
Broad Kinase Inhibition Mitigates Early Neuronal Dysfunction in Tauopathy

 , ,
, ,  , , , add
Show full author list
, , , add
Show full author list
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
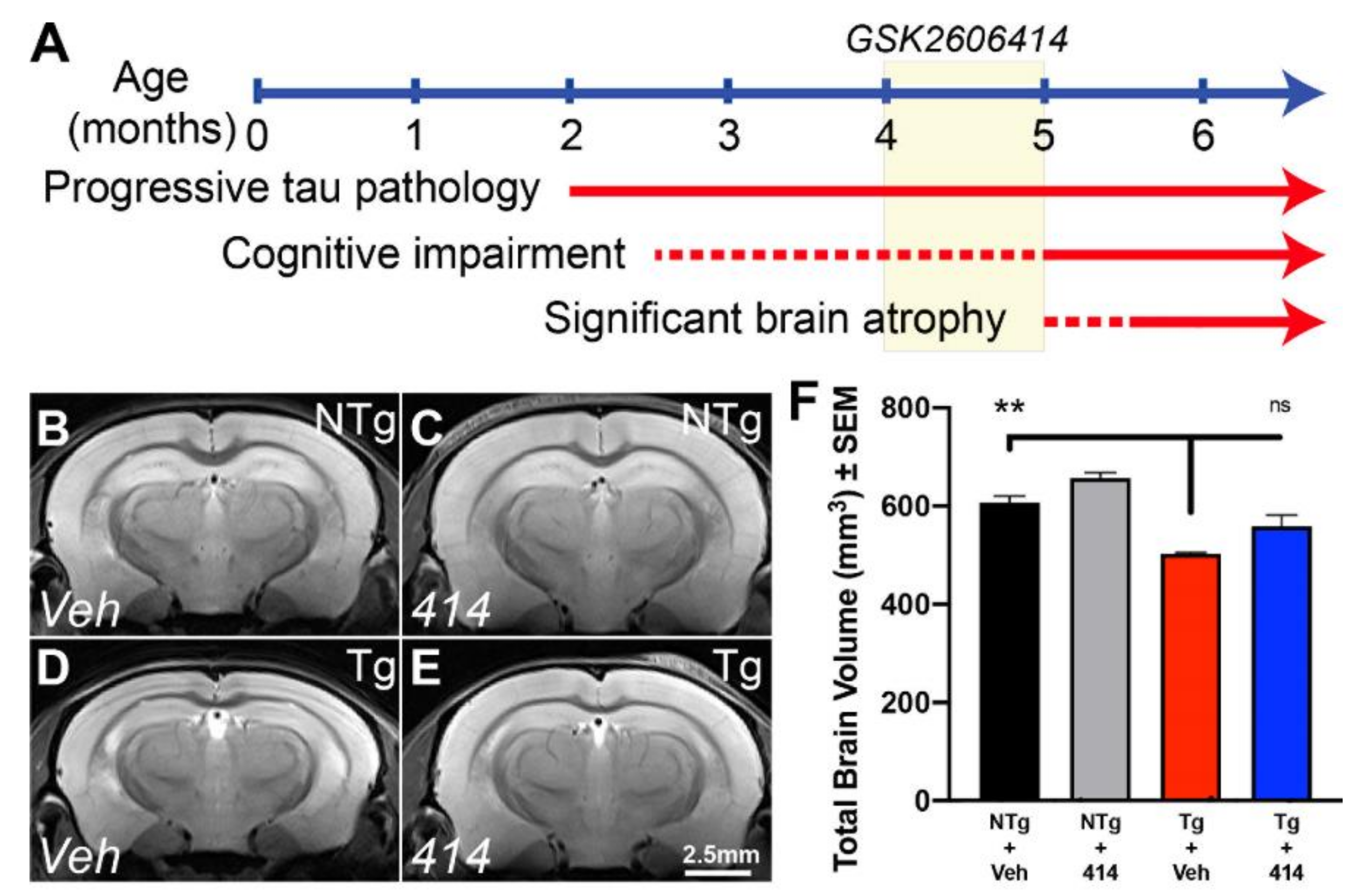
2.1. Tau Transgenic Mice Exhibit Common Neurodegenerative Features Ameliorated by Multi-Target Kinase Inhibition
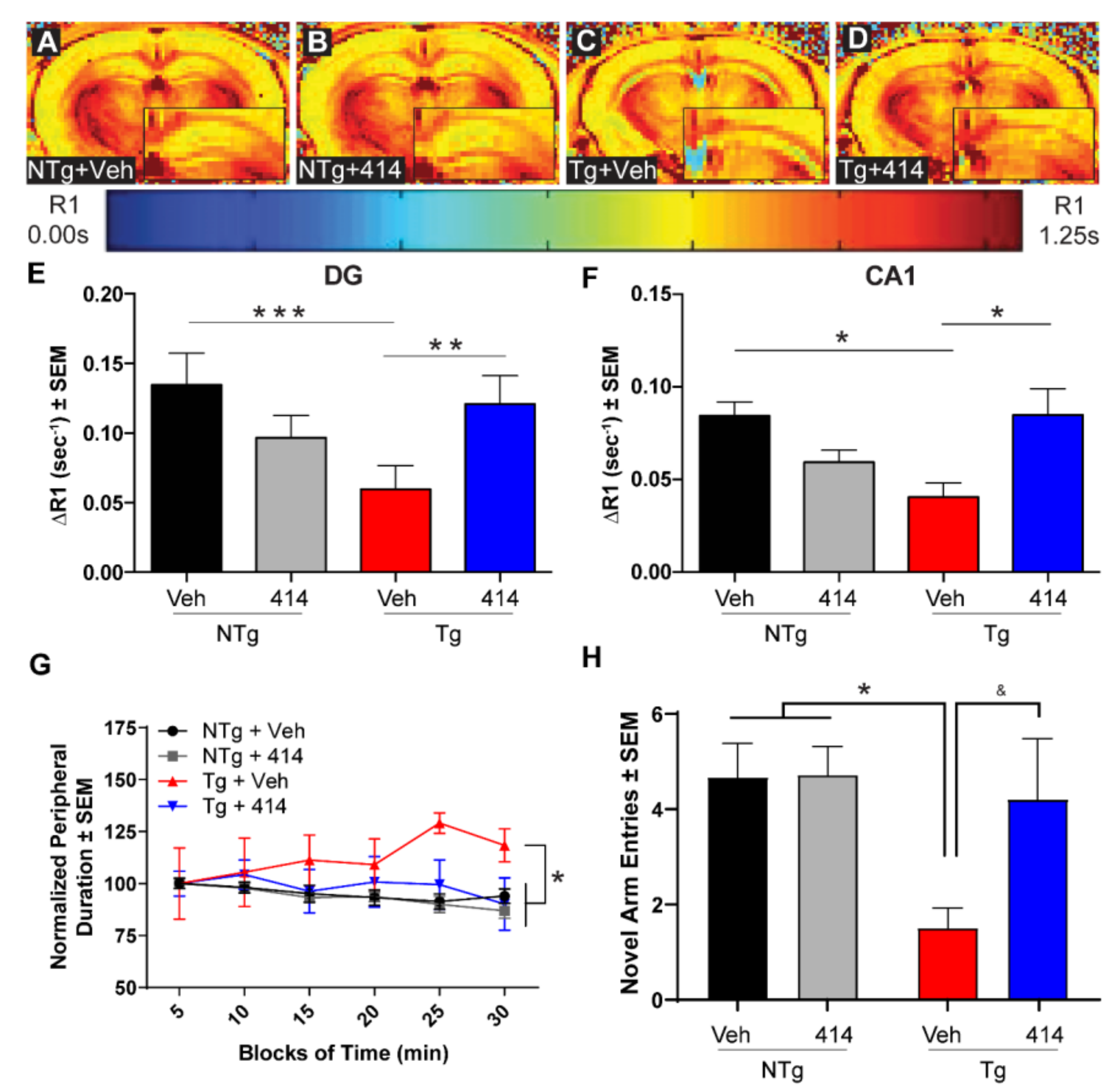
2.2. GSK2606414 Rescues Functional Deficits without Altering Tau Hyper-Phosphorylation
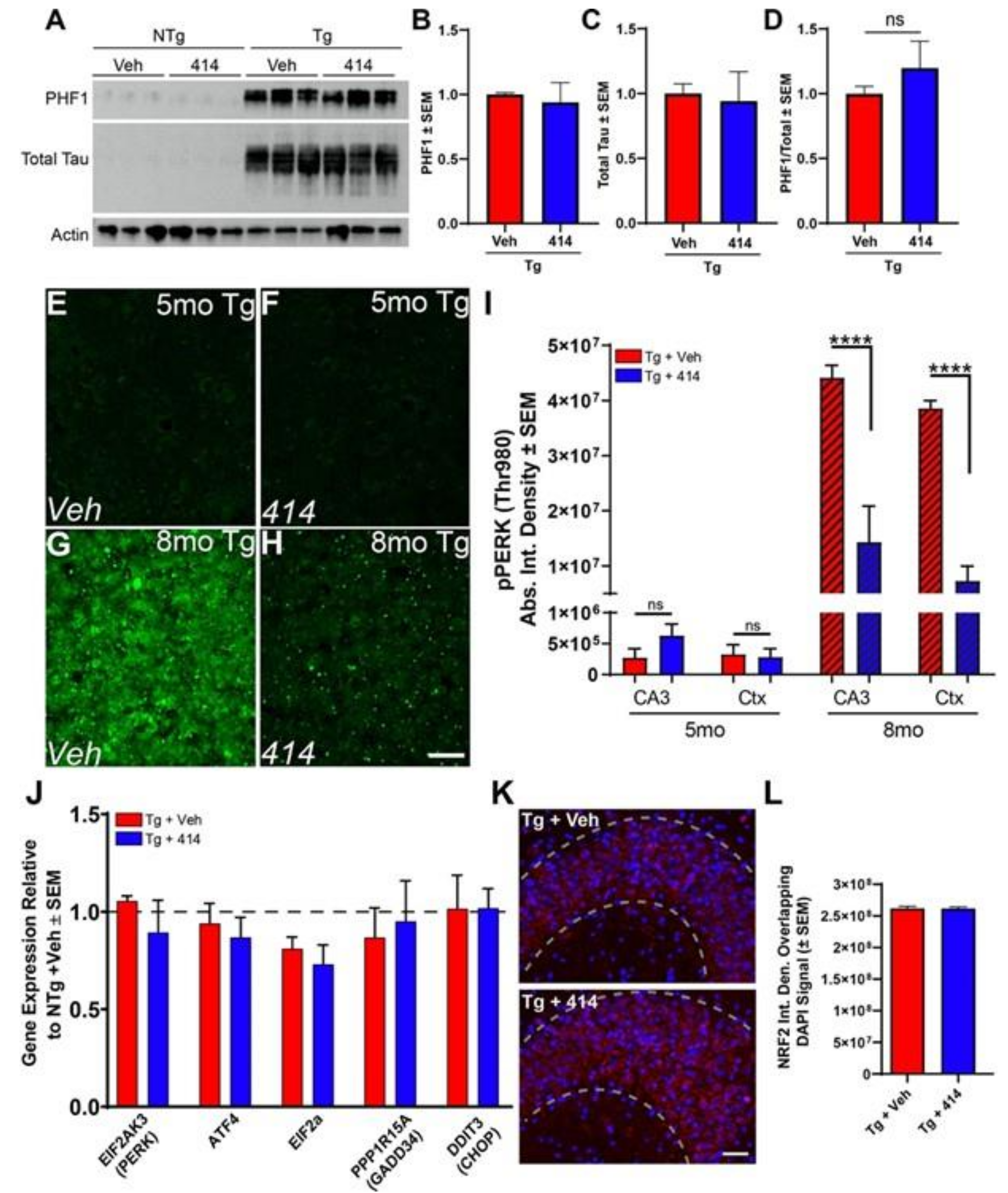
2.3. Functional Deficits of Early Stage Tau Transgenic Mice Do Not Depend on PERK/UPR Activation
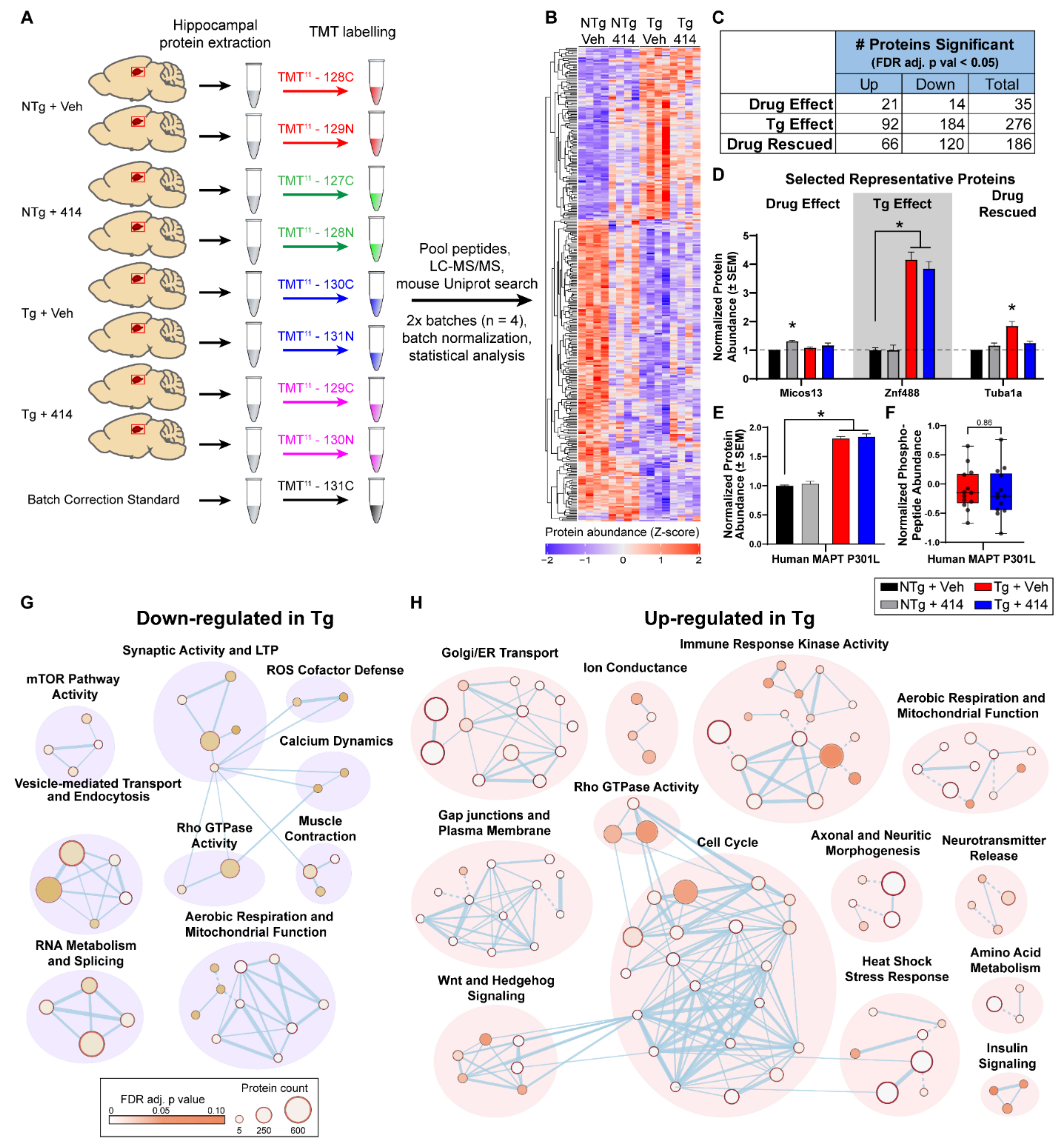
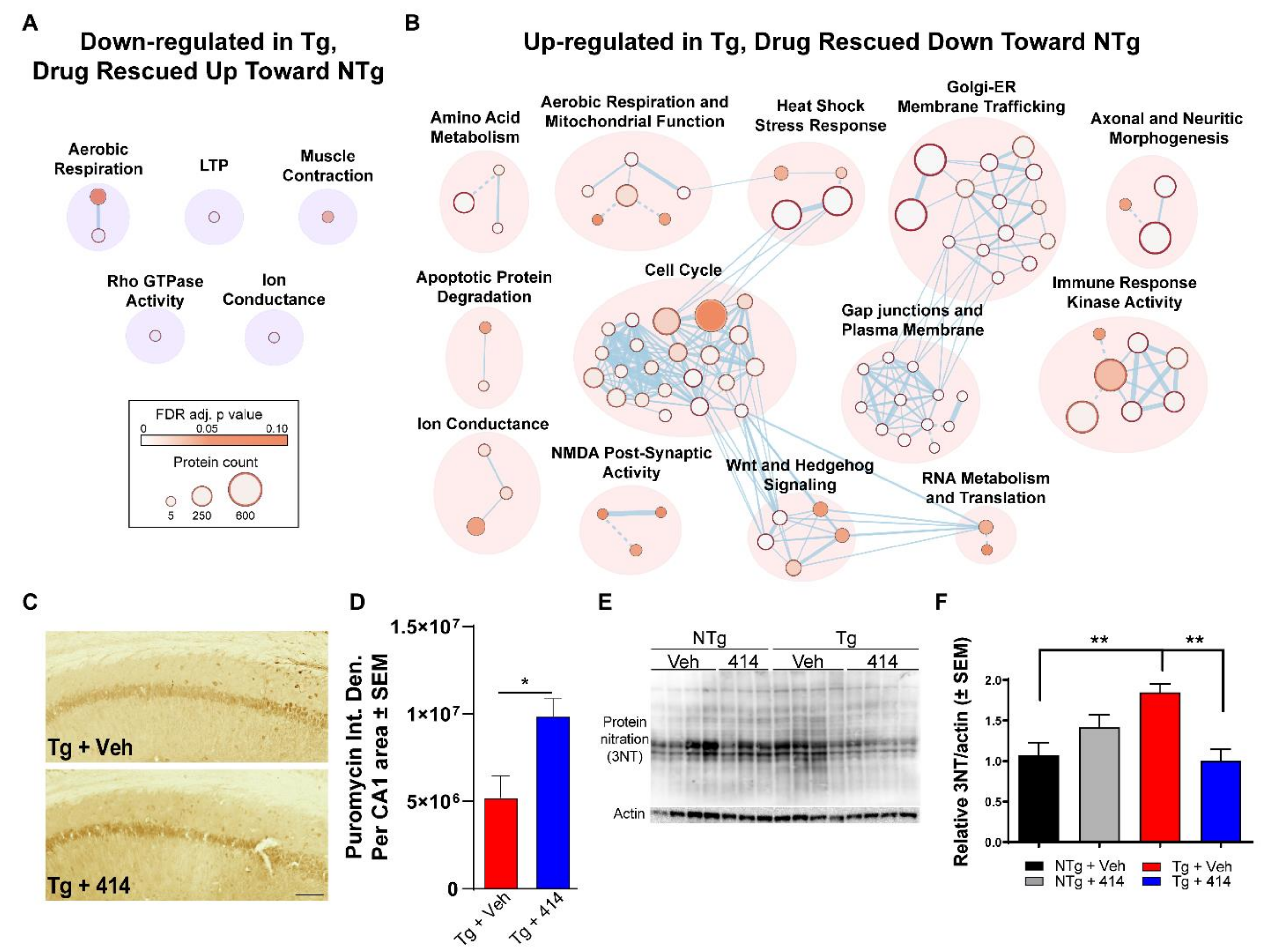
2.4. Quantitative Hippocampal Proteomics Reveals Pathways Responsible for Cognitive Decline in Early Stage Tauopathy Mice
2.5. Multi-Target Kinase Inhibition via GSK2606414 Treatment Rescues Proteomic Shifts in Tauopathy
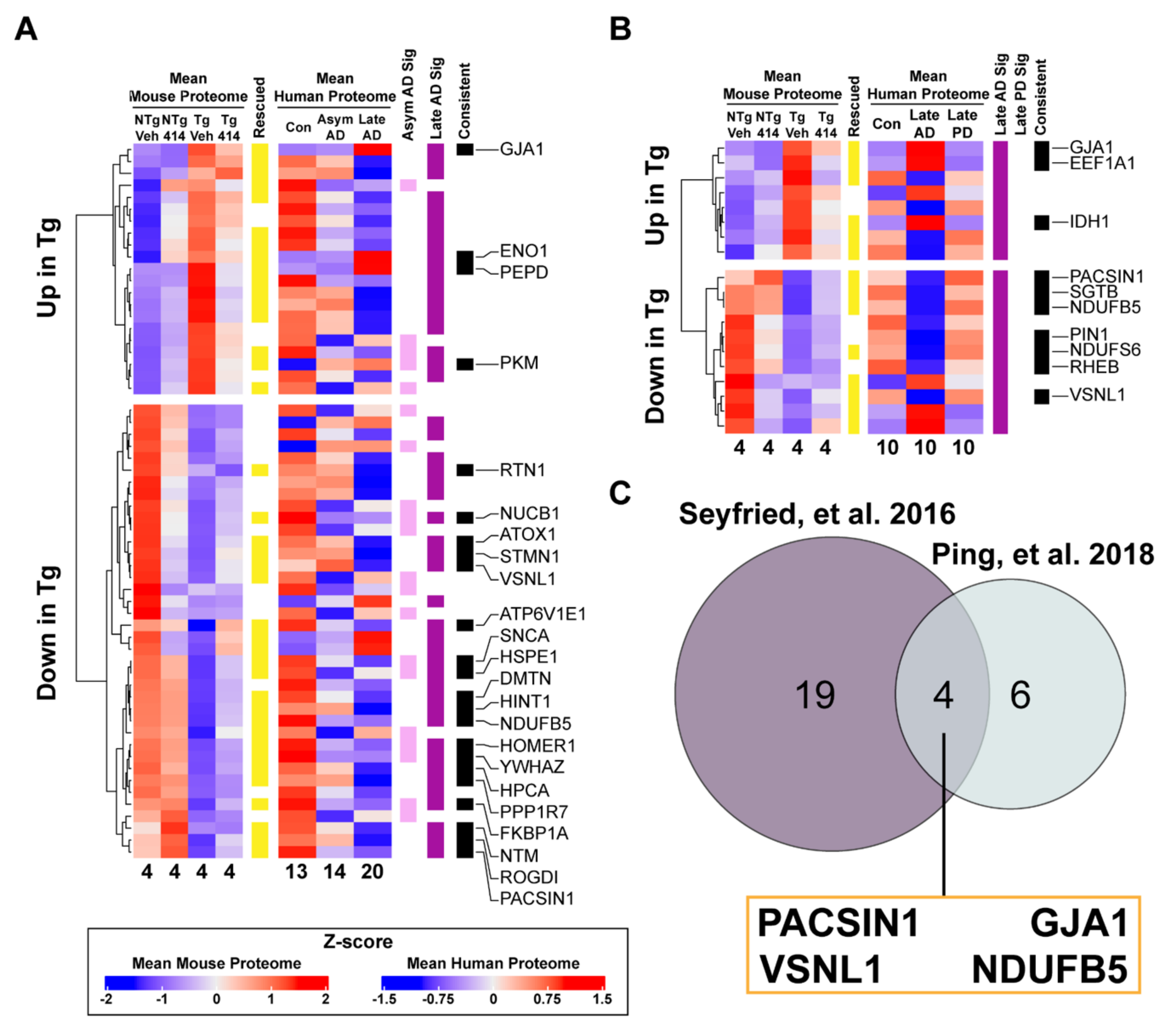
2.6. Early Stage Tauopathy Exhibits Common Protein Alterations Targetable by Broad Kinase Inhibition
3. Discussion
4. Materials and Methods
4.1. Study Design
4.2. Animals
4.3. Treatment with GSK2606414
4.4. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Khanna, M.R.; Kovalevich, J.; Lee, V.M.-Y.; Trojanowski, J.Q.; Brunden, K.R. Therapeutic Strategies for the Treatment of Tauopathies: Hopes and Challenges. Alzheimers Dement 2016, 12, 1051–1065. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jadhav, S.; Avila, J.; Schöll, M.; Kovacs, G.G.; Kövari, E.; Skrabana, R.; Evans, L.D.; Kontsekova, E.; Malawska, B.; de Silva, R.; et al. A walk through tau therapeutic strategies. Acta Neuropathol. Commun. 2019, 7, 22. [Google Scholar] [CrossRef]
- Huang, L.-K.; Chao, S.-P.; Hu, C.-J. Clinical trials of new drugs for Alzheimer disease. J. Biomed. Sci. 2020, 27, 18. [Google Scholar] [CrossRef] [PubMed]
- Boxer, A.L.; Lang, A.E.; Grossman, M.; Knopman, D.S.; Miller, B.L.; Schneider, L.S.; Doody, R.S.; Lees, A.; Golbe, L.I.; Williams, D.R.; et al. Davunetide in patients with progressive supranuclear palsy: A randomised, double-blind, placebo-controlled phase 2/3 trial. Lancet Neurol. 2014, 13, 676–685. [Google Scholar] [CrossRef] [Green Version]
- Ramsay, R.R.; Popovic-Nikolic, M.R.; Nikolic, K.; Uliassi, E.; Bolognesi, M.L. A perspective on multi-target drug discovery and design for complex diseases. Clin. Transl. Med. 2018, 7, 3. [Google Scholar] [CrossRef] [Green Version]
- Johnson, E.C.B.; Dammer, E.B.; Duong, D.M.; Ping, L.; Zhou, M.; Yin, L.; Higginbotham, L.A.; Guajardo, A.; White, B.; Troncoso, J.C.; et al. Large-scale proteomic analysis of Alzheimer’s disease brain and cerebrospinal fluid reveals early changes in energy metabolism associated with microglia and astrocyte activation. Nat. Med. 2020, 26, 769–780. [Google Scholar] [CrossRef]
