
Role of Site-Specific Glycosylation in the I-Like Domain of Integrin β1 in Small Extracellular Vesicle-Mediated Malignant Behavior and FAK Activation



Abstract
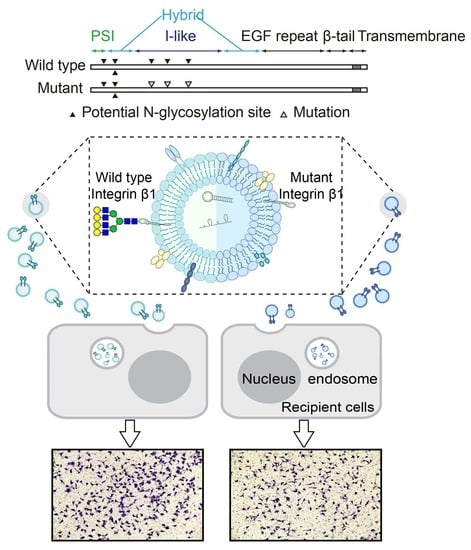
:
1. Introduction
2. Results
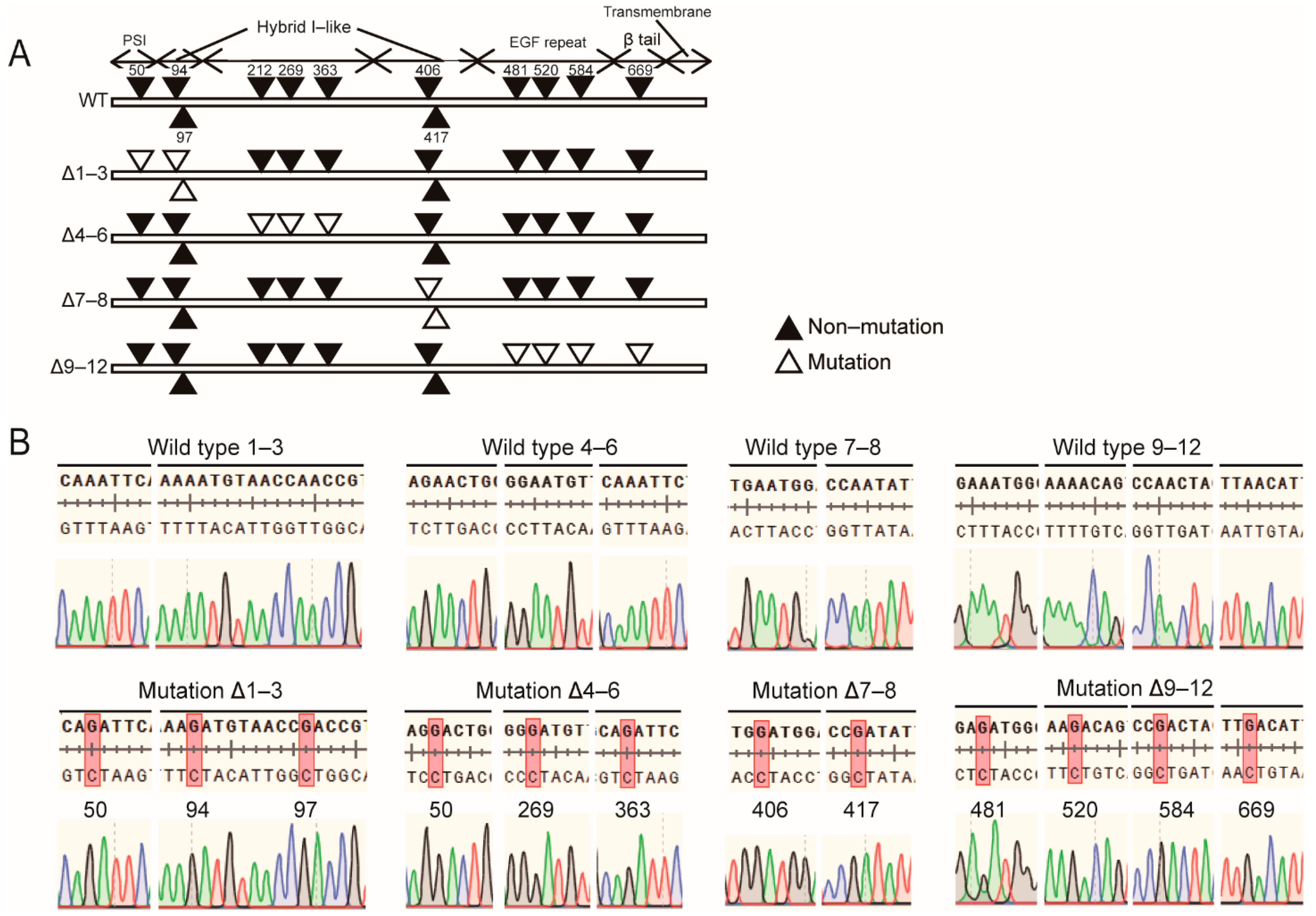
2.1. N-Glycosylation Site Mutation of Integrin β1
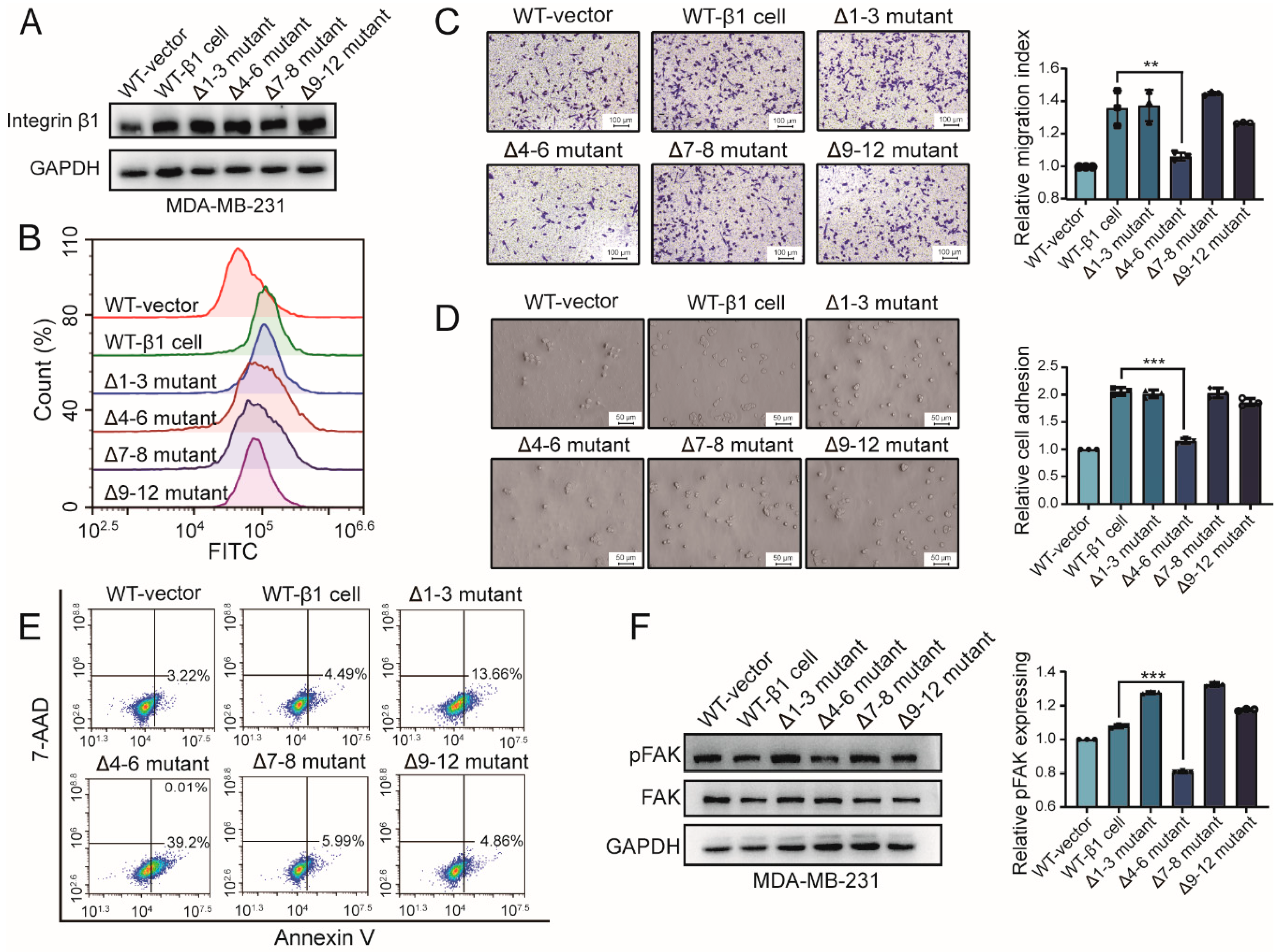
2.2. Influence of Integrin β1 Mutants on the Cell Adhesion, Mobility, and Signaling Pathway
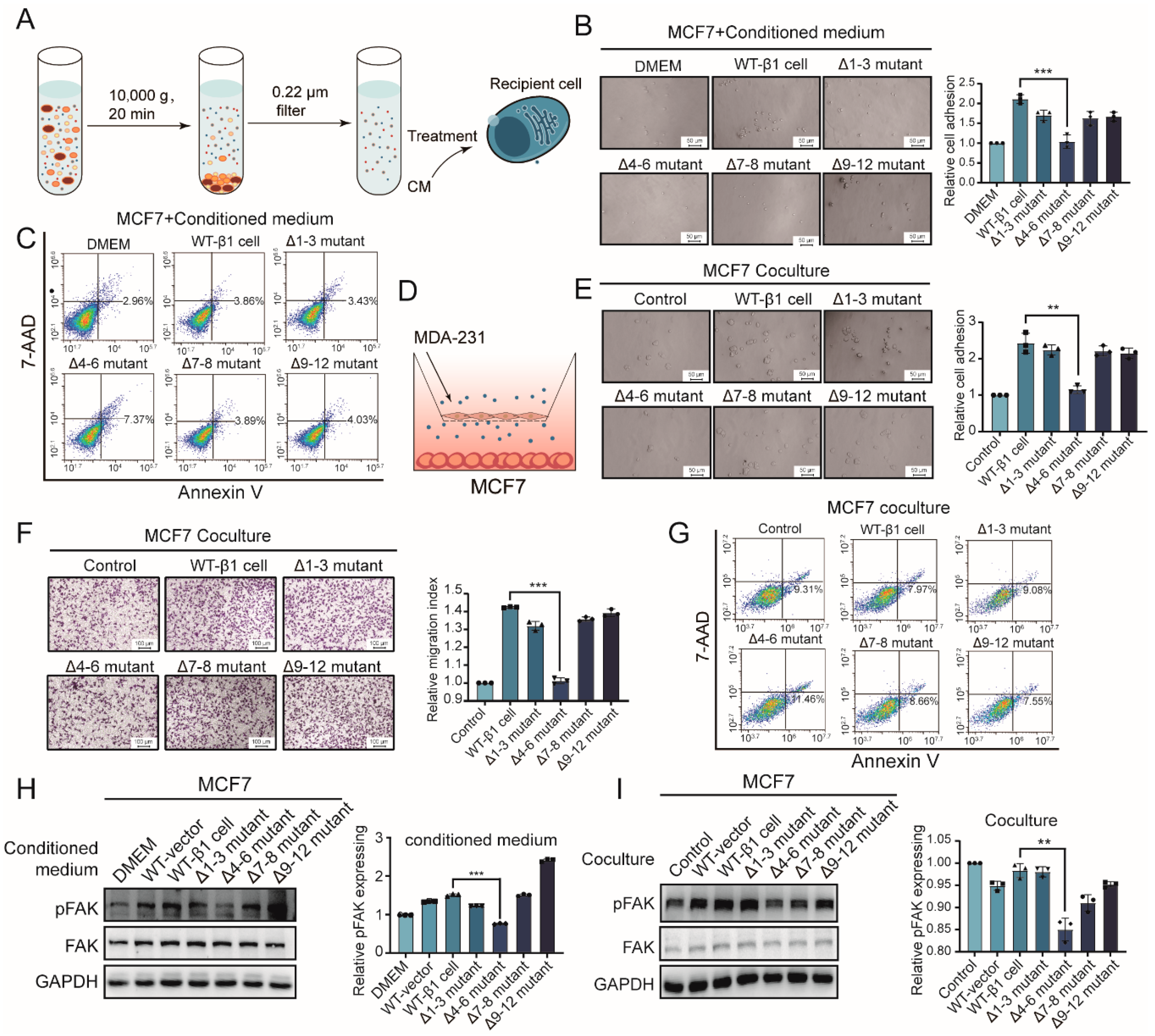
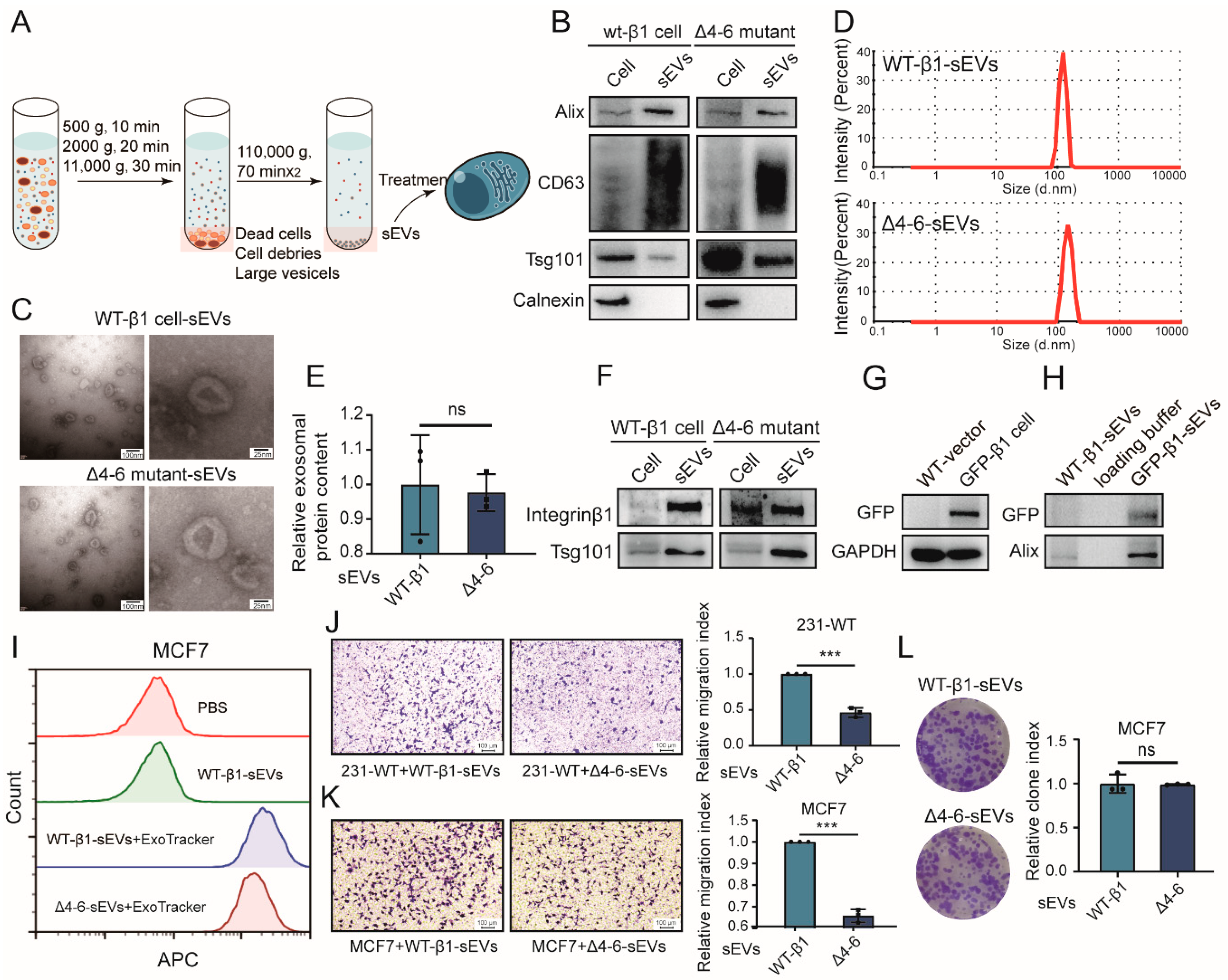
2.3. Effects of Secreted Components from β1 Mutants on Behavior of Recipient Cells
2.4. Effects of β1 Mutants on Biological Function of sEVs
3. Discussion
4. Materials and Methods
4.1. Cell Culture
4.2. Plasmid Construction
4.3. Stable Transfection of WT or Mutant Integrin β1
4.4. Whole-Cell Lysate Extraction and Western Blotting
4.5. Cell Apoptosis Assay
4.6. Cell Adhesion
4.7. Cell Migration by Transwell Assay
4.8. Conditioned Medium Extraction, sEV Isolation, and sEV-Free FBS Preparation
4.9. Co-Culture System
4.10. Nanoparticle Tracking Analysis
4.11. Transmission Electron Microscopy (TEM)
4.12. sEV Uptake by Flow Cytometry
4.13. Data Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| sEVs | Small extracellular vesicles |
| ECM | Extracellular matrix |
| FAK | Focal adhesion kinase |
| AKT | protein kinase B |
| PSI | Plexin-semaphorin-integrin domain |
| I-EGF | Integrin-epidermal growth factor domain |
| FN | Fibronectin |
| CM | Conditioned medium |
