
Advances in De Novo Drug Design: From Conventional to Machine Learning Methods

 , , , , , , , and
, , , , , , , and 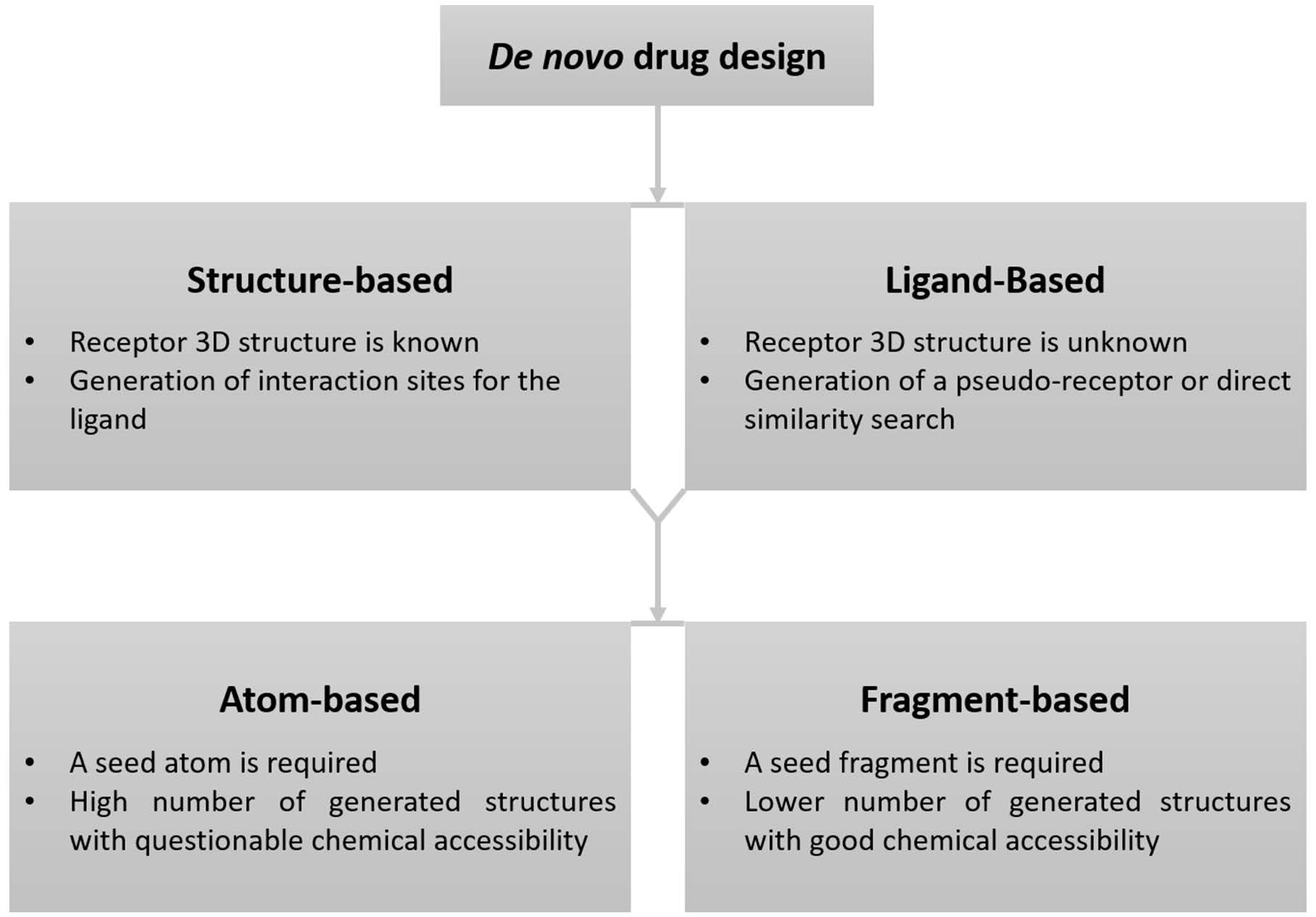{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. De Novo Drug Design Methodology
2.1. Structure-Based De Novo Drug Design
2.2. Ligand-Based De Novo Drug Design
2.3. Sampling Methods in De Novo Drug Design
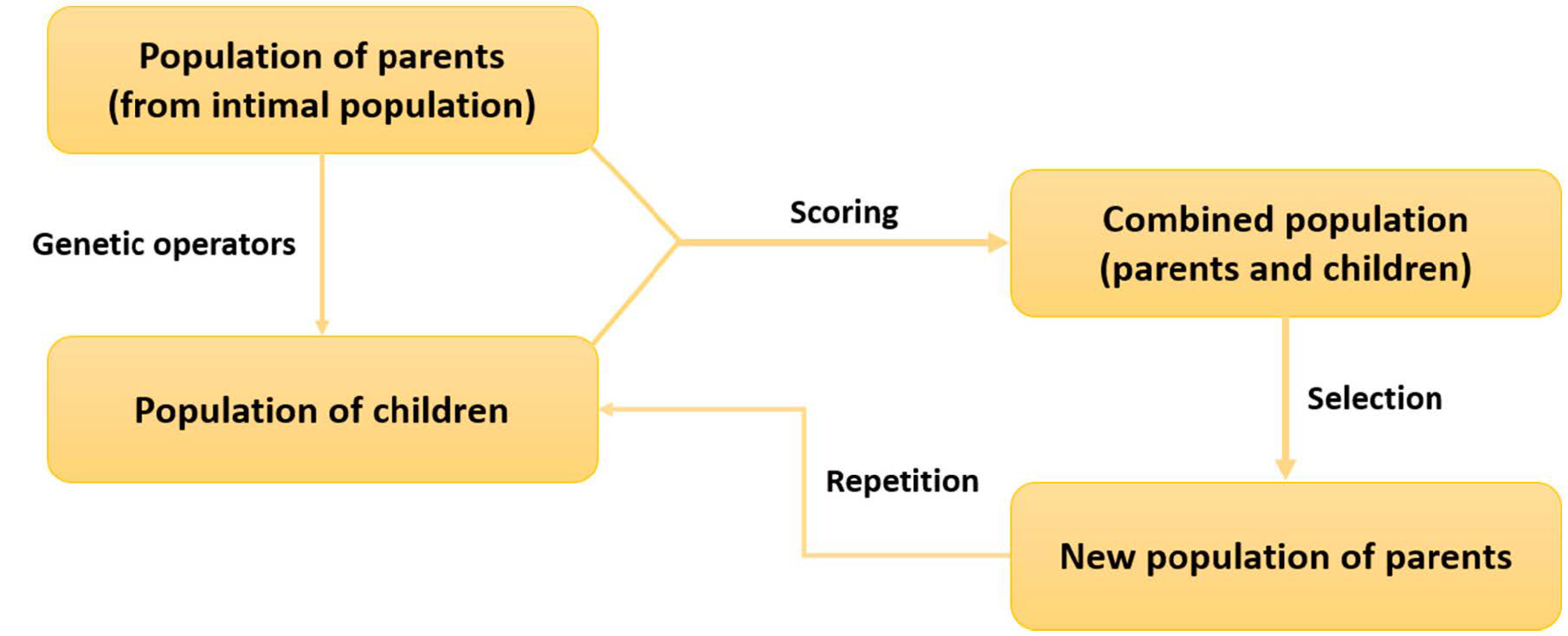
3. Evolutionary Algorithms in De Novo Drug Design
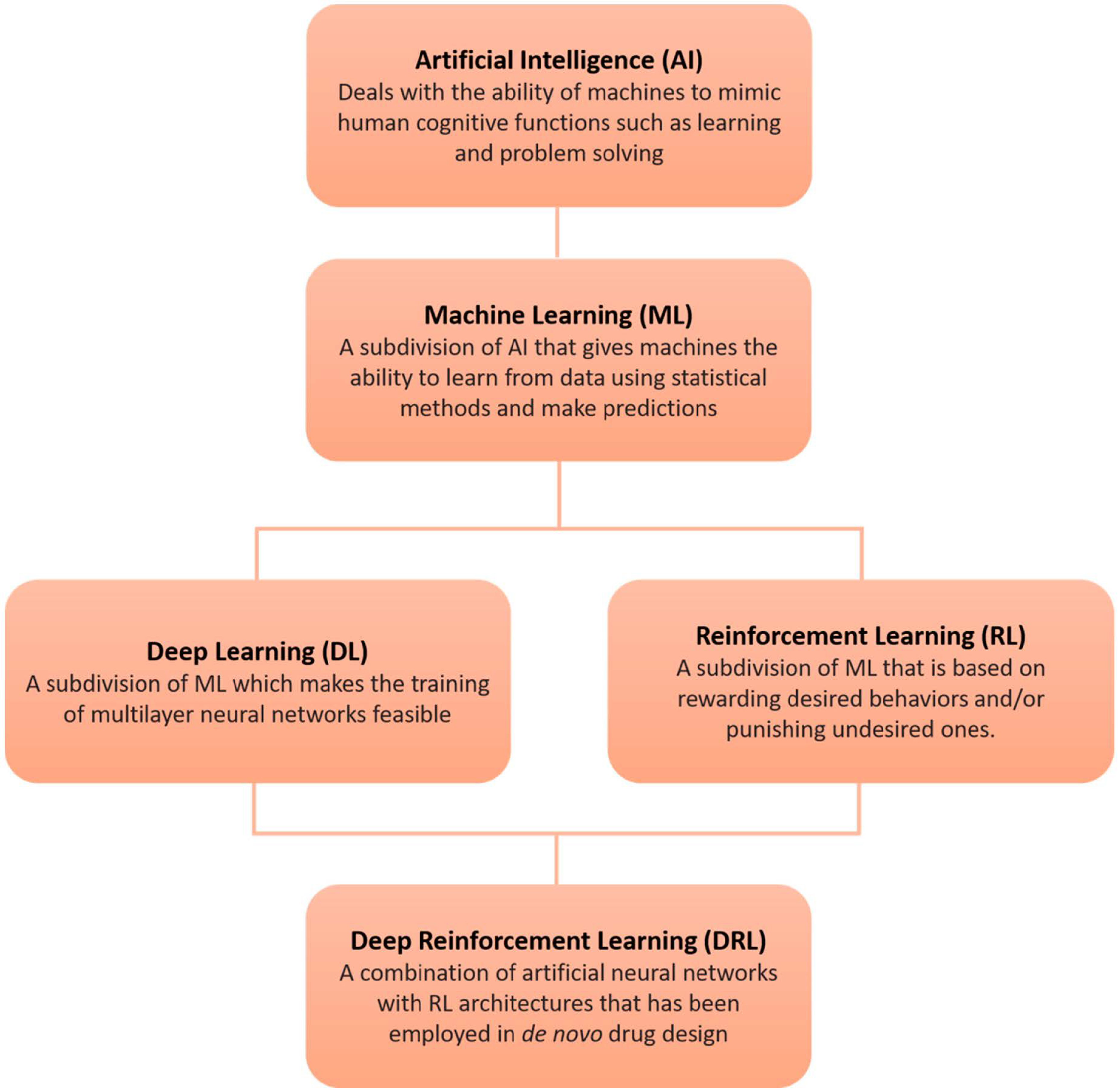
4. Artificial Intelligence in De Novo Drug Design
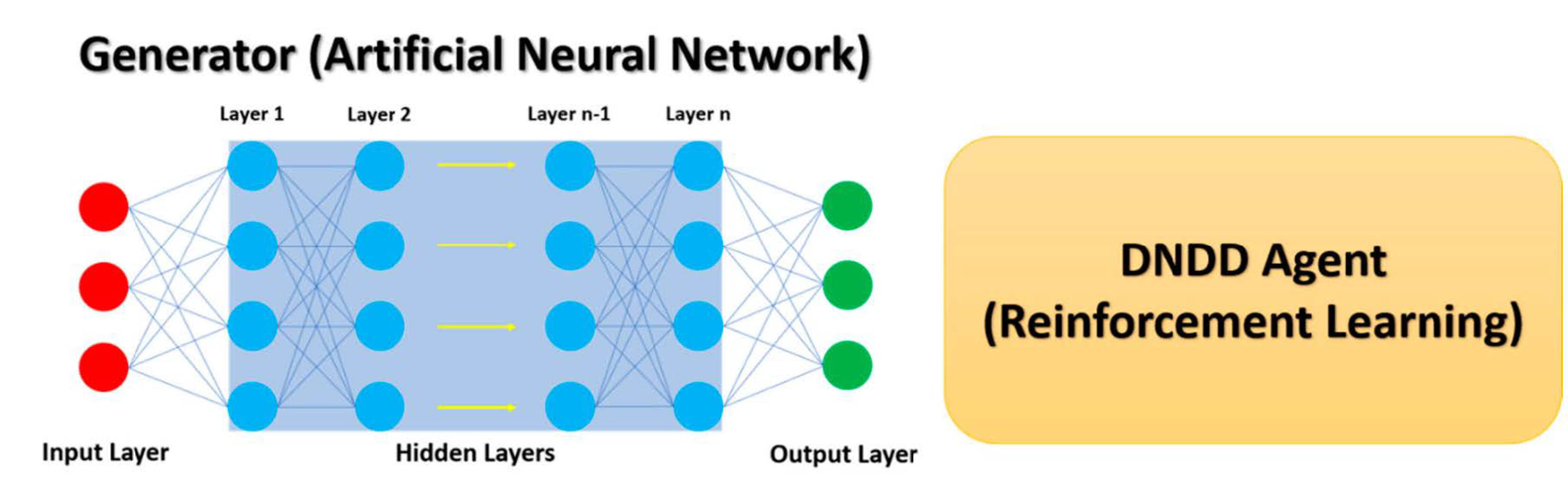
Deep Reinforcement Learning (DRL) in De Novo Drug Design
5. Examples of DRL in De Novo Drug Design
5.1. Recurrent Neural Networks (RNN)
5.2. Convolutional Neural Networks (CNN)
5.3. Generative Adversarial Networks (GAN)
5.4. Autoencoders (AE)
5.4.1. Variational Autoencoder (VAE)
5.4.2. Sequence-to-Sequence Autoencoder (seq2seq AE)
5.4.3. Adversarial Autoencoder (AAE)
6. Particle Swarm Optimization for De Novo Drug Design
7. Evaluation Criteria
7.1. Diversity and Novelty
7.2. Desired Properties
7.3. Synthetic Feasibility
8. Bridging Toxicogenomics and Molecular Design
9. De Novo Drug Design for COVID-19
10. Building Community and Regulatory Acceptance of DL Methods for De Novo Drug Design
11. Concluding Remarks
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| CADD | Computer-aided drug design |
| QSAR | Quantitative structure–activity relationships |
| NMR | Nuclear magnetic resonance |
| DNDD | De novo drug design |
| MCSS | Multiple copy simultaneous search |
| ChEMBL | Chemical database of bioactive molecules with drug-like properties |
| ADMET | Absorption, distribution, metabolism, excretion, and toxicity |
| AI | Artificial intelligence |
| ML | Machine learning |
| DL | Deep learning |
| RNN | Recurrent neural networks |
| CNN | Convolutional neural networks |
| GAN | Generative adversarial networks |
| AE | Autoencoders |
| RL | Reinforcement learning |
| DRL | Deep reinforcement learning |
| SMILES | Simplified molecular-input line-entry system |
| ReLeaSE | Reinforcement learning for structural evolution |
| TL | Transfer learning |
| LSTM | Long short-term memory |
| nll | |
| 2D | Two-dimensional |
| DNN | Deep neural network |
| RANC | Reinforced adversarial neural computer |
| ATNC | Adversarial threshold neural computer |
| IDC | Internal diversity clustering |
| VAE | Variational autoencoder |
| 3D | Three-dimensional |
| MW | Molecular weight |
| LogP | Octanol-water partition coefficient |
| HBD | Hydrogen-bond donor |
| HBA | Hydrogen-bond acceptor |
| TPSA | Topological polar surface area |
| seq2seq AE | Sequence to sequence autoencoder |
| GRU | Gated recurrent unit |
| AAE | Adversarial autoencoder |
| PSO | Particle swarm optimization |
| OECD | Organization’s for the Economic Cooperation and Development |
| SA | Synthetic accessibility |
| SC | Synthetic complexity |
| MOA | Mechanism-of-action |
| COVID-19 | Coronavirus disease 2019 |
| SARS-CoV-2 | Severe acute respiratory syndrome coronavirus 2 |
| Mpro | Main protease |
| ACE-2 | Angiotensin II |
| WGAN | Wasserstein GAN |
| US FDA | United States food and drug administration |
| GCGR | Glucagon receptor |
| EMA | European medicines agency |
| HMA | Heads of medical agencies |
| QMRF | QSAR model report format |
| DDR1 | Discoidin domain receptor 1 |
References
- Kola, I.; Landis, J. Can the pharmaceutical industry reduce attrition rates? Nat. Rev. Drug Discov. 2004, 3, 711–716. [Google Scholar] [CrossRef]
- Torjesen, I. Drug Development: The Journey of a Medicine from Lab to Shelf. Available online: https://www.pharmaceutical-journal.com/publications/tomorrows-pharmacist/drug-development-the-journey-of-a-medicine-from-lab-to-shelf/20068196.article?firstPass=false (accessed on 10 December 2020).
- Fischer, T.; Gazzola, S.; Riedl, R. Approaching Target Selectivity by De Novo Drug Design. Expert Opin. Drug Discov. 2019, 14, 791–803. [Google Scholar] [CrossRef] [PubMed]
- Schneider, G. Automating drug discovery. Nat. Rev. Drug Discov. 2018, 17, 97–113. [Google Scholar] [CrossRef]
- Mouchlis, V.D.; Melagraki, G.; Zacharia, L.C.; Afantitis, A. Computer-Aided Drug Design of beta-Secretase, gamma-Secretase and Anti-Tau Inhibitors for the Discovery of Novel Alzheimer’s Therapeutics. Int. J. Mol. Sci. 2020, 21, 703. [Google Scholar] [CrossRef] [Green Version]