- Johnson, E.C.B.; Dammer, E.B.; Duong, D.M.; Yin, L.; Thambisetty, M.; Troncoso, J.C.; Lah, J.J.; Levey, A.I.; Seyfried, N.T. Deep proteomic network analysis of Alzheimer’s disease brain reveals alterations in RNA binding proteins and RNA splicing associated with disease. Mol. Neurodegener. 2018, 13, 52. [Google Scholar] [CrossRef]
- Bai, B.; Wang, X.; Li, Y.; Chen, P.-C.; Yu, K.; Dey, K.K.; Yarbro, J.M.; Han, X.; Lutz, B.M.; Rao, S.; et al. Deep Multilayer Brain Proteomics Identifies Molecular Networks in Alzheimer’s Disease Progression. Neuron 2020, 105, 975–991.e7. [Google Scholar] [CrossRef]
- Perluigi, M.; Barone, E.; Di Domenico, F.; Butterfield, D.A. Aberrant protein phosphorylation in Alzheimer disease brain disturbs pro-survival and cell death pathways. Biochim. Biophys. Acta 2016, 1862, 1871–1882. [Google Scholar] [CrossRef]
- Axten, J.M.; Medina, J.R.; Feng, Y.; Shu, A.; Romeril, S.P.; Grant, S.W.; Li, W.H.H.; Heerding, D.A.; Minthorn, E.; Mencken, T.; et al. Discovery of 7-methyl-5-(1-{[3-(trifluoromethyl)phenyl]acetyl}-2,3-dihydro-1H-indol-5-yl)-7H-pyrrolo[2,3-d]pyrimidin-4-amine (GSK2606414), a potent and selective first-in-class inhibitor of protein kinase R (PKR)-like endoplasmic reticulum kinase (PERK). J. Med. Chem. 2012, 55, 7193–7207. [Google Scholar] [CrossRef]
- Radford, H.; Moreno, J.A.; Verity, N.; Halliday, M.; Mallucci, G.R. PERK inhibition prevents tau-mediated neurodegeneration in a mouse model of frontotemporal dementia. Acta Neuropathol. 2015, 130, 633–642. [Google Scholar] [CrossRef] [Green Version]
- Moreno, J.A.; Halliday, M.; Molloy, C.; Radford, H.; Verity, N.; Axten, J.M.; Ortori, C.A.; Willis, A.E.; Fischer, P.M.; Barrett, D.A.; et al. Oral treatment targeting the unfolded protein response prevents neurodegeneration and clinical disease in prion-infected mice. Sci. Transl. Med. 2013, 5, 206ra138. [Google Scholar] [CrossRef]
- Meng, C.; Zhang, J.; Dang, B.; Li, H.; Shen, H.; Li, X.; Wang, Z. PERK Pathway Activation Promotes Intracerebral Hemorrhage Induced Secondary Brain Injury by Inducing Neuronal Apoptosis Both in Vivo and in Vitro. Front. Neurosci. 2018, 12, 111. [Google Scholar] [CrossRef] [PubMed]
