Grube, M.; Popa-Wagner, A.; Kroemer, H.K.; Jedlitschky, G.; Kessler, C. Differential regulation of transport proteins in the periinfarct region following reversible middle cerebral artery occlusion in rats. Neuroscience 2006, 142, 1071–1079. [Google Scholar] [CrossRef]
- Wang, X.; Campos, C.R.; Peart, J.C.; Smith, L.K.; Boni, J.L.; Cannon, R.E.; Miller, D.S. Nrf2 upregulates ATP binding cassette transporter expression and activity at the blood-brain and blood-spinal cord barriers. J. Neurosci. 2014, 34, 8585–8593. [Google Scholar] [CrossRef] [Green Version]
- Do, T.M.; Noel-Hudson, M.S.; Ribes, S.; Besengez, C.; Smirnova, M.; Cisternino, S.; Buyse, M.; Calon, F.; Chimini, G.; Chacun, H.; et al. ABCG2- and ABCG4-mediated efflux of amyloid-β peptide 1-40 at the mouse blood-brain barrier. J. Alzheimers Dis. 2012, 30, 155–166. [Google Scholar] [CrossRef] [PubMed]
- Candela, P.; Gosselet, F.; Saint-Pol, J.; Sevin, E.; Boucau, M.C.; Boulanger, E.; Cecchelli, R.; Fenart, L. Apical-to-basolateral transport of amyloid-β peptides through blood-brain barrier cells is mediated by the receptor for advanced glycation end-products and is restricted by P-glycoprotein. J. Alzheimers Dis. 2010, 22, 849–859. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kilic, E.; Spudich, A.; Kilic, U.; Rentsch, K.M.; Vig, R.; Matter, C.M.; Wunderli-Allenspach, H.; Fritschy, J.M.; Bassetti, C.L.; Hermann, D.M. ABCC1: A gateway for pharmacological compounds to the ischaemic brain. Brain 2008, 131, 2679–2689. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hofrichter, J.; Krohn, M.; Schumacher, T.; Lange, C.; Feistel, B.; Walbroel, B.; Heinze, H.J.; Crockett, S.; Sharbel, T.F.; Pahnke, J. Reduced Alzheimer’s disease pathology by St. John’s Wort treatment is independent of hyperforin and facilitated by ABCC1 and microglia activation in mice. Curr. Alzheimer Res. 2013, 10, 1057–1069. [Google Scholar] [CrossRef] [PubMed]
- van Vliet, E.A.; Redeker, S.; Aronica, E.; Edelbroek, P.M.; Gorter, J.A. Expression of multidrug transporters MRP1, MRP2, and BCRP shortly after status epilepticus, during the latent period, and in chronic epileptic rats. Epilepsia 2005, 46, 1569–1580. [Google Scholar] [CrossRef] [PubMed]
- Sisodiya, S.M.; Lin, W.R.; Harding, B.N.; Squier, M.V.; Thom, M. Drug resistance in epilepsy: Expression of drug resistance proteins in common causes of refractory epilepsy. Brain 2002, 125, 22–31. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alzforum. ABCA7. Available online: https://www.alzforum.org/alzpedia/abca7 (accessed on 30 March 2021).
