
From Prions to Stress Granules: Defining the Compositional Features of Prion-Like Domains That Promote Different Types of Assemblies


{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Yeast Prion Sequence and Structure
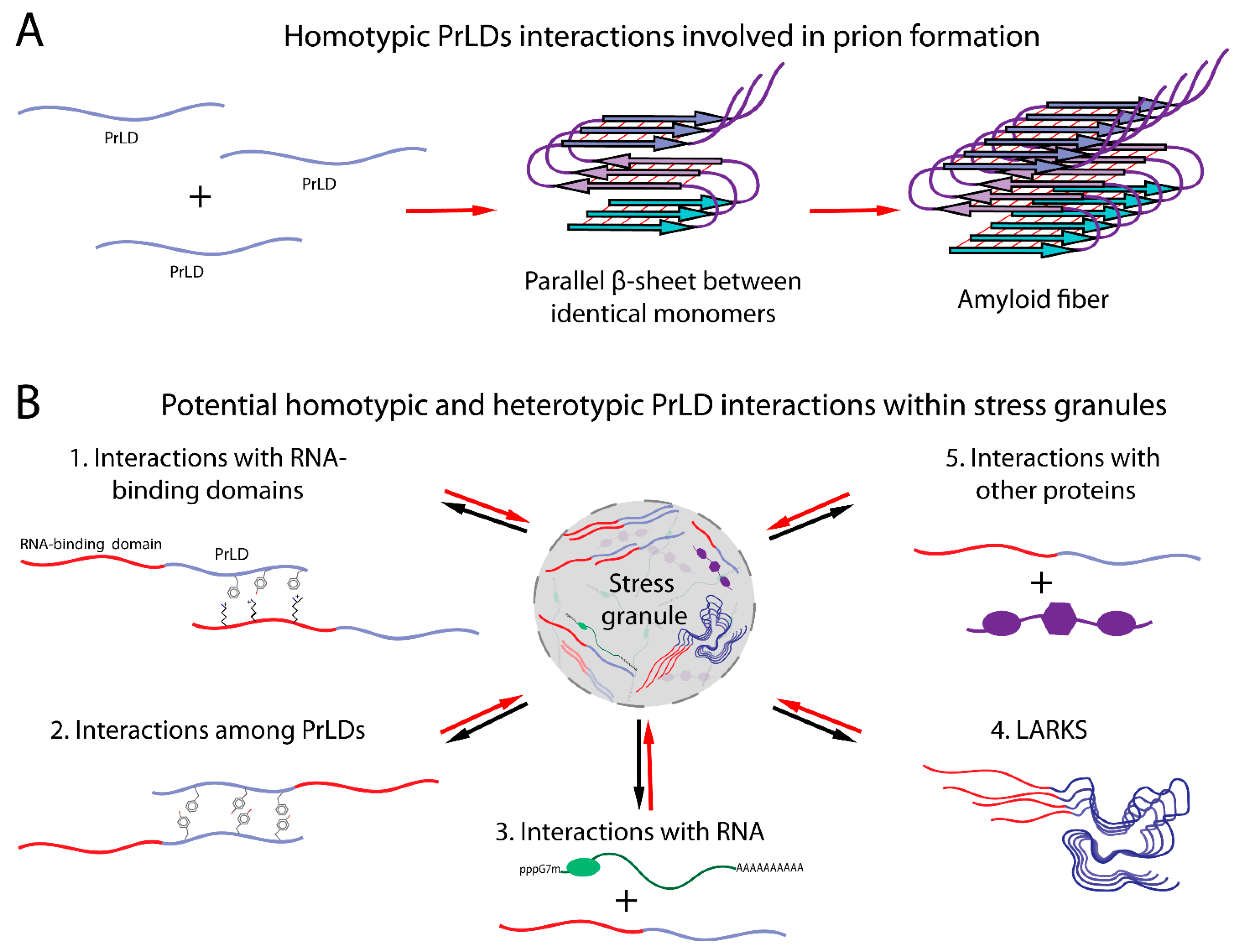
3. Cellular Interactions Influence the Sequence Requirements for Prions
4. Stress Granules
5. Possible Mechanisms of PrLD Assembly in Stress Granules
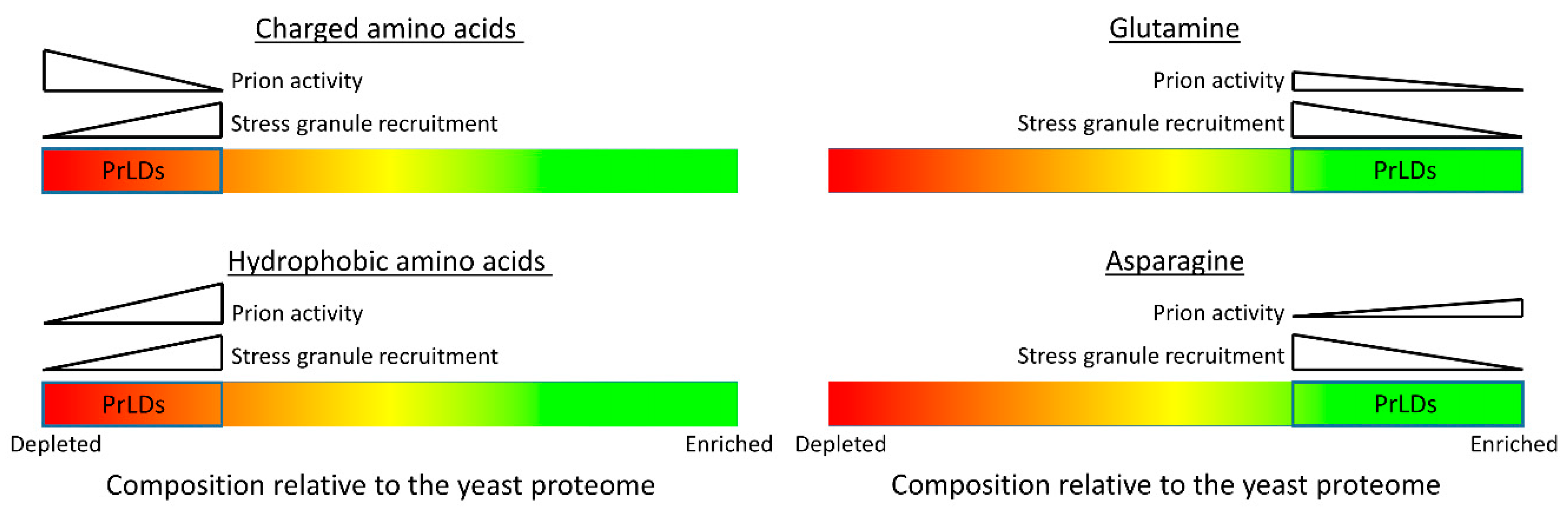
6. Sequence and Compositional Features Promoting PrLD Recruitment to Stress Granules
7. Distinct Classes of Prion-Like Domains
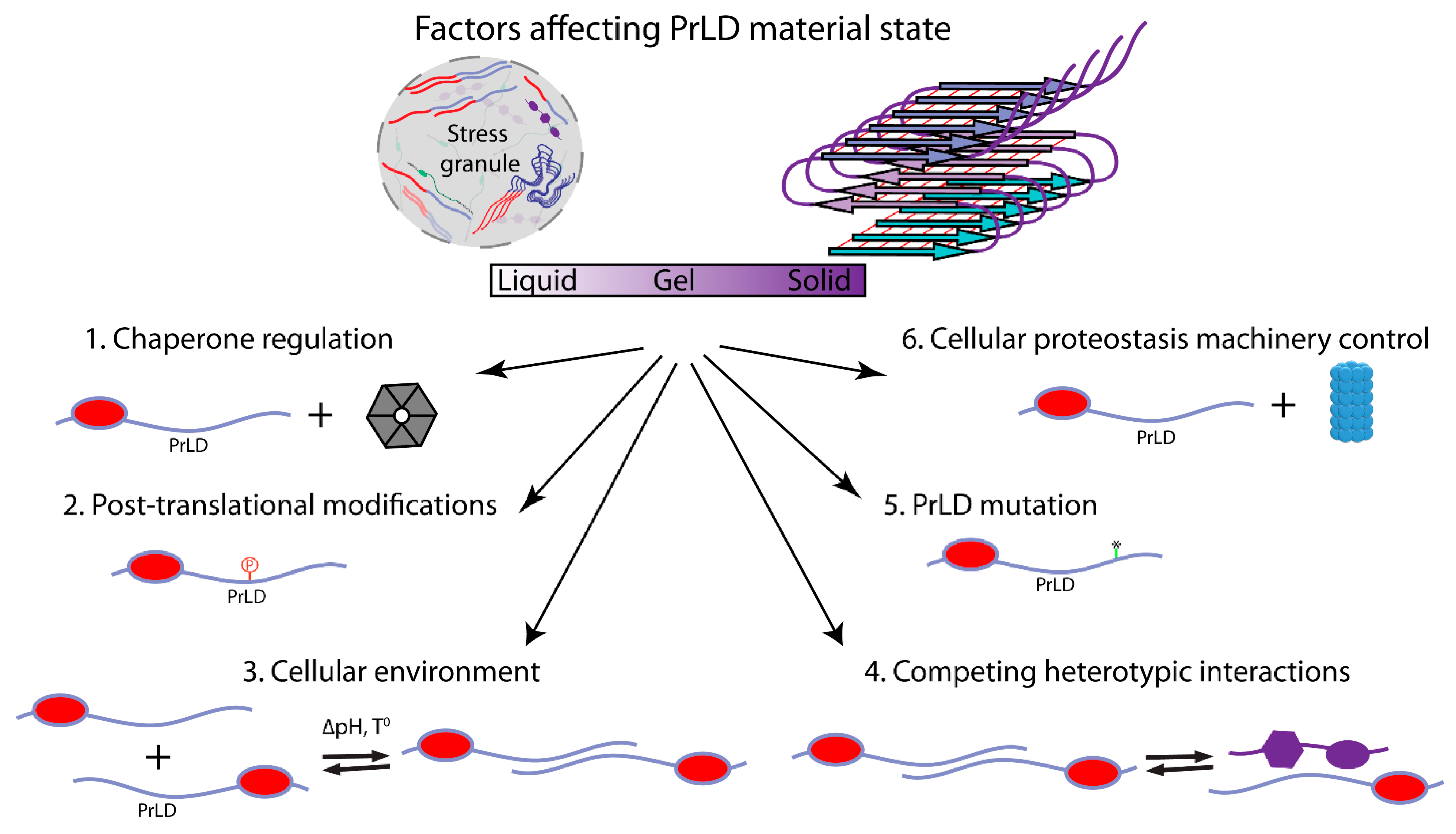
8. Stress Granule Regulation and Dysregulation
9. Conclusions and Future Perspectives
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Tycko, R.; Wickner, R.B. Molecular structures of amyloid and prion fibrils: Consensus versus controversy. Acc. Chem. Res. 2013, 46, 1487–1496. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prusiner, S.B. Novel proteinaceous infectious particles cause scrapie. Science 1982, 216, 136–144. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wickner, R.B. [URE3] as an altered URE2 protein: Evidence for a prion analog in Saccharomyces cerevisiae. Science 1994, 264, 566–569. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Coustou, V.; Deleu, C.; Saupe, S.; Begueret, J. The protein product of the het-s heterokaryon incompatibility gene of the fungus Podospora anserina behaves as a prion analog. Proc. Natl. Acad. Sci. USA 1997, 94, 9773–9778. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chakrabortee, S.; Kayatekin, C.; Newby, G.A.; Mendillo, M.L.; Lancaster, A.; Lindquist, S. Luminidependens (LD) is an Arabidopsis protein with prion behavior. Proc. Natl. Acad. Sci. USA 2016, 113, 6065–6070. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yuan, A.H.; Hochschild, A. A bacterial global regulator forms a prion. Science 2017, 355, 198–201. [Google Scholar] [CrossRef] [Green Version]
- Saupe, S.J.; Jarosz, D.F.; True, H.L. Amyloid prions in fungi. Microbiol. Spectr. 2017, 4. [Google Scholar] [CrossRef] [Green Version]
- Wickner, R.B.; Edskes, H.K.; Son, M.; Bezsonov, E.E.; DeWilde, M.; Ducatez, M. Yeast prions compared to functional prions and amyloids. J. Mol. Biol. 2018, 430, 3707–3719. [Google Scholar] [CrossRef]
- Liebman, S.W.; Chernoff, Y.O. Prions in yeast. Genetics 2012, 191, 1041. [Google Scholar] [CrossRef] [Green Version]
- Alberti, S.; Halfmann, R.; King, O.; Kapila, A.; Lindquist, S. A systematic survey identifies prions and illuminates sequence features of prionogenic proteins. Cell 2009, 137, 146–158. [Google Scholar] [CrossRef] [Green Version]
- Zambrano, R.; Conchillo-Sole, O.; Iglesias, V.; Illa, R.; Rousseau, F.; Schymkowitz, J.; Sabate, R.; Daura, X.; Ventura, S. PrionW: A server to identify proteins containing glutamine/asparagine rich prion-like domains and their amyloid cores. Nucleic Acids Res. 2015, 43, W331–W337. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Minhas, F.U.A.A.; Ross, E.D.; Ben-Hur, A. Amino acid composition predicts prion activity. PLoS Comput. Biol. 2017, 13, e1005465. [Google Scholar] [CrossRef] [Green Version]
- Harrison, P.M.; Gerstein, M. A method to assess compositional bias in biological sequences and its application to prion-like glutamine/asparagine-rich domains in eukaryotic proteomes. Genome Biol. 2003, 4, R40. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lancaster, A.K.; Nutter-Upham, A.; Lindquist, S.; King, O.D. PLAAC: A web and command-line application to identify proteins with prion-like amino acid composition. Bioinformatics 2014, 30, 2501–2502. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Espinosa Angarica, V.; Ventura, S.; Sancho, J. Discovering putative prion sequences in complete proteomes using probabilistic representations of Q/N-rich domains. BMC Genom. 2013, 14, 316. [Google Scholar] [CrossRef] [Green Version]
- March, Z.M.; King, O.D.; Shorter, J. Prion-like domains as epigenetic regulators, scaffolds for subcellular organization, and drivers of neurodegenerative disease. Brain Res. 2016, 1647, 9–18. [Google Scholar] [CrossRef] [Green Version]
- Si, K.; Lindquist, S.; Kandel, E.R. A neuronal isoform of the Aplysia CPEB has prion-like properties. Cell 2003, 115, 879–891. [Google Scholar] [CrossRef] [Green Version]
- Si, K.; Kandel, E.R. The role of functional prion-like proteins in the persistence of memory. Cold Spring Harb. Perspect. Biol. 2016, 8, a021774. [Google Scholar] [CrossRef] [Green Version]
- Caudron, F.; Barral, Y. Mnemons: Encoding memory by protein super-assembly. Microb. Cell 2014, 1, 100–102. [Google Scholar] [CrossRef]
- Caudron, F.; Barral, Y. A super-assembly of Whi3 encodes memory of deceptive encounters by single cells during yeast courtship. Cell 2013, 155, 1244–1257. [Google Scholar] [CrossRef] [Green Version]
- Banani, S.F.; Lee, H.O.; Hyman, A.A.; Rosen, M.K. Biomolecular condensates: Organizers of cellular biochemistry. Nat. Rev. Mol. Cell Biol. 2017, 18, 285–298. [Google Scholar] [CrossRef] [PubMed]
- Gilks, N.; Kedersha, N.; Ayodele, M.; Shen, L.; Stoecklin, G.; Dember, L.M.; Anderson, P. Stress granule assembly is mediated by prion-like aggregation of TIA-1. Mol. Biol. Cell 2004, 15, 5383–5398. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ivanov, P.; Kedersha, N.; Anderson, P. Stress granules and processing bodies in translational control. Cold Spring Harb. Perspect. Biol. 2019, 11, a032813. [Google Scholar] [CrossRef]