- Schneider, G.; Clark, D.E. Automated De Novo Drug Design: Are We Nearly There Yet? Angew. Chem. 2019, 58, 10792–10803. [Google Scholar] [CrossRef]
- Schneider, P.; Schneider, G. De Novo Design at the Edge of Chaos. J. Med. Chem. 2016, 59, 4077–4086. [Google Scholar] [CrossRef] [PubMed]
- Devi, R.V.; Sathya, S.S.; Coumar, M.S. Evolutionary algorithms for de novo drug design—A survey. Appl. Soft Comput. 2015, 27, 543–552. [Google Scholar] [CrossRef]
- Schneider, G.; Fechner, U. Computer-based de novo design of drug-like molecules. Nat. Rev. Drug Discov. 2005, 4, 649–663. [Google Scholar] [CrossRef] [PubMed]
- Nicolaou, C.A.; Kannas, C.; Loizidou, E. Multi-Objective Optimization Methods in De Novo Drug Design. Mini-Rev. Med. Chem. 2012, 12, 979–987. [Google Scholar] [CrossRef] [PubMed]
- Nicolaou, C.A.; Brown, N. Multi-objective optimization methods in drug design. Drug Discov. Today Technol. 2013, 10, e427–e435. [Google Scholar] [CrossRef] [PubMed]
- Cruz-Monteagudo, M.; Borges, F.; Cordeiro, M.N.D.S. Desirability-based multiobjective optimization for global QSAR studies: Application to the design of novel NSAIDs with improved analgesic, antiinflammatory, and ulcerogenic profiles. J. Comput. Chem. 2008, 29, 2445–2459. [Google Scholar] [CrossRef] [PubMed]
- Sánchez-Rodríguez, A.; Pérez-Castillo, Y.; Schürer, S.C.; Nicolotti, O.; Mangiatordi, G.F.; Borges, F.; Cordeiro, M.N.D.S.; Tejera, E.; Medina-Franco, J.L.; Cruz-Monteagudo, M. From flamingo dance to (desirable) drug discovery: A nature-inspired approach. Drug Discov. Today 2017, 22, 1489–1502. [Google Scholar] [CrossRef]
- Perez-Castillo, Y.; Sánchez-Rodríguez, A.; Tejera, E.; Cruz-Monteagudo, M.; Borges, F.; Cordeiro, M.N.D.S.; Le-Thi-Thu, H.; Pham-The, H. A desirability-based multi objective approach for the virtual screening discovery of broad-spectrum anti-gastric cancer agents. PLoS ONE 2018, 13, e0192176. [Google Scholar] [CrossRef] [Green Version]
- Berman, H.M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Weissig, H.; Shindyalov, I.N.; Bourne, P.E. The Protein Data Bank. Nucleic Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef] [Green Version]
- Burley, S.K.; Berman, H.M.; Bhikadiya, C.; Bi, C.; Chen, L.; Di Costanzo, L.; Christie, C.; Dalenberg, K.; Duarte, J.M.; Dutta, S.; et al. RCSB Protein Data Bank: Biological macromolecular structures enabling research and education in fundamental biology, biomedicine, biotechnology and energy. Nucleic Acids Res. 2019, 47, D464–D474. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Muhammed, M.T.; Aki-Yalcin, E. Homology modeling in drug discovery: Overview, current applications, and future perspectives. Chem. Biol. Drug Des. 2019, 93, 12–20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Danziger, D.J.; Dean, P.M.; Cuthbert, A.W. Automated site-directed drug design: A general algorithm for knowledge acquisition about hydrogen-bonding regions at protein surfaces. Proc. R. Soc. Lond. Ser. B Biol. Sci. 1989, 236, 101–113. [Google Scholar]
- Böhm, H.-J. LUDI: Rule-based automatic design of new substituents for enzyme inhibitor leads. J. Comput.-Aided Mol. Des. 1992, 6, 593–606. [Google Scholar] [CrossRef] [PubMed]
- Clark, D.E.; Frenkel, D.; Levy, S.A.; Li, J.; Murray, C.W.; Robson, B.; Waszkowycz, B.; Westhead, D.R. PRO_LIGAND: An approach to de novo molecular design. 1. Application to the design of organic molecules. J. Comput. Aided Mol. Des. 1995, 9, 13–32. [Google Scholar] [CrossRef]
- Waszkowycz, B.; Clark, D.E.; Frenkel, D.; Li, J.; Murray, C.W.; Robson, B.; Westhead, D.R. PRO_LIGAND: An Approach to de Novo Molecular Design. 2. Design of Novel Molecules from Molecular Field Analysis (MFA) Models and Pharmacophores. J. Med. Chem. 1994, 37, 3994–4002. [Google Scholar] [CrossRef]
- Gillet, V.J.; Myatt, G.; Zsoldos, Z.; Johnson, A.P. SPROUT, HIPPO and CAESA: Tools for de novo structure generation and estimation of synthetic accessibility. Perspect. Drug Discov. Des. 1995, 3, 34–50. [Google Scholar] [CrossRef]
- Bohacek, R.S.; McMartin, C. Multiple Highly Diverse Structures Complementary to Enzyme Binding Sites: Results of Extensive Application of a de Novo Design Method Incorporating Combinatorial Growth. J. Am. Chem. Soc. 1994, 116, 5560–5571. [Google Scholar] [CrossRef]
