
Aerobic Exercise Ameliorates Cancer Cachexia-Induced Muscle Wasting through Adiponectin Signaling

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
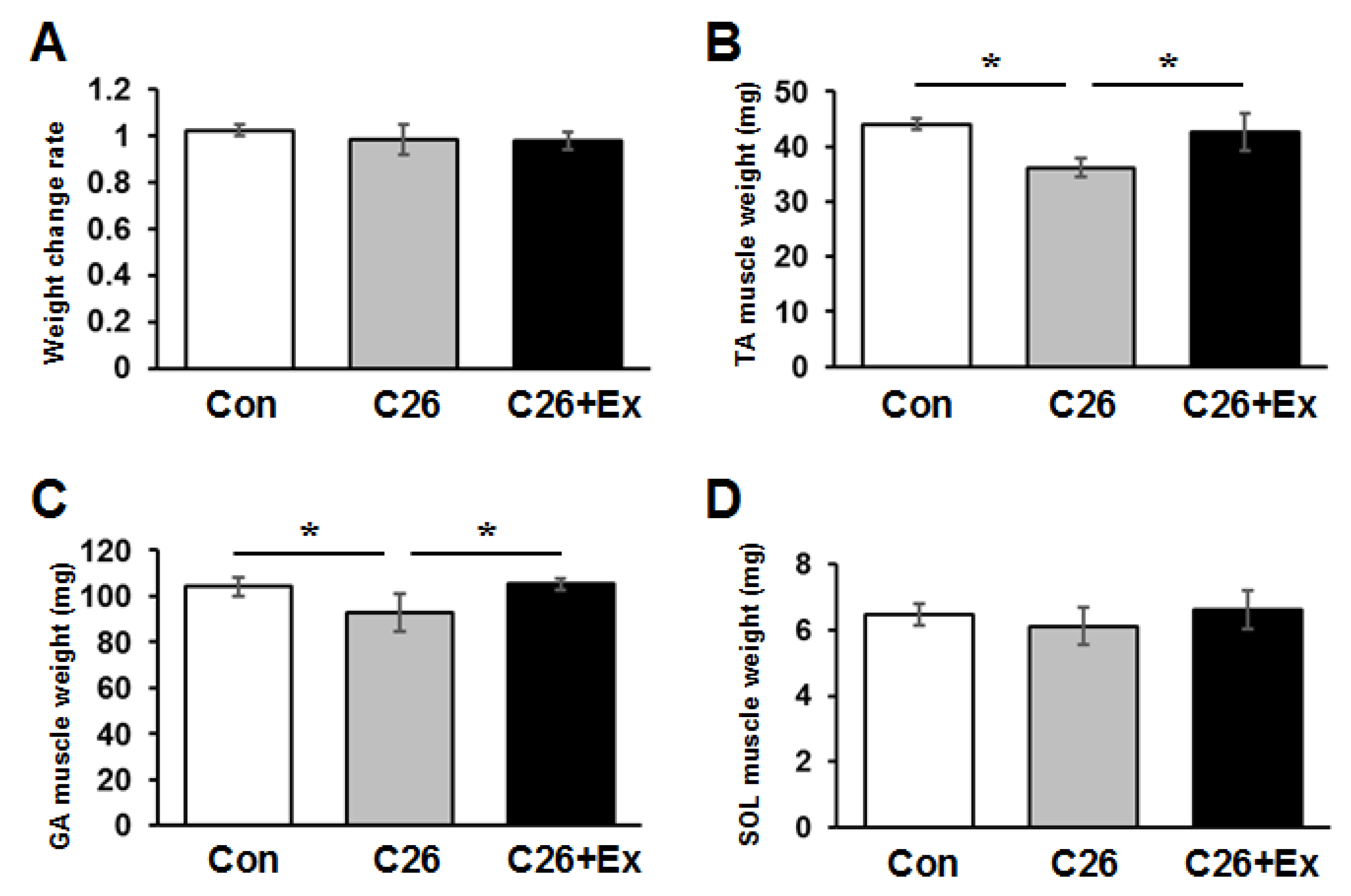
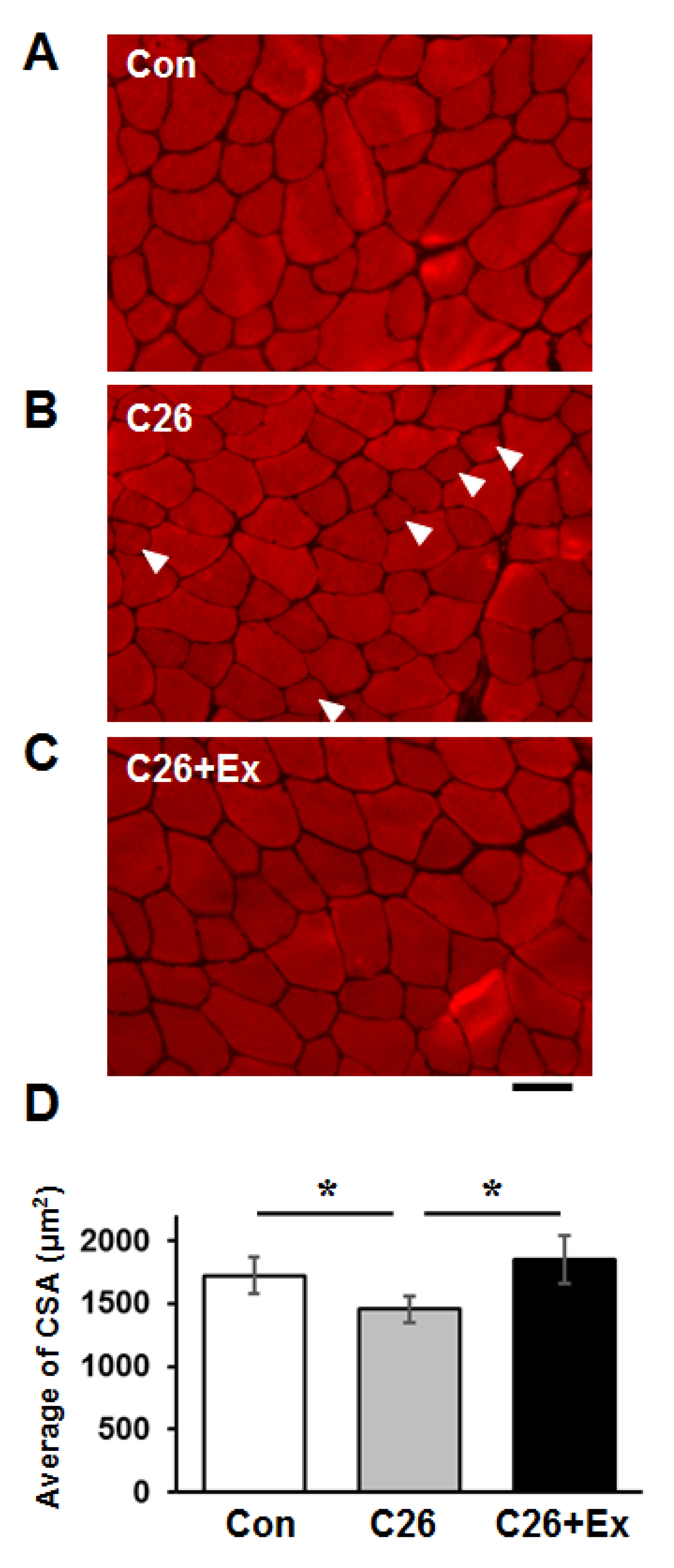
2.1. Aerobic Exercise Suppresses Cancer Cachexia-Induced Muscle Atrophy
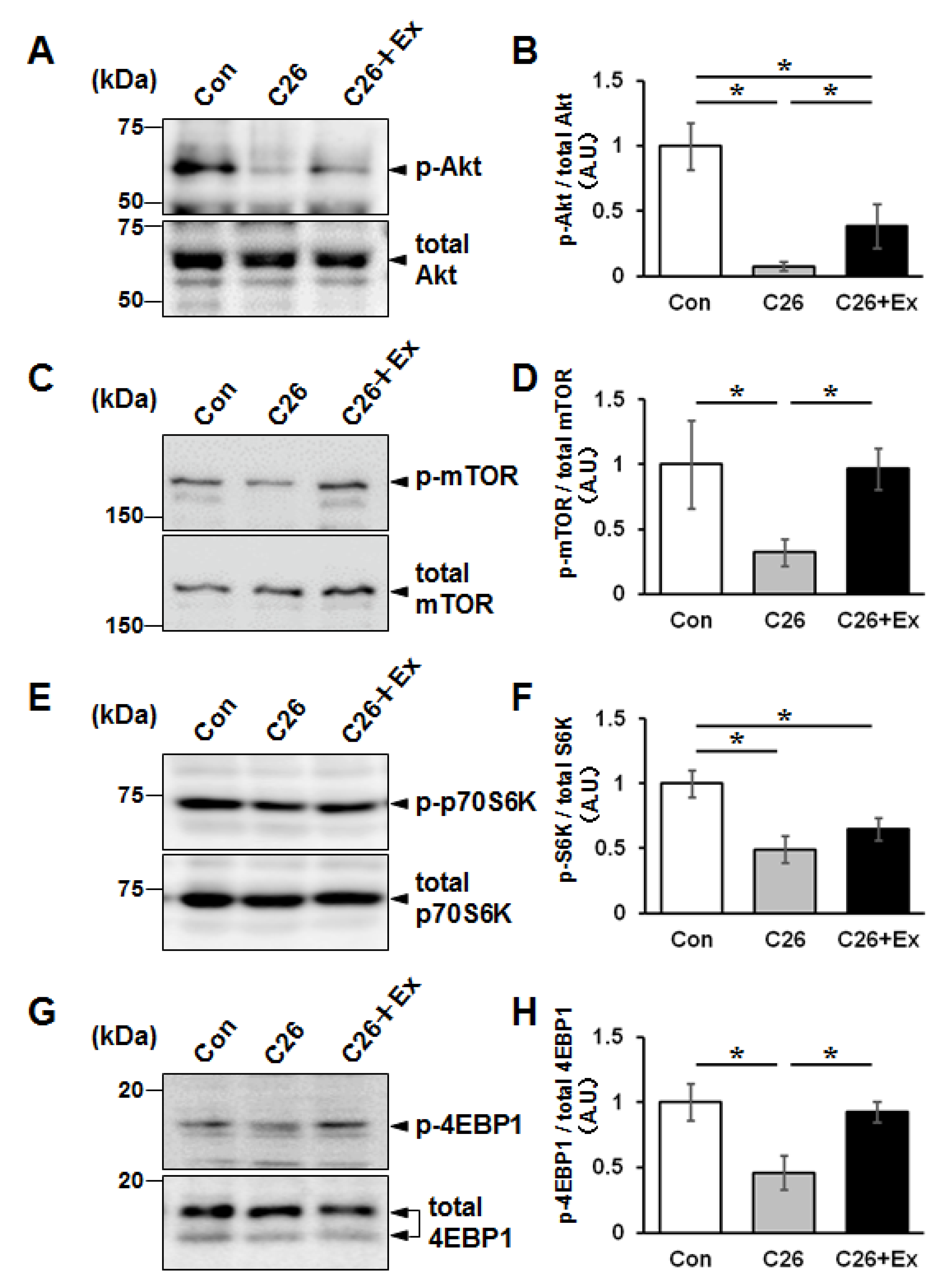
2.2. Aerobic Exercise Enhances Protein Synthesis Signaling in Cancer Cachectic Skeletal Muscle
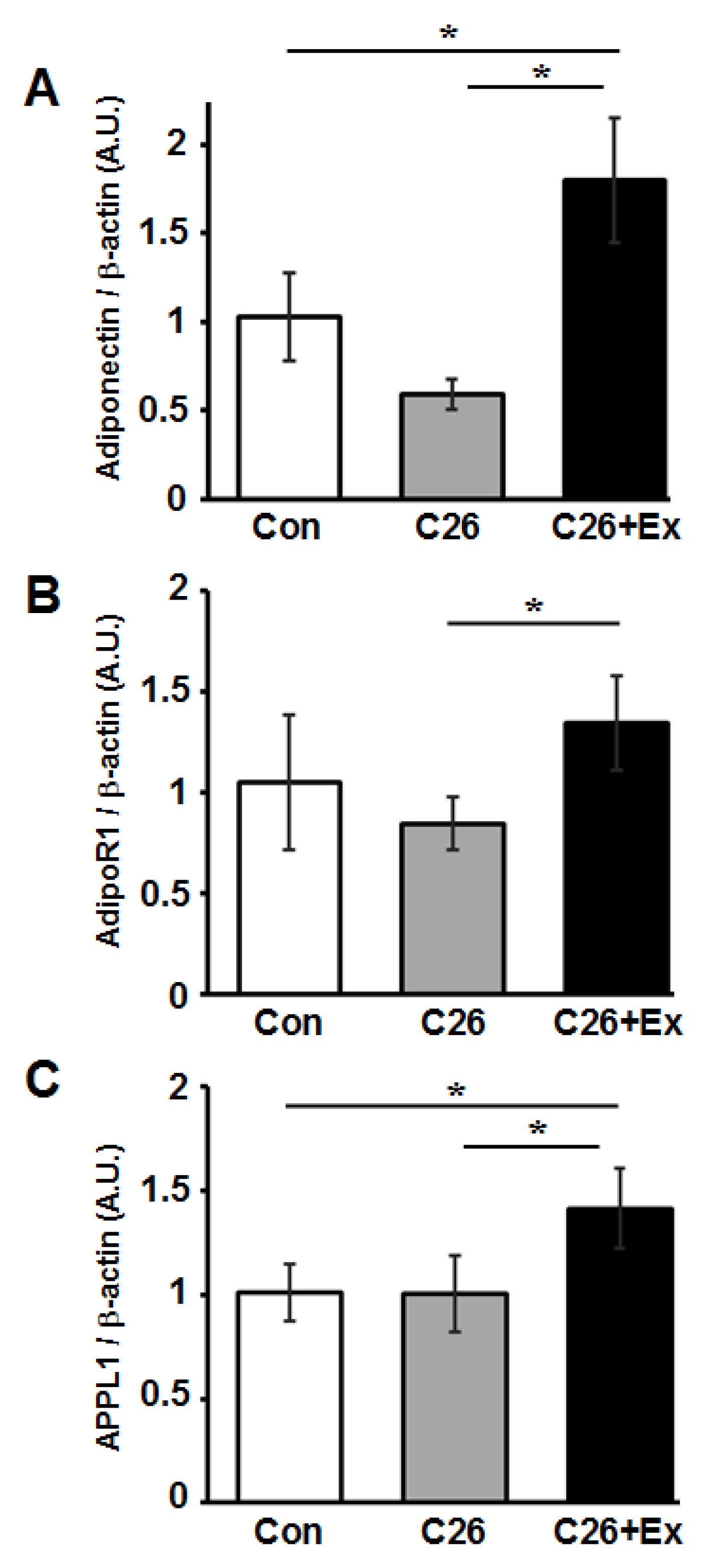
2.3. Aerobic Exercise Increases mRNA Expression of Adiponectin and Adiponectin-Related Genes in Skeletal Muscle
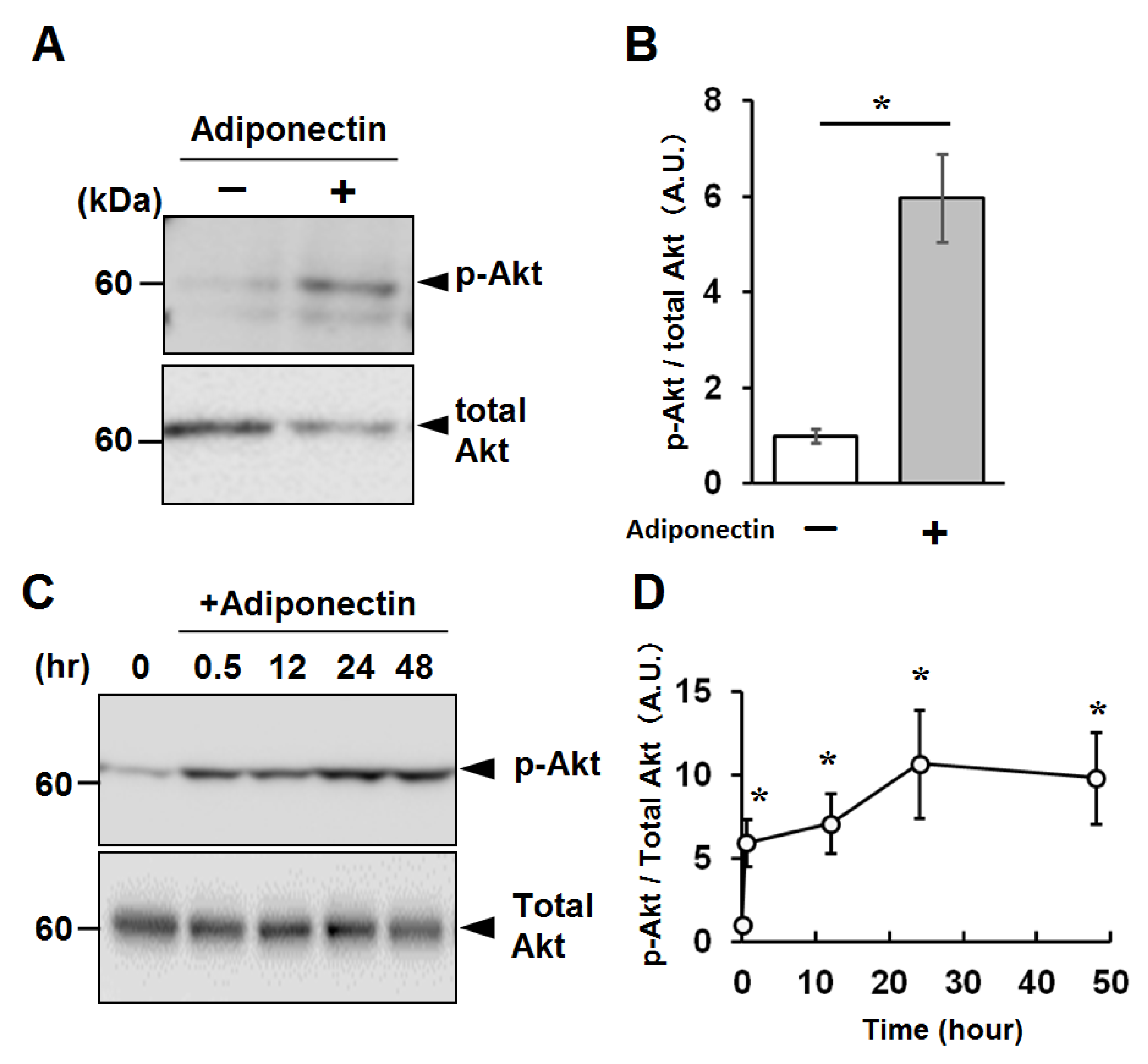
2.4. Activation of Cellular Signaling by Recombinant Adiponectin in C2C12 Myotubes
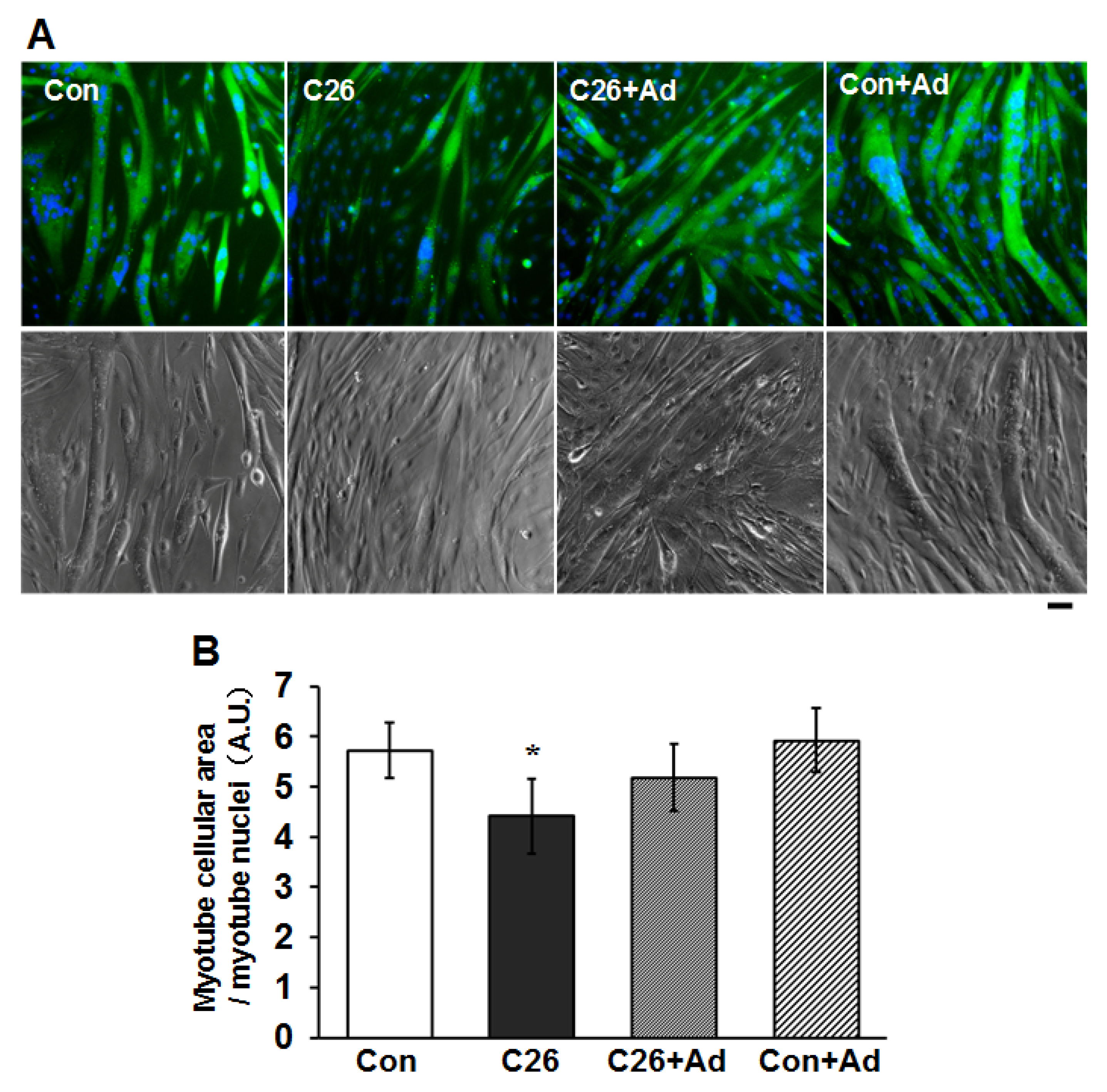
2.5. Recombinant Adiponectin Ameliorates the Atrophy of Myotubes
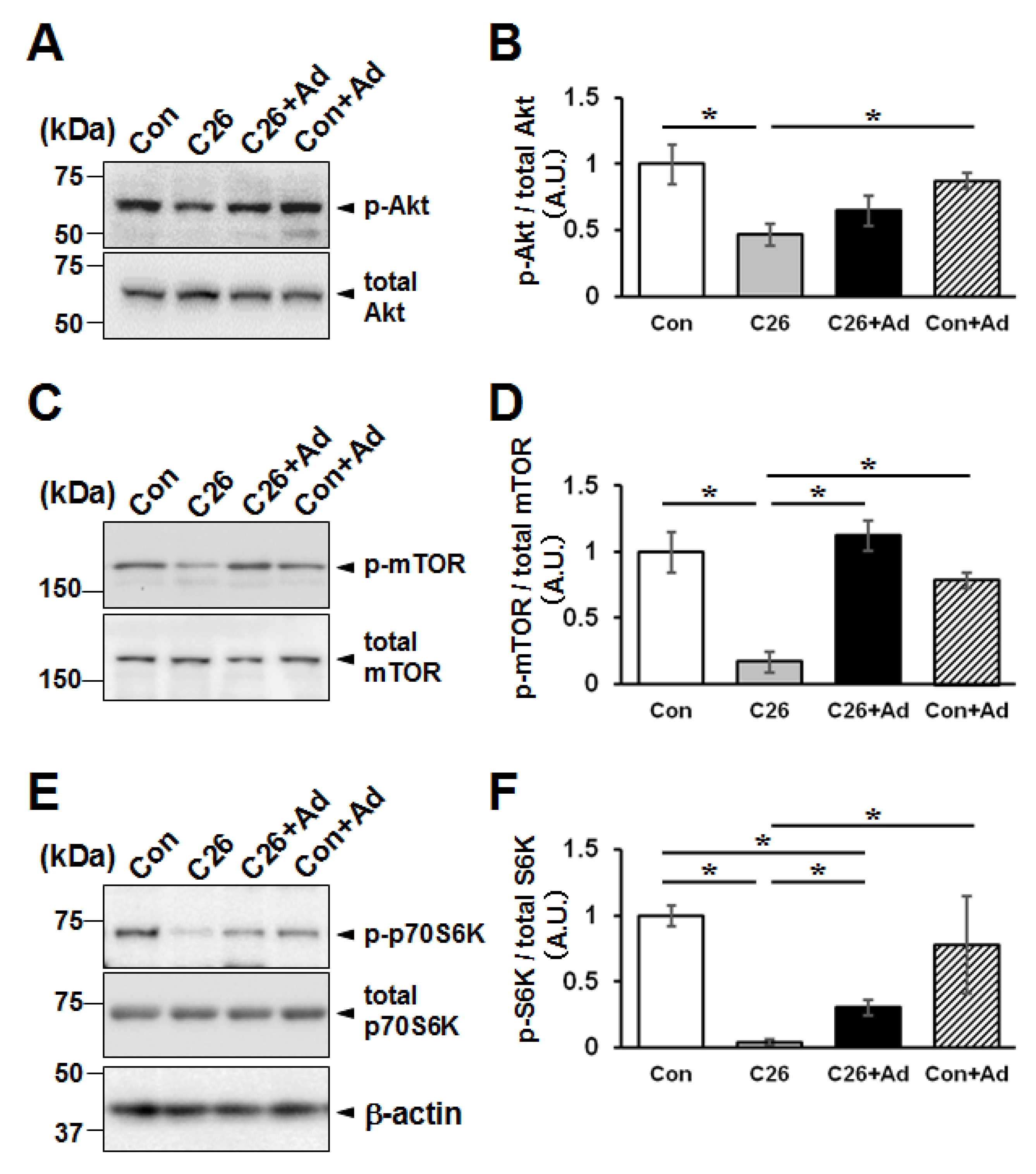
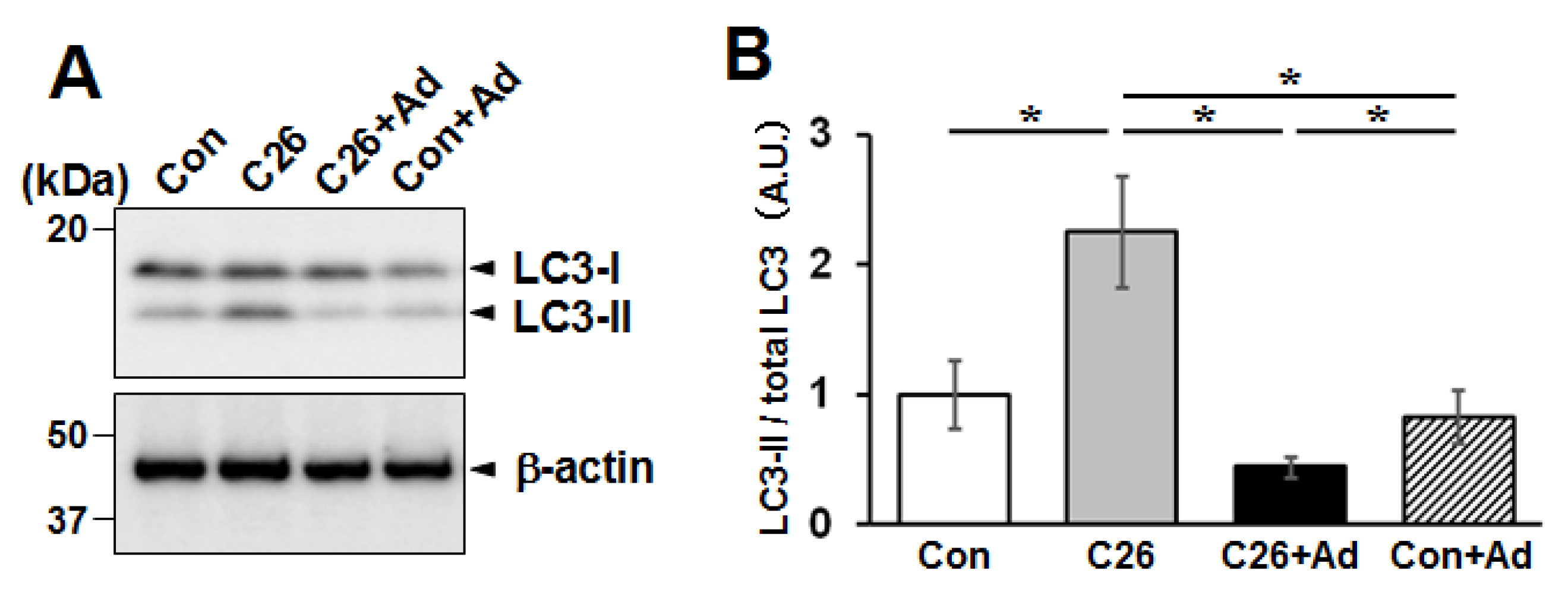
2.6. Adiponectin Treatment Alters the Protein Degradation/Synthesis Balance in Myotubes
3. Discussion
3.1. Activation of Protein Synthesis Signal by Aerobic Exercise during Cancer Cachexia
3.2. Protective Effect of Adiponectin on Cancer Cachexia-Induced Muscle Atrophy
3.3. Limitations and Conclusions
4. Materials and Methods
4.1. Animals
4.2. Cell Culture
4.3. Injection of C26 into Mice
4.4. Animal Experimental Design
4.5. Aerobic Exercise
4.6. Collection of Tissues
4.7. Analysis of Muscle Cross-Sectional Area
4.8. Western Blotting
4.9. Real-Time RT-PCR
4.10. Expression and Purification of Recombinant Adiponectin
4.11. In Vitro Cancer Cachexia-Induced Muscle Atrophy Model
4.12. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Acknowledgments
Conflicts of Interest
References
