
Molecular Alterations in Sporadic and SOD1-ALS Immortalized Lymphocytes: Towards a Personalized Therapy

 , , , , and
, , , , and 
Abstract
:
1. Introduction
2. Results
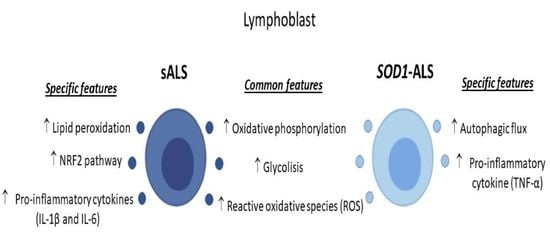
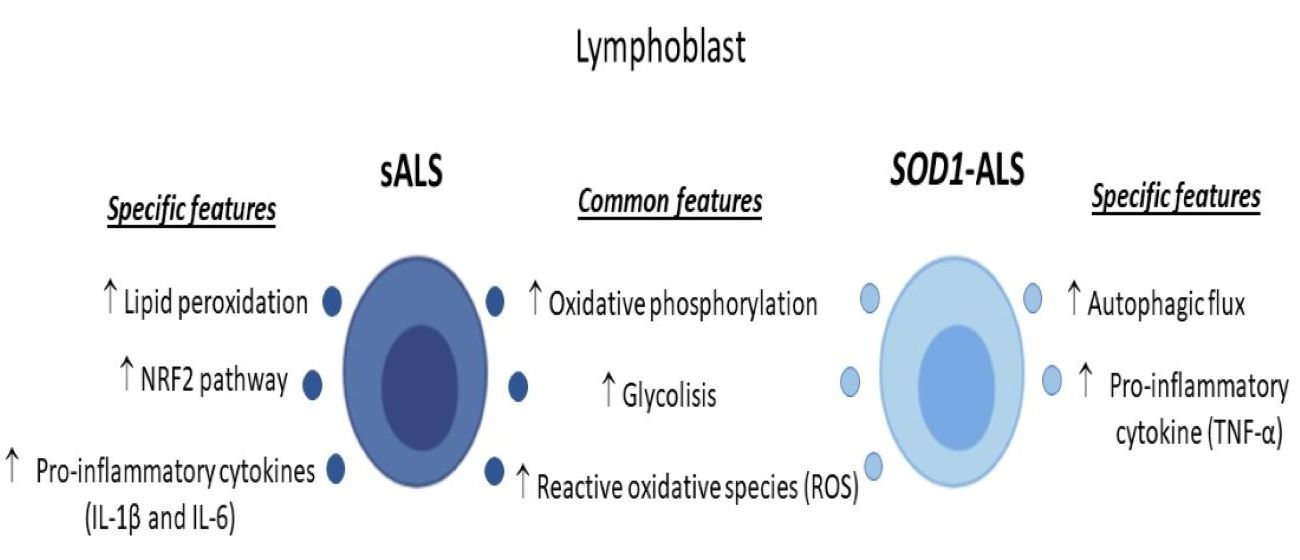
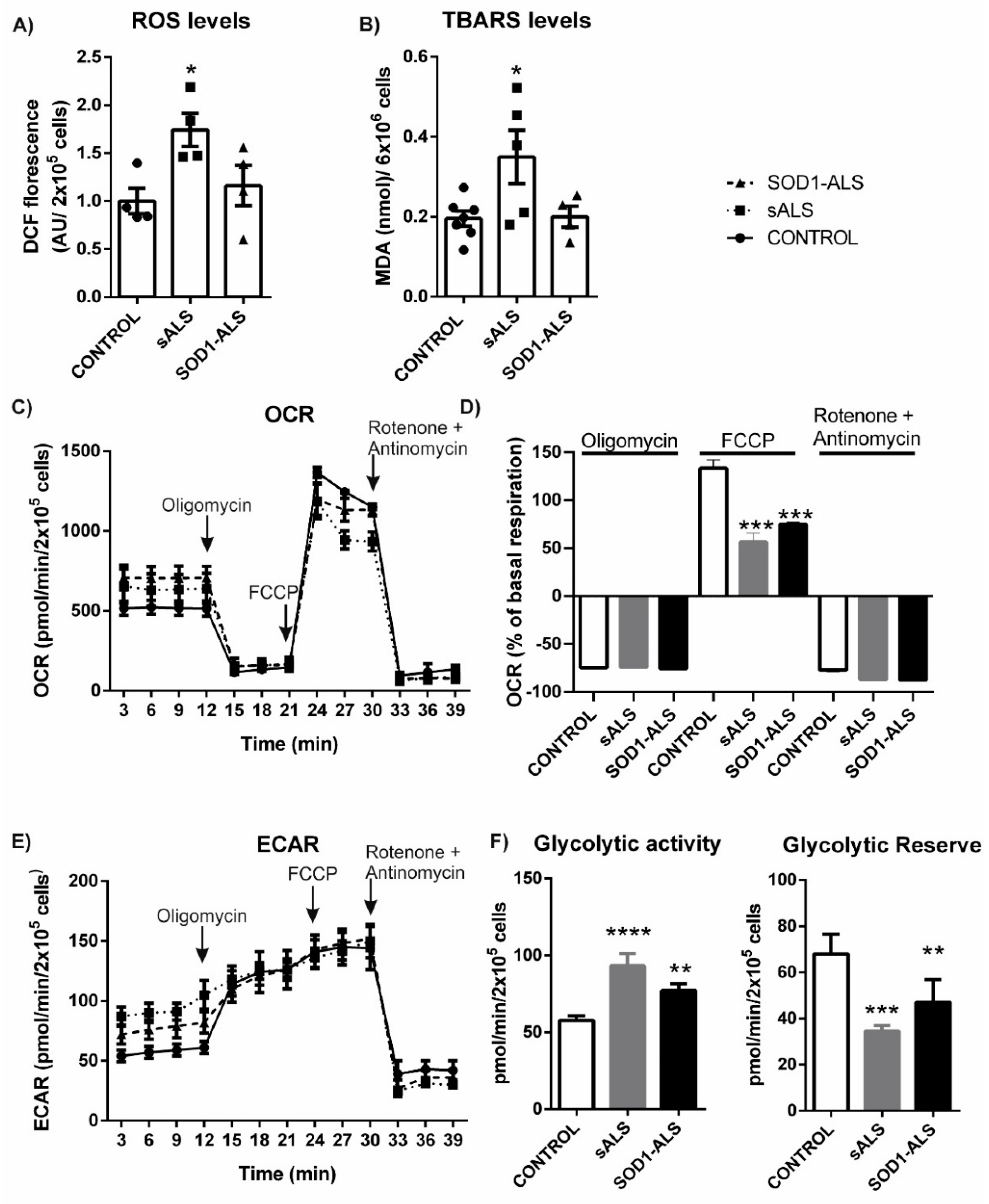
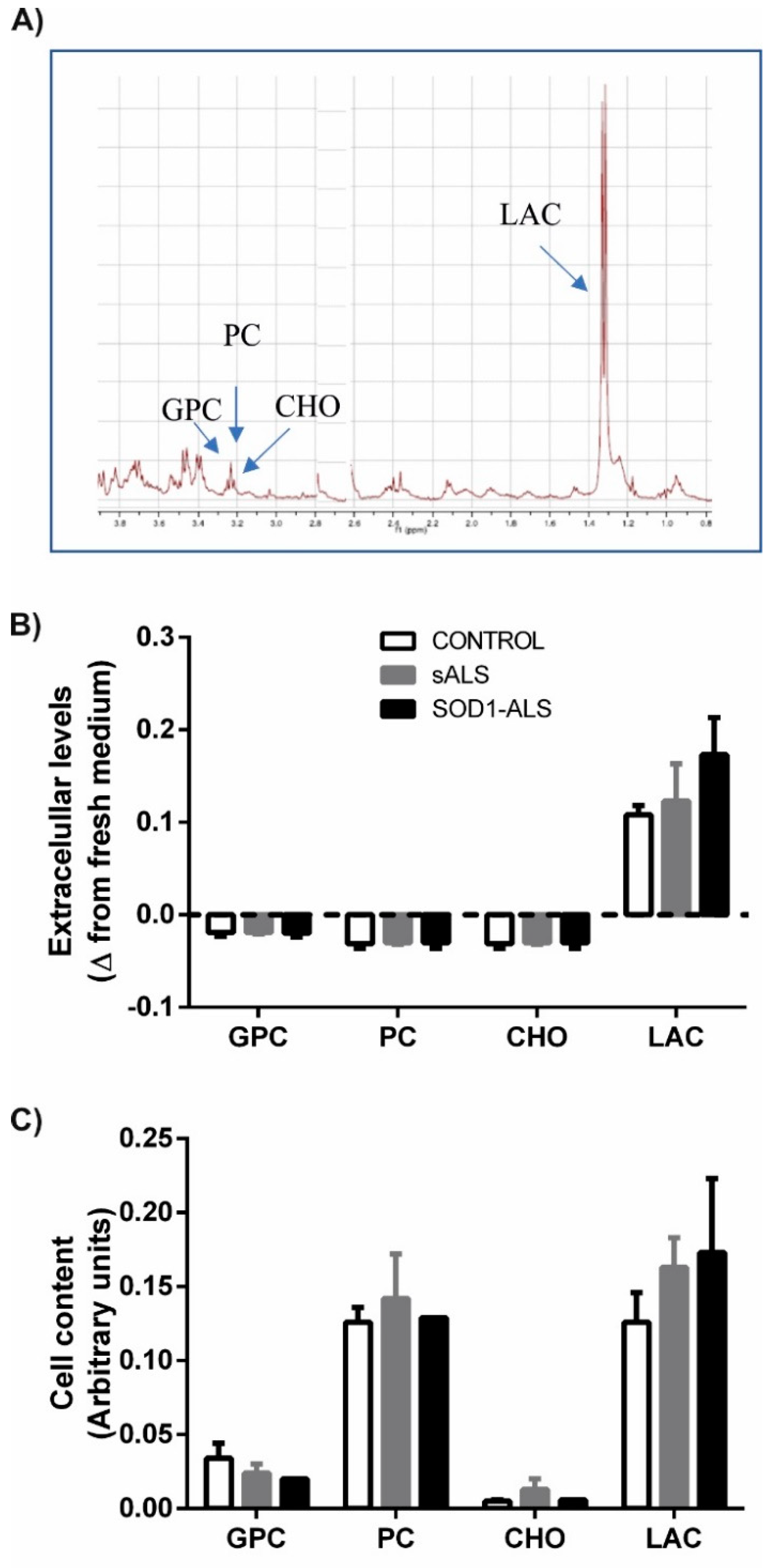
2.1. Oxidative Stress, Bioenergetic, and Metabolic Profiles of Lymphoblasts from Healthy Subjects, and Sporadic or SOD1 Mutation Carrier ALS Patients
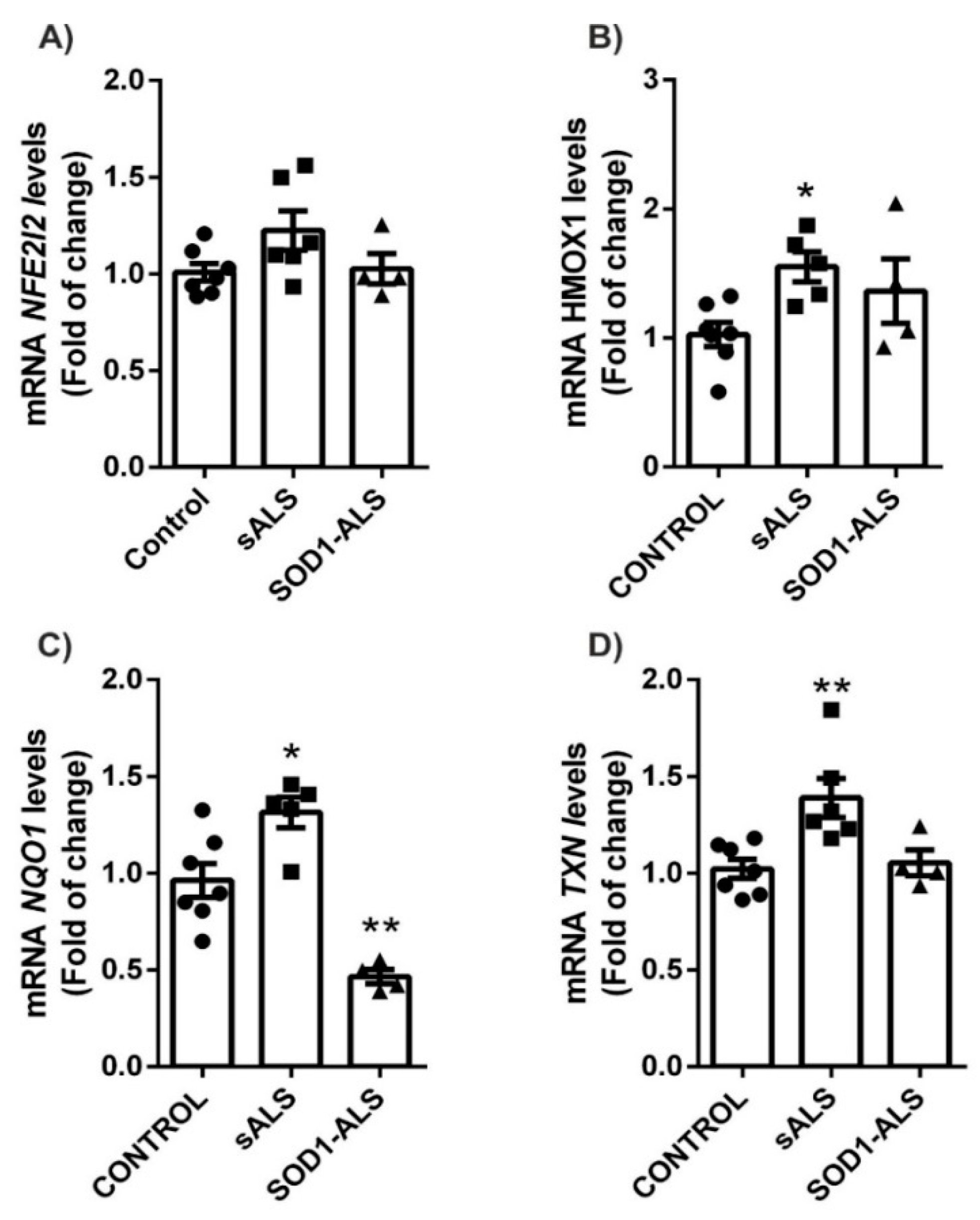
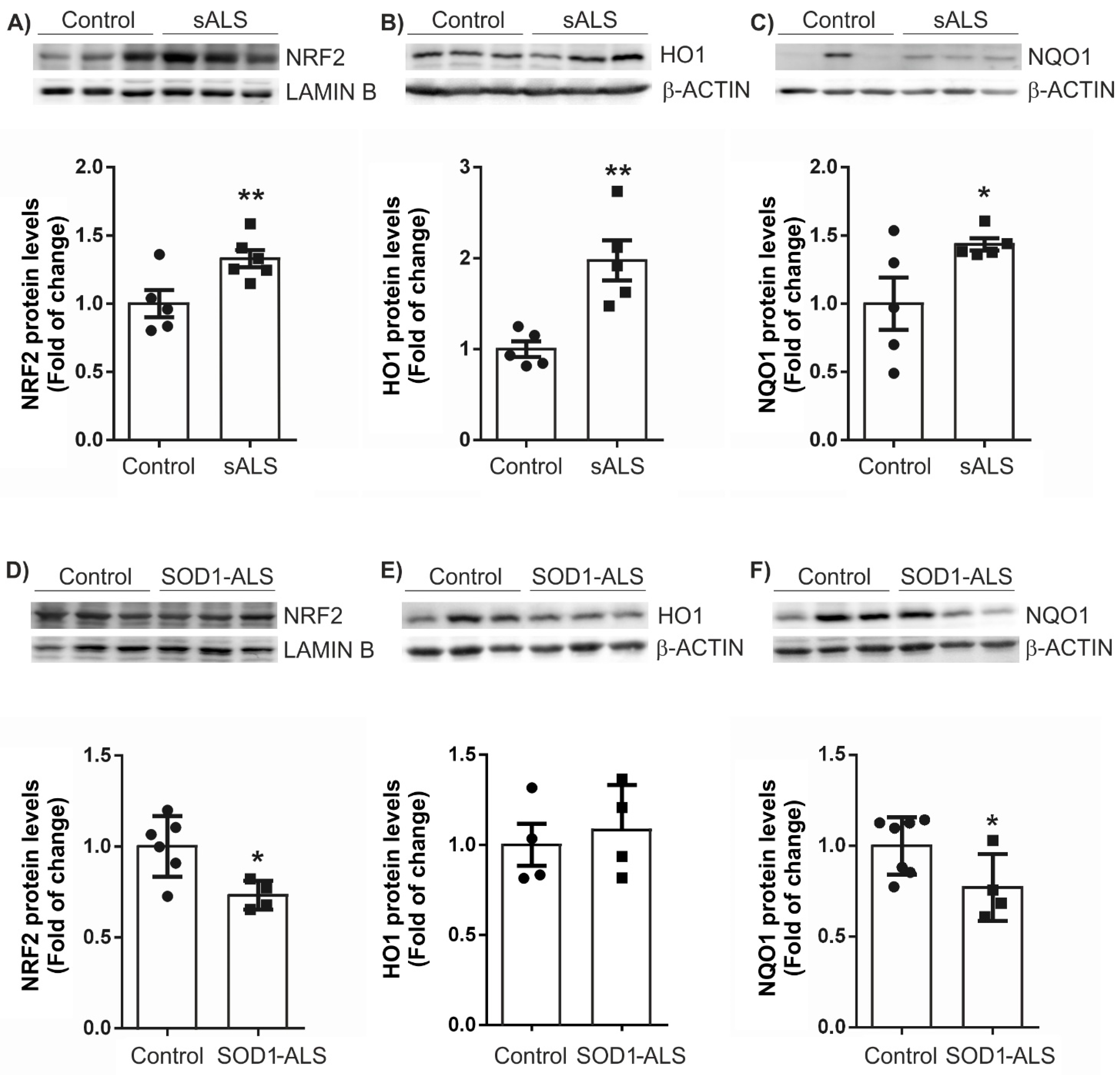
2.2. Different Profile of NRF2-Pathway between Sporadic and SOD1 Mutation Carriers ALS Patient’s Lymphoblast
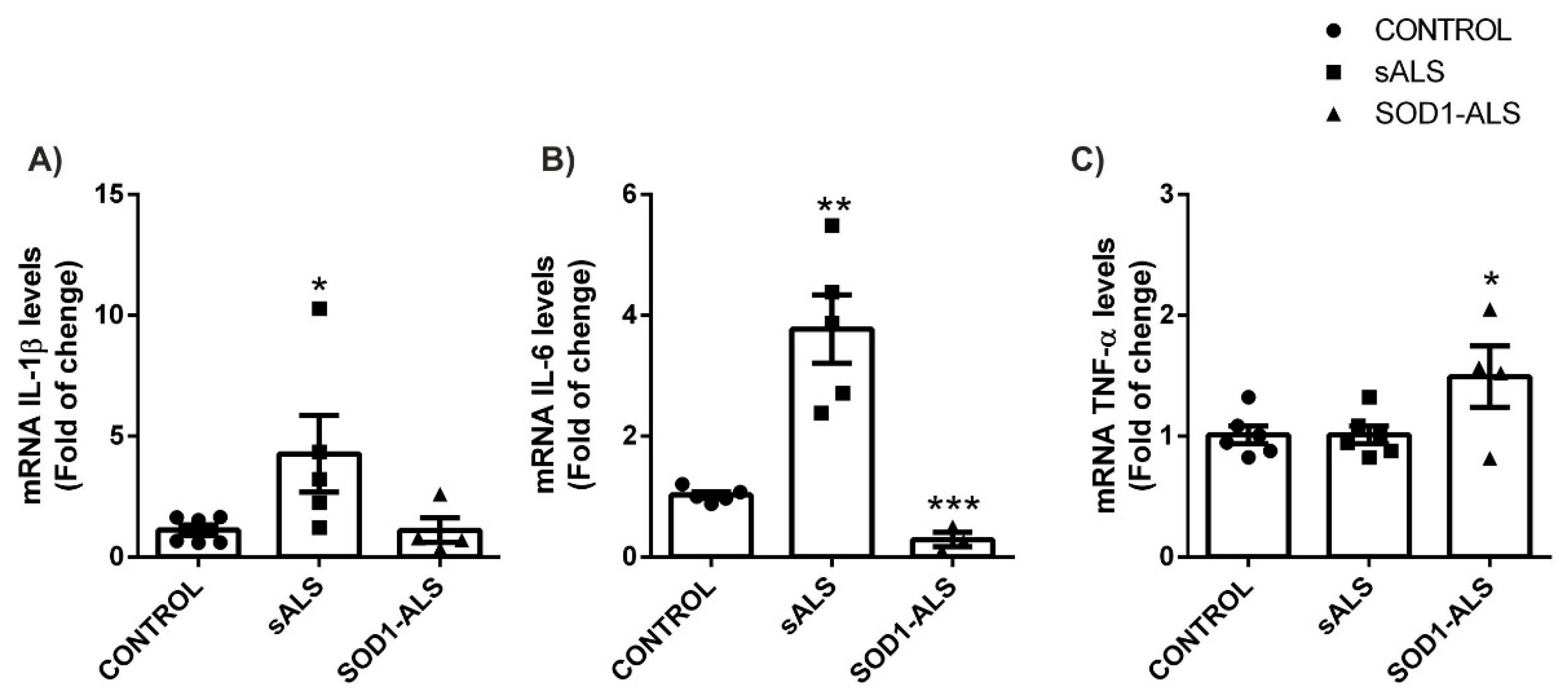
2.3. Sporadic and SOD1-Mutation Carriers ALS Patients Lymphoblast Showed Distinct Pro-Inflammatory Cytokines Profile
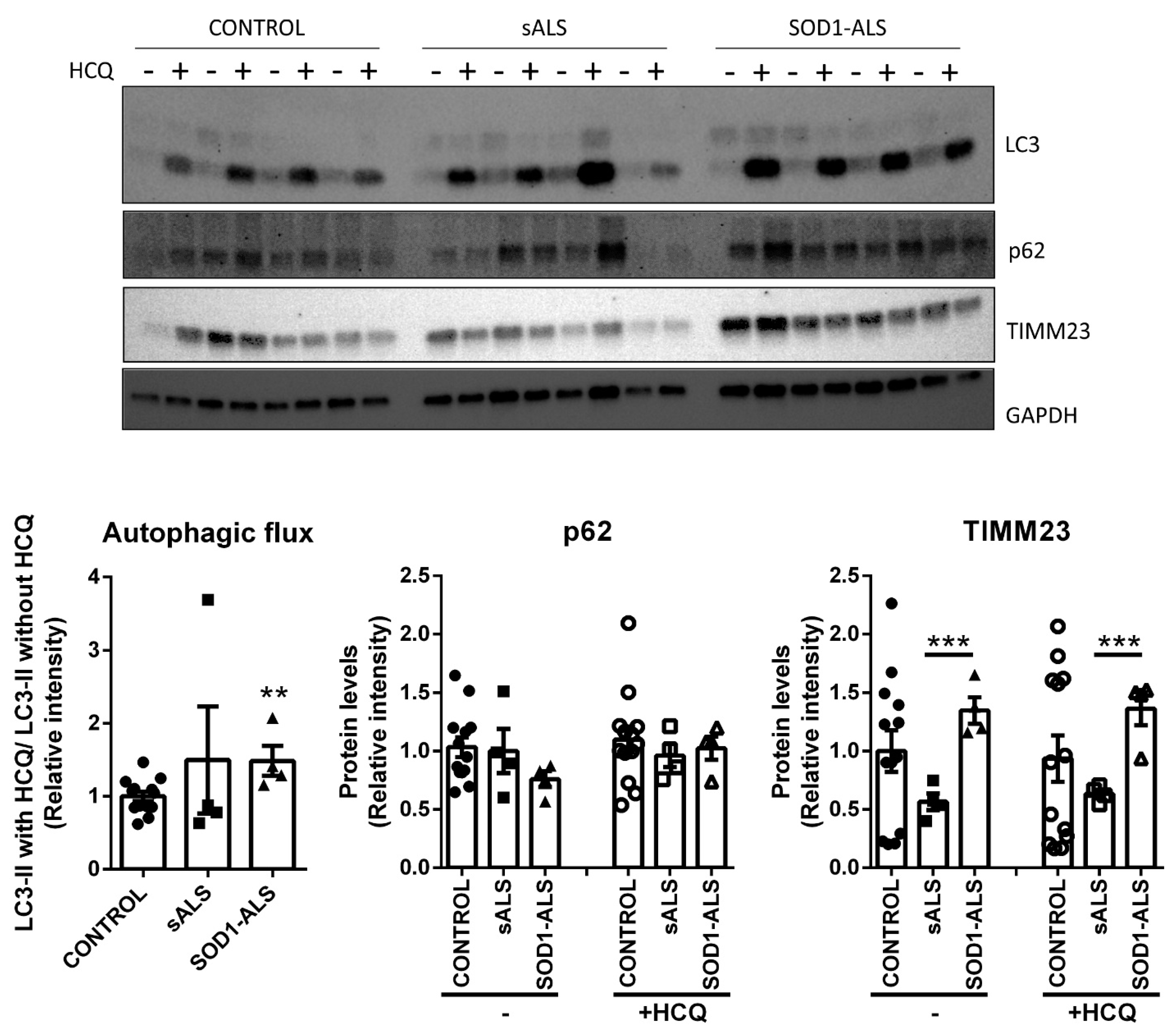
2.4. Different Autophagic Profile between Sporadic and SOD1 Mutation Carriers ALS Patient’s Lymphoblasts
3. Discussion
4. Materials and Methods
4.1. Materials
4.2. Lymphoblastic Cell Lines
4.3. Measurement of Reactive Oxygen Species (ROS) and Thiobarbituric Acid Reactive Substances (TBARs)
4.4. Oxygen Consumption and Extracellular Acidification Rates Measurements
4.5. HRMAS-NMR Acquisition and Processing
4.6. Analysis of mRNA Levels by Quantitative Real-Time PCR
4.7. Immunoblotting
4.8. Statistical Analyses
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Masrori, P.; Van Damme, P. Amyotrophic lateral sclerosis: A clinical review. Eur. J. Neurol. 2020, 27, 1918–1929. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Cheng, X.; Zhong, S.; Zhang, X.; Liu, C.; Liu, F.; Zhao, C. Peripheral and central nervous system immune response crosstalk in amyotrophic lateral sclerosis. Front. Neurosci. 2020, 14, 575. [Google Scholar] [CrossRef] [PubMed]
- Riancho, J.; Arozamena, S.; De Munaín, A.L. Dermic-derived fibroblasts for the study of amyotrophic lateral sclerosis. Neural Regen. Res. 2020, 15, 2043–2044. [Google Scholar] [CrossRef]
- De Marco, G.; Lupino, E.; Calvo, A.; Moglia, C.; Buccinnà, B.; Grifoni, S.; Ramondetti, C.; Lomartire, A.; Rinaudo, M.T.; Piccinini, M.; et al. Cytoplasmic accumulation of TDP-43 in circulating lymphomonocytes of ALS patients with and without TARDBP mutations. Acta Neuropathol. 2011, 121, 611–622. [Google Scholar] [CrossRef] [PubMed]
