
Convergence: Lactosylceramide-Centric Signaling Pathways Induce Inflammation, Oxidative Stress, and Other Phenotypic Outcomes



{kind=link}
{kind=link}
{kind=link}
Abstract
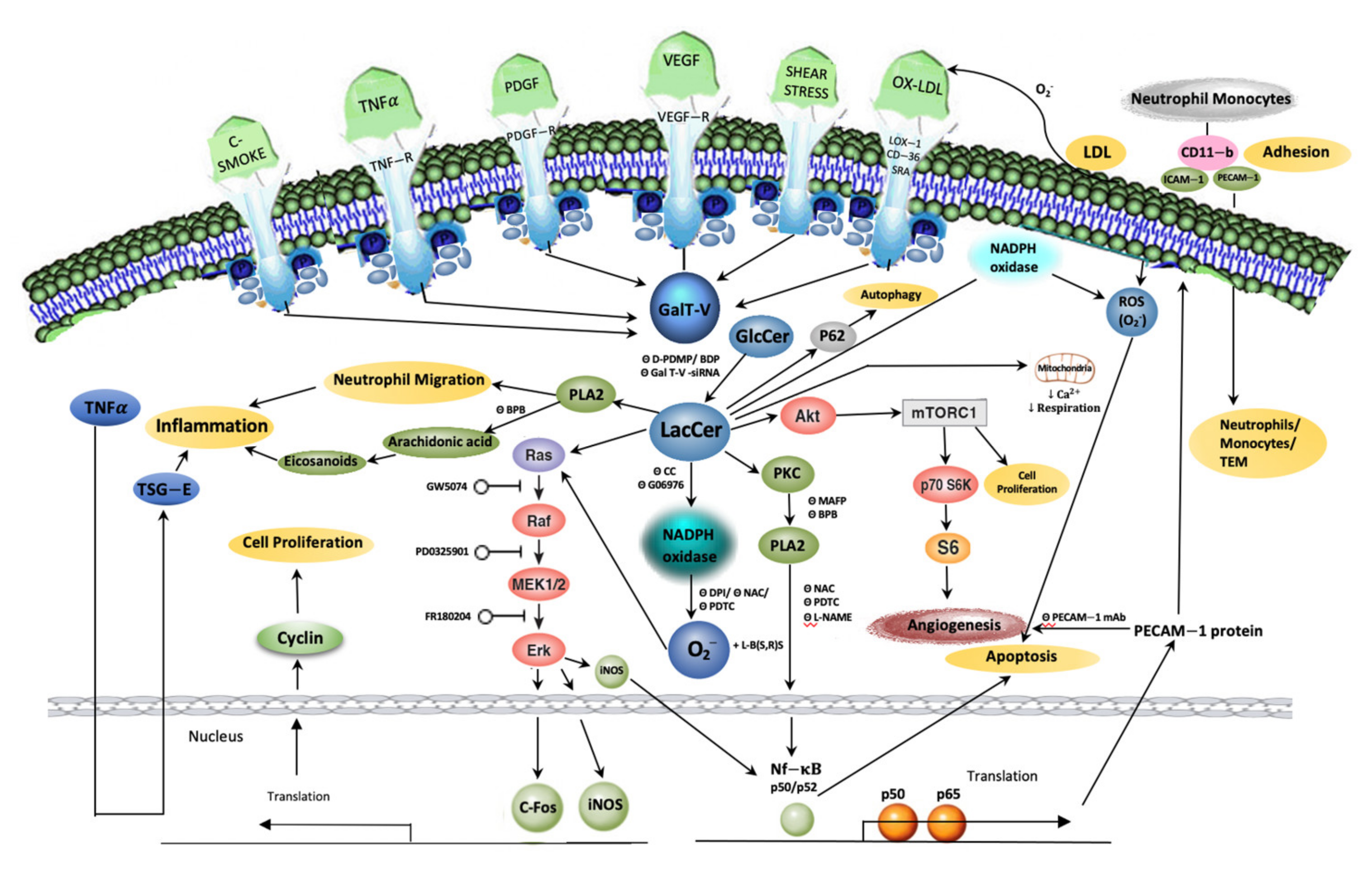
:1. Introduction
2. Lactosylceramide (LacCer) Synthesis via β-1,4 Galactosyltransferases V and VI and Physiological Implications
3. Lactosylceramide Localization
4. Lactosylceramide Synthesis via Neuraminidase, α-Galactosidase, and Sphingomyelinases
5. Role of the Low-Density Lipoprotein (LDL) Receptor in Determining the Metabolic Fate of LacCer
- (i)
- In vitro metabolism of LacCer
- (ii)
- In vivo metabolism of LacCer in man
6. Lactosylceramide Metabolomics and Cardiovascular Function
7. LacCer in Cell Proliferation: Recruitment of Reactive Oxygen Species, a Kinase Cascade, Proto-Oncogene c-Fos, and Proliferating Cell Nuclear Antigen
8. Oxidized LDL Recruits LacCer Synthase/LacCer to Induce Cell Proliferation and Atherosclerosis
9. LacCer Induces LDL Oxidation in a Cyclical Fashion: Effects of Feeding a Western Diet in ApoE−/− Mice
10. LacCer in Mitochondrial Function, Apoptosis, and Necrosis
11. LacCer-Centric Pathways Leading to Inflammation
- (i)
- LacCer mediates a signaling pathway to induce cell adhesion/migration and inflammation
- a.
- LacCer neuro-inflammation and inflammatory bowel disease
- b.
- LacCer, shear stress, and ischemia reperfusion injury
- c.
- LacCer and autophagy
- d.
- LacCer and skin inflammation
- (iii)
- LacCer recruits cytosolic phospholipase A2 pathway to induce cell adhesion and inflammation
- a.
- LacCer and phospholipase A2 in inflammation and cell adhesion in neutrophils and monocytes
- b.
- LacCer and VEGF-induced angiogenesis and inflammation in endothelial cells
12. Conclusions
- The LacCer-centric inflammatory pathway can begin to explain the pathophysiology of skin inflammation, neuro-inflammation, and hair greying/loss due to aging as well as other inflammatory diseases, e.g., COPD and inflammatory bowel disease.
- A western diet, which induced skin inflammation, hair loss, and hair discoloration, implicates the LacCer biosynthetic pathway involving neutrophil infiltration and TSG-6 expression. This was reversed by blocking LacCer synthesis and raising skin ceramide levels.