- Argilés, J.M.; Busquets, S.; Stemmler, B.; López-Soriano, F.J. Cancer cachexia: Understanding the molecular basis. Nat. Rev. Cancer 2014, 14, 754–762. [Google Scholar] [CrossRef] [PubMed]
- Tisdale, M.J. Biology of cachexia. J. Natl. Cancer Inst. 1997, 89, 1763–1773. [Google Scholar] [CrossRef]
- Esper, D.H.; Harb, W.A. The cancer cachexia syndrome: A review of metabolic and clinical manifestations. Nutr. Clin. Pract. 2005, 20, 369–376. [Google Scholar] [CrossRef] [PubMed]
- Mendes, M.C.S.; Pimentel, G.D.; Costa, F.O.; Carvalheira, J.B.C. Molecular and neuroendocrine mechanisms of cancer cachexia. J. Endocrinol. 2015, 226, R29–R43. [Google Scholar] [CrossRef] [Green Version]
- Vlassara, H.; Spiegel, R.J.; Doval, D.S.; Cerami, A. Reduced plasma lipoprotein lipase activity in patients with malignancy-associated weight loss. Horm. Metab. Res. 1986, 18, 698–703. [Google Scholar] [CrossRef] [PubMed]
- Pisters, P.W.; Pearlstone, D.B. Protein and amino acid metabolism in cancer cachexia: Investigative techniques and therapeutic interventions. Crit. Rev. Clin. Lab. Sci. 1993, 30, 223–272. [Google Scholar] [CrossRef]
- Argilés, J.M.; López-Soriano, F.J. The ubiquitin-dependent proteolytic pathway in skeletal muscle: Its role in pathological states. Trends Pharmacol. Sci. 1996, 17, 223–226. [Google Scholar] [CrossRef]
- Yuan, L.; Han, J.; Meng, Q.; Xi, Q.; Zhuang, Q.; Jiang, Y.; Han, Y.; Zhang, B.; Fang, J.; Wu, G. Muscle-specific E3 ubiquitin ligases are involved in muscle atrophy of cancer cachexia: An in vitro and in vivo study. Oncol. Rep. 2015, 33, 2261–2268. [Google Scholar] [CrossRef] [Green Version]
- Penna, F.; Ballarò, R.; Martinez-Cristobal, P.; Sala, D.; Sebastian, D.; Busquets, S.; Muscaritoli, M.; Argilés, J.M.; Costelli, P.; Zorzano, A. Autophagy exacerbates muscle wasting in cancer cachexia and impairs mitochondrial function. J. Mol. Biol. 2019, 431, 2674–2686. [Google Scholar] [CrossRef]