| GFP | green fluorescent protein |
| FBS | fetal bovine serum |
| BSA | bovine serum albumin |
| PBS | phosphate buffer saline |
References
- Tan, Z.; Cao, L.; Wu, Y.; Wang, B.; Song, Z.; Yang, J.; Cheng, L.; Yang, X.; Zhou, X.; Dai, Z.; et al. Bisecting GlcNAc modification diminishes the pro-metastatic functions of small extracellular vesicles from breast cancer cells. J. Extracell. Vesicles 2020, 10, 56–72. [Google Scholar] [CrossRef]
- Raposo, G.; Stoorvogel, W. Extracellular vesicles: Exosomes, microvesicles, and friends. J. Cell Biol. 2013, 200, 373–383. [Google Scholar] [CrossRef] [Green Version]
- Van Niel, G.; d’Angelo, G.; Raposo, G. Shedding light on the cell biology of extracellular vesicles. Nat. Rev. Mol. Cell Biol. 2018, 19, 213. [Google Scholar] [CrossRef] [PubMed]
- Maia, J.; Caja, S.; Strano Moraes, M.C.; Couto, N.; Costa-Silva, B. Exosome-based cell-cell communication in the tumor microenvironment. Fron. Cell Dev. Biol. 2018, 6, 18. [Google Scholar] [CrossRef] [PubMed]
- Dörsam, B.; Reiners, K.S.; von Strandmann, E.P. Cancer-derived extracellular vesicles: Friend and foe of tumour immunosurveillance. Philos. Trans. Royal Soc. B Biol. Sci. 2018, 373, 20160481. [Google Scholar] [CrossRef]
- Zhang, H.; Deng, T.; Liu, R.; Bai, M.; Zhou, L.; Wang, X.; Li, S.; Wang, X.; Yang, H.; Li, J.; et al. Exosome-delivered EGFR regulates liver microenvironment to promote gastric cancer liver metastasis. Nat. Commun. 2017, 8, 15016. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoshino, A.; Costa-Silva, B.; Shen, T.