- Vanderweyde, T.; Apicco, D.J.; Youmans-Kidder, K.; Ash, P.E.A.; Cook, C.; da Rocha, E.L.; Jansen-West, K.; Frame, A.A.; Citro, A.; Leszyk, J.D.; et al. Interaction of tau with the RNA-Binding Protein TIA1 Regulates tau Pathophysiology and Toxicity. Cell Rep. 2016, 15, 1455–1466. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sharma, V.; Ounallah-Saad, H.; Chakraborty, D.; Hleihil, M.; Sood, R.; Barrera, I.; Edry, E.; Chandran, S.K.; de Leon, S.B.T.; Kaphzan, H.; et al. Local Inhibition of PERK Enhances Memory and Reverses Age-Related Deterioration of Cognitive and Neuronal Properties. J. Neurosci. 2018, 38, 648–658. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smith, H.L.; Freeman, O.J.; Butcher, A.J.; Holmqvist, S.; Humoud, I.; Schätzl, T.; Hughes, D.T.; Verity, N.C.; Swinden, D.P.; Hayes, J.; et al. Astrocyte Unfolded Protein Response Induces a Specific Reactivity State that Causes Non-Cell-Autonomous Neuronal Degeneration. Neuron 2020, 105, 855–866.e5. [Google Scholar] [CrossRef] [Green Version]
- Rojas-Rivera, D.; Delvaeye, T.; Roelandt, R.; Nerinckx, W.; Augustyns, K.; Vandenabeele, P.; Bertrand, M.J.M. When PERK inhibitors turn out to be new potent RIPK1 inhibitors: Critical issues on the specificity and use of GSK2606414 and GSK2656157. Cell Death Differ. 2017, 24, 1100–1110. [Google Scholar] [CrossRef]
- Mahameed, M.; Wilhelm, T.; Darawshi, O.; Obiedat, A.; Tommy, W.-S.; Chintha, C.; Schubert, T.; Samali, A.; Chevet, E.; Eriksson, L.A.; et al. The unfolded protein response modulators GSK2606414 and KIRA6 are potent KIT inhibitors. Cell Death Dis. 2019, 10, 300. [Google Scholar] [CrossRef]
- Berger, Z.; Roder, H.; Hanna, A.; Carlson, A.; Rangachari, V.; Yue, M.; Wszolek, Z.; Ashe, K.; Knight, J.; Dickson, D.; et al. Accumulation of Pathological Tau Species and Memory Loss in a Conditional Model of Tauopathy. J. Neurosci. 2007, 27, 3650–3662. [Google Scholar] [CrossRef] [Green Version]
- SantaCruz, K.; Lewis, J.; Spires, T.; Paulson, J.; Kotilinek, L.; Ingelsson, M.; Guimaraes, A.; DeTure, M.; Ramsden, M.; McGowan, E.; et al. Tau Suppression in a Neurodegenerative Mouse Model Improves Memory Function. Science 2005, 309, 476–481. [Google Scholar] [CrossRef] [Green Version]
- Bailey, R.M.; Howard, J.; Knight, J.; Sahara, N.; Dickson, D.W.; Lewis, J. Effects of the C57BL/6 strain background on tauopathy progression in the rTg4510 mouse model. Mol. Neurodegener. 2014, 9, 8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fontaine, S.N.; Ingram, A.; Cloyd, R.A.; Meier, S.E.; Miller, E.; Lyons, D.; Nation, G.K.; Mechas, E.; Weiss, B.; Lanzillotta, C.; et al. Identification of changes in neuronal function as a consequence of aging and tauopathic neurodegeneration using a novel and sensitive magnetic resonance imaging approach. Neurobiol. Aging 2017, 56, 78–86. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- LaFerla, F.M. Calcium dyshomeostasis and intracellular signalling in Alzheimer’s disease. Nat. Rev. Neurosci. 2002, 3, 862–872. [Google Scholar] [CrossRef] [PubMed]