- Nishibata, Y.; Itai, A. Automatic creation of drug candidate structures based on receptor structure. Starting point for artificial lead generation. Tetrahedron 1991, 47, 8985–8990. [Google Scholar] [CrossRef]
- Wang, R.; Gao, Y.; Lai, L. LigBuilder: A Multi-Purpose Program for Structure-Based Drug Design. Mol. Modeling Annu. 2000, 6, 498–516. [Google Scholar] [CrossRef]
- Miranker, A.; Karplus, M. Functionality maps of binding sites: A multiple copy simultaneous search method. Proteins: Struct. Funct. Bioinform. 1991, 11, 29–34. [Google Scholar] [CrossRef] [PubMed]
- Eisen, M.B.; Wiley, D.C.; Karplus, M.; Hubbard, R.E. HOOK: A program for finding novel molecular architectures that satisfy the chemical and steric requirements of a macromolecule binding site. Proteins: Struct. Funct. Bioinform. 1994, 19, 199–221. [Google Scholar] [CrossRef]
- Luo, Z.; Wang, R.; Lai, L. RASSE: A New Method for Structure-Based Drug Design. J. Chem. Inf. Comput. Sci. 1996, 36, 1187–1194. [Google Scholar] [CrossRef]
- Pearlman, D.A.; Murcko, M.A. CONCERTS: Dynamic Connection of Fragments as an Approach to de Novo Ligand Design. J. Med. Chem. 1996, 39, 1651–1663. [Google Scholar] [CrossRef]
- Liu, H.; Duan, Z.; Luo, Q.; Shi, Y. Structure-based ligand design by dynamically assembling molecular building blocks at binding site. Proteins: Struct. Funct. Bioinform. 1999, 36, 462–470. [Google Scholar] [CrossRef]
- Zhu, J.; Fan, H.; Liu, H.; Shi, Y. Structure-based ligand design for flexible proteins: Application of new F-DycoBlock. J. Comput. Aided Mol. Des. 2001, 15, 979–996. [Google Scholar] [CrossRef] [PubMed]
- Zhu, J.; Yu, H.; Fan, H.; Liu, H.; Shi, Y. Design of new selective inhibitors of cyclooxygenase-2 by dynamic assembly of molecular building blocks. J. Comput. Aided Mol. Des. 2001, 15, 447–463. [Google Scholar] [CrossRef]
- Wang, Y.; Zhao, H.; Brewer, J.T.; Li, H.; Lao, Y.; Amberg, W.; Behl, B.; Akritopoulou-Zanze, I.; Dietrich, J.; Lange, U.E.W.; et al. De Novo Design, Synthesis, and Biological Evaluation of 3,4-Disubstituted Pyrrolidine Sulfonamides as Potent and Selective Glycine Transporter 1 Competitive Inhibitors. J. Med. Chem. 2018, 61, 7486–7502. [Google Scholar] [CrossRef]
- Gaulton, A.; Hersey, A.; Nowotka, M.; Bento, A.P.; Chambers, J.; Mendez, D.; Mutowo, P.; Atkinson, F.; Bellis, L.J.; Cibrián-Uhalte, E.; et al. The ChEMBL database in 2017. Nucleic Acids Res. 2017, 45, D945–D954. [Google Scholar] [CrossRef] [PubMed]
- Wise, A.; Gearing, K.; Rees, S. Target validation of G-protein coupled receptors. Drug Discov. Today 2002, 7, 235–246. [Google Scholar] [CrossRef]
- Afantitis, A.; Melagraki, G.; Koutentis, P.A.; Sarimveis, H.; Kollias, G. Ligand-based virtual screening procedure for the prediction and the identification of novel β-amyloid aggregation inhibitors using Kohonen maps and Counterpropagation Artificial Neural Networks. Eur. J. Med. Chem. 2011, 46, 497–508. [Google Scholar] [CrossRef] [PubMed]
- Schneider, G.; Lee, M.-L.; Stahl, M.; Schneider, P. De novo design of molecular architectures by evolutionary assembly of drug-derived building blocks. J. Comput. Aided Mol. Des. 2000, 14, 487–494. [Google Scholar] [CrossRef]
- Vinkers, H.M.; de Jonge, M.R.; Daeyaert, F.F.D.; Heeres, J.; Koymans, L.M.H.; van Lenthe, J.H.; Lewi, P.J.; Timmerman, H.; Van Aken, K.; Janssen, P.A.J. SYNOPSIS: SYNthesize and OPtimize System in Silico. J. Med. Chem. 2003, 46, 2765–2773. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hartenfeller, M.; Zettl, H.; Walter, M.; Rupp, M.; Reisen, F.; Proschak, E.; Weggen, S.; Stark, H.; Schneider, G. DOGS: Reaction-Driven de novo Design of Bioactive Compounds. PLoS Comp. Biol. 2012, 8, e1002380. [Google Scholar] [CrossRef]
- Dey, F.; Caflisch, A. Fragment-Based de Novo Ligand Design by Multiobjective Evolutionary Optimization. J. Chem. Inf. Model. 2008, 48, 679–690. [Google Scholar] [CrossRef]
- Ichihara, O.; Barker, J.; Law, R.J.; Whittaker, M. Compound Design by Fragment-Linking. Mol. Inform. 2011, 30, 298–306. [Google Scholar] [CrossRef]
- Schneider, G. Future De Novo Drug Design. Mol. Inform. 2014, 33, 397–402. [Google Scholar] [CrossRef]
