
Novel Short-Chain Quinones to Treat Vision Loss in a Rat Model of Diabetic Retinopathy


 , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
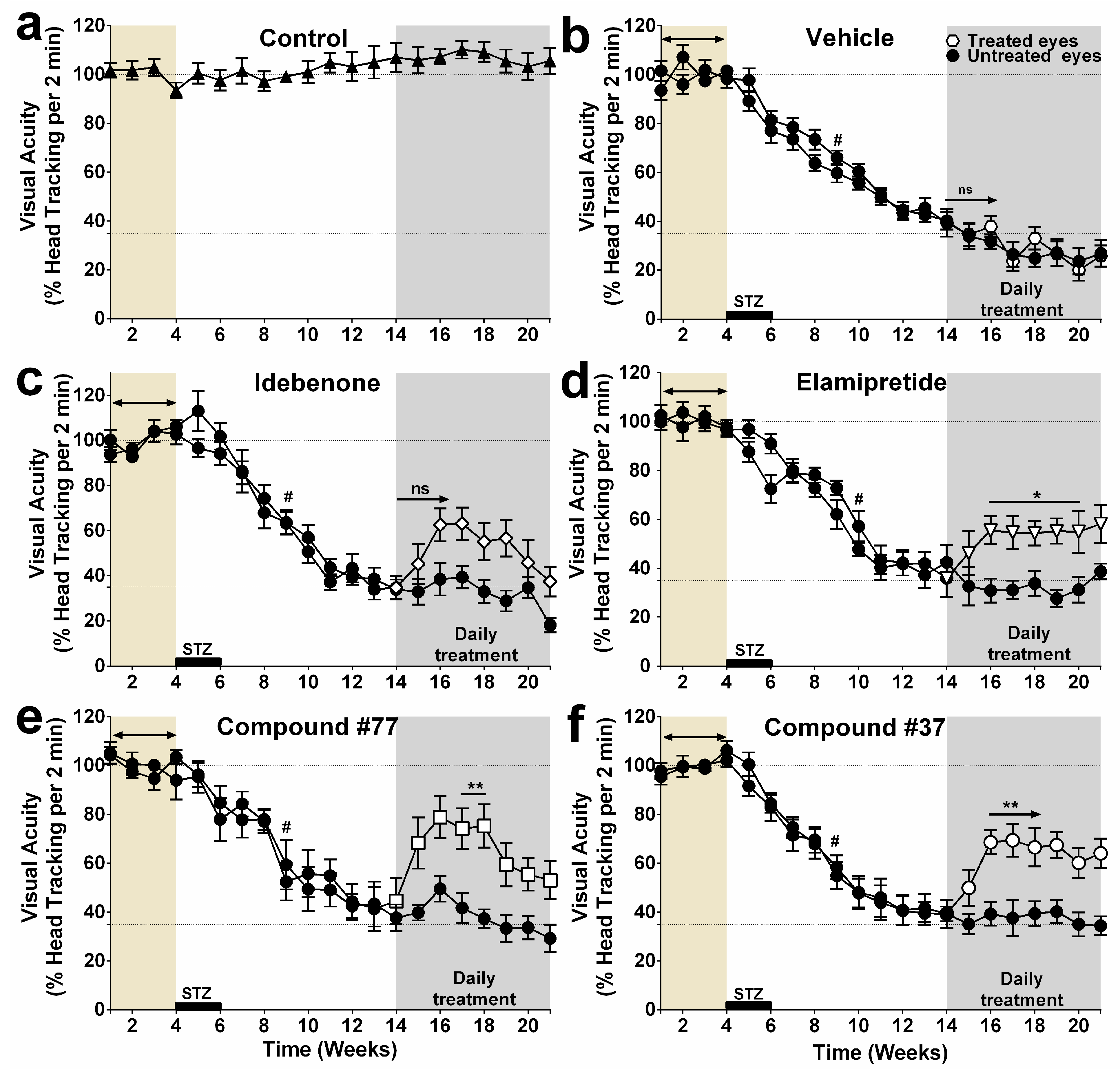
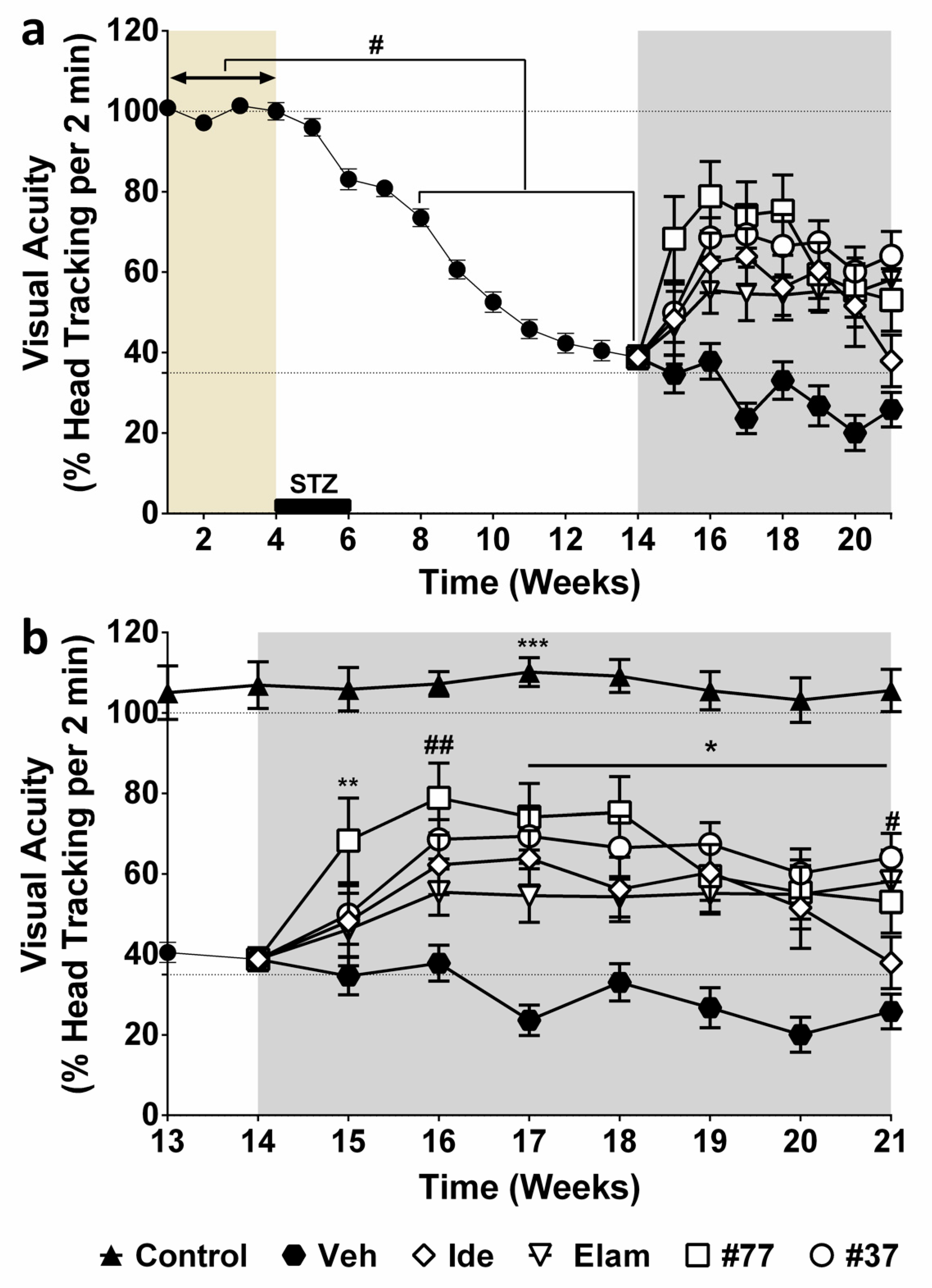
2.1. Effect of Test Compounds on Diabetes-Induced Vision Loss
