
VEGF-Independent Activation of Müller Cells by the Vitreous from Proliferative Diabetic Retinopathy Patients

 , and
, and 
Abstract
:1. Introduction
2. Results
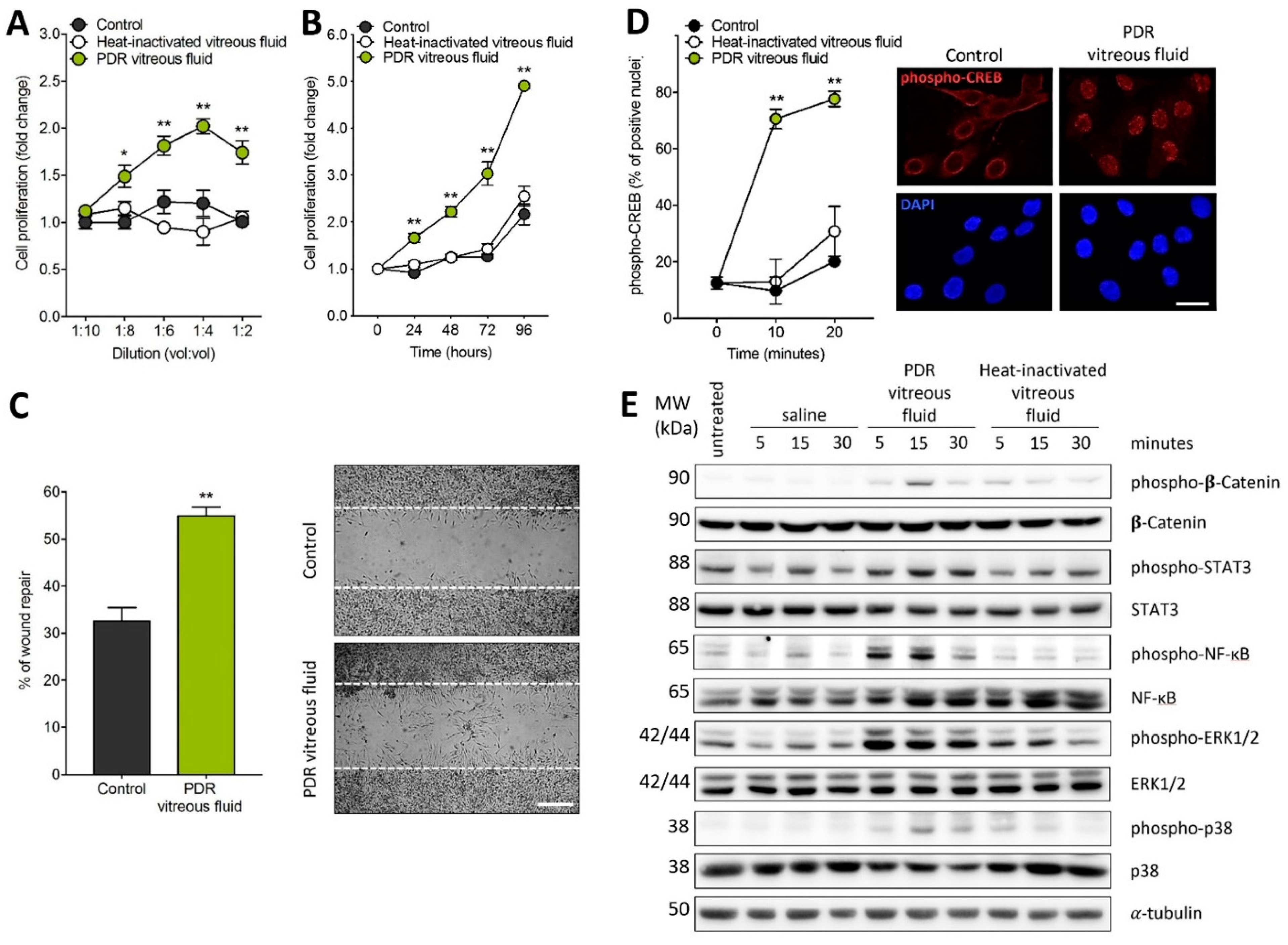
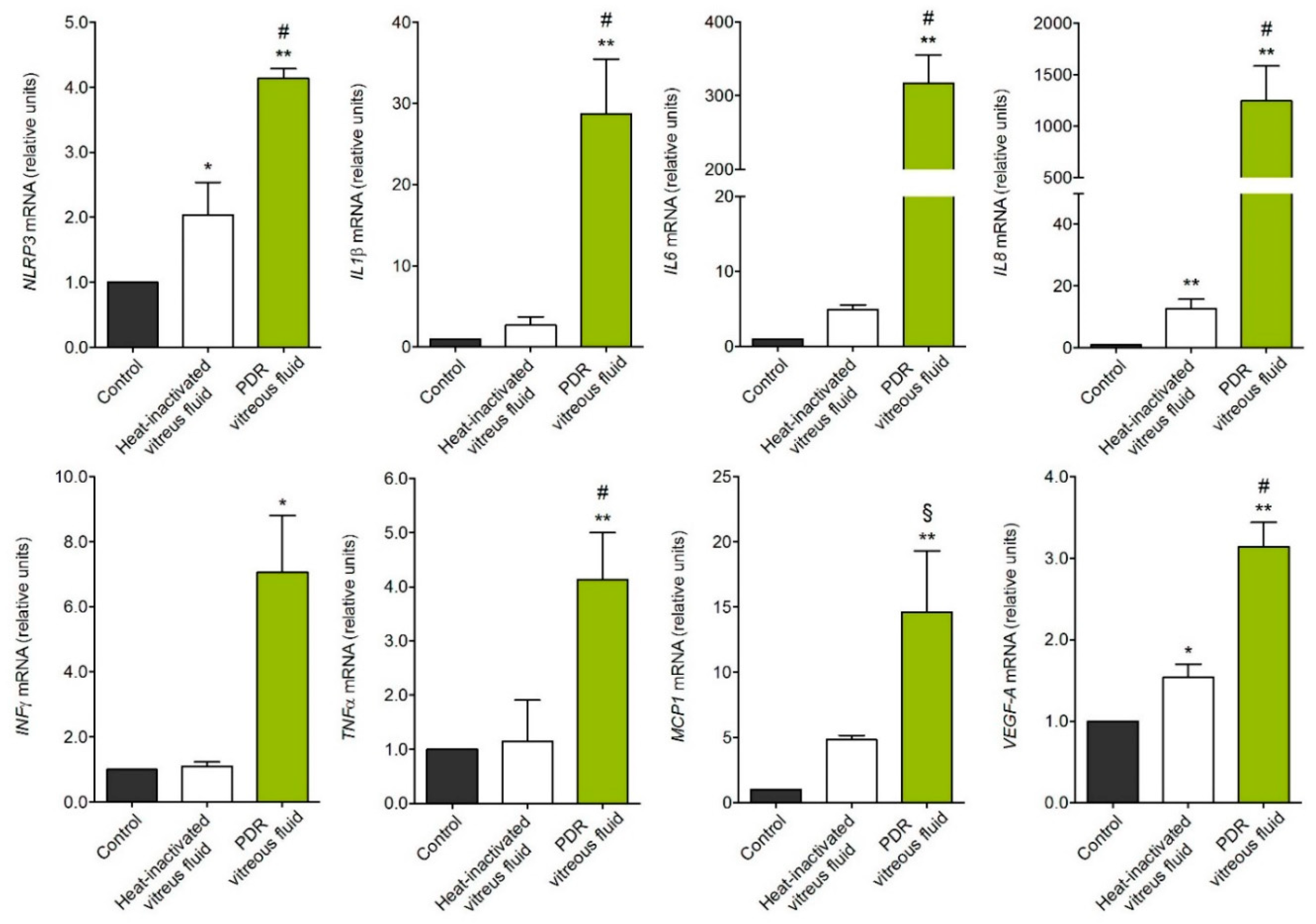
2.1. MIO-M1 Müller Cells Are Activated by PDR Vitreous
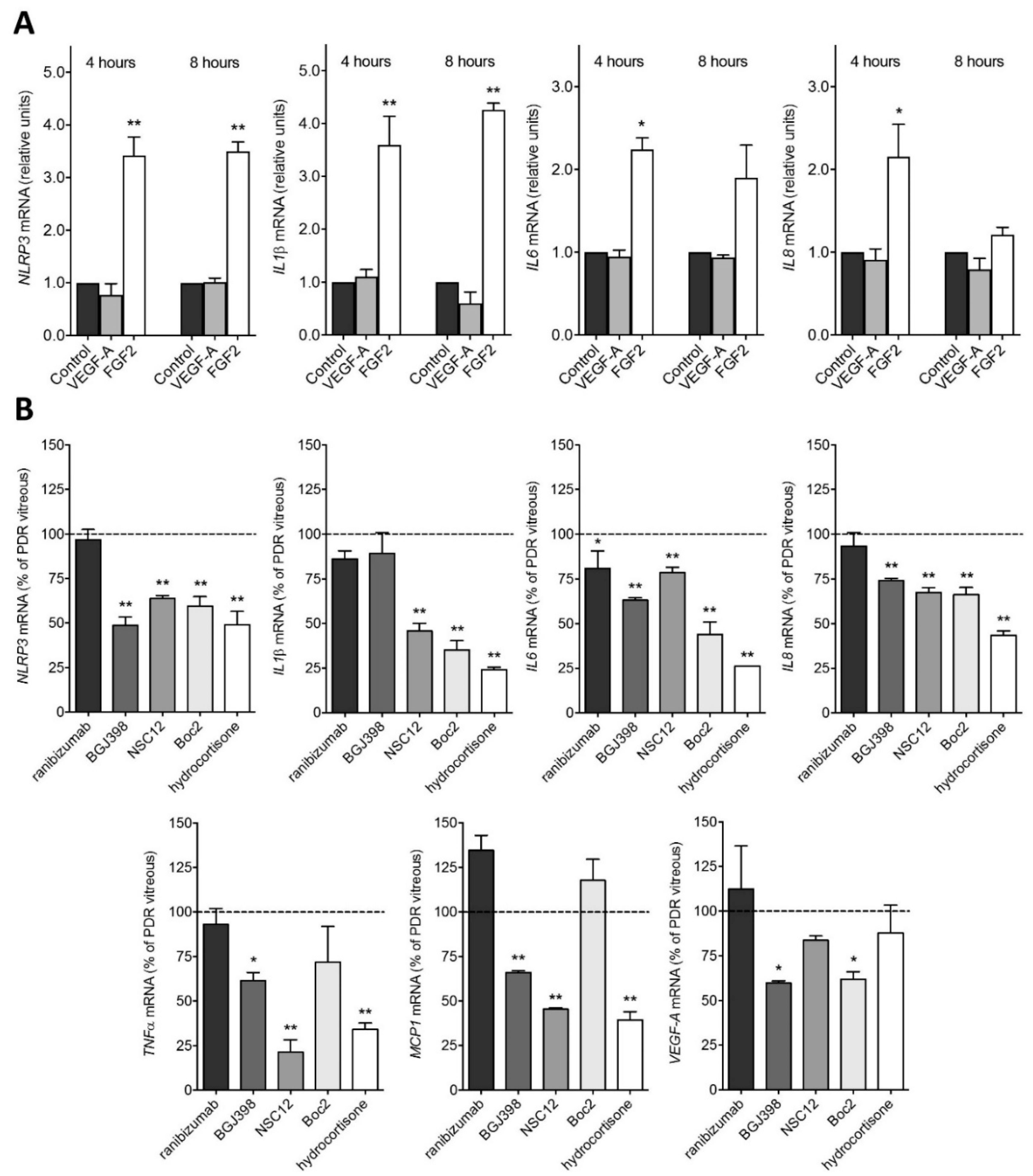
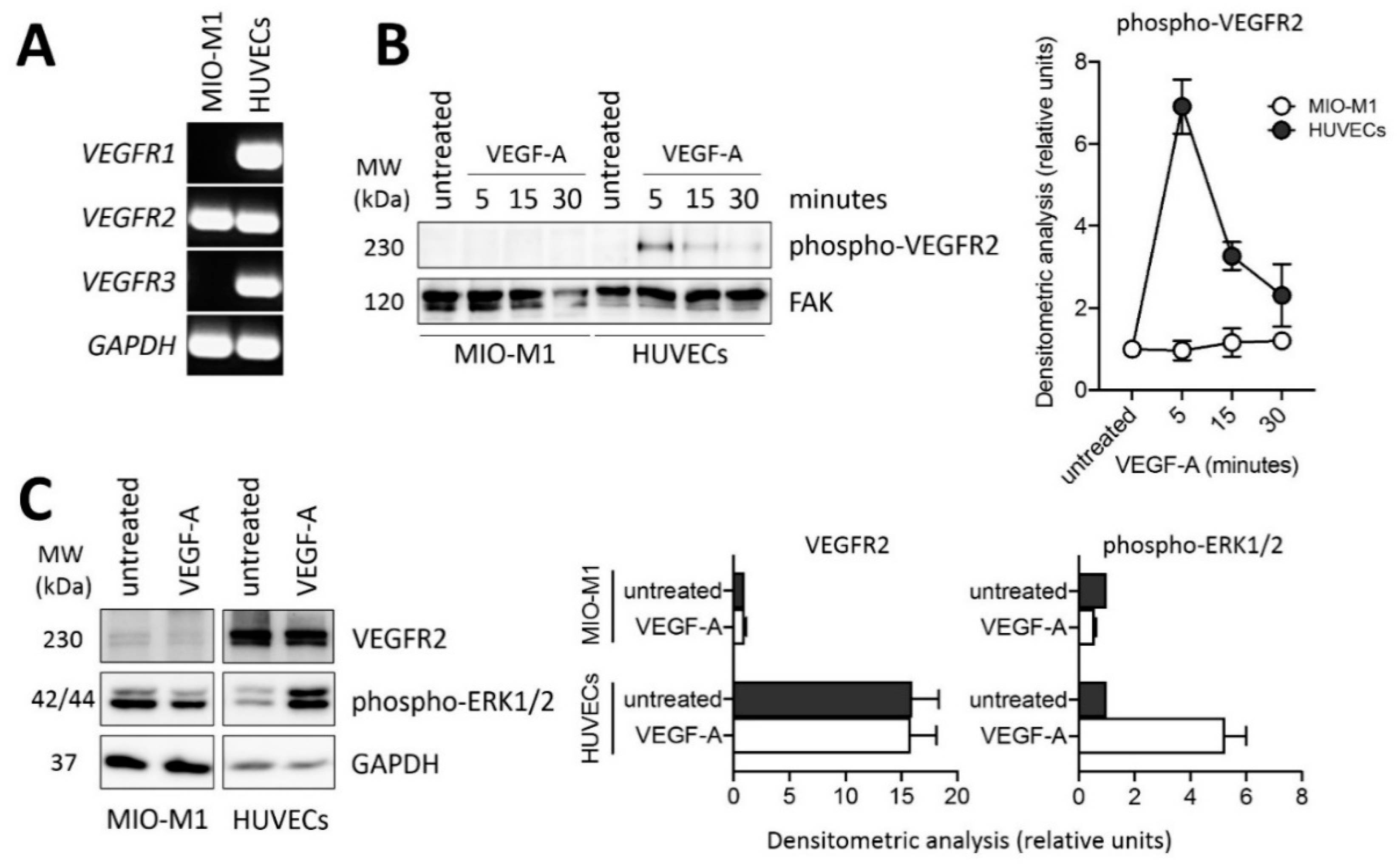
2.2. PDR Vitreous-Induced Activation of Müller Cells Is Independent from VEGF
3. Discussion
4. Materials and Methods
4.1. Reagents
4.2. Human Vitreous Fluid Samples
4.3. Cell Cultures