- Chacon-Cabrera, A.; Fermoselle, C.; Urtreger, A.J.; Mateu-Jimenez, M.; Diament, M.J.; Joffé, E.D.B.D.K.; Sandri, M.; Barreiro, E. Pharmacological strategies in lung cancer-induced cachexia: Effects on muscle proteolysis, autophagy, structure, and weakness. J. Cell. Physiol. 2014, 229, 1660–1672. [Google Scholar] [CrossRef] [Green Version]
- White, J.P.; Baynes, J.W.; Welle, S.L.; Kostek, M.C.; Matesic, L.E.; Sato, S.; Carson, J.A. The regulation of skeletal muscle protein turnover during the progression of cancer cachexia in the ApcMin/+ mouse. PLoS ONE 2011, 6, e24650. [Google Scholar] [CrossRef] [Green Version]
- Tanaka, M.; Sugimoto, K.; Fujimoto, T.; Xie, K.; Takahashi, T.; Akasaka, H.; Kurinami, H.; Yasunobe, Y.; Matsumoto, T.; Fujino, H.; et al. Preventive effects of low-intensity exercise on cancer cachexia–induced muscle atrophy. FASEB J. 2019, 33, 7852–7862. [Google Scholar] [CrossRef] [PubMed]
- Bowen, T.S.; Schuler, G.; Adams, V. Skeletal muscle wasting in cachexia and sarcopenia: Molecular pathophysiology and impact of exercise training. J. Cachexia Sarcopenia Muscle 2015, 6, 197–207. [Google Scholar] [CrossRef]
- Ábrigo, J.; Elorza, A.A.; Riedel, C.A.; Vilos, C.; Simon, F.; Cabrera, D.; Estrada, L.; Cabello-Verrugio, C. Role of oxidative stress as key regulator of muscle wasting during cachexia. Oxidative Med. Cell. Longev. 2018, 2018, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Sullivan-Gunn, M.J.; Campbell-O’Sullivan, S.P.; Tisdale, M.J.; Lewandowski, P.A. Decreased NADPH oxidase expression and antioxidant activity in cachectic skeletal muscle. J. Cachexia Sarcopenia Muscle 2011, 2, 181–188. [Google Scholar] [CrossRef] [Green Version]