- Posa, D.; Martínez-González, L.; Bartolomé, F.; Nagaraj, S.; Porras, G.; Martínez, A.; Martín-Requero, Á. Re-capitulation of pathological TDP-43 features in immortalized lymphocytes from sporadic ALS patients. Mol. Neurobiol. 2019, 56, 2424–2432. [Google Scholar] [CrossRef] [PubMed]
- Bartolomé, F.; Muñoz, U.; Esteras, N.; Esteban, J.; Bermejo-Pareja, F.; Martín-Requero, A. Distinct regulation of cell cycle and survival in lymphocytes from patients with Alzheimer’s disease and amyotrophic lateral sclerosis. Int. J. Clin. Exp. Pathol. 2009, 2, 390–398. [Google Scholar]
- Cereda, C.; Leoni, E.; Milani, P.; Pansarasa, O.; Mazzini, G.; Guareschi, S.; Alvisi, E.; Ghiroldi, A.; Diamanti, L.; Bernuzzi, S.; et al. Altered intracellular localization of SOD1 in leukocytes from patients with sporadic amyotrophic lateral sclerosis. PLoS ONE 2013, 8, e75916. [Google Scholar] [CrossRef] [Green Version]
- Nardo, G.; Pozzi, S.; Pignataro, M.; Lauranzano, E.; Spano, G.; Garbelli, S.; Mantovani, S.; Marinou, K.; Papetti, L.; Monteforte, M.; et al. Amyotrophic lateral sclerosis multiprotein biomarkers in peripheral blood mononuclear cells. PLoS ONE 2011, 6, e25545. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pansarasa, O.; Bordoni, M.; Drufuca, L.; Diamanti, L.; Sproviero, D.; Trotti, R.; Bernuzzi, S.; La Salvia, S.; Gagliardi, S.; Ceroni, M.; et al. Lymphoblastoid cell lines as a model to understand amyotrophic lateral sclerosis disease mechanisms. Dis. Model. Mech. 2018, 11, dmm031625. [Google Scholar] [CrossRef] [Green Version]
- Martínez-González, L.; Rodríguez-Cueto, C.; Cabezudo, D.; Bartolomé, F.; Andrés-Benito, P.; Ferrer, I.; Gil, C.; Martín-Requero, Á.; Fernández-Ruiz, J.; Martínez, A.; et al. Motor neuron preservation and decrease of in vivo TDP-43 phosphorylation by protein CK-1δ kinase inhibitor treatment. Sci. Rep. 2020, 10, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Vaca, G.; Martinez-Gonzalez, L.; Fernandez, A.; Rojas-Prats, E.; Porras, G.; Cuevas, E.P.; Gil, C.; Martinez, A.; Martin-Requero, Á. Therapeutic potential of novel cell division cycle kinase 7 inhibitors on TDP-43-related pathogenesis such as frontotemporal lobar degeneration (FTLD) and amyotrophic lateral sclerosis (ALS). J. Neurochem. 2021, 156, 379–390. [Google Scholar] [CrossRef]