- Buchan, J.R.; Parker, R. Eukaryotic stress granules: The ins and outs of translation. Mol. Cell 2009, 36, 932–941. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- King, O.D.; Gitler, A.D.; Shorter, J. The tip of the iceberg: RNA-binding proteins with prion-like domains in neurodegenerative disease. Brain Res. 2012, 1462, 61–80. [Google Scholar] [CrossRef] [Green Version]
- Shattuck, J.E.; Paul, K.R.; Cascarina, S.M.; Ross, E.D. The prion-like protein kinase Sky1 is required for efficient stress granule disassembly. Nat. Commun. 2019, 10, 3614. [Google Scholar] [CrossRef] [Green Version]
- Kroschwald, S.; Munder, M.C.; Maharana, S.; Franzmann, T.M.; Richter, D.; Ruer, M.; Hyman, A.A.; Alberti, S. Different material states of Pub1 condensates define distinct modes of stress adaptation and recovery. Cell Rep. 2018, 23, 3327–3339. [Google Scholar] [CrossRef]
- Iserman, C.; Desroches Altamirano, C.; Jegers, C.; Friedrich, U.; Zarin, T.; Fritsch, A.W.; Mittasch, M.; Domingues, A.; Hersemann, L.; Jahnel, M.; et al. Condensation of Ded1p promotes a translational switch from housekeeping to stress protein production. Cell 2020, 181, 818.e19–831.e19. [Google Scholar] [CrossRef]
- Vance, C.; Rogelj, B.; Hortobágyi, T.; De Vos, K.J.; Nishimura, A.L.; Sreedharan, J.; Hu, X.; Smith, B.; Ruddy, D.; Wright, P.; et al. Mutations in FUS, an RNA processing protein, cause familial amyotrophic lateral sclerosis type 6. Science 2009, 323, 1208–1211. [Google Scholar] [CrossRef] [Green Version]
- Kwiatkowski, T.J.; Bosco, D.A.; LeClerc, A.L.; Tamrazian, E.; Vanderburg, C.R.; Russ, C.; Davis, A.; Gilchrist, J.; Kasarskis, E.J.; Munsat, T.; et al. Mutations in the FUS/TLS gene on chromosome 16 cause familial amyotrophic lateral sclerosis. Science 2009, 323, 1205–1208. [Google Scholar] [CrossRef] [Green Version]
- Sreedharan, J.; Blair, I.P.; Tripathi, V.B.; Hu, X.; Vance, C.; Rogelj, B.; Ackerley, S.; Durnall, J.C.; Williams, K.L.; Buratti, E.; et al. TDP-43 mutations in familial and sporadic amyotrophic lateral sclerosis. Science 2008, 319, 1668–1672. [Google Scholar] [CrossRef] [PubMed]
- Guo, W.; Chen, Y.; Zhou, X.; Kar, A.; Ray, P.; Chen, X.; Rao, E.J.; Yang, M.; Ye, H.; Zhu, L.; et al. An ALS-associated mutation affecting TDP-43 enhances protein aggregation, fibril formation and neurotoxicity. Nat. Struct. Mol. Biol. 2011, 18, 822–830. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, H.J.; Kim, N.C.; Wang, Y.D.; Scarborough, E.A.; Moore, J.; Diaz, Z.; MacLea, K.S.; Freibaum, B.; Li, S.; Molliex, A.; et al. Mutations in prion-like domains in hnRNPA2B1 and hnRNPA1 cause multisystem proteinopathy and ALS. Nature 2013, 495, 467–473. [Google Scholar] [CrossRef] [PubMed]
- Ramaswami, M.; Taylor, J.P.; Parker, R. Altered ribostasis: RNA-protein granules in degenerative disorders. Cell 2013, 154, 727–736. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Masison, D.C.; Wickner, R.B. Prion-inducing domain of yeast Ure2p and protease resistance of Ure2p in prion-containing cells. Science 1995, 270, 93–95. [Google Scholar] [CrossRef] [Green Version]
- Ter-Avanesyan, M.D.; Kushnirov, V.V.; Dagkesamanskaya, A.R.; Didichenko, S.A.; Chernoff, Y.O.; Inge-Vechtomov, S.G.; Smirnov, V.N. Deletion analysis of the SUP35 gene of the yeast Saccharomyces cerevisiae reveals two non-overlapping functional regions in the encoded protein. Mol. Microbiol. 1993, 7, 683–692. [Google Scholar] [CrossRef]
- Michelitsch, M.D.; Weissman, J.S. A census of glutamine/asparagine-rich regions: Implications for their conserved function and the prediction of novel prions. Proc. Natl. Acad. Sci. USA 2000, 97, 11910–11915. [Google Scholar] [CrossRef] [Green Version]
- Chernova, T.A.; Kiktev, D.A.; Romanyuk, A.V.; Shanks, J.R.; Laur, O.; Ali, M.; Ghosh, A.; Kim, D.; Yang, Z.; Mang, M.; et al. Yeast short-lived actin-associated protein forms a metastable prion in response to thermal stress. Cell Rep. 2017, 18, 751–761. [Google Scholar] [CrossRef]
- Derkatch, I.L.; Bradley, M.E.; Hong, J.Y.; Liebman, S.W. Prions affect the appearance of other prions: The story of [PIN+]. Cell 2001, 106, 171–182. [Google Scholar] [CrossRef] [Green Version]
- Du, Z.; Park, K.W.; Yu, H.; Fan, Q.; Li, L. Newly identified prion linked to the chromatin-remodeling factor Swi1 in Saccharomyces cerevisiae. Nat. Genet. 2008, 40, 460–465. [Google Scholar] [CrossRef] [Green Version]
- Halfmann, R.; Wright, J.R.; Alberti, S.; Lindquist, S.; Rexach, M. Prion formation by a yeast GLFG nucleoporin. Prion 2012, 6, 391–399. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Patel, B.K.; Gavin-Smyth, J.; Liebman, S.W. The yeast global transcriptional co-repressor protein Cyc8 can propagate as a prion. Nat. Cell Biol. 2009, 11, 344–349. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sondheimer, N.; Lindquist, S. Rnq1: An epigenetic modifier of protein function in yeast. Mol. Cell 2000, 5, 163–172. [Google Scholar] [CrossRef]
- Suzuki, G.; Shimazu, N.; Tanaka, M. A yeast prion, Mod5, promotes acquired drug resistance and cell survival under environmental stress. Science 2012, 336, 355–359. [Google Scholar] [CrossRef]