4.4. MIO-M1 Proliferation Assay
4.5. MIO-M1 Wound Healing Assay
4.6. Western Blot Analysis
4.7. RT-PCR Analyses
4.8. MIO-M1 Immunofluorescence Analysis
4.9. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| ACTA2 | actin α 2 |
| ERM | epiretinal membrane |
| FGF2 | basic fibroblast growth factor |
| FGFR | fibroblast growth factor receptor |
| GAPDH | glyceraldehyde-3-phosphate dehydrogenase |
| HUVECs | human umbilical vein endothelial cells |
| IL | interleukin |
| INFγ | interferon γ |
| MCP1 | monocyte chemoattractant protein 1 |
| NLRP3 | nucleotide-binding oligomerization domain (NOD), leucine-rich repeat (LRR)-containing proteins 3 |
| PDGF | platelet derived growth factor |
| PDR | proliferative diabetic retinopathy |
| RLBP1 | retinaldehyde binding protein 1 |
| S100A4 | S100 calcium-binding protein A4 |
| TNFα | tumor necrosis factor α |
| VEGF-A | vascular endothelial growth factor-A |
| VEGFR | vascular endothelial growth factor receptor |
| VIM | vimentin |
References
- International Diabetes Federation Diabetes Atlas (9th ed.). Available online: http://www.diabetesatlas.org/ (accessed on 28 December 2020).
