
Genomic, Clinical, and Behavioral Characterization of 15q11.2 BP1-BP2 Deletion (Burnside-Butler) Syndrome in Five Families

 ,
,  ,
,
Abstract
:1. Introduction
2. Results
2.1. Cognitive and Behavioral Features
2.2. Sensorimotor Ability
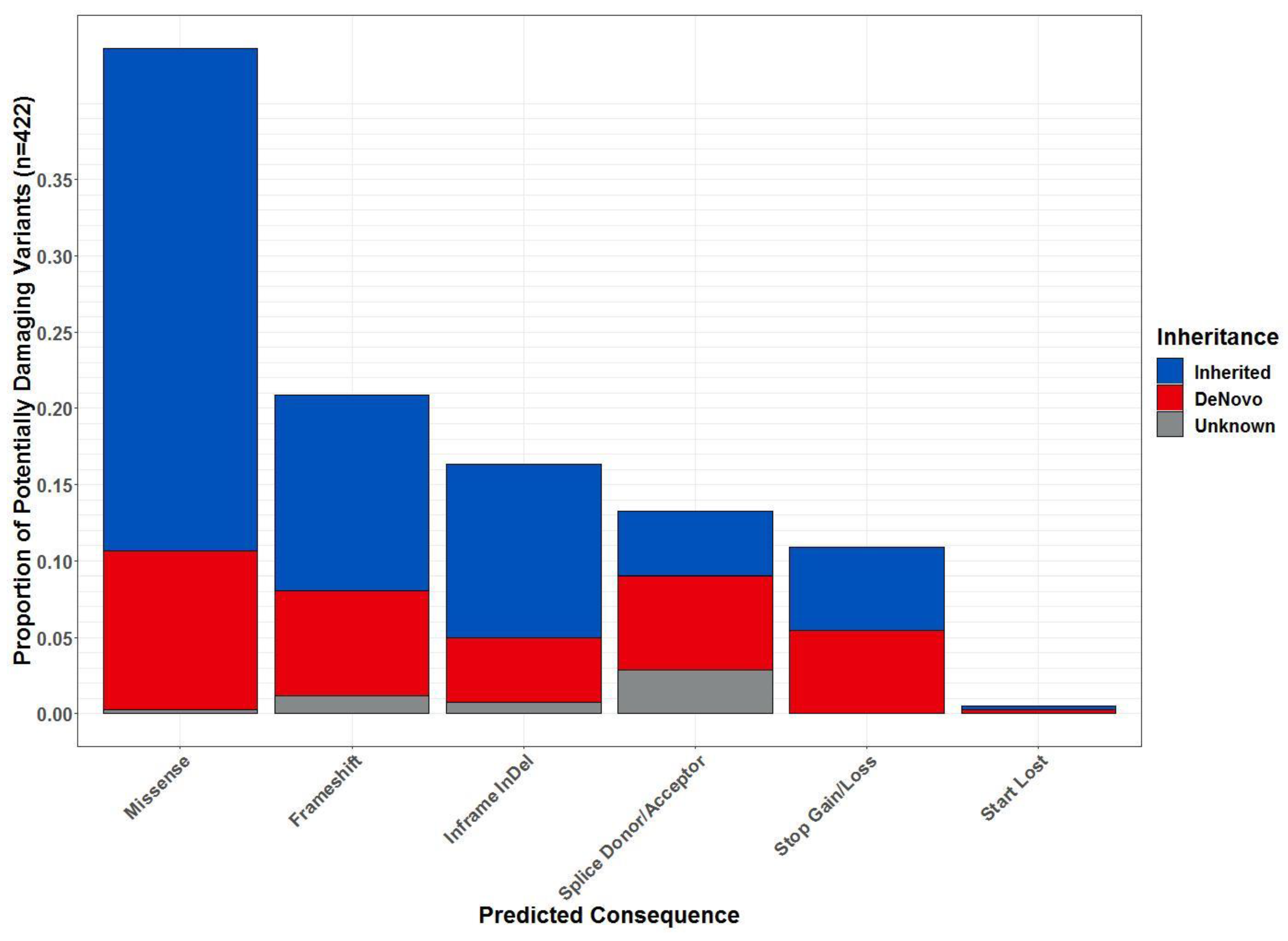
2.3. Whole-Exome Sequencing
3. Discussion
3.1. Clinical and Neuropsychiatric Behavior Developmental Findings
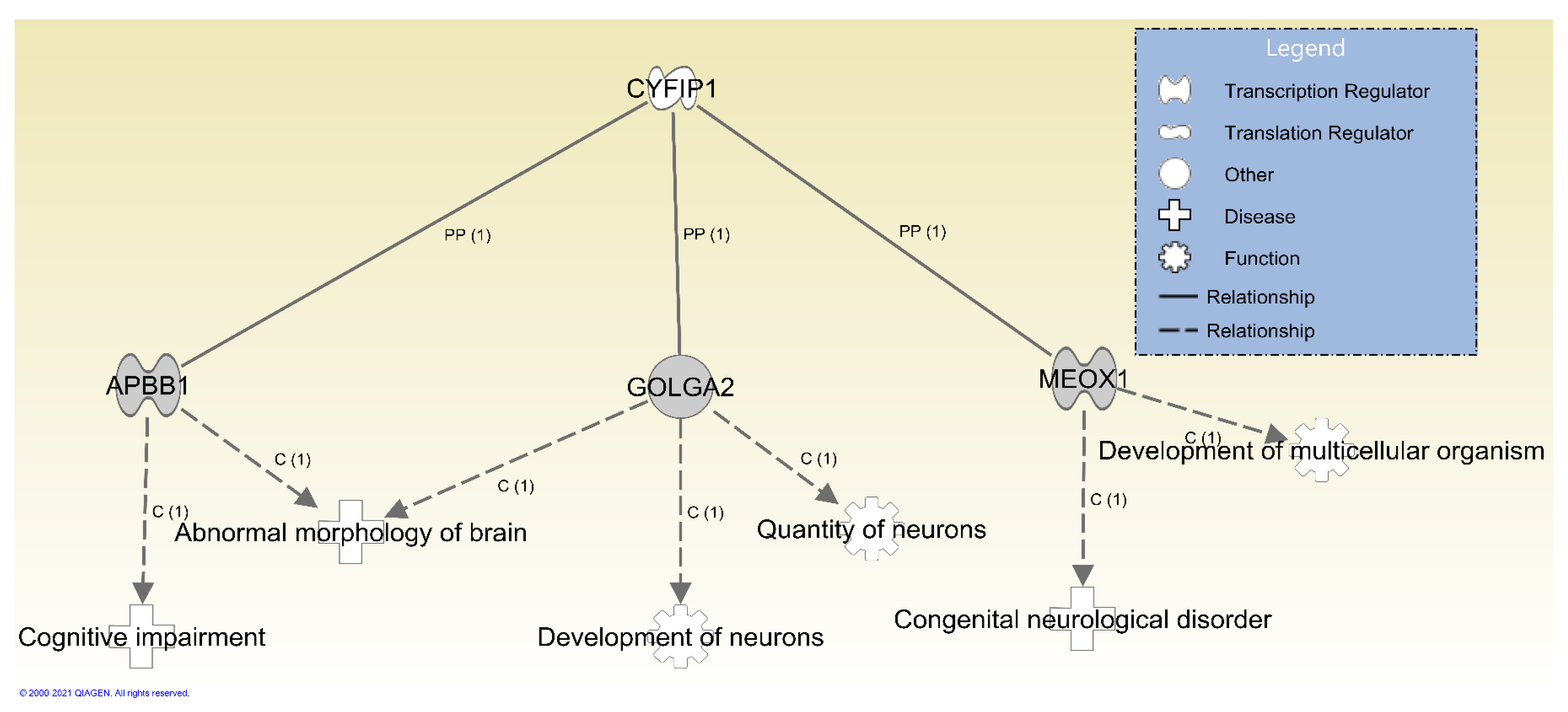
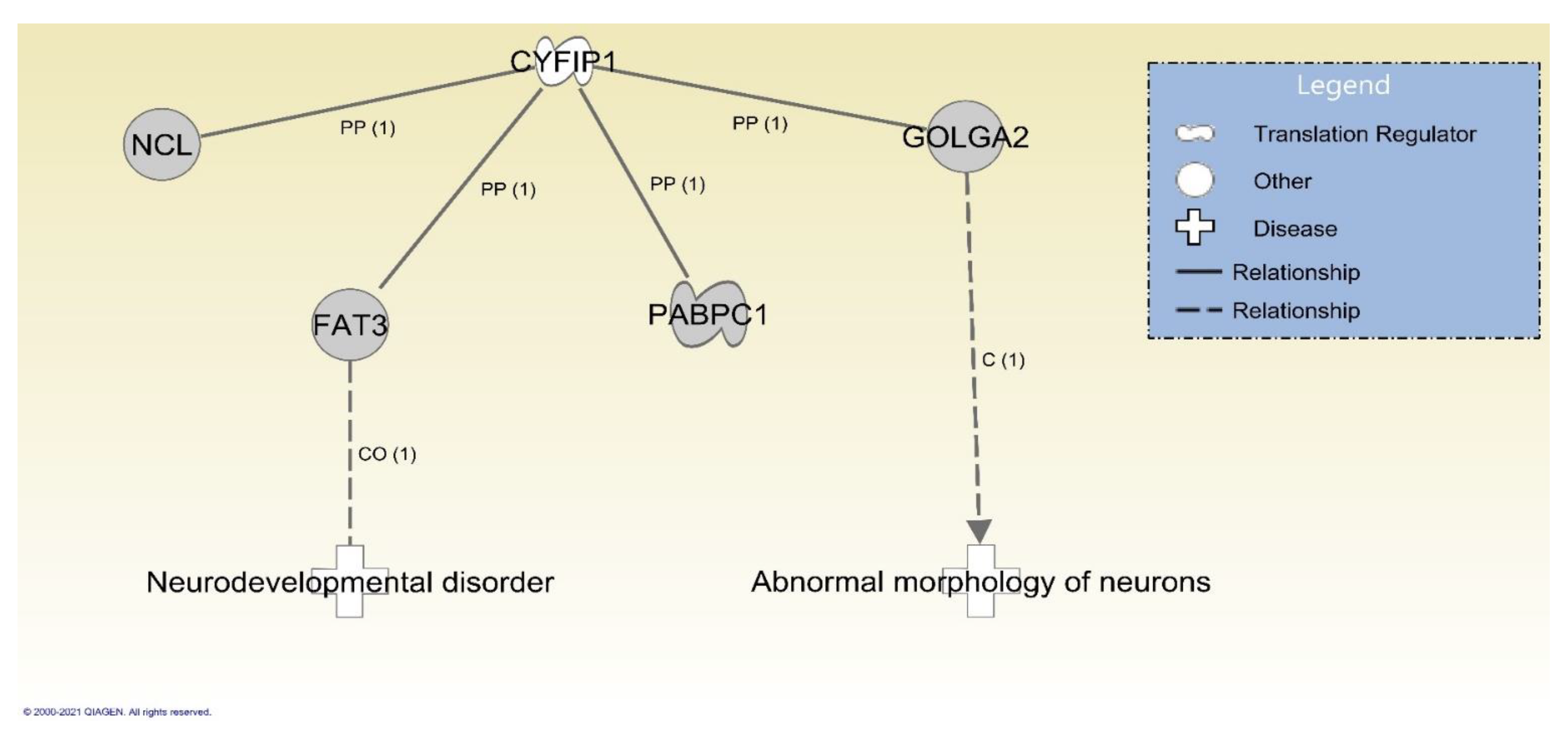
3.2. Protein–Protein Interactions and Functions Related to NIPA1, NIPA2, CYFIP1 and TUBGCP5 Genes in the 15q11.2 BP1-BP2 Region
3.3. Identified Gene Variants with Potential Clinical Significance
4. Materials and Methods
4.1. Families with 15q11.2 BP1-BP2 Deletion or Burnside-Butler Syndrome (BBS)
4.2. Cognitive and Behavioral Measures
4.3. Clinical Evaluation and Physical Examinations
4.4. Postural Control Testing
4.5. DNA Extraction
4.6. Methylation Specific-Multiplex Ligation Probe Amplification (MS-MLPA)
4.7. Whole-Exome Sequencing
4.8. Variant Calling and Quality Control Procedures
4.9. Functional and Clinical Characterization of Genes with Variants
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Nicholls, R.D.; Knoll, J.H.; Butler, M.G.; Karam, S.; Lalande, M. Genetic imprinting suggested by maternal heterodisomy in nondeletion Prader-Willi syndrome. Nature 1989, 342, 281–285. [Google Scholar] [CrossRef]
- Butler, M.G.; Manzardo, A.M.; Forster, J.L. Prader-Willi Syndrome: Clinical Genetics and Diagnostic Aspects with Treatment Approaches. Curr. Pediatr. Rev. 2016, 12, 136–166. [Google Scholar] [CrossRef]
- Butler, M.; Lee, P.D.K.; Whitman, B. Management of Prader-Willi syndrome. In Management of Prader-Willi Syndrome, 3rd ed.; Springer: Berlin/Heidelberg, Germany, 2006; pp. 1–550. [Google Scholar]
- Williams, C.A.; Driscoll, D.J.; Dagli, A.I. Clinical and genetic aspects of Angelman syndrome. Genet. Med. 2010, 12, 385–395. [Google Scholar] [CrossRef] [Green Version]
- Bittel, D.C.; Butler, M.G. Prader-Willi syndrome: Clinical genetics, cytogenetics and molecular biology. Expert Rev. Mol. Med. 2005, 7, 1–20. [Google Scholar] [CrossRef] [PubMed]
- Zarcone, J.; Napolitano, D.; Peterson, C.; Breidbord, J.; Ferraioli, S.; Caruso-Anderson, M.; Holsen, L.; Butler, M.G.; Thompson, T. The relationship between compulsive behaviour and academic achievement across the three genetic subtypes of Prader-Willi syndrome. J. Intellect. Disabil. Res. 2007, 51, 478–487. [Google Scholar] [CrossRef] [PubMed]