- Ballarò, R.; Penna, F.; Pin, F.; Gómez-Cabrera, M.C.; Viña, J.; Costelli, P. Moderate exercise improves experimental cancer cachexia by modulating the redox homeostasis. Cancers 2019, 11, 285. [Google Scholar] [CrossRef] [Green Version]
- Xu, T.; Yang, X.; Wu, C.; Qiu, J.; Fang, Q.; Wang, L.; Yu, S.; Sun, H. Pyrroloquinoline quinone attenuates cachexia-induced muscle atrophy via suppression of reactive oxygen species. J. Thorac. Dis. 2018, 10, 2752–2759. [Google Scholar] [CrossRef] [PubMed]
- Molinari, F.; Pin, F.; Gorini, S.; Chiandotto, S.; Pontecorvo, L.; Penna, F.; Rizzuto, E.; Pisu, S.; Musarò, A.; Costelli, P.; et al. The mitochondrial metabolic reprogramming agent trimetazidine as an ‘exercise mimetic’ in cachectic C26-bearing mice. J. Cachexia Sarcopenia Muscle 2017, 8, 954–973. [Google Scholar] [CrossRef]
- Hardee, J.P.; Montalvo, R.N.; Carson, J.A. Linking cancer cachexia-induced anabolic resistance to skeletal muscle oxidative metabolism. Oxidative Med. Cell. Longev. 2017, 2017, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Pigna, E.; Berardi, E.; Aulino, P.; Rizzuto, E.; Zampieri, S.; Carraro, U.; Kern, H.; Merigliano, S.; Gruppo, M.; Mericskay, M.; et al. Aerobic exercise and pharmacological treatments counteract cachexia by modulating autophagy in colon cancer. Sci. Rep. 2016, 6, 26991. [Google Scholar] [CrossRef]
- Khamoui, A.V.; Park, B.-S.; Kim, D.-H.; Yeh, M.-C.; Oh, S.-L.; Elam, M.L.; Jo, E.; Arjmandi, B.H.; Salazar, G.; Grant, S.C.; et al. Aerobic and resistance training dependent skeletal muscle plasticity in the colon-26 murine model of cancer cachexia. Metabolism 2016, 65, 685–698. [Google Scholar] [CrossRef] [Green Version]
- Ouchi, N.; Kihara, S.; Funahashi, T.; Matsuzawa, Y.; Walsh, K. Obesity, adiponectin and vascular inflammatory disease. Curr. Opin. Lipidol. 2003, 14, 561–566. [Google Scholar] [CrossRef]