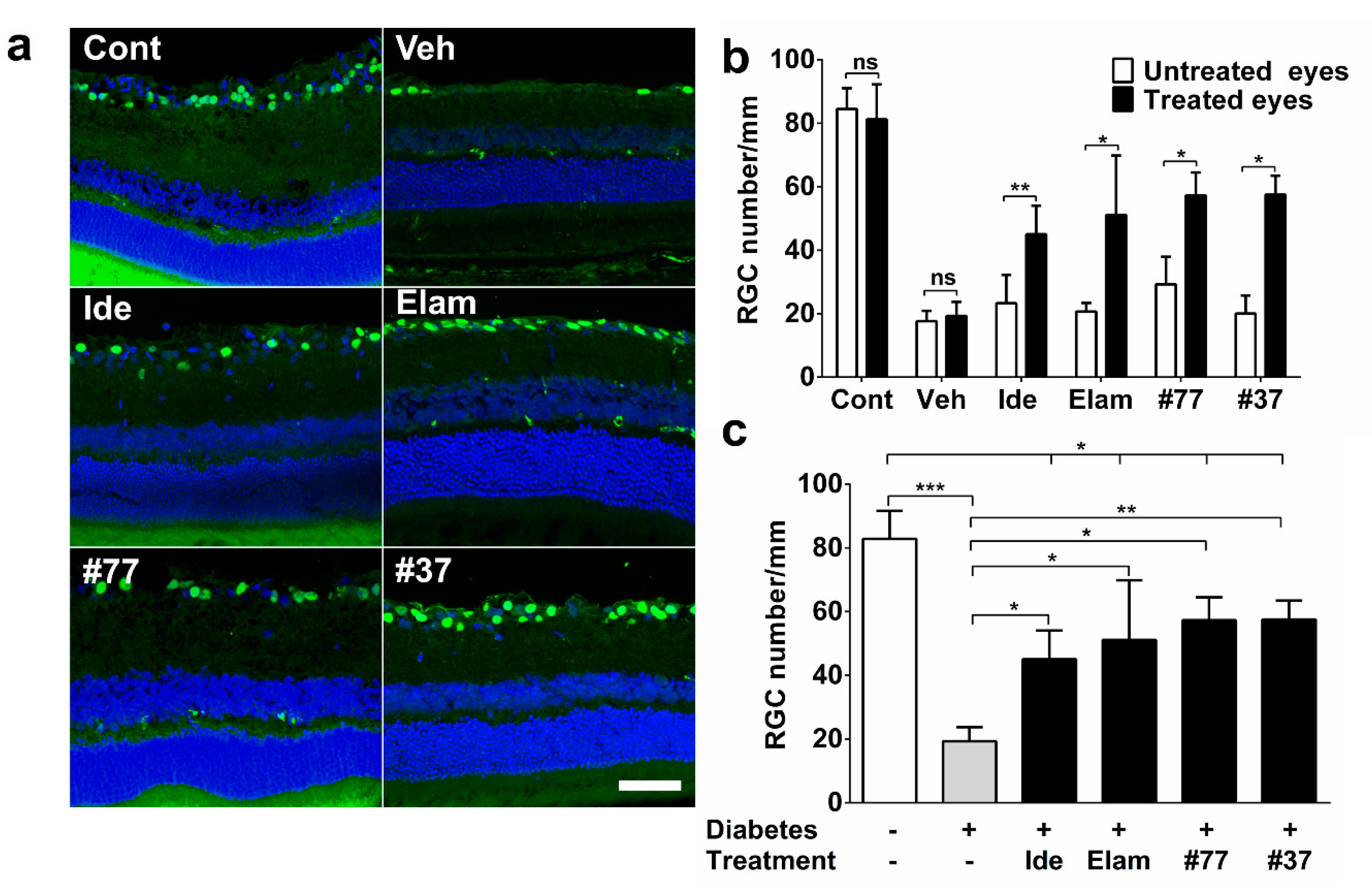
2.2. Short-Chain Quinones (SCQs) and Elamipretide Inhibit Diabetes-Induced Retinal Ganglion Cell Loss