- Muñoz, U.; Bartolomé, F.; Bermejo, F.; Martín-Requero, A. Enhanced proteasome-dependent degradation of the CDK inhibitor p27(kip1) in immortalized lymphocytes from Alzheimer’s dementia patients. Neurobiol. Aging 2008, 29, 1474–1484. [Google Scholar] [CrossRef] [Green Version]
- Bartolomé, F.; de Las Cuevas, N.; Muñoz, U.; Bermejo, F.; Martín-Requero, A. Impaired apoptosis in lymphoblasts from Alzheimer’s disease patients: Cross-talk of Ca2+/calmodulin and ERK1/2 signaling pathways. Cell. Mol. Life Sci. 2007, 64, 1437–1448. [Google Scholar] [CrossRef] [Green Version]
- Petrov, D.; Mansfield, C.; Moussy, A.; Hermine, O. ALS clinical trials review: 20 Years of failure. Are we any closer to registering a new treatment? Front. Aging Neurosci. 2017, 9, 68. [Google Scholar] [CrossRef] [Green Version]
- Riva, N.; Agosta, F.; Lunetta, C.; Filippi, M.; Quattrini, A. Recent advances in amyotrophic lateral sclerosis. J. Neurol. 2016, 263, 1241–1254. [Google Scholar] [CrossRef] [Green Version]
- Mejzini, R.; Flynn, L.L.; Pitout, I.L.; Fletcher, S.; Wilton, S.D.; Akkari, P.A. ALS genetics, mechanisms, and therapeutics: Where are we now? Front. Neurosci. 2019, 13, 1310. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taylor, P.J.; Brown, H.R.; Cleveland, W.D. Decoding ALS: From genes to mechanism. Nature 2016, 539, 197–206. [Google Scholar] [CrossRef] [Green Version]
- Kiernan, M.C.; Vucic, S.; Cheah, B.C.; Turner, M.R.; Eisen, A.; Hardiman, O.; Burrell, J.R.; Zoing, M.C. Amyotrophic lateral sclerosis. Lancet 2011, 377, 942–955. [Google Scholar] [CrossRef] [Green Version]
- Kumar, S.; Krishna Chaitanya, R.; Preedy, V.R. Chapter 20—Assessment of antioxidant potential of dietary components. In HIV/AIDS; Preedy, V.R., Watson, R.R., Eds.; Academic Press: Cambridge, MA, USA, 2018; pp. 239–253. [Google Scholar]
- Baird, L.; Yamamoto, M. The molecular mechanisms regulating the KEAP1-NRF2 pathway. Mol. Cell. Biol. 2020, 40, e00099-20. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.; Lu, Y.; Qi, F.; Su, Q.; Wang, L.; You, C.; Che, F.; Yu, J. Effect of the human SOD1-G93A gene on the Nrf2/ARE signaling pathway in NSC-34 cells. Mol. Med. Rep. 2014, 9, 2453–2458. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, J.; Wang, F. Role of neuroinflammation in amyotrophic lateral sclerosis: Cellular mechanisms and therapeutic implications. Front. Immunol. 2017, 8, 1005. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Olesen, M.N.; Wuolikainen, A.; Nilsson, A.C.; Wirenfeldt, M.; Forsberg, K.; Madsen, J.S.; Lillevang, S.T.; Brandslund, I.; Andersen, P.M.; Asgari, N. Inflammatory profiles relate to survival in subtypes of amyotrophic lateral sclerosis. Neurol. Neuroimmunol. Neuroinflamm. 2020, 7, e697. [Google Scholar] [CrossRef] [Green Version]