- Hooper, C.; Killick, R.; Lovestone, S. The GSK3 hypothesis of Alzheimer’s disease. J. Neurochem. 2008, 104, 1433–1439. [Google Scholar] [CrossRef] [Green Version]
- Bell, M.C.; Meier, S.E.; Ingram, A.L.; Abisambra, J.F. PERK-opathies: An Endoplasmic Reticulum Stress Mechanism Underlying Neurodegeneration. Curr. Alzheimer Res. 2016, 13, 150–163. [Google Scholar] [CrossRef]
- Abisambra, J.F.; Jinwal, U.K.; Blair, L.J.; O’Leary, J.C.; Li, Q.; Brady, S.; Wang, L.; Guidi, C.E.; Zhang, B.; Nordhues, B.A.; et al. Tau accumulation activates the unfolded protein response by impairing endoplasmic reticulum-associated degradation. J. Neurosci. 2013, 33, 9498–9507. [Google Scholar] [CrossRef] [Green Version]
- Koren, S.A.; Hamm, M.J.; Meier, S.E.; Weiss, B.E.; Nation, G.K.; Chishti, E.A.; Arango, J.P.; Chen, J.; Zhu, H.; Blalock, E.M.; et al. Tau drives translational selectivity by interacting with ribosomal proteins. Acta Neuropathol. 2019, 137, 571–583. [Google Scholar] [CrossRef] [Green Version]
- Cullinan, S.B.; Zhang, D.; Hannink, M.; Arvisais, E.; Kaufman, R.J.; Diehl, J.A. Nrf2 is a direct PERK substrate and effector of PERK-dependent cell survival. Mol. Cell. Biol. 2003, 23, 7198–7209. [Google Scholar] [CrossRef] [Green Version]
- Goodwin, L.O.; Splinter, E.; Davis, T.L.; Urban, R.; He, H.; Braun, R.E.; Chesler, E.J.; Kumar, V.; van Min, M.; Ndukum, J.; et al. Large-scale discovery of mouse transgenic integration sites reveals frequent structural variation and insertional mutagenesis. Genome Res. 2019, 29, 494–505. [Google Scholar] [CrossRef] [Green Version]
- Gamache, J.; Benzow, K.; Forster, C.; Kemper, L.; Hlynialuk, C.; Furrow, E.; Ashe, K.H.; Koob, M.D. Factors other than hTau overexpression that contribute to tauopathy-like phenotype in rTg4510 mice. Nat. Commun. 2019, 10, 2479. [Google Scholar] [CrossRef]
- Raudvere, U.; Kolberg, L.; Kuzmin, I.; Arak, T.; Adler, P.; Peterson, H.; Vilo, J. g:Profiler: A web server for functional enrichment analysis and conversions of gene lists (2019 update). Nucleic Acids Res. 2019, 47, W191–W198. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jassal, B.; Matthews, L.; Viteri, G.; Gong, C.; Lorente, P.; Fabregat, A.; Sidiropoulos, K.; Cook, J.; Gillespie, M.; Haw, R.; et al. The reactome pathway knowledgebase. Nucleic Acids Res. 2020, 48, D498–D503. [Google Scholar] [CrossRef] [PubMed]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef] [PubMed]
- Goodman, C.A.; Hornberger, T.A. Measuring protein synthesis with SUnSET: A valid alternative to traditional techniques? Exerc. Sport Sci. Rev. 2013, 41, 107–115. [Google Scholar] [CrossRef] [Green Version]