- Chakravarty, A.K.; Smejkal, T.; Itakura, A.K.; Garcia, D.M.; Jarosz, D.F. A Non-amyloid prion particle that activates a heritable gene expression program. Mol. Cell 2020, 77, 251.E9–265.E9. [Google Scholar] [CrossRef]
- Roberts, B.T.; Wickner, R.B. A new kind of prion: A modified protein necessary for its own modification. Cell Cycle 2004, 3, 100–103. [Google Scholar] [CrossRef] [Green Version]
- Brown, J.C.S.; Lindquist, S. A heritable switch in carbon source utilization driven by an unusual yeast prion. Genes Dev. 2009, 23, 2320–2332. [Google Scholar] [CrossRef] [Green Version]
- Chakrabortee, S.; Byers, J.S.; Jones, S.; Garcia, D.M.; Bhullar, B.; Chang, A.; She, R.; Lee, L.; Fremin, B.; Lindquist, S.; et al. Intrinsically disordered proteins drive emergence and inheritance of biological traits. Cell 2016, 167, 369.e12–381.e12. [Google Scholar] [CrossRef] [Green Version]
- Glover, J.R.; Kowal, A.S.; Schirmer, E.C.; Patino, M.M.; Liu, J.J.; Lindquist, S. Self-seeded fibers formed by Sup35, the protein determinant of [PSI+], a heritable prion-like factor of S. cerevisiae. Cell 1997, 89, 811–819. [Google Scholar] [CrossRef] [Green Version]
- Taylor, K.L.; Cheng, N.; Williams, R.W.; Steven, A.C.; Wickner, R.B. Prion domain initiation of amyloid formation in vitro from native Ure2p. Science 1999, 283, 1339–1343. [Google Scholar] [CrossRef]
- Ross, E.D.; Edskes, H.K.; Terry, M.J.; Wickner, R.B. Primary sequence independence for prion formation. Proc. Natl. Acad. Sci. USA 2005, 102, 12825–12830. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ross, E.D.; Baxa, U.; Wickner, R.B. Scrambled prion domains form prions and amyloid. Mol. Cell. Biol. 2004, 24, 7206–7213. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ross, E.D.; Minton, A.; Wickner, R.B. Prion domains: Sequences, structures and interactions. Nat. Cell Biol. 2005, 7, 1039–1044. [Google Scholar] [CrossRef] [PubMed]
- Kryndushkin, D.S.; Wickner, R.B.; Tycko, R. The core of Ure2p prion fibrils is formed by the N-terminal segment in a parallel cross-β structure: Evidence from solid-state NMR. J. Mol. Biol. 2011, 409, 263–277. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shewmaker, F.; Kryndushkin, D.; Chen, B.; Tycko, R.; Wickner, R.B. Two prion variants of Sup35p have in-register parallel β-sheet structures, independent of hydration. Biochemistry 2009, 48, 5074–5082. [Google Scholar] [CrossRef] [Green Version]
- DePace, A.H.; Santoso, A.; Hillner, P.; Weissman, J.S. A critical role for amino-terminal glutamine/asparagine repeats in the formation and propagation of a yeast prion. Cell 1998, 93, 1241–1252. [Google Scholar] [CrossRef] [Green Version]
- Danilov, L.G.; Matveenko, A.G.; Ryzhkova, V.E.; Belousov, M.V.; Poleshchuk, O.I.; Likholetova, D.V.; Sokolov, P.A.; Kasyanenko, N.A.; Kajava, A.V.; Zhouravleva, G.A.; et al. Design of a new [PSI+]-no-more mutation in SUP35 with strong inhibitory effect on the [PSI+] prion propagation. Front. Mol. Neurosci. 2019, 12, 274. [Google Scholar] [CrossRef] [Green Version]
- Toombs, J.A.; McCarty, B.R.; Ross, E.D. Compositional determinants of prion formation in yeast. Mol. Cell. Biol. 2010, 30, 319–332. [Google Scholar] [CrossRef] [Green Version]
- Gonzalez Nelson, A.C.; Paul, K.R.; Petri, M.; Flores, N.; Rogge, R.A.; Cascarina, S.M.; Ross, E.D. Increasing prion propensity by hydrophobic insertion. PLoS ONE 2014, 9, e89286. [Google Scholar] [CrossRef] [Green Version]
- Chiti, F.; Stefani, M.; Taddei, N.; Ramponi, G.; Dobson, C.M. Rationalization of the effects of mutations on peptide and protein aggregation rates. Nature 2003, 424, 805–808. [Google Scholar] [CrossRef]
- Kajava, A.V.; Baxa, U.; Wickner, R.B.; Steven, A.C. A model for Ure2p prion filaments and other amyloids: The parallel superpleated β-structure. Proc. Natl. Acad. Sci. USA 2004, 101, 7885–7890. [Google Scholar] [CrossRef] [Green Version]
- Hervas, R.; Rau, M.J.; Park, Y.; Zhang, W.; Murzin, A.G.; Fitzpatrick, J.A.J.; Scheres, S.H.W.; Si, K. Cryo-EM structure of a neuronal functional amyloid implicated in memory persistence in Drosophila. Science 2020, 367, 1230–1234. [Google Scholar] [CrossRef] [PubMed]
- Tuttle, M.D.; Comellas, G.; Nieuwkoop, A.J.; Covell, D.J.; Berthold, D.A.; Kloepper, K.D.; Courtney, J.M.; Kim, J.K.; Barclay, A.M.; Kendall, A.; et al. Solid-state NMR structure of a pathogenic fibril of full-length human α-synuclein. Nat. Struct. Mol. Biol. 2016, 23, 409–415. [Google Scholar] [CrossRef] [PubMed]
- Nelson, R.; Sawaya, M.R.; Balbirnie, M.; Madsen, A.; Riekel, C.; Grothe, R.; Eisenberg, D. Structure of the cross-β spine of amyloid-like fibrils. Nature 2005, 435, 773–778. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Batlle, C.; Fernández, M.R.; Iglesias, V.; Ventura, S. Perfecting prediction of mutational impact on the aggregation propensity of the ALS-associated hnRNPA2 prion-like protein. FEBS Lett. 2017, 591, 1966–1971. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bondarev, S.A.; Bondareva, O.V.; Zhouravleva, G.A.; Kajava, A.V. BetaSerpentine: A bioinformatics tool for reconstruction of amyloid structures. Bioinformatics 2018, 34, 599–608. [Google Scholar] [CrossRef] [Green Version]
- Goldschmidt, L.; Teng, P.K.; Riek, R.; Eisenberg, D. Identifying the amylome, proteins capable of forming amyloid-like fibrils. Proc. Natl. Acad. Sci. USA 2010, 107, 3487–3492. [Google Scholar] [CrossRef] [Green Version]