- Böhm, H.-J. The computer program LUDI: A new method for the de novo design of enzyme inhibitors. J. Comput. Aided Mol. Des. 1992, 6, 61–78. [Google Scholar] [CrossRef]
- Gillet, V.J.; Newell, W.; Mata, P.; Myatt, G.; Sike, S.; Zsoldos, Z.; Johnson, A.P. SPROUT: Recent developments in the de novo design of molecules. J. Chem. Inf. Comput. Sci. 1994, 34, 207–217. [Google Scholar] [CrossRef]
- Lipinski, C.A.; Lombardo, F.; Dominy, B.W.; Feeney, P.J. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Del. Rev. 1997, 23, 3–25. [Google Scholar] [CrossRef]
- Teague, S.J.; Davis, A.M.; Leeson, P.D.; Oprea, T. The Design of Leadlike Combinatorial Libraries. Angew. Chem. 1999, 38, 3743–3748. [Google Scholar] [CrossRef]
- Aronov, A.M. Predictive in silico modeling for hERG channel blockers. Drug Discov. Today 2005, 10, 149–155. [Google Scholar] [CrossRef]
- Goldberg, D.E. Genetic Algorithms in Search, Optimization and Machine Learning; Addison-Wesley Longman Publishing Co., Inc.: Boston, MA, USA, 1989. [Google Scholar]
- Leach, A.R. 4.05—Ligand-Based Approaches: Core Molecular Modeling. In Comprehensive Medicinal Chemistry II; Taylor, J.B., Triggle, D.J., Eds.; Elsevier: Oxford, UK, 2007; pp. 87–118. [Google Scholar]
- McGarrah, D.B.; Judson, R.S. Analysis of the genetic algorithm method of molecular conformation determination. J. Comput. Chem. 1993, 14, 1385–1395. [Google Scholar] [CrossRef]
- Clark, D.E.; Westhead, D.R. Evolutionary algorithms in computer-aided molecular design. J. Comput. Aided Mol. Des. 1996, 10, 337–358. [Google Scholar] [CrossRef] [PubMed]
- Masek, B.B.; Baker, D.S.; Dorfman, R.J.; DuBrucq, K.; Francis, V.C.; Nagy, S.; Richey, B.L.; Soltanshahi, F. Multistep Reaction Based De Novo Drug Design: Generating Synthetically Feasible Design Ideas. J. Chem. Inf. Model. 2016, 56, 605–620. [Google Scholar] [CrossRef]
- Douguet, D.; Thoreau, E.; Grassy, G. A genetic algorithm for the automated generation of small organic molecules: Drug design using an evolutionary algorithm. J. Comput. Aided Mol. Des. 2000, 14, 449–466. [Google Scholar] [CrossRef]
- Pegg, S.C.H.; Haresco, J.J.; Kuntz, I.D. A genetic algorithm for structure-based de novo design. J. Comput. Aided Mol. Des. 2001, 15, 911–933. [Google Scholar] [CrossRef]
- Nicolas, B.; Shaheen, A.; Nicolas, M.; Amedeo, C. An Evolutionary Approach for Structure-based Design of Natural and Non-natural Peptidic Ligands. Comb. Chem. High Throughput Screen. 2001, 4, 661–673. [Google Scholar]
- Douguet, D.; Munier-Lehmann, H.; Labesse, G.; Pochet, S. LEA3D: A Computer-Aided Ligand Design for Structure-Based Drug Design. J. Med. Chem. 2005, 48, 2457–2468. [Google Scholar] [CrossRef]
- Barigye, S.J.; García de la Vega, J.M.; Perez-Castillo, Y. Generative Adversarial Networks (GANs) Based Synthetic Sampling for Predictive Modeling. Mol. Inform. 2020, 39, 2000086. [Google Scholar] [CrossRef] [PubMed]
- Fechner, U.; Schneider, G. Flux (1): A Virtual Synthesis Scheme for Fragment-Based de Novo Design. J. Chem. Inf. Model. 2006, 46, 699–707. [Google Scholar] [CrossRef] [PubMed]
- Schüller, A.; Suhartono, M.; Fechner, U.; Tanrikulu, Y.; Breitung, S.; Scheffer, U.; Göbel, M.W.; Schneider, G. The concept of template-based de novo design from drug-derived molecular fragments and its application to TAR RNA. J. Comput. Aided Mol. Des. 2008, 22, 59–68. [Google Scholar] [CrossRef] [PubMed]
- Nicolaou, C.A.; Apostolakis, J.; Pattichis, C.S. De Novo Drug Design Using Multiobjective Evolutionary Graphs. J. Chem. Inf. Model. 2009, 49, 295–307. [Google Scholar] [CrossRef]
- Wong, S.S.Y.; Luo, W.; Chan, K.C.C. EvoMD: An Algorithm for Evolutionary Molecular Design. IEEE/Acm Trans. Comput. Biol. Bioinform. 2011, 8, 987–1003. [Google Scholar] [CrossRef] [PubMed]
- Chan, H.C.S.; Shan, H.; Dahoun, T.; Vogel, H.; Yuan, S. Advancing Drug Discovery via Artificial Intelligence. Trends Pharmacol. Sci. 2019, 40, 592–604. [Google Scholar] [CrossRef]
- Zhu, H. Big Data and Artificial Intelligence Modeling for Drug Discovery. Annu. Rev. Pharmacol. Toxicol. 2020, 60, 573–589. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, X.; Wang, Y.; Byrne, R.; Schneider, G.; Yang, S. Concepts of Artificial Intelligence for Computer-Assisted Drug Discovery. Chem. Rev. 2019, 119, 10520–10594. [Google Scholar] [CrossRef] [Green Version]