- Butterfield, D.A.; Reed, T.T.; Perluigi, M.; De Marco, C.; Coccia, R.; Keller, J.N.; Markesbery, W.R.; Sultana, R. Elevated Levels of 3-Nitrotyrosine in Brain From Subjects with Amnestic Mild Cognitive Impairment: Implications for the Role of Nitration in the Progression of Alzheimer’s Disease. Brain Res. 2007, 1148, 243–248. [Google Scholar] [CrossRef] [Green Version]
- Butterfield, D.A.; Reed, T.; Sultana, R. Roles of 3-nitrotyrosine- and 4-hydroxynonenal-modified brain proteins in the progression and pathogenesis of Alzheimer’s disease. Free Radic. Res. 2011, 45, 59–72. [Google Scholar] [CrossRef]
- Seyfried, N.T.; Dammer, E.B.; Swarup, V.; Nandakumar, D.; Duong, D.M.; Yin, L.; Deng, Q.; Nguyen, T.; Hales, C.M.; Wingo, T.; et al. A Multi-network Approach Identifies Protein-Specific Co-expression in Asymptomatic and Symptomatic Alzheimer’s Disease. Cell Syst. 2017, 4, 60–72.e4. [Google Scholar] [CrossRef] [Green Version]
- Ping, L.; Duong, D.M.; Yin, L.; Gearing, M.; Lah, J.J.; Levey, A.I.; Seyfried, N.T. Global quantitative analysis of the human brain proteome in Alzheimer’s and Parkinson’s Disease. Sci. Data 2018, 5, 180036. [Google Scholar] [CrossRef] [Green Version]
- Golde, T.E.; DeKosky, S.T.; Galasko, D. Alzheimer’s disease: The right drug, the right time. Science 2018, 362, 1250–1251. [Google Scholar] [CrossRef]
- Pace, M.C.; Xu, G.; Fromholt, S.; Howard, J.; Crosby, K.; Giasson, B.I.; Lewis, J.; Borchelt, D.R. Changes in proteome solubility indicate widespread proteostatic disruption in mouse models of neurodegenerative disease. Acta Neuropathol. 2018, 136, 919–938. [Google Scholar] [CrossRef]
- Modregger, J.; Ritter, B.; Witter, B.; Paulsson, M.; Plomann, M. All three PACSIN isoforms bind to endocytic proteins and inhibit endocytosis. J. Cell. Sci. 2000, 113 Pt 24, 4511–4521. [Google Scholar]
- Anggono, V.; Smillie, K.J.; Graham, M.E.; Valova, V.A.; Cousin, M.A.; Robinson, P.J. Syndapin I is the phosphorylation-regulated dynamin I partner in synaptic vesicle endocytosis. Nat. Neurosci. 2006, 9, 752–760. [Google Scholar] [CrossRef] [PubMed]
- Braun, A.; Pinyol, R.; Dahlhaus, R.; Koch, D.; Fonarev, P.; Grant, B.D.; Kessels, M.M.; Qualmann, B. EHD proteins associate with syndapin I and II and such interactions play a crucial role in endosomal recycling. Mol. Biol. Cell 2005, 16, 3642–3658. [Google Scholar] [CrossRef] [PubMed]