-L.; Rodrigues, G.; Hashimoto, A.; Tesic Mark, M.; Molina, H.; Kohsaka, S.; Di Giannatale, A.; Ceder, S.; et al. Tumour exosome integrins determine organotropic metastasis. Nature 2015, 527, 329. [Google Scholar] [CrossRef] [Green Version]
- Guo, W.; Giancotti, F.G. Integrin signalling during tumour progression. Nat. Rev. Mol. Cell Biol. 2004, 5, 816–826. [Google Scholar] [CrossRef]
- Hou, S.; Isaji, T.; Hang, Q.; Im, S.; Fukuda, T.; Gu, J. Distinct effects of β1 integrin on cell proliferation and cellular signaling in MDA-MB-231 breast cancer cells. Sci. Rep. 2016, 6, 1–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Imanishi, Y.; Hu, B.; Jarzynka, M.J.; Guo, P.; Elishaev, E.; Bar-Joseph, I.; Cheng, S.-Y. Angiopoietin-2 stimulates breast cancer metastasis through the α5β1 integrin-mediated pathway. Cancer Res. 2007, 67, 4254–4263. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, C.; Qu, L.; Zhao, C.; Shou, C. Extracellular gamma-synuclein promotes tumor cell motility by activating beta1 integrin-focal adhesion kinase signaling pathway and increasing matrix metalloproteinase-24, -2 protein secretion. J. Exp. Clin. Cancer Res. 2018, 37, 117. [Google Scholar] [CrossRef] [PubMed]
- Marsico, G.; Russo, L.; Quondamatteo, F.; Pandit, A. Glycosylation and Integrin Regulation in Cancer. Trends Cancer 2018, 4, 537–552. [Google Scholar] [CrossRef] [PubMed]