- Paul, K.R.; Molliex, A.; Cascarina, S.; Boncella, A.E.; Taylor, J.P.; Ross, E.D. Effects of mutations on the aggregation propensity of the human prion-like protein hnRNPA2B1. Mol. Cell. Biol. 2017, 37, e00652-16. [Google Scholar] [CrossRef] [Green Version]
- Scheibel, T.; Lindquist, S.L. The role of conformational flexibility in prion propagation and maintenance for Sup35p. Nat. Struct. Biol. 2001, 8, 958–962. [Google Scholar] [CrossRef]
- Rubinstein, M.; Dobrynin, A. Solutions of associative polymers. Trends Polym. Sci. (Regul. Ed.) 1997, 5, 181–186. [Google Scholar]
- Wang, J.; Choi, J.M.; Holehouse, A.S.; Lee, H.O.; Zhang, X.; Jahnel, M.; Maharana, S.; Lemaitre, R.; Pozniakovsky, A.; Drechsel, D.; et al. A molecular grammar governing the driving forces for phase separation of prion-like RNA binding proteins. Cell 2018, 174, 688.e16–699.e16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Halfmann, R.; Alberti, S.; Krishnan, R.; Lyle, N.; O’Donnell, C.W.; King, O.D.; Berger, B.; Pappu, R.V.; Lindquist, S. Opposing effects of glutamine and asparagine govern prion formation by intrinsically disordered proteins. Mol. Cell 2011, 43, 72–84. [Google Scholar] [CrossRef] [PubMed]
- Boncella, A.E.; Shattuck, J.E.; Cascarina, S.M.; Paul, K.R.; Baer, M.H.; Fomicheva, A.; Lamb, A.K.; Ross, E.D. Composition-based prediction and rational manipulation of prion-like domain recruitment to stress granules. Proc. Natl. Acad. Sci. USA 2020, 117, 5826–5835. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wickner, R.B. Anti-prion systems in yeast. J. Biol. Chem. 2019, 294, 1729–1738. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chernova, T.A.; Wilkinson, K.D.; Chernoff, Y.O. Prions, chaperones, and proteostasis in yeast. Cold Spring Harb. Perspect. Biol. 2017, 9, a023663. [Google Scholar] [CrossRef]
- Ness, F.; Ferreira, P.; Cox, B.S.; Tuite, M.F. Guanidine hydrochloride inhibits the generation of prion “seeds” but not prion protein aggregation in yeast. Mol. Cell. Biol. 2002, 22, 5593–5605. [Google Scholar] [CrossRef] [Green Version]
- Wegrzyn, R.D.; Bapat, K.; Newnam, G.P.; Zink, A.D.; Chernoff, Y.O. Mechanism of prion loss after Hsp104 inactivation in yeast. Mol. Cell. Biol. 2001, 21, 4656–4669. [Google Scholar] [CrossRef] [Green Version]
- Paushkin, S.V.; Kushnirov, V.V.; Smirnov, V.N.; Ter-Avanesyan, M.D. Propagation of the yeast prion-like [psi+] determinant is mediated by oligomerization of the SUP35-encoded polypeptide chain release factor. EMBO J. 1996, 15, 3127–3134. [Google Scholar] [CrossRef]
- Kushnirov, V.V.; Ter-Avanesyan, M.D. Structure and replication of yeast prions. Cell 1998, 94, 13–16. [Google Scholar] [CrossRef] [Green Version]
- Kryndushkin, D.S.; Alexandrov, I.M.; Ter-Avanesyan, M.D.; Kushnirov, V.V. Yeast [PSI+] prion aggregates are formed by small Sup35 polymers fragmented by Hsp104. J. Biol. Chem. 2003, 278, 49636–49643. [Google Scholar] [CrossRef] [Green Version]
- Osherovich, L.Z.; Cox, B.S.; Tuite, M.F.; Weissman, J.S. Dissection and design of yeast prions. PLoS Biol. 2004, 2, E86. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- MacLea, K.S.; Paul, K.R.; Ben-Musa, Z.; Waechter, A.; Shattuck, J.E.; Gruca, M.; Ross, E.D. Distinct amino acid compositional requirements for formation and maintenance of the [PSI+] prion in yeast. Mol. Cell. Biol. 2015, 35, 899–911. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alexandrov, A.I.; Polyanskaya, A.B.; Serpionov, G.V.; Ter-Avanesyan, M.D.; Kushnirov, V.V. The effects of amino acid composition of glutamine-rich domains on amyloid formation and fragmentation. PLoS ONE 2012, 7, e46458. [Google Scholar] [CrossRef] [PubMed]
- Helsen, C.W.; Glover, J.R. Insight into molecular basis of curing of [PSI +] prion by overexpression of 104-kDa heat shock protein (Hsp104). J. Biol. Chem. 2012, 287, 542–556. [Google Scholar] [CrossRef] [Green Version]
- Shewmaker, F.; Wickner, R.B.; Tycko, R. Amyloid of the prion domain of Sup35p has an in-register parallel β-sheet structure. Proc. Natl. Acad. Sci. USA 2006, 103, 19754–19759. [Google Scholar] [CrossRef] [Green Version]
- Ohhashi, Y.; Yamaguchi, Y.; Kurahashi, H.; Kamatari, Y.O.; Sugiyama, S.; Uluca, B.; Piechatzek, T.; Komi, Y.; Shida, T.; Müller, H.; et al. Molecular basis for diversification of yeast prion strain conformation. Proc. Natl. Acad. Sci. USA 2018, 115, 2389–2394. [Google Scholar] [CrossRef] [Green Version]
- Dergalev, A.A.; Alexandrov, A.I.; Ivannikov, R.I.; Ter-Avanesyan, M.D.; Kushnirov, V.V. Yeast sup35 Prion structure: Two types, four parts, many variants. Int. J. Mol. Sci. 2019, 20, 2633. [Google Scholar] [CrossRef] [Green Version]
- Balchin, D.; Hayer-Hartl, M.; Hartl, F.U. In vivo aspects of protein folding and quality control. Science 2016, 353, aac4354. [Google Scholar] [CrossRef]
- Samant, R.S.; Livingston, C.M.; Sontag, E.M.; Frydman, J. Distinct proteostasis circuits cooperate in nuclear and cytoplasmic protein quality control. Nature 2018, 563, 407–411. [Google Scholar] [CrossRef]
- Sontag, E.M.; Samant, R.S.; Frydman, J. Mechanisms and functions of spatial protein quality control. Annu. Rev. Biochem. 2017, 86, 97–122. [Google Scholar] [CrossRef]