- Grimm-Günter, E.-M.S.; Milbrandt, M.; Merkl, B.; Paulsson, M.; Plomann, M. PACSIN proteins bind tubulin and promote microtubule assembly. Exp. Cell Res. 2008, 314, 1991–2003. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Lv, K.; Li, Z.; Yu, A.C.H.; Chen, J.; Teng, J. PACSIN1, a Tau-interacting protein, regulates axonal elongation and branching by facilitating microtubule instability. J. Biol. Chem. 2012, 287, 39911–39924. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goodenough, D.A.; Paul, D.L. Beyond the gap: Functions of unpaired connexon channels. Nat. Rev. Mol. Cell Biol. 2003, 4, 285–294. [Google Scholar] [CrossRef]
- He, J.-T.; Li, X.-Y.; Yang, L.; Zhao, X. Astroglial connexins and cognition: Memory formation or deterioration? Biosci. Rep. 2020, 40, 1. [Google Scholar] [CrossRef] [PubMed]
- Mayorquin, L.C.; Rodriguez, A.V.; Sutachan, J.-J.; Albarracín, S.L. Connexin-Mediated Functional and Metabolic Coupling Between Astrocytes and Neurons. Front. Mol. Neurosci. 2018, 11, A822. [Google Scholar] [CrossRef]
- Nagy, J.I.; Li, W.; Hertzberg, E.L.; Marotta, C.A. Elevated connexin43 immunoreactivity at sites of amyloid plaques in Alzheimer’s disease. Brain Res. 1996, 717, 173–178. [Google Scholar] [CrossRef]
- Ren, R.; Zhang, L.; Wang, M. Specific deletion connexin43 in astrocyte ameliorates cognitive dysfunction in APP/PS1 mice. Life Sci. 2018, 208, 175–191. [Google Scholar] [CrossRef]
- Mei, X.; Ezan, P.; Giaume, C.; Koulakoff, A. Astroglial connexin immunoreactivity is specifically altered at β-amyloid plaques in β-amyloid precursor protein/presenilin1 mice. Neuroscience 2010, 171, 92–105. [Google Scholar] [CrossRef] [PubMed]
- Kajiwara, Y.; Wang, E.; Wang, M.; Sin, W.C.; Brennand, K.J.; Schadt, E.; Naus, C.C.; Buxbaum, J.; Zhang, B. GJA1 (connexin43) is a key regulator of Alzheimer’s disease pathogenesis. Acta Neuropathol. Commun. 2018, 6, 144. [Google Scholar] [CrossRef] [PubMed]
- Brackmann, M.; Schuchmann, S.; Anand, R.; Braunewell, K.-H. Neuronal Ca2+ sensor protein VILIP-1 affects cGMP signalling of guanylyl cyclase B by regulating clathrin-dependent receptor recycling in hippocampal neurons. J. Cell. Sci. 2005, 118, 2495–2505. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burgoyne, R.D. Neuronal calcium sensor proteins: Generating diversity in neuronal Ca2+ signalling. Nat. Rev. Neurosci. 2007, 8, 182–193. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Braunewell, K.-H.; Klein-Szanto, A.J. Visinin-like proteins (VSNLs): Interaction partners and emerging functions in signal transduction of a subfamily of neuronal Ca2+-sensor proteins. Cell Tissue Res. 2009, 335, 301–316. [Google Scholar] [CrossRef] [PubMed]
- Groblewska, M.; Muszyński, P.; Wojtulewska-Supron, A.; Kulczyńska-Przybik, A.; Mroczko, B. The Role of Visinin-Like Protein-1 in the Pathophysiology of Alzheimer’s Disease. J. Alzheimers Dis. 2015, 47, 17–32. [Google Scholar] [CrossRef] [PubMed]