- Isaji, T.; Sato, Y.; Fukuda, T.; Gu, J. N-glycosylation of the I-like domain of beta1 integrin is essential for beta1 integrin expression and biological function: Identification of the minimal N-glycosylation requirement for alpha5beta1. J. Biol. Chem. 2009, 284, 12207–12216. [Google Scholar] [CrossRef] [Green Version]
- Hou, S.; Hang, Q.; Isaji, T.; Lu, J.; Fukuda, T.; Gu, J. Importance of membrane-proximal N-glycosylation on integrin beta1 in its activation and complex formation. FASEB J. 2016, 30, 4120–4131. [Google Scholar] [CrossRef] [Green Version]
- Xu, Z.; Isaji, T.; Fukuda, T.; Wang, Y.; Gu, J. O-GlcNAcylation regulates integrin-mediated cell adhesion and migration via formation of focal adhesion complexes. J. Biol. Chem. 2019, 294, 3117–3124. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seales, E.C.; Shaikh, F.M.; Woodard-Grice, A.V.; Aggarwal, P.; McBrayer, A.C.; Hennessy, K.M.; Bellis, S.L. A protein kinase C/Ras/ERK signaling pathway activates myeloid fibronectin receptors by altering beta1 integrin sialylation. J. Biol. Chem. 2005, 280, 37610–37615. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, H.; Ozaki, I.; Mizuta, T.; Matsuhashi, S.; Yoshimura, T.; Hisatomi, A.; Tadano, J.; Sakai, T.; Yamamoto, K. Beta 1-integrin protects hepatoma cells from chemotherapy induced apoptosis via a mitogen-activated protein kinase dependent pathway. Cancer 2002, 95, 896–906. [Google Scholar] [CrossRef] [PubMed]
- Mitra, S.K.; Schlaepfer, D.D. Integrin-regulated FAK-Src signaling in normal and cancer cells. Curr. Opin. Cell Biol. 2006, 18, 516–523. [Google Scholar] [CrossRef]