- Amsen, D.; de Visser, K.E.; Town, T. Approaches to determine expression of inflammatory cytokines. Methods Mol. Biol. 2009, 511, 107–142. [Google Scholar] [PubMed] [Green Version]
- Cohen-Kaplan, V.; Livneh, I.; Avni, N.; Fabre, B.; Ziv, T.; Kwon, Y.T.; Ciechanover, A. p62- and ubiquitin-dependent stress-induced autophagy of the mammalian 26S proteasome. Proc. Natl. Acad. Sci. USA 2016, 113, E7490–E7499. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klionsky, D.J.; Abdelmohsen, K.; Abe, A.; Abedin, M.J.; Abeliovich, H.; Acevedo Arozena, A.; Adachi, H.; Adams, C.M.; Adams, P.D.; Adeli, K.; et al. Guidelines for the use and interpretation of assays for monitoring autophagy. Autophagy 2016, 12, 1–222. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barber, S.C.; Shaw, P.J. Oxidative stress in ALS: Key role in motor neuron injury and therapeutic target. Free Radic. Biol. Med. 2010, 48, 629–641. [Google Scholar] [CrossRef]
- Su, L.-J.; Zhang, J.-H.; Gomez, H.; Murugan, R.; Hong, X.; Xu, D.; Jiang, F.; Peng, Z.-Y. Reactive oxygen species-induced lipid peroxidation in apoptosis, autophagy, and ferroptosis. Oxid. Med. Cell. Longev. 2019, 2019, 5080843. [Google Scholar] [CrossRef] [Green Version]
- Que, X.; Hung, M.Y.; Yeang, C.; Gonen, A.; Prohaska, T.A.; Sun, X.; Diehl, C.; Määttä, A.; Gaddis, D.E.; Bowden, K.; et al. Oxidized phospholipids are proinflammatory and proatherogenic in hypercholesterolaemic mice. Nature 2018, 558, 301–306. [Google Scholar] [CrossRef]
- Konrad, C.; Kawamata, H.; Bredvik, K.G.; Arreguin, A.J.; Cajamarca, S.A.; Hupf, J.C.; Ravits, J.M.; Miller, T.M.; Maragakis, N.J.; Hales, C.M.; et al. Fibroblast bioenergetics to classify amyotrophic lateral sclerosis patients. Mol. Neurodegener. 2017, 12, 76. [Google Scholar] [CrossRef] [Green Version]
- Vallée, A.; LeCarpentier, Y.; Guillevin, R.; Vallée, J.-N. Aerobic glycolysis in amyotrophic lateral sclerosis and Huntington’s disease. Rev. Neurosci. 2018, 29, 547–555. [Google Scholar] [CrossRef]