- Laterza, O.F.; Modur, V.R.; Crimmins, D.L.; Olander, J.V.; Landt, Y.; Lee, J.-M.; Ladenson, J.H. Identification of novel brain biomarkers. Clin. Chem. 2006, 52, 1713–1721. [Google Scholar] [CrossRef] [PubMed]
- Braunewell, K.H.; Dwary, A.D.; Richter, F.; Trappe, K.; Zhao, C.; Giegling, I.; Schönrath, K.; Rujescu, D. Association of VSNL1 with schizophrenia, frontal cortical function, and biological significance for its gene product as a modulator of cAMP levels and neuronal morphology. Transl. Psychiatry 2011, 1, e22. [Google Scholar] [CrossRef]
- Lee, J.-M.; Blennow, K.; Andreasen, N.; Laterza, O.; Modur, V.; Olander, J.; Gao, F.; Ohlendorf, M.; Ladenson, J.H. The brain injury biomarker VLP-1 is increased in the cerebrospinal fluid of Alzheimer disease patients. Clin. Chem. 2008, 54, 1617–1623. [Google Scholar] [CrossRef] [Green Version]
- Tarawneh, R.; D’Angelo, G.; Macy, E.; Xiong, C.; Carter, D.; Cairns, N.J.; Fagan, A.M.; Head, D.; Mintun, M.A.; Ladenson, J.H.; et al. Visinin-like protein-1: Diagnostic and prognostic biomarker in Alzheimer disease. Ann. Neurol. 2011, 70, 274–285. [Google Scholar] [CrossRef] [Green Version]
- Tarawneh, R.; Lee, J.-M.; Ladenson, J.H.; Morris, J.C.; Holtzman, D.M. CSF VILIP-1 predicts rates of cognitive decline in early Alzheimer disease. Neurology 2012, 78, 709–719. [Google Scholar] [CrossRef] [Green Version]
- Kirkwood, C.M.; MacDonald, M.L.; Schempf, T.A.; Vatsavayi, A.V.; Ikonomovic, M.D.; Koppel, J.L.; Ding, Y.; Sun, M.; Kofler, J.K.; Lopez, O.L.; et al. Altered Levels of Visinin-Like Protein 1 Correspond to Regional Neuronal Loss in Alzheimer Disease and Frontotemporal Lobar Degeneration. J. Neuropathol. Exp. Neurol. 2016, 75, 175–182. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Keeney, P.M.; Xie, J.; Capaldi, R.A.; Bennett, J.P. Parkinson’s disease brain mitochondrial complex I has oxidatively damaged subunits and is functionally impaired and misassembled. J. Neurosci. 2006, 26, 5256–5264. [Google Scholar] [CrossRef] [PubMed]
- Andrés-Benito, P.; Gelpi, E.; Povedano, M.; Santpere, G.; Ferrer, I. Gene Expression Profile in Frontal Cortex in Sporadic Frontotemporal Lobar Degeneration-TDP. J. Neuropathol. Exp. Neurol. 2018, 77, 608–627. [Google Scholar] [CrossRef] [PubMed]