- Butler, M.G.; Bittel, D.C.; Kibiryeva, N.; Talebizadeh, Z.; Thompson, T. Behavioral differences among subjects with Prader-Willi syndrome and type I or type II deletion and maternal disomy. Pediatrics 2004, 113, 565–573. [Google Scholar] [CrossRef] [PubMed]
- Hartley, S.L.; Maclean, W.E., Jr.; Butler, M.G.; Zarcone, J.; Thompson, T. Maladaptive behaviors and risk factors among the genetic subtypes of Prader-Willi syndrome. Am. J. Med. Genet. A 2005, 136, 140–145. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burnside, R.D.; Pasion, R.; Mikhail, F.M.; Carroll, A.J.; Robin, N.H.; Youngs, E.L.; Gadi, I.K.; Keitges, E.; Jaswaney, V.L.; Papenhausen, P.R.; et al. Microdeletion/microduplication of proximal 15q11.2 between BP1 and BP2: A susceptibility region for neurological dysfunction including developmental and language delay. Hum. Genet. 2011, 130, 517–528. [Google Scholar] [CrossRef]
- Cox, D.M.; Butler, M.G. The 15q11.2 BP1-BP2 microdeletion syndrome: A review. Int. J. Mol. Sci. 2015, 16, 4068–4082. [Google Scholar] [CrossRef]
- Butler, M.G. Clinical and genetic aspects of the 15q11.2 BP1-BP2 microdeletion disorder. J. Intellect. Disabil. Res. 2017, 61, 568–579. [Google Scholar] [CrossRef]
- Ho, K.S.; Wassman, E.R.; Baxter, A.L.; Hensel, C.H.; Martin, M.M.; Prasad, A.; Twede, H.; Vanzo, R.J.; Butler, M.G. Chromosomal Microarray Analysis of Consecutive Individuals with Autism Spectrum Disorders Using an Ultra-High Resolution Chromosomal Microarray Optimized for Neurodevelopmental Disorders. Int. J. Mol. Sci. 2016, 17, 2070. [Google Scholar] [CrossRef] [Green Version]
- Rafi, S.K.; Butler, M.G. The 15q11.2 BP1-BP2 Microdeletion (Burnside–Butler) Syndrome: In Silico Analyses of the Four Coding Genes Reveal Functional Associations with Neurodevelopmental Disorders. Int. J. Mol. Sci. 2020, 21, 3296. [Google Scholar] [CrossRef]
- Delis, D.C.; Kramer, J.H.; Kaplan, E.; Ober, B.A. California Verbal Learning Test, 2nd ed.; Psychological Corporation: San Antonio, TX, USA, 2000. [Google Scholar]
- Dunn, L.M.; Dunn, D.M. Peabody Picture Vocabulary Test, 4th ed.; Pearson Education: Minneapolis, MN, USA, 2007. [Google Scholar]
- Reitan, R.M. Validity of the Trail Making Test as an Indicator of Organic Brain Damage. Percept. Mot. Skills 1958, 8, 271–276. [Google Scholar] [CrossRef]