- TNF-α, a major pro-inflammatory cytokine, recruits players in the LacCer-centric inflammatory pathway, such as cPLA2 enzyme to generate eicosanoids/prostaglandins, leading to inflammation, neutrophil migration/infiltration, and expression of neutrophil/monocyte cell adhesion molecule, Mac-1 (CD11b). This LacCer-centric inflammatory pathway is shared in neuro-inflammation, ulcerative colitis, autophagy in emphysema due to COPD and cigarette smoking.
- The LacCer-centric oxidative stress pathway may increase our understanding of atherosclerosis and lupus erythematosus via downstream generation of ROS to: (a) oxidize LDL in a cyclical fashion, raising the blood levels of oxidized LDL, (b) induce cell proliferation via activating phosphokinases Akt-1 and mTOR-C1 signaling cascade, (c) regulate cell–cell adhesion involving cell adhesion molecules, ICAM-1, PECAM-1, and Mac-1 (CD11b), (d) cause shear stress induced mechano-transduction and ischemia reperfusion injury, and (e) activate the cPLA2 enzyme and PECAM-1 gene expression to induce angiogenesis required for atherosclerotic plaque survival and cancer metastasis and tumor growth in colorectal cancer and possibly other types of cancer.
- LacCer synthase is recruited and activated by VEGF and β-FGF to induce angiogenesis in vitro and in vivo via increased expression of ICAM-1 and PECAM-1 followed by monocyte and neutrophil TEM contributing to inflammation.
- LacCer plays an important role in mitochondrial function by downregulating respiration and Ca2+ retention in diabetic rats. Both in mitochondria of diabetic mice and in N-SMase-deficient cells, ceramide generation may not be sufficient to induce apoptosis and necrosis; its glycosylation to LacCer is required.
- We are only beginning to understand the role of LacCer synthase in other inflammatory diseases such as Lupus Erythematosus. As Lupus predominantly afflicts women in their early reproductive years, this commands greater and urgent attention.
- In diabetes, the mitochondria—the powerhouse of energy production and Ca2+ metabolism—becomes dysfunctional presumably due to increased LacCer levels. Further studies, including the targeting of drugs to improve mitochondrial health, are needed in this area. Such studies, in turn, will improve cardiac health.
- Since knowledge of GlcCer and LacCer levels has increased the predictive value of atherosclerosis and diabetes disease onset and progression and cardiac function, these measurements could be prescribed as routine tests in clinical settings.
- The pre-clinical studies above have provided a wealth of information on the role of LacCer synthase in several inflammatory diseases, cancer and atherosclerosis to set the stage for future human trials using various therapeutic modalities.
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| β-1,4 GalT-V | β-1,4 Galactosyltransferases V |
| β-1,4 GalT-VI | β-1,4 Galactosyltransferases VI |
| β-FGF | β-fibroblast growth factor |
| AP-1ApoE | Activator protein 1Apolipoprotein E |
| AS | Arterial stiffness |
| BAX | Bcl-2-associated X protein |
| Cer | Ceramide |
| CD | Crohn’s disease |
| CD-36 | Cluster of differentiation 36 |
| cDNA | Complementary DNA |
| CHO | Chinese hamster ovary |
| CoA | Coenzyme A |
| COPD | Chronic obstructive pulmonary disease |
| cPLA2 | Cytosolic phospholipase A2 |
| CVD | Cardiovascular disease |
| D-PDMP | D-threo-1-phenyl-2-decanoylamino-3-morpholino-1-propanol |
| E2FELAM | E2 transcription factorEndothelial leukocyte cell adhesion molecule |
| ER | Endoplasmic reticulum |
| ERKFH | Extracellular-signal-regulated kinaseFamilial hypercholesterolemic |
| HAEC | Human arterial endothelial cells |
| HCT | Human colorectal cancer |
| HDL | High-density lipoprotein |
| HIF-1A | Hypoxia-inducible factor 1-alpha |
| GbOse3Cer | Globotriosylceramide |
| GD3 | Disialosyldihexosylceramide |
| GlcCer | Glucosylceramide |
| GM1 | Monosialotetrahexosylceramide |
| GM3 | Monosialodihexosylceramide |
| GSL | Glycosphingolipid |
| ICAM-1 | Intercellular cell adhesion molecule-1 |
| IL-23JNKKDR | Interleukin-23C-jun N-terminal kinase Kinase insert domain receptor |
| LacCer | Lactosylceramide |
| LC-MS | Liquid chromatography-mass spectrometry |
| LC-QTOFMS | Quadrupole Time-of-Flight liquid chromatography/mass spectrometry |
| LDL | Low-density lipoprotein |
| LFA-1 | Lymphocyte function-associated antigen-1 |
| LOX-1 | Lectin-like ox-LDL |
| L-NAME | Nw-nitro-L-arginine methyl ester |
| L-PDMP | L-threo-1-phenyl-2-decanoylamino-3-morpholino-1-propanol |
| MAM | Mitochrondria-associated endoplasmic reticulum membrane |
| MAPK | Mitogen-activated protein kinase |
| MEK | Mitogen-activated protein kinase kinase |
| MIP-2 | Macrophage inflammatory protein 2 |
| MMP | Metalloproteinase |
| mRNA | messenger RNA |
| MS | Mass spectrometry |
| mTORC1 | Mammalian target of rapamycin |
| NADH | Nicotinamide adenine dinucleotide hydride |
| NADPH | Nicotinamide adenine dinucleotide phosphate |
| Neu1 | Nueraminidase-1 |
| Neu3 | Neuraminidase-3 |
| NF-κB | Nuclear factor kappa B |
| N-SMase | Neutral sphingomyelinase |
| Ox-LDL | Oxidized low-density lipoprotein |
| PCNA | Proliferating cell nuclear antigen |
| PDGF | Platelet-derived growth factor |
| PDTC | Pyrrolodine-carbodithio acid |