- Martins-de-Souza, D.; Gattaz, W.F.; Schmitt, A.; Rewerts, C.; Marangoni, S.; Novello, J.C.; Maccarrone, G.; Turck, C.W.; Dias-Neto, E. Alterations in oligodendrocyte proteins, calcium homeostasis and new potential markers in schizophrenia anterior temporal lobe are revealed by shotgun proteome analysis. J. Neural. Transm. (Vienna) 2009, 116, 275–289. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Piras, I.S.; Bleul, C.; Schrauwen, I.; Talboom, J.; Llaci, L.; De Both, M.D.; Naymik, M.A.; Halliday, G.; Bettencourt, C.; Holton, J.L.; et al. Transcriptional profiling of multiple system atrophy cerebellar tissue highlights differences between the parkinsonian and cerebellar sub-types of the disease. Acta Neuropathol. Commun. 2020, 8, 1–20. [Google Scholar] [CrossRef]
- Tang, R.; Liu, H. Identification of Temporal Characteristic Networks of Peripheral Blood Changes in Alzheimer’s Disease Based on Weighted Gene Co-expression Network Analysis. Front. Aging Neurosci. 2019, 11, 83. [Google Scholar] [CrossRef]
- Wang, Z.; Yan, X.; Zhao, C. Dynamical differential networks and modules inferring disrupted genes associated with the progression of Alzheimer’s disease. Exp. Ther. Med. 2017, 14, 2969–2975. [Google Scholar] [CrossRef] [Green Version]
- Lin, A.; Giuliano, C.J.; Palladino, A.; John, K.M.; Abramowicz, C.; Yuan, M.L.; Sausville, E.L.; Lukow, D.A.; Liu, L.; Chait, A.R.; et al. Off-target toxicity is a common mechanism of action of cancer drugs undergoing clinical trials. Sci. Transl. Med. 2019, 11, eaaw8412. [Google Scholar] [CrossRef]
- Meier, S.; Bell, M.; Lyons, D.N.; Rodriguez-Rivera, J.; Ingram, A.; Fontaine, S.N.; Mechas, E.; Chen, J.; Wolozin, B.; LeVine, H.; et al. Pathological Tau Promotes Neuronal Damage by Impairing Ribosomal Function and Decreasing Protein Synthesis. J. Neurosci. 2016, 36, 1001–1007. [Google Scholar] [CrossRef] [Green Version]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Koren, S.A.; Hamm, M.J.; Cloyd, R.; Fontaine, S.N.; Chishti, E.; Lanzillotta, C.; Rodriguez-Rivera, J.; Ingram, A.; Bell, M.; Galvis-Escobar, S.M.; et al. Broad Kinase Inhibition Mitigates Early Neuronal Dysfunction in Tauopathy. Int. J. Mol. Sci. 2021, 22, 1186. https://doi.org/10.3390/ijms22031186
Koren SA, Hamm MJ, Cloyd R, Fontaine SN, Chishti E, Lanzillotta C, Rodriguez-Rivera J, Ingram A, Bell M, Galvis-Escobar SM, et al. Broad Kinase Inhibition Mitigates Early Neuronal Dysfunction in Tauopathy. International Journal of Molecular Sciences. 2021; 22(3):1186. https://doi.org/10.3390/ijms22031186
Chicago/Turabian StyleKoren, Shon A., Matthew J. Hamm, Ryan Cloyd, Sarah N. Fontaine, Emad Chishti, Chiara Lanzillotta, Jennifer Rodriguez-Rivera, Alexandria Ingram, Michelle Bell, Sara M. Galvis-Escobar, and et al. 2021. "Broad Kinase Inhibition Mitigates Early Neuronal Dysfunction in Tauopathy" International Journal of Molecular Sciences 22, no. 3: 1186. https://doi.org/10.3390/ijms22031186