- Gotham, K.; Pickles, A.; Lord, C. Standardizing ADOS scores for a measure of severity in autism spectrum disorders. J. Autism Dev. Disord. 2009, 39, 693–705. [Google Scholar] [CrossRef] [Green Version]
- Lord, C.; Luyster, R.; Guthrie, W.; Pickles, A. Patterns of developmental trajectories in toddlers with autism spectrum disorder. J. Consult. Clin. Psychol. 2012, 80, 477–489. [Google Scholar] [CrossRef] [Green Version]
- Sparrow, S.S.; Ciccheti, D.V.; Saulnier, C.A. Vineland Adaptive Behavior Scales, 3rd ed.; Pearson Education: London, UK, 2016. [Google Scholar]
- Esbensen, A.J.; Seltzer, M.M.; Lam, K.S.; Bodfish, J.W. Age-related differences in restricted repetitive behaviors in autism spectrum disorders. J. Autism Dev. Disord. 2009, 39, 57–66. [Google Scholar] [CrossRef] [Green Version]
- Hurley, R.S.; Losh, M.; Parlier, M.; Reznick, J.S.; Piven, J. The broad autism phenotype questionnaire. J. Autism Dev. Disord. 2007, 37, 1679–1690. [Google Scholar] [CrossRef] [PubMed]
- Sasson, N.J.; Lam, K.S.L.; Childress, D.; Parlier, M.; Daniels, J.L.; Piven, J. The broad autism phenotype questionnaire: Prevalence and diagnostic classification. Autism Res. 2013, 6, 134–143. [Google Scholar] [CrossRef] [PubMed]
- Bodfish, J.W.; Symons, F.J.; Parker, D.E.; Lewis, M.H. Varieties of repetitive behavior in autism: Comparisons to mental retardation. J. Autism Dev. Disord. 2000, 30, 237–243. [Google Scholar] [CrossRef]
- Quamme, G.A. Molecular identification of ancient and modern mammalian magnesium transporters. Am. J. Physiol. Cell Physiol. 2010, 298, C407–C429. [Google Scholar] [CrossRef] [Green Version]
- Rainier, S.; Chai, J.-H.; Tokarz, D.; Nicholls, R.D.; Fink, J.K. NIPA1 Gene Mutations Cause Autosomal Dominant Hereditary Spastic Paraplegia (SPG6). Am. J. Hum. Genet. 2003, 73, 967–971. [Google Scholar] [CrossRef] [Green Version]
- Maenner, M.J. Prevalence of Autism Spectrum Disorder Among Children Aged 8 Years—Autism and Developmental Disabilities Monitoring Network, 11 Sites, United States, 2016. MMWR Surv. Summ. 2020, 69, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Capodaglio, P.; Menegoni, F.; Vismara, L.; Cimolin, V.; Grugni, G.; Galli, M. Characterisation of balance capacity in Prader-Willi patients. Res. Dev. Disabil. 2011, 32, 81–86. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Hallac, R.R.; Conroy, K.C.; White, S.P.; Kane, A.A.; Collinsworth, A.L.; Sweeney, J.A.; Mosconi, M.W. Postural orientation and equilibrium processes associated with increased postural sway in autism spectrum disorder (ASD). J. Neurodev. Disord. 