| PECAM-1 | Platelet-endothelial cell adhesion molecule-1 |
| PGE2 | Prostaglandin E2 |
| PI-3 | Phosphoinositide 3 |
| PKC | Protein kinase C |
| PLA2 | Phospholipase A2 |
| POVPC | 1-palmitoyl-2-(5-oxovaleroyl)-sn-glycero-3-phosphatidylcholine |
| PWV | Pulse-wave velocity |
| RBK-Jk | Recombination signal binding protein for immunoglobulin kappa j region |
| ROS | Reactive oxygen species |
| S1P | Sphingosine-1-phosphate |
| shRNA | Short hairpin ribonucleic acid |
| SOD | Superoxide dismutase |
| SRA-1 | Scavenger receptor-A-1 |
| TEM | Trans-endothelial migration |
| TNF-α | Tumor necrosis factor-alpha |
| TSG-6 | TNF-α inducible factor |
| UC | Ulcerative colitis |
| UDP | Uridine diphosphate |
| VCAM-1 | Vascular cell adhesion molecule-1 |
| VEGF | Vascular endothelial growth factor |
| VLDL | Very low-density lipoprotein |
| WT | Wild-type |
References
- Basu, S.; Kaufman, B.; Roseman, S. Enzymatic synthesis of ceramide-glucose and ceramide lactose by glycosyltransferase from embryonic chick brain. J. Biol. Chem. 1968, 243, 5802–5804. [Google Scholar] [CrossRef]
- Spiegel, S. Sphingosine-1-phosphate: From insipid lipid to a key regulator. J. Biol. Chem. 2020, 295, 3371–3384. [Google Scholar] [CrossRef] [Green Version]
- Hannun, Y.A.; Obeid, L.M. Sphingolipids and their metabolism in physiology and disease. Nat. Rev. Mol. Cell. Biol. 2018, 19, 175–191. [Google Scholar] [CrossRef]
- Schömel, N.; Geisslinger, G.; Wegner, M.S. Influence of glycosphingolipids on cancer cell energy metabolism. Prog. Lipid Res. 2020, 79, 101050. [Google Scholar] [CrossRef] [PubMed]
- Chatterjee, S. Sphingolipids in atherosclerosis and vascular biology. Arterioscler. Thromb. Vasc. Biol. 1998, 18, 1523–1533. [Google Scholar] [CrossRef] [Green Version]
- Chatterjee, S.; Ghosh, N.; Khurana, S. Purification of uridine diphosphate-galactose: Glucosyl ceramide, beta 1-4 galactosyltransferase from human kidney. J. Biol. Chem. 1992, 267, 7148–7153. [Google Scholar] [CrossRef]
- Nomura, T.; Takizawa, M.; Aoki, J.; Arai, H.; Inoue, K.; Wakisaka, E.; Yoshizuka, N.; Imokawa, G.; Dohmae, N.; Takio, J.; et al. Purification, cDNA cloning, and expression of UDP-Gal: Glucosylceramide β-1,4-galactosyltransferase from rat brain. J. Biol. Chem. 1998, 273, 13570–13577. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lo, N.W.; Shaper, J.H.; Pevsner, J.; Shaper, N.L. The expanding β4-galactosyltransferase gene family: Messages from the databanks. Glycobiology 1998, 8, 517–526. [Google Scholar] [CrossRef] [PubMed]
- Yoshihara, T.; Satake, H.; Nishie, T.; Nozumu, O.; Hatta, T.; Otani, H.; Naruse, C.; Suzuki, H.; Sugihara, K.; Kamimura, E.; et al. Lactosylceramide synthases encoded by B4galt5 and 6 genes are pivotal for neuronal generation and myelin formation in mice. PLoS Genet. 2018, 14, e1007545. [Google Scholar] [CrossRef] [PubMed]
- Nishie, T.; Hikimochi, Y.; Zama, K.; Fukusumi, Y.; Ito, M.; Yokoyama, H.; Naruse, C.; Ito, M.; Asano, M. β4-Galactosyltransferase-5 is a lactosylceramide synthase essential for mouse extra-embryonic development. Glycobiology 2010, 20, 1311–1322. [Google Scholar] [CrossRef] [Green Version]
- Kumagai, T.; Sato, T.; Natsuka, S.; Kobayashi, Y.; Zhou, D.; Shinkai, T.; Hayakawa, S.; Furukawa, K. Involvement of murine β-1,4-galactosyltransferase V in lactosylceramide biosynthesis. Glycoconj. J. 2010, 27, 685–695. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kolmakova, A.; Chatterjee, S. Platelet derived growth factor recruits lactosylceramide to induce cell proliferation in UDP Gal:GlcCer: β1→4galactosyltransferase mutant Chinese hamster ovary cells. Glyconj. J. 2005, 22, 401–407. [Google Scholar] [CrossRef] [PubMed]
- Chatterjee, S.; Alsaeedi, N.; Hou, J.; Bandaru, V.V.; Wu, L.; Halushka, M.K.; Pili, R.; Ndikuyeze, G.; Haughey, N.J. Use of a glycolipid inhibitor to ameliorate renal cancer in a mouse model. PLoS ONE 2013, 8, e63726. [Google Scholar] [CrossRef] [Green Version]
- Ode, T.; Podyma-Inoue, K.A.; Terasawa, K.; Inokuchi, J.I.; Kobayashi, T.; Watabe, T.; Izumi, Y.; Hara-Yokoyama, M. PDMP, a ceramide analogue, acts as an inhibitor of mTORC1 by inducing its translocation from lysosome to endoplasmic reticulum. Exp. Cell Res. 2017, 350, 103–114. [Google Scholar] [CrossRef]
- Chatterjee, S.; Zheng, L.; Ma, S.; Bedja, D.; Bandaru, V.; Kim, G.; Rangecroft, A.B.; Iocco, D.; Campbell, S.A. Management of metabolic syndrome and reduction in body weight in type II diabetic mice by inhibiting glycosphingolipid synthesis. Biochem. Biophys. Res. Commun. 2020, 525, 455–461. [Google Scholar] [CrossRef] [PubMed]
- Nakayama, H.; Nagafuku, M.; Suzuki, A.; Iwabuchi, K.; Inokuchi, J.I. The regulatory roles of glycosphingolipid-enriched lipid rafts in immune systems. FEBS Lett. 2018, 592, 3921–3942. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chatterjee, S.; Kwiterovich, P.O., Jr.; Gupta, P.; Erozan, Y.S.; Alving, C.R.; Richards, R.L. Localization of urinary lactosylceramide in cytoplasmic vesicles of renal tubular cells in homozygous familial hypercholesterolemia. Proc. Natl. Acad. Sci. USA 1983, 80, 1313–1317. [Google Scholar] [CrossRef] [Green Version]
- Chatterjee, S.; Shi, W.Y.; Wilson, P.; Mazumdar, A. Role of lactosylceramide and MAP kinase in the proliferation of proximal tubular cells in human polycystic kidney disease. J. Lipid Res. 1996, 37, 1334–1344. [Google Scholar] [CrossRef]