- Tonelli, C.; Chio, I.I.C.; Tuveson, D.A. Transcriptional regulation by Nrf2. Antioxid. Redox Signal. 2018, 29, 1727–1745. [Google Scholar] [CrossRef] [Green Version]
- Castro-Sánchez, S.; García-Yagüe, Á.J.; Kügler, S.; Lastres-Becker, I. CX3CR1-deficient microglia shows impaired signalling of the transcription factor NRF2: Implications in tauopathies. Redox Biol. 2019, 22, 101118. [Google Scholar] [CrossRef] [PubMed]
- Lastres-Becker, I.; Innamorato, N.G.; Jaworski, T.; Rábano, A.; Kügler, S.; Van Leuven, F.; Cuadrado, A. Fractalkine activates NRF2/NFE2L2 and heme oxygenase 1 to restrain tauopathy-induced microgliosis. Brain 2013, 137, 78–91. [Google Scholar] [CrossRef]
- Lastres-Becker, I.; Ulusoy, A.; Innamorato, N.G.; Sahin, G.; Rábano, A.; Kirik, D.; Cuadrado, A. α-Synuclein expression and Nrf2 deficiency cooperate to aggravate protein aggregation, neuronal death and inflammation in early-stage Parkinson’s disease. Hum. Mol. Genet. 2012, 21, 3173–3192. [Google Scholar] [CrossRef] [Green Version]
- La Rosa, P.; Petrillo, S.; Turchi, R.; Berardinelli, F.; Schirinzi, T.; Vasco, G.; Lettieri-Barbato, D.; Fiorenza, M.T.; Bertini, E.S.; Aquilano, K.; et al. The Nrf2 induction prevents ferroptosis in Friedreich’s Ataxia. Redox Biol. 2021, 38, 101791. [Google Scholar] [CrossRef] [PubMed]
- Cuadrado, A. NRF2 in neurodegenerative diseases. Curr. Opin. Toxicol. 2016, 1, 46–53. [Google Scholar] [CrossRef]
- Sarlette, A.; Krampfl, K.; Grothe, C.; Von Neuhoff, N.; Dengler, R.; Petri, S. Nuclear erythroid 2-related factor 2-antioxidative response element signaling pathway in motor cortex and spinal cord in amyotrophic lateral sclerosis. J. Neuropathol. Exp. Neurol. 2008, 67, 1055–1062. [Google Scholar] [CrossRef] [PubMed]
- Petri, S.; Körner, S.; Kiaei, M. Nrf2/ARE signaling pathway: Key mediator in oxidative stress and potential therapeutic target in ALS. Neurol. Res. Int. 2012, 2012, 878030. [Google Scholar] [CrossRef] [PubMed]
- LoGerfo, A.; Chico, L.; Borgia, L.; Petrozzi, L.; Rocchi, A.; D’Amelio, A.; Carlesi, C.; Caldarazzo Ienco, E.; Mancuso, M.; Siciliano, G. Lack of association between nuclear factor erythroid-derived 2-like 2 promoter gene polymorphisms and oxidative stress biomarkers in amyotrophic lateral sclerosis patients. Oxid. Med. Cell. Longev. 2014, 2014, 432626. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Araujo, J.A.; Zhang, M.; Yin, F. Heme oxygenase-1, oxidation, inflammation, and atherosclerosis. Front. Pharmacol. 2012, 3, 119. [Google Scholar] [CrossRef] [Green Version]