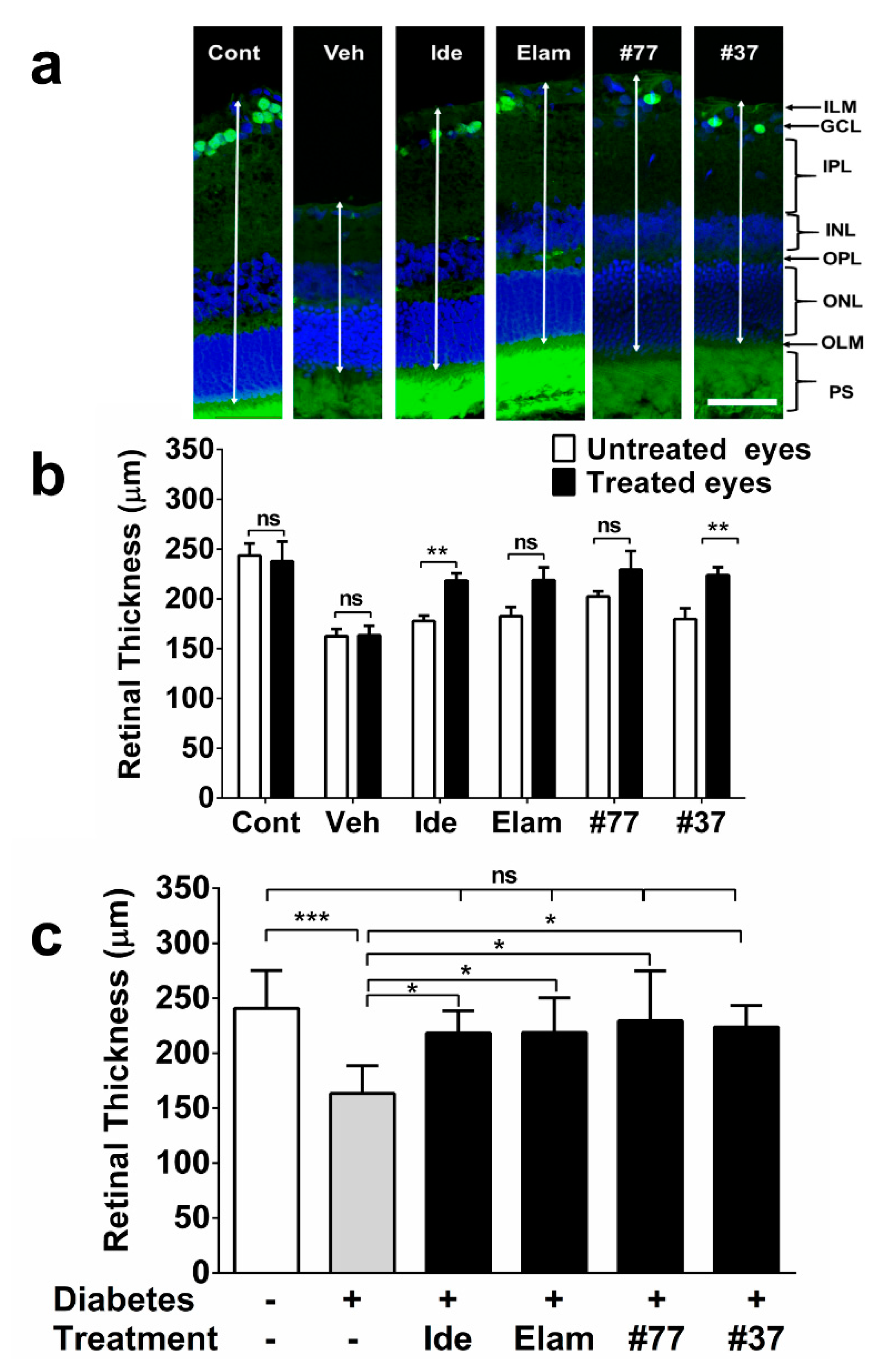
2.3. SCQs and Elamipretide Protect Against Diabetes-Induced Retinal Thinning
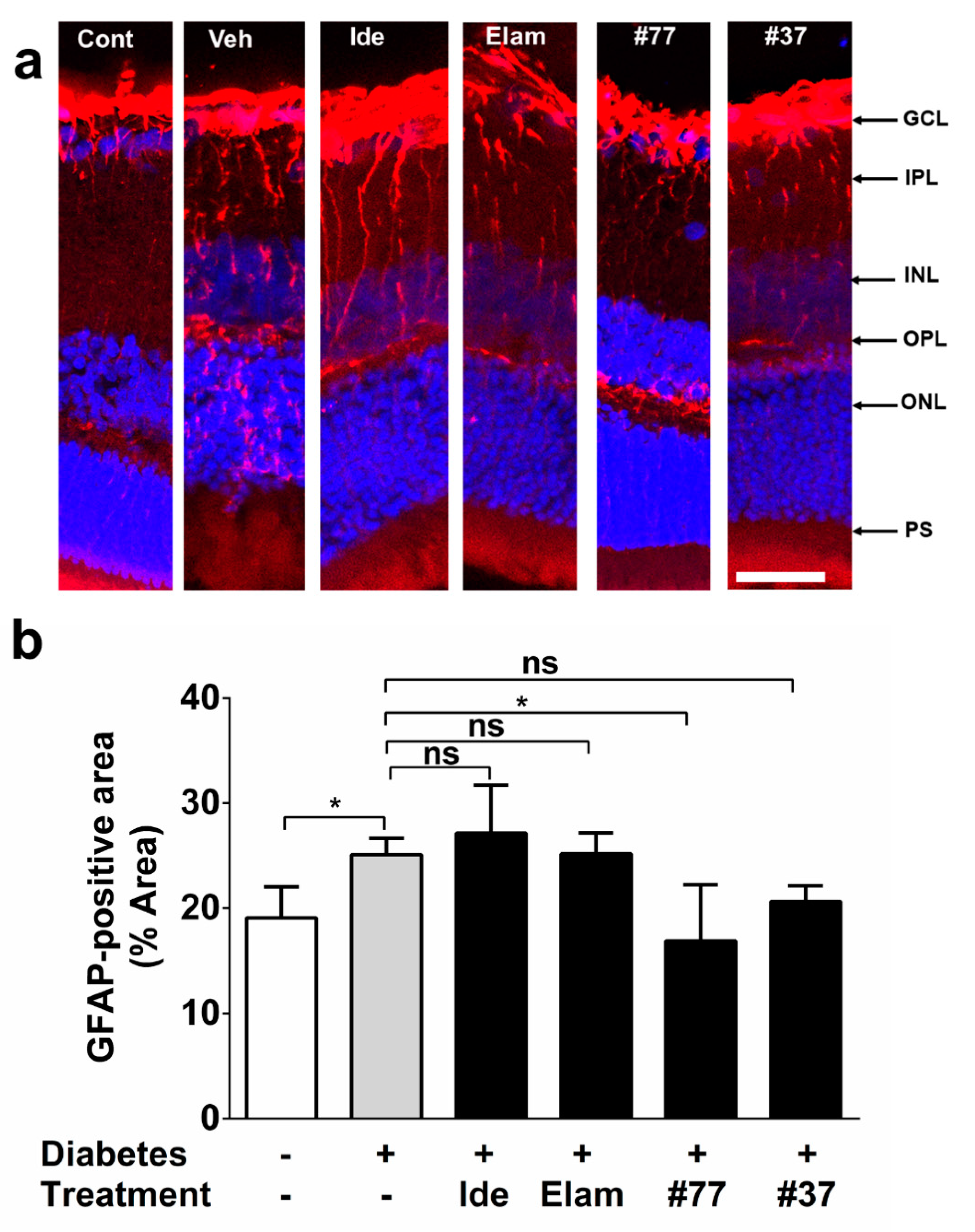