- Natoli, T.A.; Smith, L.A.; Rogers, K.A.; Wang, B.; Komarnitsky, S.; Budman, Y.; Belenky, A.; Bukanov, N.O.; Dackowski, W.R.; Husson, H.; et al. Inhibition of glucosylceramide accumulation results in effective blockade of polycystic kidney disease in mouse models. Nat. Med. 2010, 16, 788–792. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iwabuchi, K.; Masuda, H.; Kaga, N.; Nakayama, H.; Matsumoto, R.; Iwahara, C.; Yoshizaki, F.; Tamaki, Y.; Kobayashi, T.; Hayakawa, T.; et al. Properties and functions of lactosylceramide from mouse neutrophils. Glycobiology 2015, 25, 655–668. [Google Scholar] [CrossRef]
- Iwabuchi, K. Involvement of glycosphingolipid-enriched lipid rafts in inflammatory responses. Front Biosci. (Landmark Ed.) 2015, 20, 325–334. [Google Scholar] [CrossRef] [Green Version]
- Chatterjee, S.; Sekerke, C.S.; Kwiterovich, P.O., Jr. Alterations in cell surface glycosphingolipids and other lipid classes of fibroblasts in familial hypercholesterolemia. Proc. Natl. Acad. Sci. USA 1976, 73, 4339–4343. [Google Scholar] [CrossRef] [Green Version]
- Karlsson, K.A. Animal glycolipids as attachment sites for microbes. Chem. Phys. Lipids 1986, 42, 153–172. [Google Scholar] [CrossRef]
- Zimmerman, J.W.; Lindermuth, J.; Fish, P.A.; Palace, G.P.; Stevenson, T.T.; DeMong, D.E. A novel carbohydrate-glycosphingolipid interaction between a β-(1-3)-glucan immunomodulator, PGG-glucan, and lactosylceramide of human leukocytes. J. Biol. Chem. 1998, 273, 22014–22020. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chaichian, S.; Moazzami, B.; Sadoughi, F.; Haddad Kashani, H.; Zaroudi, M.; Asemi, Z. Functional activities of beta-glucans in the prevention or treatment of cervical cancer. J. Ovarian Res. 2020, 13, 24. [Google Scholar] [CrossRef] [Green Version]
- Sonnino, S.; Prinetti, A.; Mauri, L.; Chigorno, V.; Tettamanti, G. Dynamic and structural properties of sphingolipids as driving forces for the formation of membrane domains. Chem. Rev. 2006, 106, 2111–2125. [Google Scholar] [CrossRef]
- Chatterjee, S.; Pandey, A. The Yin and Yang of lactosylceramide metabolism: Implications in cell function. Biochim. Biophys. Acta 2008, 1780, 370–382. [Google Scholar] [CrossRef] [PubMed]
- Frost, R.G.; Holmes, E.W.; Norden, A.G.; O’Brien, J.S. Characterization of purified human liver acid β-d-galactosidases A2 and A3. Biochem. J. 1978, 175, 181–188. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kolter, T.; Proia, R.L.; Sandhoff, K. Combinatorial ganglioside biosynthesis. J. Biol. Chem. 2002, 277, 25859–25862. [Google Scholar] [CrossRef] [Green Version]
- Miyagi, T.; Wada, T.; Yamaguchi, K. Roles of plasma membrane-associated sialidase NEU3 in human cancers. Biochim. Biophys. Acta 2008, 1780, 532–537. [Google Scholar] [CrossRef]
- Machala, M.; Procházková, J.; Hofmanová, J.; Králiková, L.; Slavík, J.; Tylichová, Z.; Ovesná, P.; Kozubík, A.; Vondráček, J. Colon Cancer and Perturbations of the Sphingolipid Metabolism. Int. J. Mol. Sci. 2019, 20, 6051. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hocine, T.; Blaise, S.; Hachet, C.; Guillot, A.; Sartelet, H.; Maurice, P.; Bennasroune, A.; Martiny, L.; Duca, L.; Romier-Crouzet, B.; et al. Lactosylceramide induced by elastin-derived peptides decreases adipocyte differentiation. J. Physiol. Biochem. 2020, 76, 457–567. [Google Scholar] [CrossRef] [PubMed]
- Tartakoff, A.M.; Vassalli, P. Plasma cell immunoglobulin secretion: Arrest is accompanied by alterations of the golgi complex. J. Exp. Med. 1977, 146, 1332–1345. [Google Scholar] [CrossRef]
- Johnson, D.C.; Schlesinger, M.J. Vesicular stomatitis virus and sindbis virus glycoprotein transport to the cell surface is inhibited by ionophores. Virology 1980, 103, 407–424. [Google Scholar] [CrossRef]
- Basu, S.K.; Goldstein, J.L.; Anderson, R.G.; Brown, M.S. Monensin interrupts the recycling of low density lipoprotein receptors in human fibroblasts. Cell 1981, 24, 493–502. [Google Scholar] [CrossRef]
- Miller-Prodraza, H.; Fishman, P.H. Effect of drugs and temperature on biosynthesis and transport of glycosphingolipids in cultured neurotumor cells. Biochim. Biophys. Acta 1984, 804, 44–51. [Google Scholar] [CrossRef]
- Saito, M.; Saito, M.; Rosenberg, A. Influence of monovalent cation transport on anabolism of glycosphingolipids in cultured human fibroblasts. Biochemistry 1985, 24, 3054–3059. [Google Scholar] [CrossRef] [PubMed]
- Chatterjee, S. Effects of monensin on glycosphingolipid metabolism in cultured human proximal tubular cells. Indian J. Biochem. Biophys. 1993, 30, 346–352. [Google Scholar] [PubMed]
- Won, J.S.; Singh, A.K.; Singh, I. Lactosylceramide: A lipid second messenger in neuroinflammatory disease. J. Biochem. 2007, 103, 180–191. [Google Scholar] [CrossRef]
- Chatterjee, S.; Clarke, K.S.; Kwiterovich, P.O., Jr. Regulation of synthesis of lactosylceramide and long chain bases in normal and familial hypercholesterolemic cultured proximal tubular cells. J. Biol. Chem. 1986, 261, 13474–13479. [Google Scholar] [CrossRef]
- Chatterjee, S.; Ghosh, N.; Castiglione, E.; Kwiterovich, P.O., Jr. Regulation of glycosphingolipid glycosyltransferase by low density lipoprotein receptors in cultured human proximal tubular cells. J. Biol. Chem. 1988, 263, 13017–13022. [Google Scholar] [CrossRef]