- Al-Kharashi, A.S. Role of oxidative stress, inflammation, hypoxia and angiogenesis in the development of diabetic retinopathy. Saudi J. Ophthalmol. 2018, 32, 318–323. [Google Scholar] [CrossRef] [PubMed]
- Nawaz, I.M.; Rezzola, S.; Cancarini, A.; Russo, A.; Costagliola, C.; Semeraro, F.; Presta, M. Human vitreous in proliferative diabetic retinopathy: Characterization and translational implications. Prog. Retin. Eye Res. 2019, 72, 100756. [Google Scholar] [CrossRef]
- Osaadon, P.; Fagan, X.J.; Lifshitz, T.; Levy, J. A review of anti-VEGF agents for proliferative diabetic retinopathy. Eye (Lond) 2014, 28, 510–520. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, Y.; Singh, R.P. The role of anti-vascular endothelial growth factor (anti-VEGF) in the management of proliferative diabetic retinopathy. Drugs Context. 2018, 7, 212532. [Google Scholar] [CrossRef]
- Tan, G.S.; Cheung, N.; Simo, R.; Cheung, G.C.; Wong, T.Y. Diabetic macular oedema. Lancet Diabetes Endocrinol. 2017, 5, 143–155. [Google Scholar] [CrossRef]
- Brown, D.M.; Nguyen, Q.D.; Marcus, D.M.; Boyer, D.S.; Patel, S.; Feiner, L.; Schlottmann, P.G.; Rundle, A.C.; Zhang, J.; Rubio, R.G.; et al. Long-term outcomes of ranibizumab therapy for diabetic macular edema: The 36-month results from two phase III trials: RISE and RIDE. Ophthalmology 2013, 120, 2013–2022. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Semeraro, F.; Morescalchi, F.; Cancarini, A.; Russo, A.; Rezzola, S.; Costagliola, C. Diabetic retinopathy, a vascular and inflammatory disease: Therapeutic implications. Diabetes Metab. 2019, 45, 517–527. [Google Scholar] [CrossRef]
- Le, Y.Z. VEGF production and signaling in Müller glia are critical to modulating vascular function and neuronal integrity in diabetic retinopathy and hypoxic retinal vascular diseases. Vision Res. 2017, 139, 108–114. [Google Scholar] [CrossRef]
- Reichenbach, A.; Bringmann, A. New functions of Müller cells. Glia 2013, 61, 651–678. [Google Scholar] [CrossRef]
- Bringmann, A.; Iandiev, I.; Pannicke, T.; Wurm, A.; Hollborn, M.; Wiedemann, P.; Osborne, N.N.; Reichenbach, A. Cellular signaling and factors involved in Muller cell gliosis: Neuroprotective and detrimental effects. Prog. Retin. Eye Res. 2009, 28, 423–451. [Google Scholar] [CrossRef] [PubMed]
- Bringmann, A.; Pannicke, T.; Grosche, J.; Francke, M.; Wiedemann, P.; Skatchkov, S.N.; Osborne, N.N.; Reichenbach, A. Muller cells in the healthy and diseased retina. Prog. Retin. Eye Res. 2006, 25, 397–424. [Google Scholar] [CrossRef]
- Oberstein, S.Y.; Byun, J.; Herrera, D.; Chapin, E.A.; Fisher, S.K.; Lewis, G.P. Cell proliferation in human epiretinal membranes: Characterization of cell types and correlation with disease condition and duration. Mol. Vis. 2011, 17, 1794–1805. [Google Scholar]
- Roy, S.; Amin, S.; Roy, S. Retinal fibrosis in diabetic retinopathy. Exp. Eye Res. 2016, 142, 71–75. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rodrigues, M.; Xin, X.; Jee, K.; Babapoor-Farrokhran, S.; Kashiwabuchi, F.; Ma, T.; Bhutto, I.; Hassan, S.J.; Daoud, Y.; Baranano, D.; et al. VEGF secreted by hypoxic Muller cells induces MMP-2 expression and activity in endothelial cells to promote retinal neovascularization in proliferative diabetic retinopathy. Diabetes 2013, 62, 3863–3873. [Google Scholar] [CrossRef] [Green Version]
- Portillo, J.C.; Lopez Corcino, Y.; Miao, Y.; Tang, J.; Sheibani, N.; Kern, T.S.; Dubyak, G.R.; Subauste, C.S. CD40 in Retinal Muller Cells Induces P2X7-Dependent Cytokine Expression in Macrophages/Microglia in Diabetic Mice and Development of Early Experimental Diabetic Retinopathy. Diabetes 2017, 66, 483–493. [Google Scholar] [CrossRef] [Green Version]
- Rezzola, S.; Nawaz, M.I.; Cancarini, A.; Semeraro, F.; Presta, M. Vascular Endothelial Growth Factor in the Vitreous of Proliferative Diabetic Retinopathy Patients: Chasing a Hiding Prey? Diabetes Care 2019, 42, e105–e106. [Google Scholar] [CrossRef] [Green Version]
- Rezzola, S.; Corsini, M.; Chiodelli, P.; Cancarini, A.; Nawaz, I.M.; Coltrini, D.; Mitola, S.; Ronca, R.; Belleri, M.; Lista, L.; et al. Inflammation and N-formyl peptide receptors mediate the angiogenic activity of human vitreous humour in proliferative diabetic retinopathy. Diabetologia 2017, 60, 719–728. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rezzola, S.; Nawaz, I.M.; Cancarini, A.; Ravelli, C.; Calza, S.; Semeraro, F.; Presta, M. 3D endothelial cell spheroid/human vitreous humor assay for the characterization of anti-angiogenic inhibitors for the treatment of proliferative diabetic retinopathy. Angiogenesis 2017, 20, 629–640. [Google Scholar] [CrossRef]