- Ruan, H.; Dong, L.Q. Adiponectin signaling and function in insulin target tissues. J. Mol. Cell Biol. 2016, 8, 101–109. [Google Scholar] [CrossRef] [Green Version]
- Lee, S.; Park, Y.; Dellsperger, K.C.; Zhang, C. Exercise training improves endothelial function via adiponectin-dependent and independent pathways in type 2 diabetic mice. Am. J. Physiol. Heart Circ. Physiol. 2011, 301, H306–H314. [Google Scholar] [CrossRef] [Green Version]
- Yanai, H.; Yoshida, H. Beneficial effects of adiponectin on glucose and lipid metabolism and atherosclerotic progression: Mechanisms and perspectives. Int. J. Mol. Sci. 2019, 20, 1190. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Y.; Sweeney, G. Adiponectin action in skeletal muscle. Best Pract. Res. Clin. Endocrinol. Metab. 2014, 28, 33–41. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Chewchuk, S.; Lavigne, C.; Brûlé, S.; Pilon, G.; Houde, V.; Xu, A.; Marette, A.; Sweeney, G. Functional significance of skeletal muscle adiponectin production, changes in animal models of obesity and diabetes, and regulation by rosiglitazone treatment. Am. J. Physiol. Endocrinol. Metab. 2009, 297, E657–E664. [Google Scholar] [CrossRef] [PubMed]
- Yamauchi, T.; Kamon, J.; Ito, Y.; Tsuchida, A.; Yokomizo, T.; Kita, S.; Sugiyama, T.; Miyagishi, M.; Hara, K.; Tsunoda, M.; et al. Cloning of adiponectin receptors that mediate antidiabetic metabolic effects. Nature 2003, 423, 762–769. [Google Scholar] [CrossRef]
- Hug, C.; Wang, J.; Ahmad, N.S.; Bogan, J.S.; Tsao, T.-S.; Lodish, H.F. T-cadherin is a receptor for hexameric and high-molecular-weight forms of Acrp30/adiponectin. Proc. Natl. Acad. Sci. USA 2004, 101, 10308–10313. [Google Scholar] [CrossRef] [Green Version]