- Chatterjee, S. Regulation of synthesis of lactosylceramide in normal and tumor proximal tubular cells. Biochim. Biophys. Acta 1993, 1167, 339–344. [Google Scholar] [CrossRef]
- Chatterjee, S.; Kwiterovich, P.O., Jr. Lipid Storage Disorders: Biological and Medical Aspects; Plenum Publishing Corp: New York, NY, USA, 1987; pp. 613–623. [Google Scholar]
- Chatterjee, S.; Sekerke, C.S.; Kwiterovich, P.O., Jr. Increased urinary excretion of glycosphingolipids in familial hypercholesterolemia. J. Lipid Res. 1982, 23, 513–522. [Google Scholar] [CrossRef]
- Chatterjee, S.B.; Hou, J.; Bandaru, V.; Pezhouh, M.K.; Syed-Rifat-Mannan, A.A.; Sharma, R. Lactosylceramide synthase β-1,4-GalT-V: A novel target for the diagnosis and therapy of human colorectal cancer. Biochem. Biophys. Res. Commun. 2019, 508, 380–386. [Google Scholar] [CrossRef] [PubMed]
- Chatterjee, S.; Bedja, D.; Mishra, S.; Amuzie, C.; Avolio, A.; Kass, D.A.; Berkowitz, D.; Renehan, M. Inhibition of glycosphingolipid synthesis ameliorates atherosclerosis and arterial stiffness in apolipoprotein E-/- mice and rabbits fed a high-fat and -cholesterol diet. Circulation 2014, 129, 2403–2413. [Google Scholar] [CrossRef] [Green Version]
- Kim, M.; Jung, S.; Lee, S.H.; Lee, J.H. Association between arterial stiffness and serum L-octanoylcarnitine and lactosylceramide in overweight middle-aged subjects: 3-year follow-up study. PLoS ONE 2015, 10, e0119519. [Google Scholar] [CrossRef]
- Alshehry, Z.H.; Mundra, P.A.; Barlow, C.K.; Mellett, N.A.; Wong, G.; McConville, M.J.; Simes, J.; Tonkin, A.M.; Sullivan, D.R.; Barnes, E.H.; et al. Plasma Lipidomic Profiles Improve on Traditional Risk Factors for the Prediction of Cardiovascular Events in Type 2 Diabetes Mellitus. Circulation 2016, 134, 1637–1650. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Diaz, S.O.; Sánchez-Quesada, J.L.; de Freitas, V.; Leite-Moreira, A.; Barros, A.S.; Reis, A. Exploratory analysis of large-scale lipidome in large cohorts: Are we any closer of finding lipid-based markers suitable for CVD risk stratification and management? Anal. Chim. Acta 2021, 1142, 189–200. [Google Scholar] [CrossRef]
- Chatterjee, S. Lactosylceramide stimulates aortic smooth muscle cell proliferation. Biochem. Biophys. Res. Commun. 1991, 181, 554–561. [Google Scholar] [CrossRef]
- Chatterjee, S.B.; Dey, S.; Shi, W.Y.; Thomas, K.; Hutchins, G.M. Accumulation of glycosphingolipids in human atherosclerotic plaque and unaffected aorta tissues. Glycobiology 1997, 7, 57–65. [Google Scholar] [CrossRef] [Green Version]
- Bhunia, A.K.; Han, H.; Snowden, A.; Chatterjee, S. Lactosylceramide stimulates Ras-GTP loading, kinases (MEK, Raf), p44 mitogen-activated protein kinase, and c-fos expression in human aortic smooth muscle cells. J. Biol. Chem. 1996, 271, 10660–10666. [Google Scholar] [CrossRef] [Green Version]
- Chatterjee, S.; Bhunia, A.; Snowden, A.; Han, H. Oxidized low density lipoproteins stimulate galactosyltransferase activity; ras activation, p44 mitogen activated protein kinase and c-fos expression in aortic smooth muscle cells. Glycobiology 1997, 7, 703–710. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lau, L.F.; Nathans, D. Expression of a set of growth-related immediate early genes in BALB/c 3T3 cells: Coordinate regulation with c-fos or c-myc. Proc. Natl. Acad. Sci. USA 1987, 84, 1182–1186. [Google Scholar] [CrossRef] [Green Version]
- Bhunia, A.K.; Han, H.; Snowden, A.; Chatterjee, S. Redox-regulated signaling by lactosylceramide in the proliferation of human aortic smooth muscle cells. J. Biol. Chem. 1997, 272, 15642–15649. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martin, S.F.; Williams, N.; Chatterjee, S. Lactosylceramide is required in apoptosis induced by N-Smase. Glycoconj. J. 2006, 23, 147–157. [Google Scholar] [CrossRef]
- Belambri, S.A.; Rolas, L.; Raad, H.; Hurtado-Nedelec, M.; Dang, P.M.; El-Benna, J. NADPH oxidase activation in neutrophils: Role of the phosphorylation of its subunits. Eur. J. Clin. Investig. 2018, 48 (Suppl. 2), e12951. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abate, C.; Patel, L.; Rauscher, F.J., 3rd; Curran, T. Redox regulation of fos and jun DNA-binding activity in vitro. Science 1990, 249, 1157–1161. [Google Scholar] [CrossRef]
- McCarthy, G.A. Autoantibodies, 2nd ed.; Elsevier Science: Amsterdam, The Netherlands, 2007; pp. 205–210. [Google Scholar]
- Chatterjee, S. Oxidized low density lipoproteins and lactosylceramide both stimulate the expression of proliferating cell nuclear antigen and the proliferation of aortic smooth muscle cells. Indian J. Biochem. Biophys. 1997, 34, 56–60. [Google Scholar]
- Navab, M.; Berliner, J.A.; Watson, A.D.; Hama, S.Y.; Territo, M.C.; Lusis, A.J.; Shih, D.M.; Van Lenten, B.J.; Frank, J.S.; Demer, L.L.; et al. The Yin and Yang of oxidation in the development of the fatty streak. A review based on the 1994 George Lyman Duff Memorial Lecture. Arterioscler. Thromb. Vasc. Biol. 1996, 16, 831–842. [Google Scholar] [CrossRef] [PubMed]
- Chatterjee, S. Pathophysiology and Vascular Biology in Atherosclerosis. In Johns Hopkins University Textbook of Dyslipidemia, 1st ed.; Kwiterovich, M.D., Peter, O., Jr., Eds.; Lippincott, Williams & Wilkins: Baltimore, MD, USA, 2010; pp. 48–57. [Google Scholar]