2016, 8, 43. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lim, Y.H.; Partridge, K.; Girdler, S.; Morris, S.L. Standing Postural Control in Individuals with Autism Spectrum Disorder: Systematic Review and Meta-analysis. J. Autism Dev. Disord 2017, 47, 2238–2253. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Khemani, P.; Schmitt, L.M.; Lui, S.; Mosconi, M.W. Static and dynamic postural control deficits in aging fragile X mental retardation 1 (FMR1) gene premutation carriers. J. Neurodev. Disord. 2019, 11, 2. [Google Scholar] [CrossRef] [Green Version]
- Errico, A.; Ballabio, A.; Rugarli, E.I. Spastin, the protein mutated in autosomal dominant hereditary spastic paraplegia, is involved in microtubule dynamics. Hum. Mol. Genet. 2002, 11, 153–163. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Evans, K.J.; Gomes, E.R.; Reisenweber, S.M.; Gundersen, G.G.; Lauring, B.P. Linking axonal degeneration to microtubule remodeling by Spastin-mediated microtubule severing. J. Cell Biol. 2005, 168, 599–606. [Google Scholar] [CrossRef] [PubMed]
- Alber, B.; Pernauer, M.; Schwan, A.; Rothmund, G.; Hoffmann, K.T.; Brummer, D.; Sperfeld, A.D.; Uttner, I.; Binder, H.; Epplen, J.T.; et al. Spastin related hereditary spastic paraplegia with dysplastic corpus callosum. J. Neurol. Sci. 2005, 236, 9–12. [Google Scholar] [CrossRef]
- Munhoz, R.P.; Kawarai, T.; Teive, H.A.; Raskin, S.; Sato, C.; Liang, Y.; St George-Hyslop, P.H.; Rogaeva, E. Clinical and genetic study of a Brazilian family with spastic paraplegia (SPG6 locus). Mov. Disord. 2006, 21, 279–281. [Google Scholar] [CrossRef]
- Matthews, A.M.; Tarailo-Graovac, M.; Price, E.M.; Blydt-Hansen, I.; Ghani, A.; Drögemöller, B.I.; Robinson, W.P.; Ross, C.J.; Wasserman, W.W.; Siden, H.; et al. A de novo mosaic mutation in SPAST with two novel alternative alleles and chromosomal copy number variant in a boy with spastic paraplegia and autism spectrum disorder. Eur. J. Med. Genet. 2017, 60, 548–552. [Google Scholar] [CrossRef]
- Henkhaus, R.S.; Kim, S.J.; Kimonis, V.E.; Gold, J.A.; Dykens, E.M.; Driscoll, D.J.; Butler, M.G. Methylation-specific multiplex ligation-dependent probe amplification and identification of deletion genetic subtypes in Prader-Willi syndrome. Genet. Test Mol. Biomark. 2012, 16, 178–186. [Google Scholar] [CrossRef] [Green Version]