- Afantitis, A. Nanoinformatics: Artificial Intelligence and Nanotechnology in the New Decade. Comb. Chem. High Throughput Screen. 2020, 23, 4–5. [Google Scholar] [CrossRef]
- von Lilienfeld, O.A. Quantum Machine Learning in Chemical Compound Space. Angew. Chem. 2018, 57, 4164–4169. [Google Scholar] [CrossRef] [PubMed]
- Tkatchenko, A. Machine learning for chemical discovery. Nat. Commun. 2020, 11, 4125. [Google Scholar] [CrossRef] [PubMed]
- Klambauer, G.; Hochreiter, S.; Rarey, M. Machine Learning in Drug Discovery. J. Chem. Inf. Model. 2019, 59, 945–946. [Google Scholar] [CrossRef] [Green Version]
- Chen, H.; Engkvist, O.; Wang, Y.; Olivecrona, M.; Blaschke, T. The rise of deep learning in drug discovery. Drug Discov. Today 2018, 23, 1241–1250. [Google Scholar] [CrossRef]
- Han, M.; May, R.; Zhang, X.; Wang, X.; Pan, S.; Yan, D.; Jin, Y.; Xu, L. A review of reinforcement learning methodologies for controlling occupant comfort in buildings. Sustain. Cities Soc. 2019, 51, 101748. [Google Scholar] [CrossRef]
- Lavecchia, A. Deep learning in drug discovery: Opportunities, challenges and future prospects. Drug Discov. Today 2019, 24, 2017–2032. [Google Scholar] [CrossRef] [PubMed]
- Olivecrona, M.; Blaschke, T.; Engkvist, O.; Chen, H. Molecular de-novo design through deep reinforcement learning. J. Cheminform. 2017, 9, 48. [Google Scholar] [CrossRef] [Green Version]
- Graves, A.; Eck, D.; Beringer, N.; Schmidhuber, J. Biologically Plausible Speech Recognition with LSTM Neural Nets; Springer: Berlin/Heidelberg, Germany, 2004; pp. 127–136. [Google Scholar]
- Gers, F.A.; Schmidhuber, E. LSTM recurrent networks learn simple context-free and context-sensitive languages. IEEE Trans. Neural Netw. 2001, 12, 1333–1340. [Google Scholar] [CrossRef] [Green Version]
- Srivastava, N.; Mansimov, E.; Salakhutdinov, R. Unsupervised learning of video representations using LSTMs. In Proceedings of the 32nd International Conference on International Conference on Machine Learning—Volume 37; JMLR.org: Lille, France, 2015; pp. 843–852. [Google Scholar]
- Eck, D.; Schmidhuber, J. Finding temporal structure in music: Blues improvisation with LSTM recurrent networks. In Proceedings of the 12th IEEE Workshop on Neural Networks for Signal Processing, Martigny, Switzerland, 6 September 2002; pp. 747–756. [Google Scholar]
- LeCun, Y.; Bengio, Y.; Hinton, G. Deep learning. Nature 2015, 521, 436–444. [Google Scholar] [CrossRef]
- Mamoshina, P.; Vieira, A.; Putin, E.; Zhavoronkov, A. Applications of Deep Learning in Biomedicine. Mol. Pharm. 2016, 13, 1445–1454. [Google Scholar] [CrossRef]
- Miotto, R.; Wang, F.; Wang, S.; Jiang, X.; Dudley, J.T. Deep learning for healthcare: Review, opportunities and challenges. Brief. Bioinform. 2018, 19, 1236–1246. [Google Scholar] [CrossRef]
- Cherkasov, A.; Muratov, E.N.; Fourches, D.; Varnek, A.; Baskin, I.I.; Cronin, M.; Dearden, J.; Gramatica, P.; Martin, Y.C.; Todeschini, R.; et al. QSAR Modeling: Where Have You Been? Where Are You Going To? J. Med. Chem. 2014, 57, 4977–5010. [Google Scholar] [CrossRef] [Green Version]
- Ekins, S. The Next Era: Deep Learning in Pharmaceutical Research. Pharm. Res. 2016, 33, 2594–2603. [Google Scholar] [CrossRef]
- Lenselink, E.B.; ten Dijke, N.; Bongers, B.; Papadatos, G.; van Vlijmen, H.W.T.; Kowalczyk, W.; Ijzerman, A.P.; van Westen, G.J.P. Beyond the hype: Deep neural networks outperform established methods using a ChEMBL bioactivity benchmark set. J. Cheminform. 2017, 9, 45. [Google Scholar] [CrossRef] [Green Version]
- Liu, X.; Ye, K.; van Vlijmen, H.W.T.; Ijzerman, A.P.; van Westen, G.J.P. An exploration strategy improves the diversity of de novo ligands using deep reinforcement learning: A case for the adenosine A2A receptor. J. Cheminform. 2019, 11, 35. [Google Scholar] [CrossRef]
- David, L.; Thakkar, A.; Mercado, R.; Engkvist, O. Molecular representations in AI-driven drug discovery: A review and practical guide. J. Cheminform. 2020, 12, 56. [Google Scholar] [CrossRef]
- Weininger, D. SMILES, a chemical language and information system. 1. Introduction to methodology and encoding rules. J. Chem. Inf. Comput. Sci. 1988, 28, 31–36. [Google Scholar] [CrossRef]
- Pineda, F.J. Generalization of back-propagation to recurrent neural networks. Phys. Rev. Lett. 1987, 59, 2229–2232. [Google Scholar] [CrossRef]