- Mao, X.; Kikani, C.K.; Riojas, R.A.; Langlais, P.; Wang, L.; Ramos, F.J.; Fang, Q.; Christ-Roberts, C.Y.; Hong, J.Y.; Kim, R.-Y.; et al. APPL1 binds to adiponectin receptors and mediates adiponectin signalling and function. Nat. Cell Biol. 2006, 8, 516–523. [Google Scholar] [CrossRef] [PubMed]
- Fiaschi, T.; Cirelli, D.; Comito, G.; Gelmini, S.; Ramponi, G.; Serio, M.; Chiarugi, P. Globular adiponectin induces differentiation and fusion of skeletal muscle cells. Cell Res. 2009, 19, 584–597. [Google Scholar] [CrossRef] [PubMed]
- Singh, A.K.; Shree, S.; Chattopadhyay, S.; Kumar, S.; Gurjar, A.; Kushwaha, S.; Kumar, H.; Trivedi, A.K.; Chattopadhyay, N.; Maurya, R.; et al. Small molecule adiponectin receptor agonist GTDF protects against skeletal muscle atrophy. Mol. Cell. Endocrinol. 2017, 439, 273–285. [Google Scholar] [CrossRef]
- Tanaka, Y.; Kita, S.; Nishizawa, H.; Fukuda, S.; Fujishima, Y.; Obata, Y.; Nagao, H.; Masuda, S.; Nakamura, Y.; Shimizu, Y.; et al. Adiponectin promotes muscle regeneration through binding to T-cadherin. Sci. Rep. 2019, 9, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Jortay, J.; Senou, M.; Abou-Samra, M.; Noel, L.; Robert, A.; Many, M.C.; Brichard, S.M. Adiponectin and skeletal muscle: Pathophysiological implications in metabolic stress. Am. J. Pathol. 2012, 181, 245–256. [Google Scholar] [CrossRef] [PubMed]
- Krause, M.P.; Liu, Y.; Vu, V.; Chan, L.; Xu, A.; Riddell, M.C.; Sweeney, G.; Hawke, T.J. Adiponectin is expressed by skeletal muscle fibers and influences muscle phenotype and function. Am. J. Physiol. Cell Physiol. 2008, 295, C203–C212. [Google Scholar] [CrossRef]