- Ross, D.; Siegel, D. NQO1 in protection against oxidative stress. Curr. Opin. Toxicol. 2018, 7, 67–72. [Google Scholar] [CrossRef]
- Lu, J.; Holmgren, A. The thioredoxin antioxidant system. Free Radic. Biol. Med. 2014, 66, 75–87. [Google Scholar] [CrossRef] [PubMed]
- Vicencio, E.; Beltrán, S.; Labrador, L.; Manque, P.; Nassif, M.; Woehlbier, U. Implications of selective autophagy dysfunction for ALS pathology. Cells 2020, 9, 381. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Evans, C.S.; Holzbaur, E.L. Autophagy and mitophagy in ALS. Neurobiol. Dis. 2019, 122, 35–40. [Google Scholar] [CrossRef]
- Rusten, T.E.; Stenmark, H. p62, an autophagy hero or culprit? Nat. Cell Biol. 2010, 12, 207–209. [Google Scholar] [CrossRef]
- Araujo, B.G.; E Silva, L.F.S.; Torresi, J.L.D.B.; Siena, A.; Valerio, B.C.O.; Brito, M.D.; Rosenstock, T.R. Decreased mitochondrial function, biogenesis, and degradation in peripheral blood mononuclear cells from amyotrophic lateral sclerosis patients as a potential tool for biomarker research. Mol. Neurobiol. 2020, 57, 5084–5102. [Google Scholar] [CrossRef]
- Morán, M.; Delmiro, A.; Blázquez, A.; Ugalde, C.; Arenas, J.; Martín, M.A. Bulk autophagy, but not mitophagy, is increased in cellular model of mitochondrial disease. Biochim. Biophys. Acta Mol. Basis Dis. 2014, 1842, 1059–1070. [Google Scholar] [CrossRef] [Green Version]
- Watanabe, T.; Tahara, M.; Todo, S. The novel antioxidant Edaravone: From bench to bedside. Cardiovasc. Ther. 2008, 26, 101–114. [Google Scholar] [CrossRef]
- Chang, M.C.; Kwak, S.G.; Park, J.-S.; Park, D. The effectiveness of nonsteroidal anti-inflammatory drugs and acetaminophen in reduce the risk of amyotrophic lateral sclerosis? A meta-analysis. Sci. Rep. 2020, 10, 1–7. [Google Scholar] [CrossRef]
- Carlström, K.E.; Ewing, E.; Granqvist, M.; Gyllenberg, A.; Aeinehband, S.; Enoksson, S.L.; Checa, A.; Badam, T.V.S.; Huang, J.; Gomez-Cabrero, D.; et al. Therapeutic efficacy of dimethyl fumarate in relapsing-remitting multiple sclerosis associates with ROS pathway in monocytes. Nat. Commun. 2019, 10, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Lastres-Becker, I.; Garcia-Yague, A.J.; Scannevin, R.H.; Casarejos, M.J.; Kugler, S.; Rabano, A.; Cuadrado, A. Repurposing the NRF2 activator dimethyl fumarate as therapy against synucleinopathy in Parkinson’s Disease. Antioxid. Redox Signal. 2016, 25, 61–77. [Google Scholar] [CrossRef] [Green Version]