- Liu, C.; Mei, M.; Li, Q.; Roboti, P.; Pang, Q.; Ying, Z.; Gao, F.; Lowe, M.; Bao, S. Loss of the golgin GM130 causes Golgi disruption, Purkinje neuron loss, and ataxia in mice. Proc. Natl. Acad. Sci. USA 2017, 114, 346–351. [Google Scholar] [CrossRef] [Green Version]
- Matsuki, T.; Matthews, R.T.; Cooper, J.A.; van der Brug, M.P.; Cookson, M.R.; Hardy, J.A.; Olson, E.C.; Howell, B.W. Reelin and stk25 have opposing roles in neuronal polarization and dendritic Golgi deployment. Cell 2010, 143, 826–836. [Google Scholar] [CrossRef] [Green Version]
- Futreal, P.A.; Cochran, C.; Rosenthal, J.; Miki, Y.; Swenson, J.; Hobbs, M.; Bennett, L.M.; Haugen-Strano, A.; Marks, J.; Barrett, J.C.; et al. Isolation of a diverged homeobox gene, MOX1, from the BRCA1 region on 17q21 by solution hybrid capture. Hum. Mol. Genet. 1994, 3, 1359–1364. [Google Scholar] [CrossRef]
- Mankoo, B.S.; Skuntz, S.; Harrigan, I.; Grigorieva, E.; Candia, A.; Wright, C.V.; Arnheiter, H.; Pachnis, V. The concerted action of Meox homeobox genes is required upstream of genetic pathways essential for the formation, patterning and differentiation of somites. Development 2003, 130, 4655–4664. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ricard-Blum, S. The collagen family. Cold Spring Harb. Perspect. Biol. 2011, 3, a004978. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fitzgerald, J.; Holden, P.; Hansen, U. The expanded collagen VI family: New chains and new questions. Connect. Tissue Res. 2013, 54, 345–350. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mukhopadhyay, A.; Kramer, J.M.; Merkx, G.; Lugtenberg, D.; Smeets, D.F.; Oortveld, M.A.; Blokland, E.A.; Agrawal, J.; Schenck, A.; van Bokhoven, H.; et al. CDK19 is disrupted in a female patient with bilateral congenital retinal folds, microcephaly and mild mental retardation. Hum. Genet. 2010, 128, 281–291. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wechsler, D.; Zhou, X. WASI-II: Wechsler Abbreviated Scale of Intelligence; The Pychological Corporation: San Antonio, TX, USA, 2011. [Google Scholar]
- Wilkinson, G.S.; Robertson, G.J. WRAT 4: Wide Range Achievement Test; Psychological Assessment Resources: Lutz, FL, USA, 2006. [Google Scholar]
- Cohen, J. Statistical Power Analysis for the Behavioral Sciences, 2nd ed.; L. Erlbaum Associates: Hillsdale, NJ, USA, 1988. [Google Scholar]
- Andrews, S. FastQC: A Quality Control Tool for High Throughput Sequence Data. Available online: http://www.bioinformatics.babraham.ac.uk/projects/fastqc2010 (accessed on 10 June 2020).