- Lipton, Z.C.; Berkowitz, J.; Elkan, C. A critical review of recurrent neural networks for sequence learning. arXiv 2015, arXiv:1506.00019. [Google Scholar]
- Merk, D.; Friedrich, L.; Grisoni, F.; Schneider, G. De Novo Design of Bioactive Small Molecules by Artificial Intelligence. Mol. Inform. 2018, 37, 1700153. [Google Scholar] [CrossRef] [Green Version]
- Segler, M.H.S.; Kogej, T.; Tyrchan, C.; Waller, M.P. Generating Focused Molecule Libraries for Drug Discovery with Recurrent Neural Networks. Acs. Cent. Sci. 2018, 4, 120–131. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maragakis, P.; Nisonoff, H.; Cole, B.; Shaw, D.E. A Deep-Learning View of Chemical Space Designed to Facilitate Drug Discovery. J. Chem. Inf. Model. 2020, 60, 4487–4496. [Google Scholar] [CrossRef]
- Popova, M.; Isayev, O.; Tropsha, A. Deep reinforcement learning for de novo drug design. Sci. Adv. 2018, 4, eaap7885. [Google Scholar] [CrossRef] [Green Version]
- Ståhl, N.; Falkman, G.; Karlsson, A.; Mathiason, G.; Boström, J. Deep Reinforcement Learning for Multiparameter Optimization in de novo Drug Design. J. Chem. Inf. Model. 2019, 59, 3166–3176. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yasonik, J. Multiobjective de novo drug design with recurrent neural networks and nondominated sorting. J. Cheminform. 2020, 12, 14. [Google Scholar] [CrossRef] [Green Version]
- Pan, S.J.; Yang, Q. A Survey on Transfer Learning. IEEE Trans. Knowl. Data Eng. 2010, 22, 1345–1359. [Google Scholar] [CrossRef]
- Gupta, A.; Müller, A.T.; Huisman, B.J.H.; Fuchs, J.A.; Schneider, P.; Schneider, G. Generative Recurrent Networks for De Novo Drug Design. Mol. Inform. 2018, 37, 1700111. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kotsias, P.-C.; Arús-Pous, J.; Chen, H.; Engkvist, O.; Tyrchan, C.; Bjerrum, E.J. Direct steering of de novo molecular generation with descriptor conditional recurrent neural networks. Nat. Mach. Intell. 2020, 2, 254–265. [Google Scholar] [CrossRef]
- Kusner, M.J.; Paige, B.; Hernández-Lobato, J.M. Grammar Variational Autoencoder; ICML: Sydney, Australia, 2017. [Google Scholar]
- Grisoni, F.; Moret, M.; Lingwood, R.; Schneider, G. Bidirectional Molecule Generation with Recurrent Neural Networks. J. Chem. Inf. Mod. 2020, 60, 1175–1183. [Google Scholar] [CrossRef]
- Rifaioglu, A.S.; Nalbat, E.; Atalay, V.; Martin, M.J.; Cetin-Atalay, R.; Doğan, T. DEEPScreen: High performance drug–target interaction prediction with convolutional neural networks using 2-D structural compound representations. Chem. Sci. 2020, 11, 2531–2557. [Google Scholar] [CrossRef] [Green Version]
- Lecun, Y.; Bottou, L.; Bengio, Y.; Haffner, P. Gradient-based learning applied to document recognition. Proc. IEEE 1998, 86, 2278–2324. [Google Scholar] [CrossRef] [Green Version]
- Sun, M.; Zhao, S.; Gilvary, C.; Elemento, O.; Zhou, J.; Wang, F. Graph convolutional networks for computational drug development and discovery. Brief. Bioinform. 2020, 21, 919–935. [Google Scholar] [CrossRef]
- Li, Y.; Hu, J.; Wang, Y.; Zhou, J.; Zhang, L.; Liu, Z. DeepScaffold: A Comprehensive Tool for Scaffold-Based De Novo Drug Discovery Using Deep Learning. J. Chem. Inf. Model. 2020, 60, 77–91. [Google Scholar] [CrossRef]
- Khemchandani, Y.; O’Hagan, S.; Samanta, S.; Swainston, N.; Roberts, T.J.; Bollegala, D.; Kell, D.B. DeepGraphMolGen, a multi-objective, computational strategy for generating molecules with desirable properties: A graph convolution and reinforcement learning approach. J. Cheminform. 2020, 12, 53. [Google Scholar] [CrossRef]
- Li, Y.; Zhang, L.; Liu, Z. Multi-objective de novo drug design with conditional graph generative model. J. Cheminform. 2018, 10, 33. [Google Scholar] [CrossRef] [Green Version]
- Yi, X.; Walia, E.; Babyn, P. Generative adversarial network in medical imaging: A review. Med. Image Anal. 2019, 58, 101552. [Google Scholar] [CrossRef] [Green Version]
- Gui, J.; Sun, Z.; Wen, Y.; Tao, D.; Ye, J. A review on generative adversarial networks: Algorithms, theory, and applications. arXiv 2020, arXiv:2001.06937. [Google Scholar]
- Vanhaelen, Q.; Lin, Y.-C.; Zhavoronkov, A. The Advent of Generative Chemistry. Acs. Med. Chem. Lett. 2020, 11, 1496–1505. [Google Scholar] [CrossRef] [PubMed]