- Meisinger, C.; Baumert, J.; Khuseyinova, N.; Loewel, H.; Koenig, W. Plasma oxidized low-density lipoprotein; a strong predictor for acute coronary heart disease events in apparently healthy; middle-aged men from the general population. Circulation 2005, 112, 651–657. [Google Scholar] [CrossRef] [Green Version]
- Ahmad, H.M.; Sarhan, E.M.; Komber, U. Higher circulating levels of OxLDL % of LDL are associated with subclinical atherosclerosis in female patients with systemic lupus erythematosus. Rheumatol. Int. 2014, 34, 617–623. [Google Scholar] [CrossRef]
- Chatterjee, S.; Kolmakova, A.; Rajesh, M. Regulation of lactosylceramide synthase (glucosylceramide β1→4 galactosyltransferase); implication as a drug target. Curr. Drug. Targets 2008, 9, 272–281. [Google Scholar] [CrossRef] [PubMed]
- Chatterjee, S.; Berliner, J.A.; Subbanagounder, G.G.; Bhunia, A.K.; Koh, S. Identification of a biologically active component in minimally oxidized low density lipoprotein (MM-LDL) responsible for aortic smooth muscle cell proliferation. Glycoconj. J. 2004, 20, 331–338. [Google Scholar] [CrossRef] [PubMed]
- Trayssac, M.; Hannun, Y.A.; Obeid, L.M. Role of sphingolipids in senescence: Implication in aging and age-related diseases. J. Clin. Investig. 2018, 128, 2702–2712. [Google Scholar] [CrossRef]
- Mishra, S.; Bedja, D.; Amuzie, C.; Avolio, A.; Chatterjee, S. Prevention of cardiac hypertrophy by the use of a glycosphingolipid synthesis inhibitor in ApoE-/- mice. Biochem. Biophys. Res. Commun. 2015, 465, 159–164. [Google Scholar] [CrossRef] [Green Version]
- Sadras, V.; Petri, M.A.; Jones, S.R.; Peterlin, B.L.; Chatterjee, S. Glycosphingolipid-associated β-1,4 galactosyltransferase is elevated in patients with systemic lupus erythematosus. Lupus Sci. Med. 2020, 7, e000368. [Google Scholar] [CrossRef] [PubMed]
- Elloumi, N.; Ben-Mansour, R.; Marzouk, S.; Mseddi, M.; Fakhfakh, R.; Gargouri, B.; Masmoudi, H.; Lassoued, S. Differential reactive oxygen species production of neutrophils and their oxidative damage in patients with active and inactive systemic lupus erythematosus. Immunol. Lett. 2017, 184, 1–6. [Google Scholar] [CrossRef]
- Schömel, N.; Gruber, L.; Alexopoulos, S.J.; Trautmann, S.; Olzomer, E.M.; Byrne, F.L.; Hoehn, K.L.; Gurke, R.; Thomas, D.; Ferreirós, N.; et al. UGCG overexpression leads to increased glycolysis and increased oxidative phosphorylation of breast cancer cells. Sci. Rep. 2020, 10, 8182. [Google Scholar] [CrossRef] [PubMed]
- Chatterjee, S. Role of oxidized human plasma low density lipoproteins in atherosclerosis: Effects on smooth muscle cell proliferation. Mol. Cell Biochem. 1992, 111, 143–147. [Google Scholar] [CrossRef]
- Chatterjee, S.; Gakenheimer, K.; Han, H. Oxidized-LDL stimulates apoptosis via the activation of a neutral sphingomyelinase in human arterial smooth muscle cells. In Sphingolipid Metabolizing Enzymes; Research Signpost: Trivandrum, India, 2004; pp. 101–106. [Google Scholar]
- Salvayre, R.; Auge, N.; Benoist, H.; Negre-Salvayre, A. Oxidized low-density lipoprotein-induced apoptosis. Biochim. Biophys. Acta 2002, 1585, 213–221. [Google Scholar] [CrossRef]
- García-Ruiz, C.; Colell, A.; París, R.; Fernández-Checa, J.C. Direct interaction of GD3 ganglioside with mitochondria generates reactive oxygen species followed by mitochondrial permeability transition; cytochrome c release; and caspase activation. FASEB J. 2000, 14, 847–858. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Novgorodov, S.A.; Riley, C.L.; Yu, J.; Keffler, J.A.; Clarke, C.J.; van Laer, A.O.; Baicu, C.F.; Zile, M.R.; Gudz, T.I. Lactosylceramide contributes to mitochondrial dysfunction in diabetes. J. Lipid Res. 2016, 57, 546–562. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roca, F.J.; Whitworth, L.J.; Redmond, S.; Jones, A.A.; Ramakrishnan, L. TNF Induces Pathogenic Programmed Macrophage Necrosis in Tuberculosis through a Mitochondrial-Lysosomal-Endoplasmic Reticulum Circuit. Cell 2019, 178, 1344–1361. [Google Scholar] [CrossRef] [Green Version]
- Kim, M.J.; Wainwright, H.C.; Locketz, M.; Bekker, L.G.; Walther, G.B.; Dittrich, C.; Visser, A.; Wang, W.; Hsu, F.F.; Wiehart, U.; et al. Caseation of human tuberculosis granulomas correlates with elevated host lipid metabolism. EMBO Mol. Med. 2010, 2, 258–274. [Google Scholar] [CrossRef] [PubMed]
- Jovinge, S.; Ares, M.P.; Kallin, B.; Nilsson, J. Human monocytes/macrophages release TNF-alpha in response to Ox-LDL. Arterioscler. Thromb. Vasc. Biol. 1996, 16, 1573–1579. [Google Scholar] [CrossRef] [PubMed]
- Bevilacqua, M.P.; Stengelin, S.; Gimbrone, M.A., Jr.; Seed, B. Endothelial leukocyte adhesion molecule 1: An inducible receptor for neutrophils related to complement regulatory proteins and lectins. Science 1989, 243, 1160–1165. [Google Scholar] [CrossRef]
- Bhunia, A.K.; Arai, T.; Bulkley, G.; Chatterjee, S. Lactosylceramide mediates tumor necrosis factor-alpha-induced intercellular adhesion molecule-1 (ICAM-1) expression and the adhesion of neutrophil in human umbilical vein endothelial cells. J. Biol. Chem. 1998, 273, 34349–34357. [Google Scholar] [CrossRef] [Green Version]