- Frisch, S.M.; Vuori, K.; Ruoslahti, E.; Chan-Hui, P.Y. Control of adhesion-dependent cell survival by focal adhesion kinase. J. Cell Biol. 1996, 134, 793–799. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zomer, A.; Maynard, C.; Verweij, F.J.; Kamermans, A.; Schafer, R.; Beerling, E.; Schiffelers, R.M.; de Wit, E.; Berenguer, J.; Ellenbroek, S.I.J.; et al. In Vivo imaging reveals extracellular vesicle-mediated phenocopying of metastatic behavior. Cell 2015, 161, 1046–1057. [Google Scholar] [CrossRef] [Green Version]
- Huet-Calderwood, C.; Rivera-Molina, F.; Iwamoto, D.V.; Kromann, E.B.; Toomre, D.; Calderwood, D.A. Novel ecto-tagged integrins reveal their trafficking in live cells. Nat. Commun. 2017, 8, 570. [Google Scholar] [CrossRef] [Green Version]
- Barkan, D.; Chambers, A.F. beta1-integrin: A potential therapeutic target in the battle against cancer recurrence. Clin. Cancer Res. 2011, 17, 7219–7223. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Pan, D.; Bellis, S.L.; Song, Y. Effect of altered glycosylation on the structure of the I-like domain of beta1 integrin: A molecular dynamics study. Proteins 2008, 73, 989–1000. [Google Scholar] [CrossRef] [PubMed]
- DeRita, R.M.; Sayeed, A.; Garcia, V.; Krishn, S.R.; Shields, C.D.; Sarker, S.; Friedman, A.; McCue, P.; Molugu, S.K.; Rodeck, U.; et al. Tumor-Derived Extracellular Vesicles Require beta1 Integrins to Promote Anchorage-Independent Growth. iScience 2019, 14, 199–209. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jeppesen, D.K.; Fenix, A.M.; Franklin, J.L.; Higginbotham, J.N.; Zhang, Q.; Zimmerman, L.J.; Liebler, D.C.; Ping, J.; Liu, Q.; Evans, R.; et al. Reassessment of Exosome Composition. Cell 2019, 177, 428–445.e418. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.; Zeng, C.; Zhan, Y.; Wang, H.; Jiang, X.; Li, W. Aberrant low expression of p85alpha in stromal fibroblasts promotes breast cancer cell metastasis through exosome-mediated paracrine Wnt10b. Oncogene 2017, 36, 4692–4705. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ji, Q.; Zhou, L.; Sui, H.; Yang, L.; Wu, X.; Song, Q.; Jia, R.; Li, R.; Sun, J.; Wang, Z.; et al. Primary tumors release ITGBL1-rich extracellular vesicles to promote distal metastatic tumor growth through fibroblast-niche formation. Nat. Commun. 2020, 11, 1211. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bajc Cesnik, A.; Darovic, S.; Prpar Mihevc, S.; Stalekar, M.; Malnar, M.; Motaln, H.; Lee, Y.B.; Mazej, J.; Pohleven, J.; Grosch, M.; et al. Nuclear RNA foci from C9ORF72 expansion mutation form paraspeckle-like bodies. J. Cell Sci. 2019, 132. [Google Scholar] [CrossRef] [Green Version]
- Cheng, J.; Huo, D.H.; Kuang, D.M.; Yang, J.; Zheng, L.; Zhuang, S.M. Human macrophages promote the motility and invasiveness of osteopontin-knockdown tumor cells. Cancer Res. 2007, 67, 5141–5147. [Google Scholar] [CrossRef] [Green Version]
- Lobb, R.J.; Becker, M.; Wen, S.W.; Wong, C.S.; Wiegmans, A.P.; Leimgruber, A.; Möller, A. Optimized exosome isolation protocol for cell culture supernatant and human plasma. J. Extracell. Vesicles 2015, 4, 27031. [Google Scholar] [CrossRef]