- Putin, E.; Asadulaev, A.; Ivanenkov, Y.; Aladinskiy, V.; Sanchez-Lengeling, B.; Aspuru-Guzik, A.; Zhavoronkov, A. Reinforced Adversarial Neural Computer for de Novo Molecular Design. J. Chem. Inf. Model. 2018, 58, 1194–1204. [Google Scholar] [CrossRef]
- Putin, E.; Asadulaev, A.; Vanhaelen, Q.; Ivanenkov, Y.; Aladinskaya, A.V.; Aliper, A.; Zhavoronkov, A. Adversarial Threshold Neural Computer for Molecular de Novo Design. Mol. Pharm. 2018, 15, 4386–4397. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prykhodko, O.; Johansson, S.V.; Kotsias, P.-C.; Arús-Pous, J.; Bjerrum, E.J.; Engkvist, O.; Chen, H. A de novo molecular generation method using latent vector based generative adversarial network. J. Cheminform. 2019, 11, 74. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Girin, L.; Leglaive, S.; Bie, X.; Diard, J.; Hueber, T.; Alameda-Pineda, X. Dynamical Variational Autoencoders: A Comprehensive Review. arXiv 2020, arXiv:2008.12595. [Google Scholar]
- Gómez-Bombarelli, R.; Wei, J.N.; Duvenaud, D.; Hernández-Lobato, J.M.; Sánchez-Lengeling, B.; Sheberla, D.; Aguilera-Iparraguirre, J.; Hirzel, T.D.; Adams, R.P.; Aspuru-Guzik, A. Automatic Chemical Design Using a Data-Driven Continuous Representation of Molecules. Acs. Cent. Sci. 2018, 4, 268–276. [Google Scholar] [CrossRef] [PubMed]
- Skalic, M.; Jiménez, J.; Sabbadin, D.; De Fabritiis, G. Shape-Based Generative Modeling for de Novo Drug Design. J. Chem. Inf. Model. 2019, 59, 1205–1214. [Google Scholar] [CrossRef] [PubMed]
- Lim, J.; Ryu, S.; Kim, J.W.; Kim, W.Y. Molecular generative model based on conditional variational autoencoder for de novo molecular design. J. Cheminform. 2018, 10, 31. [Google Scholar] [CrossRef] [Green Version]
- Cho, K.; Van Merriënboer, B.; Gulcehre, C.; Bahdanau, D.; Bougares, F.; Schwenk, H.; Bengio, Y. Learning phrase representations using RNN encoder-decoder for statistical machine translation. arXiv 2014, arXiv:1406.1078. [Google Scholar]
- Hochreiter, S.; Schmidhuber, J. Long Short-Term Memory. Neural Comput. 1997, 9, 1735–1780. [Google Scholar] [CrossRef]
- Sutskever, I.; Vinyals, O.; Le, Q.V. Sequence to sequence learning with neural networks. In Proceedings of the 27th International Conference on Neural Information Processing Systems—Volume 2; MIT Press: Montreal, QC, Canada, 2014; pp. 3104–3112. [Google Scholar]
- Gao, K.; Nguyen, D.D.; Tu, M.; Wei, G.-W. Generative Network Complex for the Automated Generation of Drug-like Molecules. J. Chem. Inf. Model. 2020, 60, 5682–5698. [Google Scholar] [CrossRef] [PubMed]
- Bjerrum, E.J.; Sattarov, B. Improving chemical autoencoder latent space and molecular de novo generation diversity with heteroencoders. Biomolecules 2018, 8, 131. [Google Scholar] [CrossRef] [Green Version]
- Makhzani, A.; Shlens, J.; Jaitly, N.; Goodfellow, I.; Frey, B. Adversarial autoencoders. arXiv 2015, arXiv:1511.05644. [Google Scholar]
- Kadurin, A.; Aliper, A.; Kazennov, A.; Mamoshina, P.; Vanhaelen, Q.; Khrabrov, K.; Zhavoronkov, A. The cornucopia of meaningful leads: Applying deep adversarial autoencoders for new molecule development in oncology. Oncotarget 2017, 8, 10883–10890. [Google Scholar] [CrossRef] [Green Version]
- Kadurin, A.; Nikolenko, S.; Khrabrov, K.; Aliper, A.; Zhavoronkov, A. druGAN: An Advanced Generative Adversarial Autoencoder Model for de Novo Generation of New Molecules with Desired Molecular Properties in Silico. Mol. Pharm. 2017, 14, 3098–3104. [Google Scholar] [CrossRef]
- Hartenfeller, M.; Proschak, E.; Schüller, A.; Schneider, G. Concept of Combinatorial De Novo Design of Drug-like Molecules by Particle Swarm Optimization. Chem. Biol. Drug Des. 2008, 72, 16–26. [Google Scholar] [CrossRef] [PubMed]
- Winter, R.; Montanari, F.; Steffen, A.; Briem, H.; Noé, F.; Clevert, D.-A. Efficient multi-objective molecular optimization in a continuous latent space. Chem. Sci. 2019, 10, 8016–8024. [Google Scholar] [CrossRef]
- Metz, L.; Poole, B.; Pfau, D.; Sohl-Dickstein, J. Unrolled generative adversarial networks. arXiv 2016, arXiv:1611.02163. [Google Scholar]
- Rupp, M.; Schneider, G. Graph Kernels for Molecular Similarity. Mol. Inform. 2010, 29, 266–273. [Google Scholar] [CrossRef] [PubMed]
- OECD, Validation of (Q)SAR Models. Available online: https://www.oecd.org/chemicalsafety/risk-assessment/validationofqsarmodels.htm. (accessed on 8 November 2019).