- Betof, A.S.; Lascola, C.D.; Weitzel, D.; Landon, C.; Scarbrough, P.M.; Devi, G.R.; Palmer, G.; Jones, L.W.; Dewhirst, M.W. Modulation of murine breast tumor vascularity, hypoxia, and chemotherapeutic response by exercise. J. Natl. Cancer Inst. 2015, 107, 040. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, C.; Mao, X.; Wang, L.; Liu, M.; Wetzel, M.D.; Guan, K.L.; Dong, L.Q.; Liu, F. Adiponectin sensitizes insulin signaling by reducing p70 S6 Kinase-mediated serine phosphorylation of IRS-1. J. Biol. Chem. 2007, 282, 7991–7996. [Google Scholar] [CrossRef] [Green Version]
- Mihaylova, M.M.; Shaw, R.J. The AMPK signalling pathway coordinates cell growth, autophagy and metabolism. Nat. Cell Biol. 2011, 13, 1016–1023. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, E.K.; Clavarino, G.; Ceppi, M.; Pierre, P. SUnSET, a nonradioactive method to monitor protein synthesis. Nat. Methods 2009, 6, 275–277. [Google Scholar] [CrossRef]
- Kabeya, Y.; Mizushima, N.; Ueno, T.; Yamamoto, A.; Kirisako, T.; Noda, T.; Kominami, E.; Ohsumi, Y.; Yoshimori, T. LC3, a mammalian homologue of yeast Apg8p, is localized in autophagosome membranes after processing. EMBO J. 2000, 19, 5720–5728. [Google Scholar] [CrossRef] [PubMed]
- Tsoli, M.; Schweiger, M.; Vanniasinghe, A.S.; Painter, A.; Zechner, R.; Clarke, S.; Robertson, G. Depletion of white adipose tissue in cancer cachexia syndrome is associated with inflammatory signaling and disrupted circadian regulation. PLoS ONE 2014, 9, e92966. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Pessin, J.E. Mechanisms for fiber-type specificity of skeletal muscle atrophy. Curr. Opin. Clin. Nutr. Metab. Care 2013, 16, 243–250. [Google Scholar] [CrossRef]
- Nikooie, R.; Moflehi, D.; Zand, S. Lactate regulates autophagy through ROS-mediated activation of ERK1/2/m-TOR/p-70S6K pathway in skeletal muscle. J. Cell Commun. Signal. 2021, 15, 107–123. [Google Scholar] [CrossRef]