- Pahnke, J.; Walker, L.C.; Schroeder, E.; Vogelgesang, S.; Stausske, D.; Walther, R.; Warzok, R.W. Cerebral beta-amyloid deposition is augmented by the -491AA promoter polymorphism in non-demented elderly individuals bearing the apolipoprotein E epsilon4 allele. Acta Neuropathol. 2003, 105, 25–29. [Google Scholar] [CrossRef] [PubMed]
- Vasquez, J.B.; Fardo, D.W.; Estus, S. ABCA7 expression is associated with Alzheimer’s disease polymorphism and disease status. Neurosci. Lett. 2013, 556, 58–62. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aikawa, T.; Holm, M.-L.; Kanekiyo, T. ABCA7 and Pathogenic Pathways of Alzheimer’s Disease. Brain Sci. 2018, 8, 27. [Google Scholar] [CrossRef] [Green Version]
- Lamartinière, Y.; Boucau, M.-C.; Dehouck, L.; Krohn, M.; Pahnke, J.; Candela, P.; Gosselet, F.; Fenart, L. ABCA7 Downregulation Modifies Cellular Cholesterol Homeostasis and Decreases Amyloid-β Peptide Efflux in an in vitro Model of the Blood-Brain Barrier. J. Alzheimer’s Dis. 2018, 64, 1195–1211. [Google Scholar] [CrossRef]
- Ochiai, Y.; Uchida, Y.; Ohtsuki, S.; Tachikawa, M.; Aizawa, S.; Terasaki, T. The blood-brain barrier fatty acid transport protein 1 (FATP1/SLC27A1) supplies docosahexaenoic acid to the brain, and insulin facilitates transport. J. Neurochem. 2017, 141, 400–412. [Google Scholar] [CrossRef] [Green Version]
- Ochiai, Y.; Uchida, Y.; Tachikawa, M.; Couraud, P.O.; Terasaki, T. Amyloid beta(25-35) impairs docosahexaenoic acid efflux by down-regulating fatty acid transport protein 1 (FATP1/SLC27A1) protein expression in human brain capillary endothelial cells. J. Neurochem. 2019, 150, 385–401. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Transporter | BBB Expression | Function | Disease Impacted and Key Findings | References |
|---|---|---|---|---|
| ATP-binding cassette (ABC) transporters | ||||
| ABCB1 (P-gp) | Luminal (apical) | Efflux |
| [122,185,188,199,200,203,204,205,206,207,208,209,210,211,212,213,214,215,216] |
| ABCG2 (BCRP) | Luminal (apical) | Efflux |
| [128,199,211,216,217,218,219] |
| ABCC1 (MRP1) | Luminal (apical) and basolateral | Efflux |
| [127,199,220,221,222,223] |
| ABCC2 (MRP2) | Luminal (apical) | Efflux |
| [137,199,222] |
| ABCC4 (MRP4) | Luminal (apical) and basolateral | Efflux |
| [207,222] |
| ABCC5 (MRP5) | Luminal (apical) | Efflux |
| [137,216] |
| ABCA7 | Not clear | Efflux |
| [132,224,225,226,227,228] |
| Organic Anion Transporting Polypeptides | ||||
| SLCO1A2 (OATP1A2) | Luminal (apical) | Uptake |
| [31,66,70] |
| SLCO2B1 (OATP2B1) | Luminal (apical) | Uptake |
| [120] |
| Others | ||||
| SLC27A1 (FATP1) | Basolateral | Efflux |
| [229,230] |
| SLC29A1 (ENT1) | Not clear | Efflux/uptake? |
| [120] |
| SLC2A1 (GLUT1) | Luminal (apical) and basolateral | Uptake |
| [141] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Al Rihani, S.B.; Darakjian, L.I.; Deodhar, M.; Dow, P.; Turgeon, J.; Michaud, V. Disease-Induced Modulation of Drug Transporters at the Blood–Brain Barrier Level. Int. J. Mol. Sci. 2021, 22, 3742. https://doi.org/10.3390/ijms22073742
Al Rihani SB, Darakjian LI, Deodhar M, Dow P, Turgeon J, Michaud V. Disease-Induced Modulation of Drug Transporters at the Blood–Brain Barrier Level. International Journal of Molecular Sciences. 2021; 22(7):3742. https://doi.org/10.3390/ijms22073742
Chicago/Turabian StyleAl Rihani, Sweilem B., Lucy I. Darakjian, Malavika Deodhar, Pamela Dow, Jacques Turgeon, and Veronique Michaud. 2021. "Disease-Induced Modulation of Drug Transporters at the Blood–Brain Barrier Level" International Journal of Molecular Sciences 22, no. 7: 3742. https://doi.org/10.3390/ijms22073742