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef] [Green Version]
- Van der Auwera, G.A.; Carneiro, M.O.; Hartl, C.; Poplin, R.; Del Angel, G.; Levy-Moonshine, A.; Jordan, T.; Shakir, K.; Roazen, D.; Thibault, J.; et al. From FastQ data to high confidence variant calls: The Genome Analysis Toolkit best practices pipeline. Curr. Protoc. Bioinform. 2013, 43, 11.10.1–11.10.33. [Google Scholar] [CrossRef]
- Cingolani, P.; Platts, A.; Wang, L.L.; Coon, M.; Nguyen, T.; Wang, L.; Land, S.J.; Lu, X.; Ruden, D.M. A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w1118; iso-2; iso-3. Fly 2012, 6, 80–92. [Google Scholar] [CrossRef] [Green Version]
- McLaren, W.; Gil, L.; Hunt, S.E.; Riat, H.S.; Ritchie, G.R.; Thormann, A.; Flicek, P.; Cunningham, F. The Ensembl Variant Effect Predictor. Genome Biol. 2016, 17, 122. [Google Scholar] [CrossRef] [Green Version]
- Ng, P.C.; Henikoff, S. SIFT: Predicting amino acid changes that affect protein function. Nucleic Acids Res. 2003, 31, 3812–3814. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Adzhubei, I.; Jordan, D.M.; Sunyaev, S.R. Predicting functional effect of human missense mutations using PolyPhen-2. Curr. Protoc. Hum. Genet. 2013, 76, 7.20.1–7.20.41. [Google Scholar] [CrossRef] [Green Version]
- Grantham, R. Amino acid difference formula to help explain protein evolution. Science 1974, 185, 862–864. [Google Scholar] [CrossRef] [PubMed]
- Krämer, A.; Green, J.; Pollard, J., Jr.; Tugendreich, S. Causal analysis approaches in Ingenuity Pathway Analysis. Bioinformatics 2014, 30, 523–530. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Corominas, R.; Lin, G.N. De novo Mutations From Whole Exome Sequencing in Neurodevelopmental and Psychiatric Disorders: From Discovery to Application. Front. Genet. 2019, 10, 258. [Google Scholar] [CrossRef] [Green Version]
- Davis, K.W.; Serrano, M.; Loddo, S.; Robinson, C.; Alesi, V.; Dallapiccola, B.; Novelli, A.; Butler, M.G. Parent-of-Origin Effects in 15q11.2 BP1-BP2 Microdeletion (Burnside-Butler) Syndrome. Int. J. Mol. Sci. 2019, 20, 1459. [Google Scholar] [CrossRef] [PubMed] [Green Version]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Family A | Family B | Family C | Family D | Family E | |||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Tests | 52 yr Mother (Subject1) | 15 yr Male (Subject2) | 6 yr Male (Subject3) | 54 yr Mother (Subject4) | 16 yr Female (Subject5) | 33 yr Mother (Subject6) | 10 yr Male (Subject7) | 37 yr Mother (Subject8) | 9 yr Female (Subject9) | 33 yr Father (Subject10) | 6 yr Female (Subject11) |
| WASI-II IQ | |||||||||||
| Perceptual | 104 | 105 | 79 | 142 | 116 | 112 | 89 | 97 | 83 | 92 | NT |
| Verbal | 96 | 93 | 94 | 111 | 125 | 94 | 99 | 101 | 102 | 109 | NT |
| Full Scale | 100 | 99 | 84 | 128 | 124 | 103 | 93 | 99 | 92 | 101 | NT |
| WRAT-IV | |||||||||||
| Reading | 92 | 103 | 75 | 98 | 115 | 95 | 99 | 99 | 112 | 128 | NT |
| Math | 96 | 82 | 88 | 124 | 116 | 97 | 72 | 90 | 100 | 105 | NT |
| Spelling | 96 | 75 | 79 | 125 | 134 | 109 | 85 | 106 | 87 | 102 | NT |
| CVLT-II/CVLT-C | |||||||||||
| Short Delay Free Recall | −1.5 | 0.5 | −1.0 | 1.0 | −0.5 | 0.5 | 0.0 | 1.0 | −0.5 | −0.5 | NT |
| Short Delay Cued Recall | −1.5 | 0.5 | −1.5 | 1.0 | 0.5 | 1.0 | −1.0 | 0.5 | −1.5 | −0.5 | NT |
| Long Delay Free Recall | −1.5 | 0.0 | −2.5 | 1.0 | 0.5 | 1.0 | −1.0 | 0.0 | −0.5 | −1.5 | NT |
| Long Delay Cued Recall | −1.5 | 0.5 | −2.5 | 1.0 | 0.5 | 0.5 | −0.5 | 0.0 | −1.0 | −0.5 | NT |
| Recognition | −2.5 | 0.5 | 0.5 | 0.0 | −0.5 | 0.0 | 1.5 | 0.0 | 1.0 | −3 | NT |
| PPVT-4 | |||||||||||
| Total Score | 99 | 119 | 120 | 107 | 130 | 106 | 107 | 97 | 104 | 104 | NT |
| Trail Making Test | |||||||||||
| Part A | 130 | 129 | 92 | 126 | 99 | 110 | 88 | 111 | 107 | 119 | NT |
| Part B | 126 | 117 | 104 | 75 | 82 | 106 | 71 | 103 | 95 | 104 | NT |
| Family A | Family B | Family C | Family D | Family E | |||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Behavioral Tests | 52 yr Mother (Subject1) | 15 yr Male (Subject2) | 6 yr Male (Subject3) | 54 yr Mother (Subject4) | 16 yr Female (Subject5) | 33 yr Mother (Subject6) | 10 yr Male (Subject7) | 37 yr Mother (Subject8) | 9 yr Female (Subject9) | 33 yr Father (Subject10) | 6 yr Female (Subject11) |