- Nawaz, I.M.; Chiodelli, P.; Rezzola, S.; Paganini, G.; Corsini, M.; Lodola, A.; Lodola, A.; Di Ianni, A.; Mor, M.; Presta, M. N-tert-butyloxycarbonyl-Phe-Leu-Phe-Leu-Phe (BOC2) inhibits the angiogenic activity of heparin-binding growth factors. Angiogenesis 2018, 21, 47–59. [Google Scholar] [CrossRef]
- Rezzola, S.; Dal Monte, M.; Belleri, M.; Bugatti, A.; Chiodelli, P.; Corsini, M.; Cammalleri, M.; Cancarini, A.; Morbidelli, L.; Oreste, P.; et al. Therapeutic Potential of Anti-Angiogenic Multitarget, N,O-Sulfated E. Coli K5 Polysaccharide in Diabetic Retinopathy. Diabetes 2015, 64, 2581–2592. [Google Scholar] [CrossRef] [Green Version]
- Rezzola, S.; Loda, A.; Corsini, M.; Semeraro, F.; Annese, T.; Presta, M.; Ribatti, D. Angiogenesis-Inflammation Cross Talk in Diabetic Retinopathy: Novel Insights from the Chick Embryo Chorioallantoic Membrane/Human Vitreous Platform. Front. Immunol. 2020, 11, 581288. [Google Scholar] [CrossRef]
- Dal Monte, M.; Rezzola, S.; Cammalleri, M.; Belleri, M.; Locri, F.; Morbidelli, L.; Corsini, M.; Paganini, G.; Semeraro, F.; Cancarini, A.; et al. Antiangiogenic Effectiveness of the Urokinase Receptor-Derived Peptide UPARANT in a Model of Oxygen-Induced Retinopathy. Investig. Ophthalmol. Vis. Sci. 2015, 56, 2392–2407. [Google Scholar] [CrossRef]
- Presta, M.; Dell’Era, P.; Mitola, S.; Moroni, E.; Ronca, R.; Rusnati, M. Fibroblast growth factor/fibroblast growth factor receptor system in angiogenesis. Cytokine Growth Factor Rev. 2005, 16, 159–178. [Google Scholar] [CrossRef] [Green Version]
- Guagnano, V.; Furet, P.; Spanka, C.; Bordas, V.; Le Douget, M.; Stamm, C.; Brueggen, J.; Jensen, M.R.; Schnell, C.; Schmid, H.; et al. Discovery of 3-(2,6-dichloro-3,5-dimethoxy-phenyl)-1-{6-[4-(4-ethyl-piperazin-1-yl)-phenylamin o]-pyrimidin-4-yl}-1-methyl-urea (NVP-BGJ398), a potent and selective inhibitor of the fibroblast growth factor receptor family of receptor tyrosine kinase. J. Med. Chem. 2011, 54, 7066–7083. [Google Scholar] [CrossRef] [PubMed]
- Ronca, R.; Giacomini, A.; Di Salle, E.; Coltrini, D.; Pagano, K.; Ragona, L.; Matarazzo, S.; Rezzola, S.; Maiolo, D.; Torrella, R.; et al. Long-Pentraxin 3 Derivative as a Small-Molecule FGF Trap for Cancer Therapy. Cancer Cell 2015, 28, 225–239. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bressler, S.B.; Liu, D.; Glassman, A.R.; Blodi, B.A.; Castellarin, A.A.; Jampol, L.M.; Kaufman, P.L.; Melia, M.; Singh, H.; Wells, J.A.; et al. Change in Diabetic Retinopathy Through 2 Years: Secondary Analysis of a Randomized Clinical Trial Comparing Aflibercept, Bevacizumab, and Ranibizumab. JAMA Ophthalmol. 2017, 135, 558–568. [Google Scholar] [CrossRef] [PubMed]
- Hollborn, M.; Jahn, K.; Limb, G.A.; Kohen, L.; Wiedemann, P.; Bringmann, A. Characterization of the basic fibroblast growth factor-evoked proliferation of the human Muller cell line, MIO-M1. Graefes Arch. Clin. Exp. Ophthalmol. 2004, 242, 414–422. [Google Scholar] [CrossRef]
- Toniolo, C.; Bonora, G.M.; Showell, H.; Freer, R.J.; Becker, E.L. Structural requirements for formyl homooligopeptide chemoattractants. Biochemistry 1984, 23, 698–704. [Google Scholar] [CrossRef]
- Liu, X.; Ye, F.; Xiong, H.; Hu, D.; Limb, G.A.; Xie, T.; Peng, L.; Yang, W.; Sun, Y.; Zhou, M.; et al. IL-1beta Upregulates IL-8 Production in Human Muller Cells Through Activation of the p38 MAPK and ERK1/2 Signaling Pathways. Inflammation 2014, 37, 1486–1495. [Google Scholar] [CrossRef]
- Liu, X.; Ye, F.; Xiong, H.; Hu, D.N.; Limb, G.A.; Xie, T.; Peng, L.; Zhang, P.; Wei, Y.; Zhang, W.; et al. IL-1beta induces IL-6 production in retinal Muller cells predominantly through the activation of p38 MAPK/NF-kappaB signaling pathway. Exp. Cell Res. 2015, 331, 223–231. [Google Scholar] [CrossRef]
- Nelson, C.M.; Ackerman, K.M.; O’Hayer, P.; Bailey, T.J.; Gorsuch, R.A.; Hyde, D.R. Tumor necrosis factor-alpha is produced by dying retinal neurons and is required for Muller glia proliferation during zebrafish retinal regeneration. J. Neurosci. 2013, 33, 6524–6539. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ghaseminejad, F.; Kaplan, L.; Pfaller, A.M.; Hauck, S.M.; Grosche, A. The role of Muller cell glucocorticoid signaling in diabetic retinopathy. Graefes Arch. Clin. Exp. Ophthalmol. 2020, 258, 221–230. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zong, H.; Ward, M.; Madden, A.; Yong, P.H.; Limb, G.A.; Curtis, T.M.; Stitt, A.W. Hyperglycaemia-induced pro-inflammatory responses by retinal Muller glia are regulated by the receptor for advanced glycation end-products (RAGE). Diabetologia 2010, 53, 2656–2666. [Google Scholar] [CrossRef] [Green Version]