- Vucic, S.; Ryder, J.; Mekhael, L.; Rd, H.; Mathers, S.; Needham, M.; Dw, S.; Mc, K. Phase 2 randomized placebo controlled double blind study to assess the efficacy and safety of tecfidera in patients with amyotrophic lateral sclerosis (TEALS Study): Study protocol clinical trial (SPIRIT Compliant). Medicine 2020, 99, e18904. [Google Scholar] [CrossRef]
- Mandrioli, J.; D’Amico, R.; Zucchi, E.; Gessani, A.; Fini, N.; Fasano, A.; Caponnetto, C.; Chiò, A.; Dalla Bella, E.; Lunetta, C.; et al. Rapamycin treatment for amyotrophic lateral sclerosis: Protocol for a phase II randomized, double-blind, placebo-controlled, multicenter, clinical trial (RAP-ALS trial). Medicine 2018, 97, e11119. [Google Scholar] [CrossRef] [PubMed]
- Brooks, B.R.; Miller, R.G.; Swash, M.; Munsat, T.L. El Escorial revisited: Revised criteria for the diagnosis of amyotrophic lateral sclerosis. Amyotroph. Lateral Scler. 2000, 1, 293–299. [Google Scholar] [CrossRef]
- de las Cuevas, N.; Munoz, U.; Bartolome, F.; Esteras, N.; Alquezar, C.; Martin-Requero, A. Cell cycle and Alzheimer’s disease: Studies in non-neuronal cells. J. Appl. Biomed. 2010, 8, 121–130. [Google Scholar] [CrossRef] [Green Version]
- Garcia, E.; Limon, D.; Perez-De La Cruz, V.; Giordano, M.; Diaz-Muñoz, M.; Maldonado, P.D.; Herre-ra-Mundo, M.N.; Pedraza-Chaverri, J.; Santamaria, A. Lipid peroxidation, mitochondrial dysfunction and neurochemical and behavioural deficits in different neurotoxic models: Protective role of S-allylcysteine. Free Radic. Res. 2008, 42, 892–902. [Google Scholar] [CrossRef]
- Chen, J.-H.; Sambol, E.B.; Kennealey, P.T.; O’Connor, R.B.; DeCarolis, P.L.; Cory, D.G.; Singer, S. Water suppression without signal loss in HR-MAS 1H NMR of cells and tissues. J. Magn. Reson. 2004, 171, 143–150. [Google Scholar] [CrossRef]
- Cuadrado, A.; Kugler, S.; Lastres-Becker, I. Pharmacological targeting of GSK-3 and NRF2 provides neuro-protection in a preclinical model of tauopathy. Redox Biol. 2018, 14, 522–534. [Google Scholar] [CrossRef] [PubMed]