2.4. Novel SCQ Protects Against Diabetes-Induced Gliosis
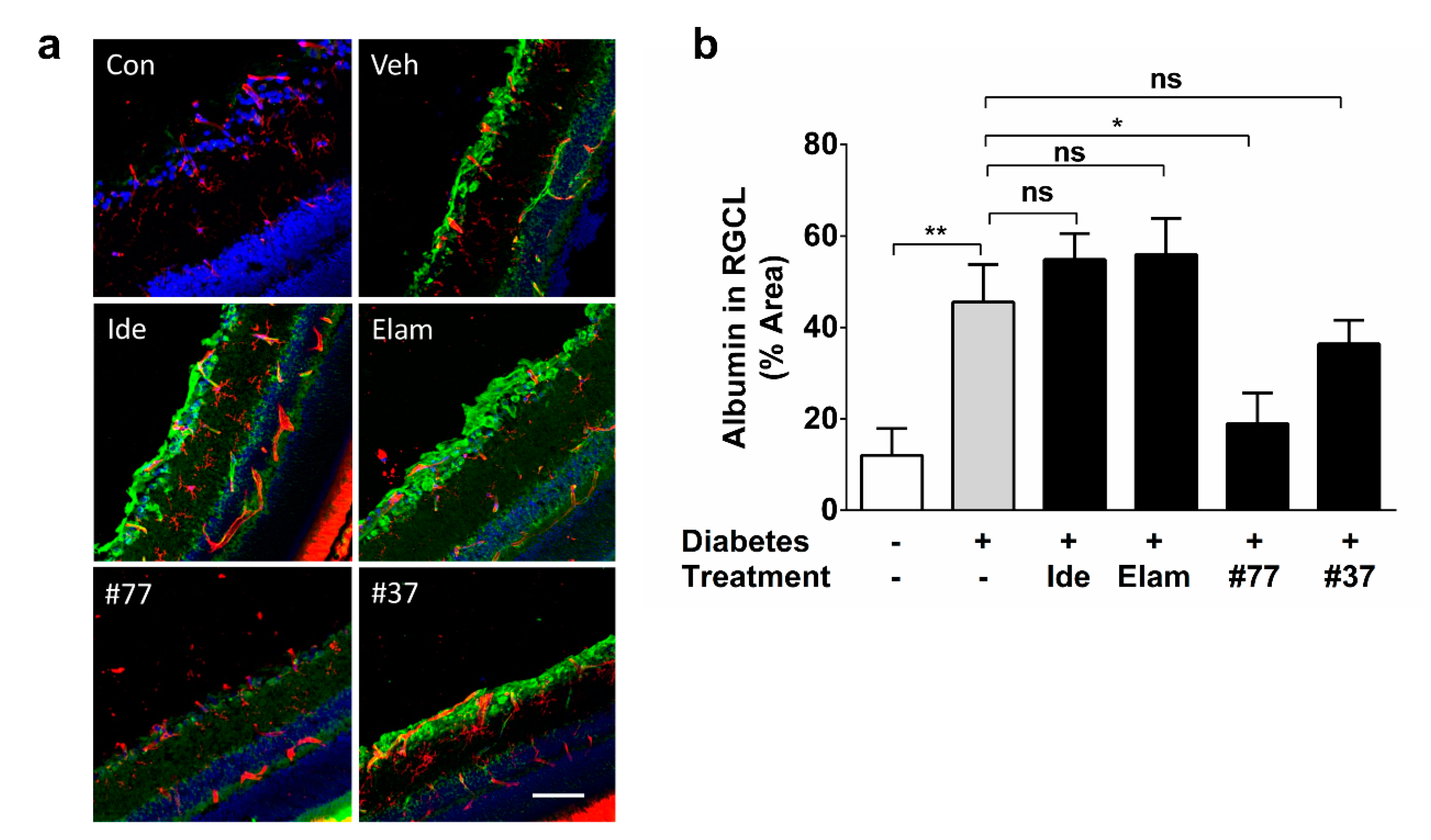
2.5. Novel SCQ Protects Against Diabetes-Induced Vascular Leakage