- Mu, H.; Zhang, X.M.; Liu, J.J.; Dong, L.; Feng, Z.L. Effect of high glucose concentration on VEGF and PEDF expression in cultured retinal Muller cells. Mol. Biol Rep. 2009, 36, 2147–2151. [Google Scholar] [CrossRef]
- Wang, J.; Xu, X.; Elliott, M.H.; Zhu, M.; Le, Y.Z. Muller cell-derived VEGF is essential for diabetes-induced retinal inflammation and vascular leakage. Diabetes 2010, 59, 2297–2305. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guidry, C. The role of Muller cells in fibrocontractive retinal disorders. Prog. Retin. Eye Res. 2005, 24, 75–86. [Google Scholar] [CrossRef] [PubMed]
- Chen, Q.; Liu, Z.X. Idiopathic Macular Hole: A Comprehensive Review of Its Pathogenesis and of Advanced Studies on Metamorphopsia. J. Ophthalmol. 2019, 2019, 7294952. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bu, S.C.; Kuijer, R.; van der Worp, R.J.; Postma, G.; Renardel de Lavalette, V.W.; Li, X.R.; Hooymans, J.M.M.; Los, L.I. Immunohistochemical Evaluation of Idiopathic Epiretinal Membranes and In Vitro Studies on the Effect of TGF-beta on Muller Cells. Investig. Ophthalmol. Vis. Sci. 2015, 56, 6506–6514. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mayr, B.; Montminy, M. Transcriptional regulation by the phosphorylation-dependent factor CREB. Nat. Rev. Mol. Cell Biol. 2001, 2, 599–609. [Google Scholar] [CrossRef]
- Fischer, A.J.; Scott, M.A.; Tuten, W. Mitogen-activated protein kinase-signaling stimulates Muller glia to proliferate in acutely damaged chicken retina. Glia 2009, 57, 166–181. [Google Scholar] [CrossRef] [Green Version]
- Swanson, K.V.; Deng, M.; Ting, J.P. The NLRP3 inflammasome: Molecular activation and regulation to therapeutics. Nat. Rev. Immunol. 2019, 19, 477–489. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Biarnes Costa, M.; Gerhardinger, C. IL-1beta is upregulated in the diabetic retina and retinal vessels: Cell-specific effect of high glucose and IL-1beta autostimulation. PLoS ONE 2012, 7, e36949. [Google Scholar] [CrossRef] [Green Version]
- Chaurasia, S.S.; Lim, R.R.; Parikh, B.H.; Wey, Y.S.; Tun, B.B.; Wong, T.Y.; Luu, C.D.; Agrawal, R.; Ghosh, A.; Mortellaro, A.; et al. The NLRP3 Inflammasome May Contribute to Pathologic Neovascularization in the Advanced Stages of Diabetic Retinopathy. Sci Rep. 2018, 8, 2847. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, L.P.; Sun, H.L.; Wu, L.M.; Guo, X.J.; Dou, H.L.; Tso, M.O.; Zhao, L.; Li, S. Baicalein reduces inflammatory process in a rodent model of diabetic retinopathy. Investig. Ophthalmol. Vis. Sci. 2009, 50, 2319–2327. [Google Scholar] [CrossRef] [Green Version]
- Loukovaara, S.; Piippo, N.; Kinnunen, K.; Hytti, M.; Kaarniranta, K.; Kauppinen, A. NLRP3 inflammasome activation is associated with proliferative diabetic retinopathy. Acta Ophthalmol. 2017, 95, 803–808. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, H.; Zhang, X.; Liao, N.; Mi, L.; Peng, Y.; Liu, B.; Zhang, S.; Wen, F. Enhanced Expression of NLRP3 Inflammasome-Related Inflammation in Diabetic Retinopathy. Investig. Ophthalmol. Vis. Sci. 2018, 59, 978–985. [Google Scholar] [CrossRef]
- He, Y.; Hara, H.; Nunez, G. Mechanism and Regulation of NLRP3 Inflammasome Activation. Trends Biochem. Sci. 2016, 41, 1012–1021. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chung, I.C.; Yuan, S.N.; OuYang, C.N.; Lin, H.C.; Huang, K.Y.; Chen, Y.J.; Chung, A.K.; Chu, C.L.; Ojcius, D.M.; Chang, Y.S.; et al. Src-family kinase-Cbl axis negatively regulates NLRP3 inflammasome activation. Cell Death Dis. 2018, 9, 1109. [Google Scholar] [CrossRef] [PubMed]
- Fogli, S.; Del Re, M.; Rofi, E.; Posarelli, C.; Figus, M.; Danesi, R. Clinical pharmacology of intravitreal anti-VEGF drugs. Eye (Lond) 2018, 32, 1010–1020. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fu, S.; Dong, S.; Zhu, M.; Sherry, D.M.; Wang, C.; You, Z.; Haigh, J.J.; Le, Y.Z. Muller Glia Are a Major Cellular Source of Survival Signals for Retinal Neurons in Diabetes. Diabetes 2015, 64, 3554–3563. [Google Scholar] [CrossRef] [Green Version]