- Cianciaruso, C.; Phelps, E.A.; Pasquier, M.; Hamelin, R.; Demurtas, D.; Alibashe Ahmed, M.; Piemonti, L.; Hirosue, S.; Swartz, M.A.; De Palma, M.; et al. Primary Human and Rat beta-Cells Release the Intracellular Autoantigens GAD65, IA-2, and Proinsulin in Exosomes Together With Cytokine-Induced Enhancers of Immunity. Diabetes 2017, 66, 460–473. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, X.; Zhang, J.; Song, Z.; Lu, S.; Yu, Y.; Tian, J.; Li, X.; Guan, F. ExoTracker: A low-pH-activatable fluorescent probe for labeling exosomes and monitoring endocytosis and trafficking. Chem. Commun. 2020, 56, 14869–14872. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Oligonucleotide (Integrin β1) | Primer Sequence (5′-3′) | |
|---|---|---|
| Full length-sense: | CCGGAATTCATGAATTTACAACCAATTTTCTGGATTGGACT | |
| Full length-antisense: | GCTCTAGATCATTTTCCCTCATACTTCGGATTGAC | |
| Δ1–3 mutant | 50-sense: | TGTTGAATCTGTGCACCACCCACAATTTGG |
| 50-anti-sense | TGGTGCACAGATTCAACATTTTTACAGGAAGG | |
| 94 and 97-sense: | GCTACGGTCGGTTACATCTTTATTTTTCTTTAT | |
| 94 and 97-anti-sense: | AATAAAGATGTAACCGACCGTAGCAAAGGAAC | |
| Δ4–6 mutant | 212-sense: | CTGGTGCAGTCCTGTTCACTTGTGC |
| 212-anti-sense: | TGAAC AGGAC TGCAC CAGCC CATT | |
| 269-sense: | TGTAACATCCCTCCAGCCAATCAGTG | |
| 269-anti-sense: | TGGAGGGATGTTACACGGCTGC | |
| 363-sense: | CATTGCTAGAATCTGCAGATAATGTTCC | |
| 363-anti-sense: | TATCTGCAGATTCTAGCAATGTAATTCAGTT | |
| Δ7–8 mutant | 406-sense: | TTCCATCCACCCCGTTCTTGCAGTAAG |