- Arai, T.; Bhunia, A.K.; Chatterjee, S.; Bulkley, G.B. Lactosylceramide stimulates human neutrophils to upregulate Mac-1; adhere to endothelium; and generate reactive oxygen metabolites in vitro. Circ. Res. 1998, 82, 540–547. [Google Scholar] [CrossRef] [Green Version]
- Ross, R. The pathogenesis of atherosclerosis: A perspective for the 1990s. Nature 1993, 362, 801–809. [Google Scholar] [CrossRef]
- Hansson, G.K. Inflammation; atherosclerosis; and coronary artery disease. N. Engl. J. Med. 2005, 352, 1685–1695. [Google Scholar] [CrossRef] [Green Version]
- Pannu, R.; Won, J.S.; Khan, M.; Singh, A.K.; Singh, I. A novel role of lactosylceramide in the regulation of lipopolysaccharide/interferon-gamma-mediated inducible nitric oxide synthase gene expression: Implications for neuroinflammatory diseases. J. Neurosci. 2004, 24, 5942–5954. [Google Scholar] [CrossRef] [Green Version]
- Pannu, R.; Singh, A.K.; Singh, I. A novel role of lactosylceramide in the regulation of tumor necrosis factor alpha-mediated proliferation of rat primary astrocytes. Implications for astrogliosis following neurotrauma. J. Biol. Chem. 2005, 280, 13742–13751. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Asada, M.; Furukawa, K.; Segawa, K.; Endo, T.; Kobata, A. Increased expression of highly branched N-glycans at cell surface is correlated with the malignant phenotypes of mouse tumor cells. Cancer Res. 1997, 57, 1073–1080. [Google Scholar] [PubMed]
- Dennis, J.W.; Granovsky, M.; Warren, C.E. Glycoprotein glycosylation and cancer progression. Biochim. Biophys. Acta 1999, 1473, 21–34. [Google Scholar] [CrossRef]
- Stevens, C.R.; Oberholzer, V.G.; Walker-Smith, J.A.; Phillips, A.D. Lactosylceramide in inflammatory bowel disease: A biochemical study. Gut 1988, 29, 580–587. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Daniluk, U.; Daniluk, J.; Kucharski, R.; Kowalczyk, T.; Pietrowska, K.; Samczuk, P.; Filimoniuk, A.; Kretowski, A.; Lebensztejn, D.; Ciborowski, M. Untargeted Metabolomics and Inflammatory Markers Profiling in Children With Crohn’s Disease and Ulcerative Colitis-A Preliminary Study. Inflamm. Bowel Dis. 2019, 25, 1120–1128. [Google Scholar] [CrossRef] [PubMed]
- Filimoniuk, A.; Blachnio-Zabielska, A.; Imierska, M.; Lebensztejn, D.M.; Daniluk, U. Sphingolipid Analysis Indicate Lactosylceramide as a Potential Biomarker of Inflammatory Bowel Disease in Children. Biomolecules 2020, 10, 1083. [Google Scholar] [CrossRef] [PubMed]
- Yeh, L.H.; Kinsey, A.M.; Chatterjee, S.; Alevriadou, B.R. Lactosylceramide mediates shear-induced endothelial superoxide production and intercellular adhesion molecule-1 expression. J. Vasc. Res. 2001, 38, 551–559. [Google Scholar] [CrossRef]
- Novgorodov, S.A.; Gudz, T.I. Ceramide and mitochondria in ischemia/reperfusion. J. Cardiovasc. Pharmacol. 2009, 53, 198–208. [Google Scholar] [CrossRef] [Green Version]
- Martin, S.F.; Chatterjee, S.; Parinandi, N.; Alevriadou, B.R. Rac1 inhibition protects against hypoxia/reoxygenation-induced lipid peroxidation in human vascular endothelial cells. Vascul. Pharmacol. 2005, 43, 148–156. [Google Scholar] [CrossRef] [PubMed]
- Uhlig, S.; Gulbins, E. Sphingolipids in the lungs. Am. J. Respir. Crit. Care Med. 2008, 178, 1100–1114. [Google Scholar] [CrossRef]
- Petrache, I.; Natarajan, V.; Zhen, L.; Medler, T.R.; Richter, A.T.; Cho, C.; Hubbard, W.C.; Berdyshev, E.V.; Tuder, R.M. Ceramide upregulation causes pulmonary cell apoptosis and emphysema-like disease in mice. Nat. Med. 2005, 11, 491–498. [Google Scholar] [CrossRef] [Green Version]
- Bodas, M.; Min, T.; Mazur, S.; Vij, N. Critical modifier role of membrane-cystic fibrosis transmembrane conductance regulator-dependent ceramide signaling in lung injury and emphysema. J. Immunol. 2011, 186, 602–613. [Google Scholar] [CrossRef]
- MacNee, W. Oxidants and COPD. Curr. Drug Targets Inflamm. Allergy 2005, 4, 627–641. [Google Scholar] [CrossRef]
- Bodas, M.; Min, T.; Vij, N. Lactosylceramide-accumulation in lipid-rafts mediate aberrant-autophagy; inflammation and apoptosis in cigarette smoke induced emphysema. Apoptosis 2015, 20, 725–739. [Google Scholar] [CrossRef]
- Elias, P.M.; Menon, G.K. Structural and lipid biochemical correlates of the epidermal permeability barrier. Adv. Lipid Res. 1991, 24, 1–26. [Google Scholar] [CrossRef] [PubMed]
- Feingold, K.R. The regulation and role of epidermal lipid synthesis. Adv. Lipid Res. 1991, 24, 57–82. [Google Scholar] [CrossRef] [PubMed]
- Bedja, D.; Yan, W.; Lad, V.; Iocco, D.; Sivakumar, N.; Bandaru, V.; Chatterjee, S. Inhibition of glycosphingolipid synthesis reverses skin inflammation and hair loss in ApoE-/- mice fed western diet. Sci. Rep. 2018, 8, 11463. [Google Scholar] [CrossRef] [PubMed]
- Feingold, K.R.; Elias, P.M.; Mao-Qiang, M.; Fartasch, M.; Zhang, S.H.; Maeda, N. Apolipoprotein E deficiency leads to cutaneous foam cell formation in mice. J. Investig. Dermatol. 1995, 104, 246–250. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Greenwald, I. LIN-12/Notch signaling: Lessons from worms and flies. Genes Dev. 1998, 12, 1751–1762. [Google Scholar] [CrossRef] [Green Version]
- Schouwey, K.; Delmas, V.; Larue, L.; Zimber-Strobl, U.; Strobl, L.J.; Radtke, F.; Beermann, F. Notch1 and Notch2 receptors influence progressive hair graying in a dose-dependent manner. Dev. Dyn. 2007, 236, 282–289. [Google Scholar] [CrossRef]