- Cascarina, S.M.; Paul, K.R.; Machihara, S.; Ross, E.D. Sequence features governing aggregation or degradation of prion-like proteins. PLoS Genet. 2018, 14, e1007517. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Okamoto, A.; Hosoda, N.; Tanaka, A.; Newnam, G.P.; Chernoff, Y.O.; Hoshino, S.I. Proteolysis suppresses spontaneous prion generation in yeast. J. Biol. Chem. 2017, 292, 20113–20124. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taylor, J.P.; Brown, R.H.; Cleveland, D.W. Decoding ALS: From genes to mechanism. Nature 2016, 539, 197–206. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gall, J.G.; Bellini, M.; Wu, Z.; Murphy, C. Assembly of the nuclear transcription and processing machinery: Cajal bodies (coiled bodies) and transcriptosomes. Mol. Biol. Cell 1999, 10, 4385–4402. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thiry, M.; Lafontaine, D.L.J. Birth of a nucleolus: The evolution of nucleolar compartments. Trends Cell Biol. 2005, 15, 194–199. [Google Scholar] [CrossRef] [PubMed]
- Sheth, U.; Parker, R. Decapping and decay of messenger RNA occur in cytoplasmic processing bodies. Science 2003, 300, 805–808. [Google Scholar] [CrossRef] [Green Version]
- Micklem, D.R. mRNA localisation during development. Dev. Biol. 1995, 172, 377–395. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kedersha, N.; Anderson, P. Stress granules: Sites of mRNA triage that regulate mRNA stability and translatability. Biochem. Soc. Trans. 2002, 30, 963–969. [Google Scholar] [CrossRef] [Green Version]
- Protter, D.S.W.; Parker, R. Principles and properties of stress granules. Trends Cell Biol. 2016, 26, 668–679. [Google Scholar] [CrossRef] [Green Version]
- Kedersha, N.; Ivanov, P.; Anderson, P. Stress granules and cell signaling: More than just a passing phase? Trends Biochem. Sci. 2013, 38, 494–506. [Google Scholar] [CrossRef] [Green Version]
- Jain, S.; Wheeler, J.R.; Walters, R.W.; Agrawal, A.; Barsic, A.; Parker, R. ATPase-modulated stress granules contain a diverse proteome and substructure. Cell 2016, 164, 487–498. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kroschwald, S.; Maharana, S.; Mateju, D.; Malinovska, L.; Nüske, E.; Poser, I.; Richter, D.; Alberti, S. Promiscuous interactions and protein disaggregases determine the material state of stress-inducible RNP granules. eLife 2015, 4, e06807. [Google Scholar] [CrossRef] [PubMed]
- Patel, A.; Lee, H.O.; Jawerth, L.; Maharana, S.; Jahnel, M.; Hein, M.Y.; Stoynov, S.; Mahamid, J.; Saha, S.; Franzmann, T.M.; et al. A liquid-to-solid phase transition of the ALS protein FUS accelerated by disease mutation. Cell 2015, 162, 1066–1077. [Google Scholar] [CrossRef] [Green Version]
- Shin, Y.; Berry, J.; Pannucci, N.; Haataja, M.P.; Toettcher, J.E.; Brangwynne, C.P. Spatiotemporal control of intracellular phase transitions using light-activated optodroplets. Cell 2017, 168, 159–171. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, P.; Fan, B.; Yang, P.; Temirov, J.; Messing, J.; Kim, H.J.; Taylor, J.P. Chronic optogenetic induction of stress granules is cytotoxic and reveals the evolution of ALS-FTD pathology. eLife 2019, 8, e39578. [Google Scholar] [CrossRef]
- Khong, A.; Matheny, T.; Jain, S.; Mitchell, S.F.; Wheeler, J.R.; Parker, R. The stress granule transcriptome reveals principles of mRNA accumulation in stress granules. Mol. Cell 2017, 68, 808.e5–820.e5. [Google Scholar] [CrossRef]
- Padrón, A.; Iwasaki, S.; Ingolia, N.T. Proximity RNA Labeling by APEX-Seq Reveals the Organization of Translation Initiation Complexes and Repressive RNA Granules. Mol. Cell 2019, 75, 875–887. [Google Scholar] [CrossRef]
- Markmiller, S.; Soltanieh, S.; Server, K.L.; Mak, R.; Jin, W.; Fang, M.Y.; Luo, E.C.; Krach, F.; Yang, D.; Sen, A.; et al. Context-dependent and disease-specific diversity in protein interactions within stress granules. Cell 2018, 172, 590.e13–604.e13. [Google Scholar] [CrossRef] [Green Version]
- Youn, J.Y.; Dunham, W.H.; Hong, S.J.; Knight, J.D.R.; Bashkurov, M.; Chen, G.I.; Bagci, H.; Rathod, B.; MacLeod, G.; Eng, S.W.M.; et al. High-density proximity mapping reveals the subcellular organization of mRNA-associated granules and bodies. Mol. Cell 2018, 69, 517.e11–532.e11. [Google Scholar] [CrossRef]
- Matsuki, H.; Takahashi, M.; Higuchi, M.; Makokha, G.N.; Oie, M.; Fujii, M. Both G3BP1 and G3BP2 contribute to stress granule formation. Genes Cells 2013, 18, 135–146. [Google Scholar] [CrossRef]
- Kedersha, N.; Panas, M.D.; Achorn, C.A.; Lyons, S.; Tisdale, S.; Hickman, T.; Thomas, M.; Lieberman, J.; McInerney, G.M.; Ivanov, P.; et al. G3BP-Caprin1-USP10 complexes mediate stress granule condensation and associate with 40S subunits. J. Cell Biol. 2016, 212, 845–860. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guillén-Boixet, J.; Kopach, A.; Holehouse, A.S.; Wittmann, S.; Jahnel, M.; Schlüßler, R.; Kim, K.; Trussina, I.R.E.A.; Wang, J.; Mateju, D.; et al. RNA-induced conformational switching and clustering of G3BP drive stress granule assembly by condensation. Cell 2020, 181, 346.e17–361.e17. [Google Scholar] [CrossRef]
- Yang, P.; Mathieu, C.; Kolaitis, R.M.; Zhang, P.; Messing, J.; Yurtsever, U.; Yang, Z.; Wu, J.; Li, Y.; Pan, Q.; et al. G3BP1 is a tunable switch that triggers phase separation to assemble stress granules. Cell 2020, 181, 325.e28–345.e28. [Google Scholar] [CrossRef] [PubMed]