| ADOS-2 | |||||||||||
| RRB | NT | 1 | 3 | NT | 0 | NT | 0 | NT | 4 | NT | 3 * |
| Social Affect | NT | 11 | 3 | NT | 3 | NT | 4 | NT | 5 | NT | 19 * |
| Total (ASD Cutoff = 7) | NT | 12 | 6 | NT | 3 | NT | 4 | NT | 9 | NT | 22 * |
| Classification | NT | Autism | Non-Autism | NT | Non-Autism | NT | Non-Autism | NT | Autism | NT | Autism |
| BAP-Q | |||||||||||
| Total Score (cut-offs: M = 3.55, F = 3.17) | 5.33 | NT | NT | 1.11 | NT | 3.67 | NT | 0.89 | NT | 3.28 | NT |
| VABS-III (Standard Scores) | |||||||||||
| Communication | NT | 54 | 77 | NT | 97 | NT | 70 | NT | 69 | NT | 36 |
| Daily Living | NT | 60 | 82 | NT | 98 | NT | 92 | NT | 105 | NT | 46 |
| Socialization | NT | 40 | 82 | NT | 82 | NT | 80 | NT | 80 | NT | 51 |
| Adaptive Behavior Composite | NT | 55 | 78 | NT | 90 | NT | 59 | NT | 82 | NT | 48 |
| RBS-R | |||||||||||
| Overall Score | 10 | 39 | 9 | 2 | 18 | 9 | 21 | 3 | 15 | 8 | 31 |
| Children | Adults | |||||
|---|---|---|---|---|---|---|
| Postural Control Variables | BBS (n = 6) | Control (n = 6) | BBS (n = 5) | Control (n = 4) | ||
| Mean (SD) | Mean (SD) | Cohen’s d | Mean (SD) | Mean (SD) | Cohen’s d | |
| COP Length (cm) | 35.74 (21.37) | 23.81 (15.36) | −0.641 | 19.45 (11.15) | 11.95 (1.36) | −0.944 |
| ML SD (log) | −0.49 (0.34) | −0.75 (0.40) | −0.702 | −1.02 (0.31) | −1.06 (0.13) | −0.166 |
| AP SD (log) | −0.32 (0.31) | −0.42 (0.15) | −0.383 | −0.41 (0.32) | −0.57 (0.19) | −0.606 |
| Medical History | Family A | Family B | Family C | Family D | Family E | ||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| 52 yr Mother (Subject1) | 15 yr Male (Subject2) | 6 yr Male (Subject3) | 54 yr Mother (Subject4) | 16 yr Female (Subject5) | 33 yr Mother (Subject6) | 10 yr Male (Subject7) | 37 yr Mother (Subject8) | 9 yr Female (Subject9) | 33 yr Father (Subject10) | 6 yr Female (Subject11) | |
| Prenatal | Mother age 46 yr | Pre-eclampsia | Gestational diabetes | ||||||||
| Birth | C-section | C-section | C-section | C-section | |||||||
| Birth weight | 3.4 kg (30th%) | 3.6 kg (50th%) | 3.7 kg (55th%) | 3.8 kg (75th%) | |||||||
| Neuro-develop-mental | Learning difficulties | Autism spectrum disorder | Global develop-mental delay | Dyslexia, regression | Learning difficulties | Regression at 12mo | |||||
| Neuro- psychiatric | Depression | ADHD, OCD, anxiety | ADHD, GAD, OCD, social phobia, selective mutism | ADHD, OCD, ODD | Sleep disturbance | ||||||
| Neurological | Non-essential tremor | Right calf paresthesia | Epilepsy | Hypotonia, fine and gross motor delay | Epilepsy (Lennox-Gastaut syndrome) | ||||||
| Eye | Astigmatism | Left strabismus | No retinal folds | ||||||||
| Musculo- skeletal | Leg braces, ankle instability | Herniated disc lumbar 5 | Kyphosis, hyper-flexible | Scoliosis | Scoliosis | ||||||
| Motor | Weak postural control, walked at 16 mo | Fine and gross motor delay, walked at 25 mo | Walked at 12 months | Crawled at 20 months, walked at 26 months | Sat at 13mo, crawled at 18mo, poor balance and coordination | ||||||
| Cardio- vascular | Hyper-tension Hyper-lipidemia | Patent foramen ovale, Postural hypotension | Hyper-tension | ||||||||
| Respiratory | Asthma | ||||||||||
| Gastro- intestinal | Consti-pation | Consti-pation | |||||||||
| Skin | Easy bruising, delayed healing | Eczema, delayed healing | |||||||||
| Endocrine | Diabetes mellitus | Post-partum thyroiditis | Delayed bone age, delayed growth | Gestational diabetes | |||||||
| Reproductive | PCOS | ||||||||||
| Immunologic | Thymic hyper-trophy | Anaphy-laxis | |||||||||
| Physical Exam | Family A | Family B | Family C | Family D | Family E | ||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| 52 yr Mother (Subject1) | 15 yr Male (Subject2) | 6 yr Male (Subject3) | 54 yr Mother (Subject4) | 16 yr Female (Subject5) | 33 yr Mother (Subject6) | 10 yr Male (Subject7) | 37 yr Mother (Subject8) | 9 yr Female (Subject9) | 33 yr Father (Subject10) | 6 yr Female (Subject11) | |
| Head circumference | 57.3 cm (75th%) | 56.6 cm (75th%) | 51.5 cm (50th%) | 56.8 cm (90th%) | 53.3 cm (25th%) | 56.2 cm (50th%) | 54.2 cm (85th%) | 55.5 cm (50th%) | 55.7 cm (>97th%) | 57.8 cm (75th%) | 49.5 cm (25th%) |