| 406-anti-sense: | CGGGGTGGATGGAACAGGGGAAAAT | |
| 417-sense: | GGAAATATCGGAACATTTTCTTCC | |
| 417-anti-sense: | TGTTCCGATATTTCCATTGGAGATGAGG | |
| Δ9–12 mutant primer | 481-sense: | AATGTCCCATCTCCTTCATGACACTT |
| 481-anti-sense: | CATGAAGGAGATGGGACATTTGAGTG | |
| 520-sense: | CTGAACTGTCTTCTTTCCTGCAGTAAGC | |
| 520-anti-sense: | GGAAAGAAGACAGTTCAGAAATCT | |
| 584-sense: | GTGTAGTCGGGGTTGCACTCACAC | |
| 584-anti-sense: | GCAACCCCGACTACACTGGC | |
| 669-sense: | CTACCTTGGTAATGTCAAAATAGGAACATTC | |
| 669-anti-sense: | CCTATTTTGACATTACCAAGGTAGAAAGT | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cao, L.; Wu, Y.; Wang, X.; Li, X.; Tan, Z.; Guan, F. Role of Site-Specific Glycosylation in the I-Like Domain of Integrin β1 in Small Extracellular Vesicle-Mediated Malignant Behavior and FAK Activation. Int. J. Mol. Sci. 2021, 22, 1770. https://doi.org/10.3390/ijms22041770
Cao L, Wu Y, Wang X, Li X, Tan Z, Guan F. Role of Site-Specific Glycosylation in the I-Like Domain of Integrin β1 in Small Extracellular Vesicle-Mediated Malignant Behavior and FAK Activation. International Journal of Molecular Sciences. 2021; 22(4):1770. https://doi.org/10.3390/ijms22041770
Chicago/Turabian StyleCao, Lin, Yurong Wu, Xiuxiu Wang, Xiang Li, Zengqi Tan, and Feng Guan. 2021. "Role of Site-Specific Glycosylation in the I-Like Domain of Integrin β1 in Small Extracellular Vesicle-Mediated Malignant Behavior and FAK Activation" International Journal of Molecular Sciences 22, no. 4: 1770. https://doi.org/10.3390/ijms22041770