- Cuadrado, A.; Martín-Moldes, Z.; Ye, J.; Lastres-Becker, I. Transcription factors NRF2 and NF-κB are coordinated effectors of the Rho family, GTP-binding protein RAC1 during inflammation. J. Biol. Chem. 2014, 289, 15244–15258. [Google Scholar] [CrossRef] [Green Version]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Patient ID | Clinic Presentation | Affected Motoneurons | Age | Gender | Mutation |
|---|---|---|---|---|---|
| C-34 | n/a | 88 | Female | ---- | |
| C-35 | n/a | 63 | Male | ---- | |
| C-37 | n/a | 57 | Female | ---- | |
| C39 | n/a | 53 | Female | ---- | |
| C105 | n/a | 54 | Female | ---- | |
| C106 | n/a | 67 | Female | ---- | |
| C107 | n/a | 58 | Female | ---- | |
| C108 | n/a | 68 | Female | ---- | |
| C110 | n/a | 75 | Male | ---- | |
| C112 | n/a | 71 | Male | ---- | |
| C122 | n/a | 77 | Male | ---- | |
| C123 | n/a | 55 | Male | ---- | |
| C132 | n/a | 52 | Male | ---- | |
| E2 | Bulbar | MNS | 76 | Female | * |
| E4 | bulbar | MNS & MNI | 54 | Female | * |
| E5 | Spinal | MNS | 54 | Female | * |
| E6 | Bulbar | MNI | 79 | Male | * |
| E8 | Respiratory | MNI | 55 | Male | * |
| E10’ | Bulbar | MNS | 68 | Male | * |
| E11 | Spinal | MNI | 64 | Male | SOD1 het N65S |
| E18-5 | Spinal | MNI | 46 | Male | SOD1 het p.Leu117Val |
| E18-6 | Spinal | MNS & MNI | 58 | Male | SOD1 het p.ASn139His |
| E18-7 | Spinal | MNI | 59 | Male | SOD1 het p.Leu117Val |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lastres-Becker, I.; Porras, G.; Arribas-Blázquez, M.; Maestro, I.; Borrego-Hernández, D.; Boya, P.; Cerdán, S.; García-Redondo, A.; Martínez, A.; Martin-Requero, Á. Molecular Alterations in Sporadic and SOD1-ALS Immortalized Lymphocytes: Towards a Personalized Therapy. Int. J. Mol. Sci. 2021, 22, 3007. https://doi.org/10.3390/ijms22063007
Lastres-Becker I, Porras G, Arribas-Blázquez M, Maestro I, Borrego-Hernández D, Boya P, Cerdán S, García-Redondo A, Martínez A, Martin-Requero Á. Molecular Alterations in Sporadic and SOD1-ALS Immortalized Lymphocytes: Towards a Personalized Therapy. International Journal of Molecular Sciences. 2021; 22(6):3007. https://doi.org/10.3390/ijms22063007
Chicago/Turabian StyleLastres-Becker, Isabel, Gracia Porras, Marina Arribas-Blázquez, Inés Maestro, Daniel Borrego-Hernández, Patricia Boya, Sebastián Cerdán, Alberto García-Redondo, Ana Martínez, and Ángeles Martin-Requero. 2021. "Molecular Alterations in Sporadic and SOD1-ALS Immortalized Lymphocytes: Towards a Personalized Therapy" International Journal of Molecular Sciences 22, no. 6: 3007. https://doi.org/10.3390/ijms22063007