- Sanders, D.W.; Kedersha, N.; Lee, D.S.W.; Strom, A.R.; Drake, V.; Riback, J.A.; Bracha, D.; Eeftens, J.M.; Iwanicki, A.; Wang, A.; et al. Competing protein-RNA interaction networks control multiphase intracellular organization. Cell 2020, 181, 306.e28–324.e28. [Google Scholar] [CrossRef] [PubMed]
- Brengues, M.; Parker, R. Accumulation of polyadenylated mRNA, Pab1p, eIF4E, and eIF4G with P-bodies in Saccharomyces cerevisiae. Mol. Biol. Cell 2007, 18, 2592–2602. [Google Scholar] [CrossRef] [Green Version]
- Buchan, J.R.; Muhlrad, D.; Parker, R. P bodies promote stress granule assembly in Saccharomyces cerevisiae. J. Cell Biol. 2008, 183, 441–455. [Google Scholar] [CrossRef]
- Dumetz, A.C.; Chockla, A.M.; Kaler, E.W.; Lenhoff, A.M. Protein phase behavior in aqueous solutions: Crystallization, liquid-liquid phase separation, gels, and aggregates. Biophys. J. 2008, 94, 570–583. [Google Scholar] [CrossRef] [Green Version]
- Van Treeck, B.; Protter, D.S.W.; Matheny, T.; Khong, A.; Link, C.D.; Parker, R. RNA self-assembly contributes to stress granule formation and defining the stress granule transcriptome. Proc. Natl. Acad. Sci. USA 2018, 115, 2734–2739. [Google Scholar] [CrossRef] [Green Version]
- Jain, A.; Vale, R.D. RNA phase transitions in repeat expansion disorders. Nature 2017, 546, 243–247. [Google Scholar] [CrossRef] [Green Version]
- Gomes, E.; Shorter, J. The molecular language of membraneless organelles. J. Biol. Chem. 2019, 294, 7115–7127. [Google Scholar] [CrossRef] [Green Version]
- Alberti, S.; Gladfelter, A.; Mittag, T. Considerations and challenges in studying liquid-liquid phase separation and biomolecular condensates. Cell 2019, 176, 419–434. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hyman, A.A.; Weber, C.A.; Jülicher, F. Liquid-liquid phase separation in biology. Annu. Rev. Cell Dev. Biol. 2014, 30, 39–58. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shelkovnikova, T.A.; Robinson, H.K.; Troakes, C.; Ninkina, N.; Buchman, V.L. Compromised paraspeckle formation as a pathogenic factor in FUSopathies. Hum. Mol. Genet. 2014, 23, 2298–2312. [Google Scholar] [CrossRef] [PubMed]
- Decker, C.J.; Teixeira, D.; Parker, R. Edc3p and a glutamine/asparagine-rich domain of Lsm4p function in processing body assembly in Saccharomyces cerevisiae. J. Cell Biol. 2007, 179, 437–449. [Google Scholar] [CrossRef] [Green Version]
- Hennig, S.; Kong, G.; Mannen, T.; Sadowska, A.; Kobelke, S.; Blythe, A.; Knott, G.J.; Iyer, S.S.; Ho, D.; Newcombe, E.A.; et al. Prion-like domains in RNA binding proteins are essential for building subnuclear paraspeckles. J. Cell Biol. 2015, 210, 529–539. [Google Scholar] [CrossRef]
- Molliex, A.; Temirov, J.; Lee, J.; Coughlin, M.; Kanagaraj, A.P.; Kim, H.J.; Mittag, T.; Taylor, J.P. Phase separation by low complexity domains promotes stress granule assembly and drives pathological fibrillization. Cell 2015, 163, 123–133. [Google Scholar] [CrossRef] [Green Version]
- Zhu, M.; Kuechler, E.R.; Zhang, J.; Matalon, O.; Dubreuil, B.; Hofmann, A.; Loewen, C.; Levy, E.D.; Gsponer, J.; Mayor, T. Proteomic analysis reveals the direct recruitment of intrinsically disordered regions to stress granules in S. cerevisiae. J. Cell Sci. 2020, 133, jcs244657. [Google Scholar] [CrossRef]
- Kato, M.; Han, T.W.; Xie, S.; Shi, K.; Du, X.; Wu, L.C.; Mirzaei, H.; Goldsmith, E.J.; Longgood, J.; Pei, J.; et al. Cell-free formation of RNA granules: Low complexity sequence domains form dynamic fibers within hydrogels. Cell 2012, 149, 753–767. [Google Scholar] [CrossRef] [Green Version]
- Xiang, S.; Kato, M.; Wu, L.C.; Lin, Y.; Ding, M.; Zhang, Y.; Yu, Y.; McKnight, S.L. The LC Domain of hnRNPA2 adopts similar conformations in hydrogel polymers, liquid-like droplets, and nuclei. Cell 2015, 163, 829–839. [Google Scholar] [CrossRef] [Green Version]
- Lin, Y.; Protter, D.S.W.; Rosen, M.K.; Parker, R. Formation and maturation of phase-separated liquid droplets by RNA-binding proteins. Mol. Cell 2015, 60, 208–219. [Google Scholar] [CrossRef] [Green Version]
- Protter, D.S.W.; Rao, B.S.; Van Treeck, B.; Lin, Y.; Mizoue, L.; Rosen, M.K.; Parker, R. Intrinsically disordered regions can contribute promiscuous interactions to RNP granule assembly. Cell Rep. 2018, 22, 1401–1412. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bracha, D.; Walls, M.T.; Wei, M.T.; Zhu, L.; Kurian, M.; Avalos, J.L.; Toettcher, J.E.; Brangwynne, C.P. Mapping local and global liquid phase behavior in living cells using photo-oligomerizable seeds. Cell 2018, 175, 1467.e13–1480.e13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Choi, J.M.; Dar, F.; Pappu, R.V. LASSI: A lattice model for simulating phase transitions of multivalent proteins. PLoS Comput. Biol. 2019, 15, e1007028. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jonas, S.; Izaurralde, E. The role of disordered protein regions in the assembly of decapping complexes and RNP granules. Genes Dev. 2013, 27, 2628–2641. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Franzmann, T.M.; Alberti, S. Prion-like low-complexity sequences: Key regulators of protein solubility and phase behavior. J. Biol. Chem. 2019, 294, 7128–7136. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Franzmann, T.M.; Jahnel, M.; Pozniakovsky, A.; Mahamid, J.; Holehouse, A.S.; Nüske, E.; Richter, D.; Baumeister, W.; Grill, S.W.; Pappu, R.V.; et al. Phase separation of a yeast prion protein promotes cellular fitness. Science 2018, 359, eaao5654. [Google Scholar] [CrossRef] [Green Version]