| Inner canthal distance | 3.3 cm (75th%) | 2.9 cm (50th%) | 2.7 cm (50th%) | 3.2 cm (75th%) | 3.2 cm (75th%) | 2.5 cm (3rd%) | 3.2 cm (75th%) | 3.1 cm (50th%) | 3.0 cm (50th%) | ||
| Outer canthal distance | 8.3 cm (25th%) | 8.6 cm (50th%) | 7.3 cm (3rd%) | 8.4 cm (25th%) | 8.3 cm (25th%) | 8.0 cm (3rd%) | 8.4 cm (50th%) | 9.2 cm (75th%) | 8.7 cm (75th%) | ||
| Right hand length | 19.2 cm (97th%) | 20.2 cm (97th%) | 15.6 cm (97th%) | 16.3 cm (3rd%) | 17.0 cm (25th%) | 18.2 cm (75th%) | 15.6 cm (25th%) | 16.6 cm (25th%) | 15.8 cm (75th%) | 19.0 cm (75th%) | 13.3 cm (50th%) |
| Right middle finger length | 8.3 cm (75th%) | 8.8 cm (97th%) | 6.7 cm (97th%) | 7.2 cm (3rd%) | 7.6 cm (25th%) | 7.6 cm (25th%) | 6.9 cm (50th%) | 7.2 cm (3rd%) | 6.8 cm (75th%) | 8.1 cm (75th%) | 5.2 cm (25th%) |
| Right ear length | 7.4 cm (75th%) | 6.7 cm (75th%) | 6.2 cm (75th%) | 6.8 cm (75th%) | 6.1 cm (50th%) | 6.8 cm (75th%) | 6.4 cm (75th%) | 6.2 cm (50th%) | 5.7 cm (25th%) | NA | 5.7 cm (50th%) |
| Weight | 110 kg (>95th%) | 67.1 kg (80th%) | 26.3 kg (90th%) | 67.1 kg (75th%) | 45.8 kg (10th%) | 89.1 kg (>95th%) | 32.5 kg (50th%) | 79.3 kg (90th%) | 35.5 kg (80th%) | 123.2 kg (>95th%) | 27.6 kg (>95th%) |
| Height | 176 cm (>95th%) | 182 cm (95th%) | 137 cm (>95th%) | 158 cm (20th%) | 158 cm (20th%) | 158.5 cm (20th%) | 139 cm (50th%) | 159 cm (25th%) | 142 cm (90th%) | 178 cm (50th%) | NA |
| BMI | 35.5 (>95th%) | 24.9 (90th%) | 14.0 (5th%) | 26.9 (85th%) | 18.3 (20th%) | 35.5 (>95th%) | 16.8 (50th%) | 31.4 (95th%) | 17.6 (70th%) | 38.9 (>95th%) | NA |
| Head/Facial features | Soft ears, fold easily | Broad, soft ears | Small upper incisor (#10) | Small upper incisor (#10) | Flat occiput, fleshy ears, thin upper lip, and flat philtrum | Right ear overfolded | Broad, round face | Broad, round face, broad nose, full lips, prominent jaw, forehead, and ears, depressed nasal bridge | |||
| Eyes | Pigment changes, Sensitive to light | Pigment changes, Sensitive to light | Left pupil larger than right | Mild left ptosis | Mild bilateral ptosis (L > R) | Myopia, right sided ptosis | |||||
| Back | No scoliosis | No scoliosis | Right shoulder higher than left, no scoliosis | Mild scoliosis | Kyphosis, right shoulder droop | Mild scoliosis | |||||
| Upper extremities | Hyper-extensibility: Beighton score 6/9 | Hyper-extensibility: Beighton score 7/9, cubitus valgus | Broad hands | Soft fleshy, broad hands | |||||||
| Lower extremities | Second toes overlap third, plantar creases, flat feet, ankle instability on pronation | Left leg longer than right, knee-buttock asymmetry | Knee-buttock asymmetry, flat feet | Left leg longer than right | Flat feet | Flat feet | |||||
| Other musculo- skeletal | Pectus carinatum | Large body size | |||||||||
| Skin | Birth mark on thigh | Soft, fleshy | Loose, soft, velvety | Eczema | Soft with freckles | Soft | Birth marks on thigh and forehead | ||||
| Neurological | Increased deep tendon reflexes | Decreased sensation right lateral calf | Decreased deep tendon reflexes | Decreased muscle tone and reflexes in all extremities | Toe walking, poor coordination and balance | ||||||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Baldwin, I.; Shafer, R.L.; Hossain, W.A.; Gunewardena, S.; Veatch, O.J.; Mosconi, M.W.; Butler, M.G. Genomic, Clinical, and Behavioral Characterization of 15q11.2 BP1-BP2 Deletion (Burnside-Butler) Syndrome in Five Families. Int. J. Mol. Sci. 2021, 22, 1660. https://doi.org/10.3390/ijms22041660
Baldwin I, Shafer RL, Hossain WA, Gunewardena S, Veatch OJ, Mosconi MW, Butler MG. Genomic, Clinical, and Behavioral Characterization of 15q11.2 BP1-BP2 Deletion (Burnside-Butler) Syndrome in Five Families. International Journal of Molecular Sciences. 2021; 22(4):1660. https://doi.org/10.3390/ijms22041660
Chicago/Turabian StyleBaldwin, Isaac, Robin L. Shafer, Waheeda A. Hossain, Sumedha Gunewardena, Olivia J. Veatch, Matthew W. Mosconi, and Merlin G. Butler. 2021. "Genomic, Clinical, and Behavioral Characterization of 15q11.2 BP1-BP2 Deletion (Burnside-Butler) Syndrome in Five Families" International Journal of Molecular Sciences 22, no. 4: 1660. https://doi.org/10.3390/ijms22041660