- Farias, J.; Maggi, R.; Tromm, C.; Silva, L.; Luciano, T.; Marques, S.; Lira, F.; De Souza, C.; Pinho, R. Exercise training performed simultaneously to a high-fat diet reduces the degree of insulin resistance and improves adipoR1-2/APPL1 protein levels in mice. Lipids Health Dis. 2012, 11, 134. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aapro, M.; Arends, J.; Bozzetti, F.; Fearon, K.; Grunberg, S.M.; Herrstedt, J.; Hopkinson, J.; Jacquelin-Ravel, N.; Jatoi, A.; Kaasa, S.; et al. Early recognition of malnutrition and cachexia in the cancer patient: A position paper of a European School of Oncology Task Force. Ann. Oncol. 2014, 25, 1492–1499. [Google Scholar] [CrossRef] [PubMed]
- Maddocks, M.; Hopkinson, J.; Conibear, J.; Reeves, A.; Shaw, C.; Fearon, K.C. Practical multimodal care for cancer cachexia. Curr. Opin. Support. Palliat. Care 2016, 10, 298–305. [Google Scholar] [CrossRef] [Green Version]








Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Morinaga, M.; Sako, N.; Isobe, M.; Lee-Hotta, S.; Sugiura, H.; Kametaka, S. Aerobic Exercise Ameliorates Cancer Cachexia-Induced Muscle Wasting through Adiponectin Signaling. Int. J. Mol. Sci. 2021, 22, 3110. https://doi.org/10.3390/ijms22063110
Morinaga M, Sako N, Isobe M, Lee-Hotta S, Sugiura H, Kametaka S. Aerobic Exercise Ameliorates Cancer Cachexia-Induced Muscle Wasting through Adiponectin Signaling. International Journal of Molecular Sciences. 2021; 22(6):3110. https://doi.org/10.3390/ijms22063110
Chicago/Turabian StyleMorinaga, Makoto, Naoki Sako, Mari Isobe, Sachiko Lee-Hotta, Hideshi Sugiura, and Satoshi Kametaka. 2021. "Aerobic Exercise Ameliorates Cancer Cachexia-Induced Muscle Wasting through Adiponectin Signaling" International Journal of Molecular Sciences 22, no. 6: 3110. https://doi.org/10.3390/ijms22063110