- Ryan, V.H.; Dignon, G.L.; Zerze, G.H.; Chabata, C.V.; Silva, R.; Conicella, A.E.; Amaya, J.; Burke, K.A.; Mittal, J.; Fawzi, N.L. Mechanistic view of hnRNPA2 low-complexity domain structure, interactions, and phase separation altered by mutation and arginine methylation. Mol. Cell 2018, 69, 465.e7–479.e7. [Google Scholar] [CrossRef]
- Hughes, M.P.; Sawaya, M.R.; Boyer, D.R.; Goldschmidt, L.; Rodriguez, J.A.; Cascio, D.; Chong, L.; Gonen, T.; Eisenberg, D.S. Atomic structures of low-complexity protein segments reveal kinked b sheets that assemble networks. Science 2018, 359, 698–701. [Google Scholar] [CrossRef] [Green Version]
- Luo, F.; Gui, X.; Zhou, H.; Gu, J.; Li, Y.; Liu, X.; Zhao, M.; Li, D.; Li, X.; Liu, C. Atomic structures of FUS LC domain segments reveal bases for reversible amyloid fibril formation. Nat. Struct. Mol. Biol. 2018, 25, 341–346. [Google Scholar] [CrossRef]
- Murray, D.T.; Kato, M.; Lin, Y.; Thurber, K.R.; Hung, I.; McKnight, S.L.; Tycko, R. Structure of FUS protein fibrils and its relevance to self-assembly and phase separation of low-complexity domains. Cell 2017, 171, 615.e16–627.e16. [Google Scholar] [CrossRef]
- Martin, E.W.; Holehouse, A.S.; Peran, I.; Farag, M.; Incicco, J.J.; Bremer, A.; Grace, C.R.; Soranno, A.; Pappu, R.V.; Mittag, T. Valence and patterning of aromatic residues determine the phase behavior of prion-like domains. Science 2020, 367, 694–699. [Google Scholar] [CrossRef]
- Vernon, R.M.C.; Chong, P.A.; Tsang, B.; Kim, T.H.; Bah, A.; Farber, P.; Lin, H.; Forman-Kay, J.D. Pi-Pi contacts are an overlooked protein feature relevant to phase separation. Elife 2018, 7, e31486. [Google Scholar] [CrossRef]
- Bolognesi, B.; Gotor, N.L.; Dhar, R.; Cirillo, D.; Baldrighi, M.; Tartaglia, G.G.; Lehner, B. A concentration-dependent liquid phase separation can cause toxicity upon increased protein expression. Cell Rep. 2016, 16, 222–231. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qamar, S.; Wang, G.Z.; Randle, S.J.; Ruggeri, F.S.; Varela, J.A.; Lin, J.Q.; Phillips, E.C.; Miyashita, A.; Williams, D.; Ströhl, F.; et al. FUS phase separation is modulated by a molecular chaperone and methylation of arginine cation-π interactions. Cell 2018, 173, 720.e15–734.e15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chong, P.A.; Vernon, R.M.; Forman-Kay, J.D. RGG/RG motif regions in RNA binding and phase separation. J. Mol. Biol. 2018, 430, 4650–4665. [Google Scholar] [CrossRef] [PubMed]
- Nott, T.J.; Petsalaki, E.; Farber, P.; Jervis, D.; Fussner, E.; Plochowietz, A.; Craggs, T.D.; Bazett-Jones, D.P.; Pawson, T.; Forman-Kay, J.D.; et al. Phase transition of a disordered nuage protein generates environmentally responsive membraneless organelles. Mol. Cell 2015, 57, 936–947. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pak, C.W.; Kosno, M.; Holehouse, A.S.; Padrick, S.B.; Mittal, A.; Ali, R.; Yunus, A.A.; Liu, D.R.; Pappu, R.V.; Rosen, M.K. Sequence determinants of intracellular phase separation by complex coacervation of a disordered protein. Mol. Cell 2016, 63, 72–85. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mitsuhashi, K.; Ito, D.; Mashima, K.; Oyama, M.; Takahashi, S.; Suzuki, N. De novo design of RNA-binding proteins with a prion-like domain related to ALS/FTD proteinopathies. Sci. Rep. 2017, 7, 16871. [Google Scholar] [CrossRef] [Green Version]
- Ryan, V.H.; Perdikari, T.M.; Naik, M.T.; Saueressig, C.F.; Lins, J.; Dignon, G.L.; Mittal, J.; Hart, A.C.; Fawzi, N.L. Tyrosine phosphorylation regulates hnRNPA2 granule protein partitioning and reduces neurodegeneration. EMBO J. 2020, e105001. [Google Scholar] [CrossRef]
- Cascarina, S.M.; Ross, E.D. Yeast prions and human prion-like proteins: Sequence features and prediction methods. Cell. Mol. Life Sci. 2014, 71, 2047–2063. [Google Scholar] [CrossRef] [Green Version]
- Shattuck, J.E.; Waechter, A.C.; Ross, E.D. The effects of glutamine/asparagine content on aggregation and heterologous prion induction by yeast prion-like domains. Prion 2017, 11, 249–264. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, L.; Kim, H.J.; Wang, H.; Monaghan, J.; Freyermuth, F.; Sung, J.C.; O’Donovan, K.; Fare, C.M.; Diaz, Z.; Singh, N.; et al. Nuclear-import receptors reverse aberrant phase transitions of RNA-binding proteins with prion-like domains. Cell 2018, 173, 677–690.e20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoshizawa, T.; Ali, R.; Jiou, J.; Fung, H.Y.J.; Burke, K.A.; Kim, S.J.; Lin, Y.; Peeples, W.B.; Saltzberg, D.; Soniat, M.; et al. Nuclear import receptor inhibits phase separation of FUS through binding to multiple sites. Cell 2018, 173, 693.e22–705.e22. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hofweber, M.; Hutten, S.; Bourgeois, B.; Spreitzer, E.; Niedner-Boblenz, A.; Schifferer, M.; Ruepp, M.D.; Simons, M.; Niessing, D.; Madl, T.; et al. Phase separation of FUS is suppressed by its nuclear import receptor and arginine methylation. Cell 2018, 173, 706–719.e13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maharana, S.; Wang, J.; Papadopoulos, D.K.; Richter, D.; Pozniakovsky, A.; Poser, I.; Bickle, M.; Rizk, S.; Guillén-Boixet, J.; Franzmann, T.M.; et al. RNA buffers the phase separation behavior of prion-like RNA binding proteins. Science 2018, 360, 918–921. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hofweber, M.; Dormann, D. Friend or foe-post-translational modifications as regulators of phase separation and RNP granule dynamics. J. Biol. Chem. 2019, 294, 7137–7150. [Google Scholar] [CrossRef] [Green Version]