- Liao, C.P.; Booker, R.C.; Morrison, S.J.; Le, L.Q. Identification of hair shaft progenitors that create a niche for hair pigmentation. Genes Dev. 2017, 31, 744–756. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Symington, F.W.; Hedges, D.L.; Hakomori, S. Glycolipid antigens of human polymorphonuclear neutrophils and the inducible HL-60 myeloid leukemia line. J. Immunol. 1985, 134, 2498–2506. [Google Scholar] [PubMed]
- Wakshull, E.; Brunke-Reese, D.; Lindermuth, J.; Fisette, L.; Nathans, R.S.; Crowley, J.J.; Tufts, J.C.; Zimmerman, J.; Mackin, W.; Adams, D.S. PGG-glucan; a soluble β-(1,3)-glucan; enhances the oxidative burst response; microbicidal activity, and activates an NF-κB-like factor in human PMN: Evidence for a glycosphingolipid β-(1,3)-glucan receptor. Immunopharmacology 1999, 41, 89–107. [Google Scholar] [CrossRef]
- Nakamura, H.; Moriyama, Y.; Watanabe, K.; Tomizawa, S.; Yamazaki, R.; Takahashi, H.; Murayama, T. Lactosylceramide-Induced Phosphorylation Signaling to Group IVA Phospholipase A2 via Reactive Oxygen Species in Tumor Necrosis Factor-α-Treated Cells. J. Cell Biochem. 2017, 118, 4370–4382. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, H.; Moriyama, Y.; Makiyama, T.; Emori, S.; Yamashita, H.; Yamazaki, R.; Murayama, T. Lactosylceramide interacts with and activates cytosolic phospholipase A2α. J. Biol. Chem. 2013, 288, 23264–23272. [Google Scholar] [CrossRef] [Green Version]
- Gong, N.; Wei, H.; Chowdhury, S.H.; Chatterjee, S. Lactosylceramide recruits PKCalpha/epsilon and phospholipase A2 to stimulate PECAM-1 expression in human monocytes and adhesion to endothelial cells. Proc. Natl. Acad. Sci. USA 2004, 101, 6490–6495. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Botella, L.M.; Puig-Kröger, A.; Almendro, N.; Sánchez-Elsner, T.; Muñoz, E.; Corbí, A.; Bernabéu, C. Identification of a functional NF-kappa B site in the platelet endothelial cell adhesion molecule-1 promoter. J. Immunol. 2000, 164, 1372–1378. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Folkman, J. Angiogenesis in cancer; vascular; rheumatoid and other disease. Nat. Med. 1995, 1, 27–31. [Google Scholar] [CrossRef] [PubMed]
- Ferrara, N. Vascular endothelial growth factor. Eur. J. Cancer 1996, 32, 2413–2422. [Google Scholar] [CrossRef]
- Rajesh, M.; Kolmakova, A.; Chatterjee, S. Novel role of lactosylceramide in vascular endothelial growth factor-mediated angiogenesis in human endothelial cells. Circ. Res. 2005, 98, 796–804. [Google Scholar] [CrossRef] [Green Version]
- Kolmakova, A.; Rajesh, M.; Zang, D.; Pili, R.; Chatterjee, S. VEGF recruits lactosylceramide to induce endothelial cell adhesion molecule expression and angiogenesis in vitro and in vivo. Glycoconj. J. 2009, 26, 547–558. [Google Scholar] [CrossRef] [PubMed]
- Procházková, J.; Slavík, J.; Bouchal, J.; Levková, M.; Hušková, Z.; Ehrmann, J.; Ovesná, P.; Kolář, Z.; Skalický, P.; Straková, N.; et al. Specific alterations of sphingolipid metabolism identified in EpCAM-positive cells isolated from human colon tumors. Biochim. Biophys. Acta. Mol. Cell Biol. Lipids 2020, 1865, 158742. [Google Scholar] [CrossRef] [PubMed]
- Jiang, J.; Shen, J.; Wu, T.; Wei, Y.; Chen, X.; Zong, H.; Zhang, S.; Sun, M.; Xie, J.; Kong, X.; et al. Down-regulation of β1,4-galactosyltransferase V is a critical part of etoposide-induced apoptotic process and could be mediated by decreasing Sp1 levels in human glioma cells. Glycobiology 2006, 16, 1045–1051. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wei, Y.; Zhou, F.; Ge, Y.; Chen, H.; Cui, C.; Li, Q.; Liu, D.; Yang, Z.; Wu, G.; Sun, S.; et al. β1,4-galactosyltransferase V regulates self-renewal of glioma-initiating cell. Biochem. Biophys. Res. Commun. 2010, 396, 602–607. [Google Scholar] [CrossRef]
- Wei, Y.; Zhou, F.; Ge, Y.; Chen, H.; Cui, C.; Liu, D.; Yang, Z.; Wu, G.; Shen, J.; Gu, J.; et al. Regulation of the β1,4-Galactosyltransferase I promoter by E2F1. J. Biochem. 2010, 148, 263–271. [Google Scholar] [CrossRef] [PubMed]
- Dennis, J.W.; Laferté, S.; Waghorne, C.; Breitman, M.L.; Kerbel, R.S. Beta 1-6 branching of Asn-linked oligosaccharides is directly associated with metastasis. Science 1987, 236, 582–585. [Google Scholar] [CrossRef]
- Mondal, K.; Mandal, N. Role of Bioactive Sphingolipids in Inflammation and Eye Diseases. Adv. Exp. Med. Biol. 2019, 1161, 149–167. [Google Scholar] [CrossRef]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chatterjee, S.; Balram, A.; Li, W. Convergence: Lactosylceramide-Centric Signaling Pathways Induce Inflammation, Oxidative Stress, and Other Phenotypic Outcomes. Int. J. Mol. Sci. 2021, 22, 1816. https://doi.org/10.3390/ijms22041816
Chatterjee S, Balram A, Li W. Convergence: Lactosylceramide-Centric Signaling Pathways Induce Inflammation, Oxidative Stress, and Other Phenotypic Outcomes. International Journal of Molecular Sciences. 2021; 22(4):1816. https://doi.org/10.3390/ijms22041816
Chicago/Turabian StyleChatterjee, Subroto, Amrita Balram, and Wendy Li. 2021. "Convergence: Lactosylceramide-Centric Signaling Pathways Induce Inflammation, Oxidative Stress, and Other Phenotypic Outcomes" International Journal of Molecular Sciences 22, no. 4: 1816. https://doi.org/10.3390/ijms22041816