- Muratov, E.N.; Bajorath, J.; Sheridan, R.P.; Tetko, I.V.; Filimonov, D.; Poroikov, V.; Oprea, T.I.; Baskin, I.I.; Varnek, A.; Roitberg, A.; et al. QSAR without borders. Chem. Soc. Rev. 2020, 49, 3525–3564. [Google Scholar] [CrossRef] [PubMed]
- Bickerton, G.R.; Paolini, G.V.; Besnard, J.; Muresan, S.; Hopkins, A.L. Quantifying the chemical beauty of drugs. Nat. Chem. 2012, 4, 90–98. [Google Scholar] [CrossRef] [Green Version]
- Hutter, M.C. In Silico Prediction of Drug Properties. Curr. Med. Chem. 2009, 16, 189–202. [Google Scholar] [CrossRef]
- Ertl, P.; Schuffenhauer, A. Estimation of synthetic accessibility score of drug-like molecules based on molecular complexity and fragment contributions. J. Cheminform. 2009, 1, 8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Antreas, A.; Andreas, T.; Georgia, M. Enalos Suite of Tools: Enhancing Cheminformatics and Nanoinfor-matics through KNIME. Curr. Med. Chem. 2020, 27, 6523–6535. [Google Scholar]
- Gao, W.; Coley, C.W. The Synthesizability of Molecules Proposed by Generative Models. J. Chem. Inf. Model. 2020, 60, 5714–5723. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Coley, C.W.; Rogers, L.; Green, W.H.; Jensen, K.F. SCScore: Synthetic Complexity Learned from a Reaction Corpus. J. Chem. Inf. Model. 2018, 58, 252–261. [Google Scholar] [CrossRef] [PubMed]
- Boda, K.; Johnson, A.P. Molecular Complexity Analysis of de Novo Designed Ligands. J. Med. Chem. 2006, 49, 5869–5879. [Google Scholar] [CrossRef]
- Kinaret, P.A.S.; Serra, A.; Federico, A.; Kohonen, P.; Nymark, P.; Liampa, I.; Ha, M.K.; Choi, J.-S.; Jagiello, K.; Sanabria, N. Transcriptomics in toxicogenomics, Part I: Experimental design, technologies, publicly available data, and regulatory aspects. Nanomaterials 2020, 10, 750. [Google Scholar] [CrossRef] [Green Version]
- Federico, A.; Serra, A.; Ha, M.K.; Kohonen, P.; Choi, J.S.; Liampa, I.; Nymark, P.; Sanabria, N.; Cattelani, L.; Fratello, M.; et al. Transcriptomics in Toxicogenomics, Part II: Preprocessing and Differential Expression Analysis for High Quality Data. Nanomaterials 2020, 10, 903. [Google Scholar] [CrossRef] [PubMed]
- Méndez-Lucio, O.; Baillif, B.; Clevert, D.-A.; Rouquié, D.; Wichard, J. De novo generation of hit-like molecules from gene expression signatures using artificial intelligence. Nat. Commun. 2020, 11, 10. [Google Scholar] [CrossRef] [Green Version]
- Keshavarzi Arshadi, A.; Webb, J.; Salem, M.; Cruz, E.; Calad-Thomson, S.; Ghadirian, N.; Collins, J.; Diez-Cecilia, E.; Kelly, B.; Goodarzi, H.; et al. Artificial Intelligence for COVID-19 Drug Discovery and Vaccine Development. Front. Artif. Intell. 2020, 3, 65. [Google Scholar] [CrossRef]
- Lalmuanawma, S.; Hussain, J.; Chhakchhuak, L. Applications of machine learning and artificial intelligence for Covid-19 (SARS-CoV-2) pandemic: A review. Chaossolitons Fractals 2020, 139, 110059. [Google Scholar] [CrossRef] [PubMed]
- Mohanty, S.; Harun Ai Rashid, M.; Mridul, M.; Mohanty, C.; Swayamsiddha, S. Application of Artificial Intelligence in COVID-19 drug repurposing. Diabetes Metab. Syndr Clin. Res. Rev. 2020, 14, 1027–1031. [Google Scholar] [CrossRef]
- Ton, A.-T.; Gentile, F.; Hsing, M.; Ban, F.; Cherkasov, A. Rapid Identification of Potential Inhibitors of SARS-CoV-2 Main Protease by Deep Docking of 1.3 Billion Compounds. Mol. Inform. 2020, 39, 2000028. [Google Scholar] [CrossRef] [Green Version]
- Chenthamarakshan, V.; Das, P.; Hoffman, S.; Strobelt, H.; Padhi, I.; Lim, K.W.; Hoover, B.; Manica, M.; Born, J.; Laino, T. Cogmol: Target-specific and selective drug design for covid-19 using deep generative models. arXiv 2020, arXiv:2004.01215. [Google Scholar]
- Tang, B.; He, F.; Liu, D.; Fang, M.; Wu, Z.; Xu, D. AI-aided design of novel targeted covalent inhibitors against SARS-CoV-2. bioRxiv 2020. [Google Scholar] [CrossRef] [Green Version]
- Bai, Q.; Tan, S.; Xu, T.; Liu, H.; Huang, J.; Yao, X. MolAICal: A soft tool for 3D drug design of protein targets by artificial intelligence and classical algorithm. Brief. Bioinform. 2020. [Google Scholar] [CrossRef]
- HMA HMA-EMA Joint Big Data TaskforcePhase II Report: ‘Evolving Data-Driven Regulation’. Available online: https://www.ema.europa.eu/en/documents/other/hma-ema-joint-big-data-taskforce-phase-ii-report-evolving-data-driven-regulation_en.pdf (accessed on 10 December 2020).
- OECD OECD AI Principles Overview. Available online: https://oecd.ai/ai-principles (accessed on 10 December 2020).
- OECD Using Artificial Intelligence to Help Combat COVID-19. Available online: https://read.oecd-ilibrary.org/view/?ref=130_130771-3jtyra9uoh&title=Using-artificial-intelligence-to-help-combat-COVID-19 (accessed on 10 December 2020).
- Baruffaldi, S.; Beuzekom, B.V.; Dernis, H.; Harhoff, D.; Rao, N.; Rosenfeld, D.; Squicciarini, M. Identifying and measuring developments in artificial intelligence. Oecd Sci. Technol. Ind. Work. Pap. 2020. No. 2020/05. [Google Scholar] [CrossRef]
- Wu, H.; Wang, C.; Yin, J.; Lu, K.; Zhu, L. Interpreting shared deep learning models via explicable boundary trees. arXiv 2017, arXiv:1709.03730. [Google Scholar]
- Zhao, S.; Talasila, M.; Jacobson, G.; Borcea, C.; Aftab, S.A.; Murray, J.F. Packaging and Sharing Machine Learning Models via the Acumos ai Open Platform. In Proceedings of the 2018 17th IEEE International Conference on Machine Learning and Applications (ICMLA); 2018; pp. 841–846. Available online: https://arxiv.org/ftp/arxiv/papers/1810/1810.07159.pdf (accessed on 28 January 2021).
- Tan, J.J.; Cong, X.J.; Hu, L.M.; Wang, C.X.; Jia, L.; Liang, X.-J. Therapeutic strategies underpinning the development of novel techniques for the treatment of HIV infection. Drug Discov. Today 2010, 15, 186–197. [Google Scholar] [CrossRef] [Green Version]
- Hopkins, A. All Drugs Will be Designed by Computers by 2030. The Telegraph. Available online: https://www.telegraph.co.uk/technology/2021/01/18/drugs-will-designed-ai-decades-end/#comment (accessed on 28 January 2021).
- Zhavoronkov, A.; Ivanenkov, Y.A.; Aliper, A.; Veselov, M.S.; Aladinskiy, V.A.; Aladinskaya, A.V.; Terentiev, V.A.; Polykovskiy, D.A.; Kuznetsov, M.D.; Asadulaev, A.; et al. Deep learning enables rapid identification of potent DDR1 kinase inhibitors. Nat. Biotechnol. 2019, 37, 1038–1040. [Google Scholar] [CrossRef]
- Zheng, S.; Rao, J.; Zhang, Z.; Xu, J.; Yang, Y. Predicting Retrosynthetic Reactions Using Self-Corrected Transformer Neural Networks. J. Chem. Inf. Model. 2020, 60, 47–55. [Google Scholar] [CrossRef] [PubMed]
- Brown, N.; Fiscato, M.; Segler, M.H.S.; Vaucher, A.C. GuacaMol: Benchmarking Models for de Novo Molecular Design. J. Chem. Inf. Model. 2019, 59, 1096–1108. [Google Scholar] [CrossRef] [PubMed]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mouchlis, V.D.; Afantitis, A.; Serra, A.; Fratello, M.; Papadiamantis, A.G.; Aidinis, V.; Lynch, I.; Greco, D.; Melagraki, G. Advances in De Novo Drug Design: From Conventional to Machine Learning Methods. Int. J. Mol. Sci. 2021, 22, 1676. https://doi.org/10.3390/ijms22041676
Mouchlis VD, Afantitis A, Serra A, Fratello M, Papadiamantis AG, Aidinis V, Lynch I, Greco D, Melagraki G. Advances in De Novo Drug Design: From Conventional to Machine Learning Methods. International Journal of Molecular Sciences. 2021; 22(4):1676. https://doi.org/10.3390/ijms22041676
Chicago/Turabian StyleMouchlis, Varnavas D., Antreas Afantitis, Angela Serra, Michele Fratello, Anastasios G. Papadiamantis, Vassilis Aidinis, Iseult Lynch, Dario Greco, and Georgia Melagraki. 2021. "Advances in De Novo Drug Design: From Conventional to Machine Learning Methods" International Journal of Molecular Sciences 22, no. 4: 1676. https://doi.org/10.3390/ijms22041676