- Owen, I.; Shewmaker, F. The role of post-translational modifications in the phase transitions of intrinsically disordered proteins. Int. J. Mol. Sci. 2019, 20, 5501. [Google Scholar] [CrossRef] [Green Version]
- Monahan, Z.; Ryan, V.H.; Janke, A.M.; Burke, K.A.; Rhoads, S.N.; Zerze, G.H.; O’Meally, R.; Dignon, G.L.; Conicella, A.E.; Zheng, W.; et al. Phosphorylation of the FUS low-complexity domain disrupts phase separation, aggregation, and toxicity. EMBO J. 2017, 36, 2951–2967. [Google Scholar] [CrossRef]
- Wang, A.; Conicella, A.E.; Schmidt, H.B.; Martin, E.W.; Rhoads, S.N.; Reeb, A.N.; Nourse, A.; Ramirez Montero, D.; Ryan, V.H.; Rohatgi, R.; et al. A single N-terminal phosphomimic disrupts TDP-43 polymerization, phase separation, and RNA splicing. EMBO J. 2018, 37, e97452. [Google Scholar] [CrossRef]
- Andrusiak, M.G.; Sharifnia, P.; Lyu, X.; Wang, Z.; Dickey, A.M.; Wu, Z.; Chisholm, A.D.; Jin, Y. Inhibition of axon regeneration by liquid-like TIAR-2 granules. Neuron 2019, 104, 290.e8–304.e8. [Google Scholar] [CrossRef]
- Munder, M.C.; Midtvedt, D.; Franzmann, T.; Nüske, E.; Otto, O.; Herbig, M.; Ulbricht, E.; Müller, P.; Taubenberger, A.; Maharana, S.; et al. A pH-driven transition of the cytoplasm from a fluid- to a solid-like state promotes entry into dormancy. eLife 2016, 5, e093. [Google Scholar] [CrossRef] [PubMed]
- Riback, J.A.; Katanski, C.D.; Kear-Scott, J.L.; Pilipenko, E.V.; Rojek, A.E.; Sosnick, T.R.; Drummond, D.A. Stress-triggered phase separation is an adaptive, evolutionarily tuned response. Cell 2017, 168, 1028.e19–1040.e19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoo, H.; Triandafillou, C.; Drummond, D.A. Cellular sensing by phase separation: Using the process, not just the products. J. Biol. Chem. 2019, 294, 7151–7159. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mackenzie, I.R.; Nicholson, A.M.; Sarkar, M.; Messing, J.; Purice, M.D.; Pottier, C.; Annu, K.; Baker, M.; Perkerson, R.B.; Kurti, A.; et al. TIA1 mutations in amyotrophic lateral sclerosis and frontotemporal dementia promote phase separation and alter stress granule dynamics. Neuron 2017, 25, 341–346. [Google Scholar] [CrossRef] [PubMed]
- Mathieu, C.; Pappu, R.V.; Taylor, J.P. Beyond aggregation: Pathological phase transitions in neurodegenerative disease. Science 2020, 370, 56–60. [Google Scholar] [CrossRef] [PubMed]
- Da Cruz, S.; Cleveland, D.W. Understanding the role of TDP-43 and FUS/TLS in ALS and beyond. Curr. Opin. Neurobiol. 2011, 21, 904–919. [Google Scholar] [CrossRef] [Green Version]
- Mateju, D.; Franzmann, T.M.; Patel, A.; Kopach, A.; Boczek, E.E.; Maharana, S.; Lee, H.O.; Carra, S.; Hyman, A.A.; Alberti, S. An aberrant phase transition of stress granules triggered by misfolded protein and prevented by chaperone function. EMBO J. 2017, 36, 1669–1687. [Google Scholar] [CrossRef]
- Ganassi, M.; Mateju, D.; Bigi, I.; Mediani, L.; Poser, I.; Lee, H.O.; Seguin, S.J.; Morelli, F.F.; Vinet, J.; Leo, G.; et al. A surveillance function of the HSPB8-BAG3-HSP70 chaperone complex ensures stress granule integrity and dynamism. Mol. Cell 2016, 63, 796–810. [Google Scholar] [CrossRef] [Green Version]
- Li, S.; Zhang, P.; Freibaum, B.D.; Kim, N.C.; Kolaitis, R.M.; Molliex, A.; Kanagaraj, A.P.; Yabe, I.; Tanino, M.; Tanaka, S.; et al. Genetic interaction of hnRNPA2B1 and DNAJB6 in a Drosophila model of multisystem proteinopathy. Hum. Mol. Genet. 2016, 25, 936–950. [Google Scholar] [CrossRef] [Green Version]
- Buchan, J.R.; Kolaitis, R.M.; Taylor, J.P.; Parker, R. Eukaryotic stress granules are cleared by autophagy and Cdc48/VCP function. Cell 2013, 153, 1461–1474. [Google Scholar] [CrossRef] [Green Version]
- Dao, T.P.; Kolaitis, R.M.; Kim, H.J.; O’Donovan, K.; Martyniak, B.; Colicino, E.; Hehnly, H.; Taylor, J.P.; Castañeda, C.A. Ubiquitin modulates liquid-liquid phase separation of UBQLN2 via disruption of multivalent interactions. Mol. Cell 2018, 69, 965–978. [Google Scholar] [CrossRef] [Green Version]
- Alexander, E.J.; Niaki, A.G.; Zhang, T.; Sarkar, J.; Liu, Y.; Nirujogi, R.S.; Pandey, A.; Myong, S.; Wang, J. Ubiquilin 2 modulates ALS/FTD-linked FUS–RNA complex dynamics and stress granule formation. Proc. Natl. Acad. Sci. USA 2018, 115, E11485–E11494. [Google Scholar] [CrossRef] [Green Version]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fomicheva, A.; Ross, E.D. From Prions to Stress Granules: Defining the Compositional Features of Prion-Like Domains That Promote Different Types of Assemblies. Int. J. Mol. Sci. 2021, 22, 1251. https://doi.org/10.3390/ijms22031251
Fomicheva A, Ross ED. From Prions to Stress Granules: Defining the Compositional Features of Prion-Like Domains That Promote Different Types of Assemblies. International Journal of Molecular Sciences. 2021; 22(3):1251. https://doi.org/10.3390/ijms22031251
Chicago/Turabian StyleFomicheva, Anastasia, and Eric D. Ross. 2021. "From Prions to Stress Granules: Defining the Compositional Features of Prion-Like Domains That Promote Different Types of Assemblies" International Journal of Molecular Sciences 22, no. 3: 1251. https://doi.org/10.3390/ijms22031251