- Saint-Geniez, M.; Maharaj, A.S.; Walshe, T.E.; Tucker, B.A.; Sekiyama, E.; Kurihara, T.; Darland, D.C.; Young, M.J.; D’Amore, P.A. Endogenous VEGF is required for visual function: Evidence for a survival role on muller cells and photoreceptors. PLoS ONE 2008, 3, e3554. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Caceres-Del-Carpio, J.; Moustafa, M.T.; Toledo-Corral, J.; Hamid, M.A.; Atilano, S.R.; Schneider, K.; Fukuhara, P.S.; Donato Costa, R.; Norman, J.L.; Malik, D.; et al. In vitro response and gene expression of human retinal Muller cells treated with different anti-VEGF drugs. Exp. Eye Res. 2020, 191, 107903. [Google Scholar] [CrossRef] [PubMed]
- Matsuda, M.; Krempel, P.G.; Marquezini, M.V.; Sholl-Franco, A.; Lameu, A.; Monteiro, M.L.R.; de Oliveira Miguel, N.C. Cellular stress response in human Muller cells (MIO-M1) after bevacizumab treatment. Exp. Eye Res. 2017, 160, 1–10. [Google Scholar] [CrossRef]
- Hueber, A.; Wiedemann, P.; Esser, P.; Heimann, K. Basic fibroblast growth factor mRNA, bFGF peptide and FGF receptor in epiretinal membranes of intraocular proliferative disorders (PVR and PDR). Int. Ophthalmol. 1996, 20, 345–350. [Google Scholar] [CrossRef]
- Liang, X.; Li, C.; Li, Y.; Huang, J.; Tang, S.; Gao, R.; Li, S. Platelet-derived growth factor and basic fibroblast growth factor immunolocalized in proliferative retinal diseases. Chin. Med. J. 2000, 113, 144–147. [Google Scholar]
- Coltrini, D.; Belleri, M.; Gambicorti, E.; Romano, D.; Morescalchi, F.; Krishna Chandran, A.M.; Calza, S.; Semeraro, F.; Presta, M. Gene expression analysis identifies two distinct molecular clusters of idiopatic epiretinal membranes. Biochim. Biophys. Acta Mol. Basis Dis. 2020, 1866, 165938. [Google Scholar] [CrossRef]
- Yoshida, S.; Yoshida, A.; Ishibashi, T. Induction of IL-8, MCP-1, and bFGF by TNF-alpha in retinal glial cells: Implications for retinal neovascularization during post-ischemic inflammation. Graefes Arch. Clin. Exp. Ophthalmol. 2004, 242, 409–413. [Google Scholar] [CrossRef] [PubMed]
- Cheng, T.; Cao, W.; Wen, R.; Steinberg, R.H.; LaVail, M.M. Prostaglandin E2 induces vascular endothelial growth factor and basic fibroblast growth factor mRNA expression in cultured rat Muller cells. Investig. Ophthalmol. Vis. Sci. 1998, 39, 581–591. [Google Scholar]
- Itoh, N.; Ornitz, D.M. Functional evolutionary history of the mouse Fgf gene family. Developmental. Dyn. 2008, 237, 18–27. [Google Scholar] [CrossRef]
- Beenken, A.; Mohammadi, M. The FGF family: Biology, pathophysiology and therapy. Nat. Rev. Drug Discov. 2009, 8, 235–253. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krejci, P.; Aklian, A.; Kaucka, M.; Sevcikova, E.; Prochazkova, J.; Masek, J.K.; Mikolka, P.; Pospisilova, T.; Spoustova, T.; Weis, M.; et al. Receptor tyrosine kinases activate canonical WNT/beta-catenin signaling via MAP kinase/LRP6 pathway and direct beta-catenin phosphorylation. PLoS ONE 2012, 7, e35826. [Google Scholar] [CrossRef] [PubMed]
- Huang, L.; Luo, R.; Li, J.; Wang, D.; Zhang, Y.; Liu, L.; Zhang, N.; Xu, X.; Lu, B.; Zhano, K. beta-catenin promotes NLRP3 inflammasome activation via increasing the association between NLRP3 and ASC. Mol. Immunol. 2020, 121, 186–194. [Google Scholar] [CrossRef] [PubMed]
- Kanda, A.; Noda, K.; Hirose, I.; Ishida, S. TGF-beta-SNAIL axis induces Muller glial-mesenchymal transition in the pathogenesis of idiopathic epiretinal membrane. Sci. Rep. 2019, 9, 673. [Google Scholar] [CrossRef]
- Guidry, C.; Bradley, K.M.; King, J.L. Tractional force generation by human muller cells: Growth factor responsiveness and integrin receptor involvement. Investig. Ophthalmol. Vis. Sci. 2003, 44, 1355–1363. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bringmann, A.; Wiedemann, P. Involvement of Muller glial cells in epiretinal membrane formation. Graefes Arch. Clin. Exp. Ophthalmol. 2009, 247, 865–883. [Google Scholar] [CrossRef]
- Guidry, C.; Feist, R.; Morris, R.; Hardwick, C.W. Changes in IGF activities in human diabetic vitreous. Diabetes 2004, 53, 2428–2435. [Google Scholar] [CrossRef] [Green Version]
- Romaniuk, D.; Kimsa, M.W.; Strzalka-Mrozik, B.; Kimsa, M.C.; Kabiesz, A.; Romaniuk, W.; Mazurek, U. Gene expression of IGF1, IGF1R, and IGFBP3 in epiretinal membranes of patients with proliferative diabetic retinopathy: Preliminary study. Mediat. Inflamm. 2013, 2013, 986217. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Natoli, R.; Fernando, N.; Madigan, M.; Chu-Tan, J.A.; Valter, K.; Provis, J.; Rutar, M. Microglia-derived IL-1beta promotes chemokine expression by Muller cells and RPE in focal retinal degeneration. Mol. Neurodegener. 2017, 12, 31. [Google Scholar] [CrossRef] [PubMed]
- Limb, G.A.; Salt, T.E.; Munro, P.M.; Moss, S.E.; Khaw, P.T. In vitro characterization of a spontaneously immortalized human Muller cell line (MIO-M1). Investig. Ophthalmol. Vis. Sci. 2002, 43, 864–869. [Google Scholar]
- Ravelli, C.; Grillo, E.; Corsini, M.; Coltrini, D.; Presta, M.; Mitola, S. beta3 Integrin Promotes Long-Lasting Activation and Polarization of Vascular Endothelial Growth Factor Receptor 2 by Immobilized Ligand. Arter. Thromb. Vasc. Biol. 2015, 35, 2161–2171. [Google Scholar] [CrossRef] [Green Version]
- Schindelin, J.; Arganda-Carreras, I.; Frise, E.; Kaynig, V.; Longair, M.; Pietzsch, T.; Preibisch, S.; Rueden, C.; Saalfeld, S.; Schimd, B.; et al. Fiji: An open-source platform for biological-image analysis. Nat. Methods 2012, 9, 676–682. [Google Scholar] [CrossRef] [Green Version]
- Rezzola, S.; Di Somma, M.; Corsini, M.; Leali, D.; Ravelli, C.; Polli, V.A.B.; Grillo, E.; Presta, M.; Mitola, S. VEGFR2 activation mediates the pro-angiogenic activity of BMP4. Angiogenesis 2019, 22, 521–533. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Patients/Eyes | 39/42 |
|---|---|
| Clinical features | |
| Gender (male/female) | 28/11 |
| Age (years) | 65 ± 10 |
| Type 1/type 2 diabetes | 4/35 |
| Duration of diabetes (years) | 21 ± 6 |
| Oral hypoglycemic drug treatment | 10/39 |
| Insulin treatment | 10/39 |
| Oral hypoglycemic drug + insulin treatment | 19/39 |
| Glycaemia (mg/dL) | 161 ± 56 |
| HbA1c (%) | 7.9 ± 1.1 |
| Neuropathy | 6/39 |
| Nephropathy | 13/39 |
| Cardiopathy | 15/39 |
| Hypertension | 37/39 |
| Dyslipidemia | 23/39 |
| Triglycerides (mg/dL) | 120 ± 54 |
| Cholesterol (mg/dL) | 153 ± 44 |
| Creatinine (mg/dL) | 1.4 ± 0.7 |
| Hemoglobin (g/dL) | 13.1 ± 1.6 |
| Ophthalmic features | |
| PDR | 42/42 |
| PDR with vitreous hemorrhage | 19/42 |
| PDR with macular edema | 19/42 |
| PDR with ERM | 31/38 |
| Ocular therapies | |
| Intravitreal injection of anti-VEGF blocker | 29/42 |
| Panretinal laser photocoagulation | 32/42 |
| Gene | Forward | Reverse |
|---|---|---|
| ACTA2 | 5′-AATGGCTCTGGGCTCTGTAA-3′ | 5′-TTTTGCTCTGTGCTTCGTCA-3′ |
| FGFR1 | 5′-GGGCTGGAATACTGCTACAA-3′ | 5′-GCCAAAGTCTGCTATCTTCATC-3′ |
| FGFR2 | 5′-GGATAACAACACGCCTCTCTT-3′ | 5′-GCCCAAAGCAACCTTCTC-3′ |
| FGFR3 | 5′-TGGTGTCCTGTGCCTACC-3′ | 5′-CCGTTGGTCGTCTTCTTGT-3′ |
| FGFR4 | 5′-AACCGCATTGGAGGCATT-3′ | 5′-TCTACCAGGCAGGTGTATGT-3′ |
| GAPDH | 5′-GAAGGTCGGAGTCAACGGATT-3′ | 5′-TGACGGTGCCATGGAATTTG-3′ |
| IL1β | 5′-GTGGCAATGAGGATGACTTG-3′ | 5′-GTGGTGGTCGGAGATTCGTA-3′ |
| IL6 | 5′-TGTGTGGGTCTGTTGTAGGG-3′ | 5′-CCCGTGCAATATCTAGGAAAA-3′ |
| IL8 | 5′-TGTGTGGGTCTGTTGTAGGG-3′ | 5′-CCCGTGCAATATCTAGGAAAA-3′ |
| INFγ | 5′-GCAGGTCATTCAGATGTAGCGG-3′ | 5′-CCACACTCTTTTGGATGCTCTGG-3′ |
| MCP1 | 5′-CTCAGCCAGATGCAATCAA-3′ | 5′-CACTTCTGCTTGGGGTCA-3′ |
| NLRP3 | 5′-GGACTGAAGCACCTGTTGTGCA-3′ | 5′-TCCTGAGTCTCCCAAGGCATTC-3′ |
| RLBP1 | 5′-GCTGCTGGAGAATGAGGAAA-3′ | 5′-TGGTGGATGAAGTGGATGG-3′ |
| S100A4 | 5′-CCTGGATGTGATGGTGTCC-3′ | 5′-TCGTTGTCCCTGTTGCTGT-3′ |
| TNFα | 5′-TGCTTGTTCCTCAGCCTCTT-3′ | 5′-GCTTGTCACTCGGGGTTC-3′ |
| VEGF-A | 5′-AATCGAGACCCTGGTGGAC-3′ | 5′-GGTGAGGTTTGATCCGCATA-3′ |
| VEGFR1 | 5′-AGCAGTTCCACCACTTTAGA-3′ | 5′-GAACTTTCCACAGAGCCCTT-3′ |
| VEGFR2 | 5′-GGAAATGACACTGGAGCCTA-3′ | 5′-TTTGAAATGGACCCGAGACA-3′ |
| VEGFR3 | 5′-CAACGACCTACAAAGGCTCT-3′ | 5′-GTAAAACACCTGGCCTCCTC-3′ |
| VIM | 5′-CGCCAGATGCGTGAAATG-3′ | 5′-ACCAGAGGGAGTGAATCCAGA-3′ |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rezzola, S.; Guerra, J.; Krishna Chandran, A.M.; Loda, A.; Cancarini, A.; Sacristani, P.; Semeraro, F.; Presta, M. VEGF-Independent Activation of Müller Cells by the Vitreous from Proliferative Diabetic Retinopathy Patients. Int. J. Mol. Sci. 2021, 22, 2179. https://doi.org/10.3390/ijms22042179
Rezzola S, Guerra J, Krishna Chandran AM, Loda A, Cancarini A, Sacristani P, Semeraro F, Presta M. VEGF-Independent Activation of Müller Cells by the Vitreous from Proliferative Diabetic Retinopathy Patients. International Journal of Molecular Sciences. 2021; 22(4):2179. https://doi.org/10.3390/ijms22042179
Chicago/Turabian StyleRezzola, Sara, Jessica Guerra, Adwaid Manu Krishna Chandran, Alessandra Loda, Anna Cancarini, Piergiuseppe Sacristani, Francesco Semeraro, and Marco Presta. 2021. "VEGF-Independent Activation of Müller Cells by the Vitreous from Proliferative Diabetic Retinopathy Patients" International Journal of Molecular Sciences 22, no. 4: 2179. https://doi.org/10.3390/ijms22042179