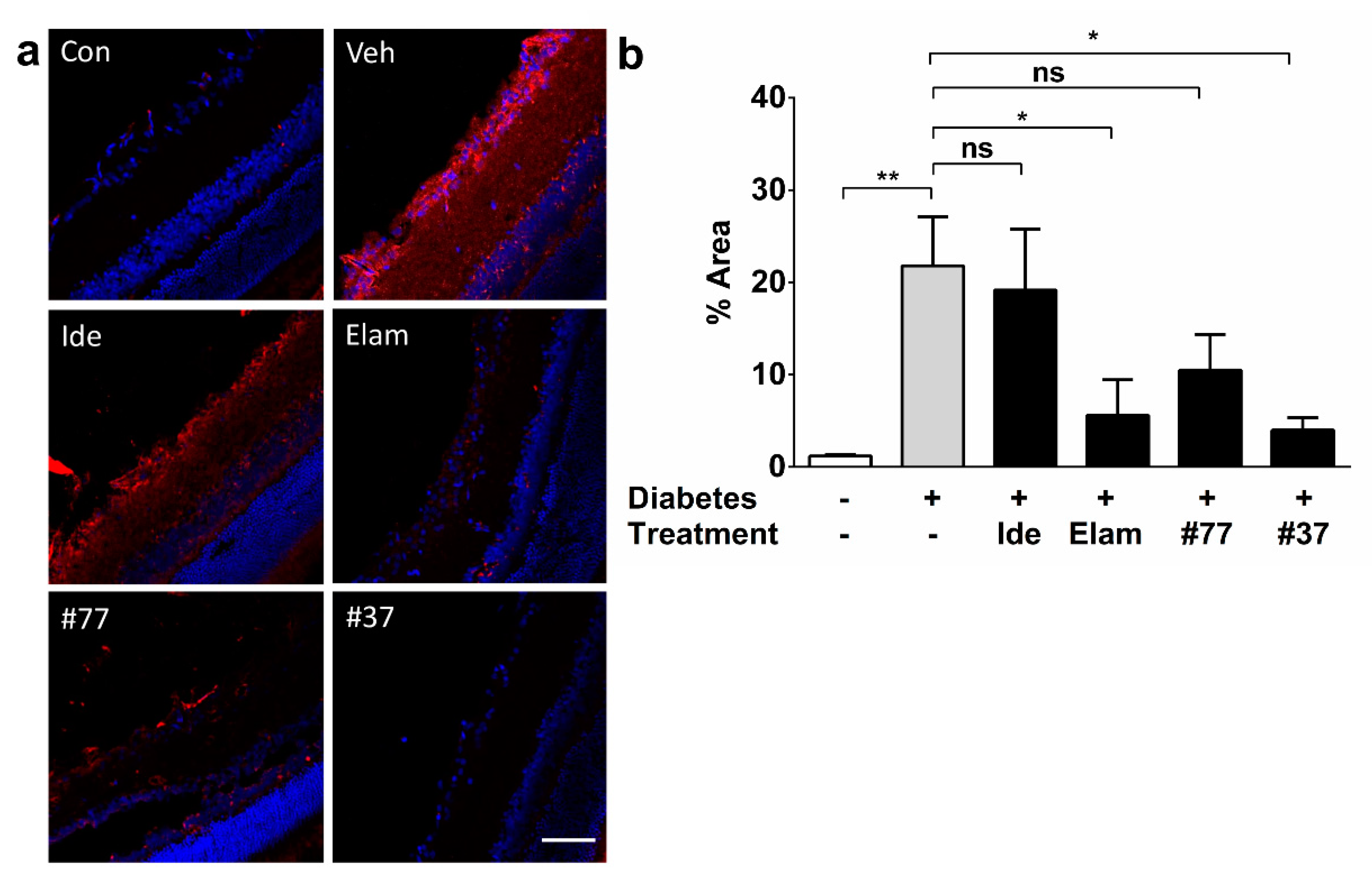
2.6. Novel SCQs and Elamipretide Inhibit Diabetes-Induced Oxidative Stress
3. Discussion
4. Materials and Methods
4.1. Test Compounds and Formulation
4.2. Animals
4.3. Measurement of Food, Water-Intake, and Body Weight
4.4. Blood Glucose Measurement
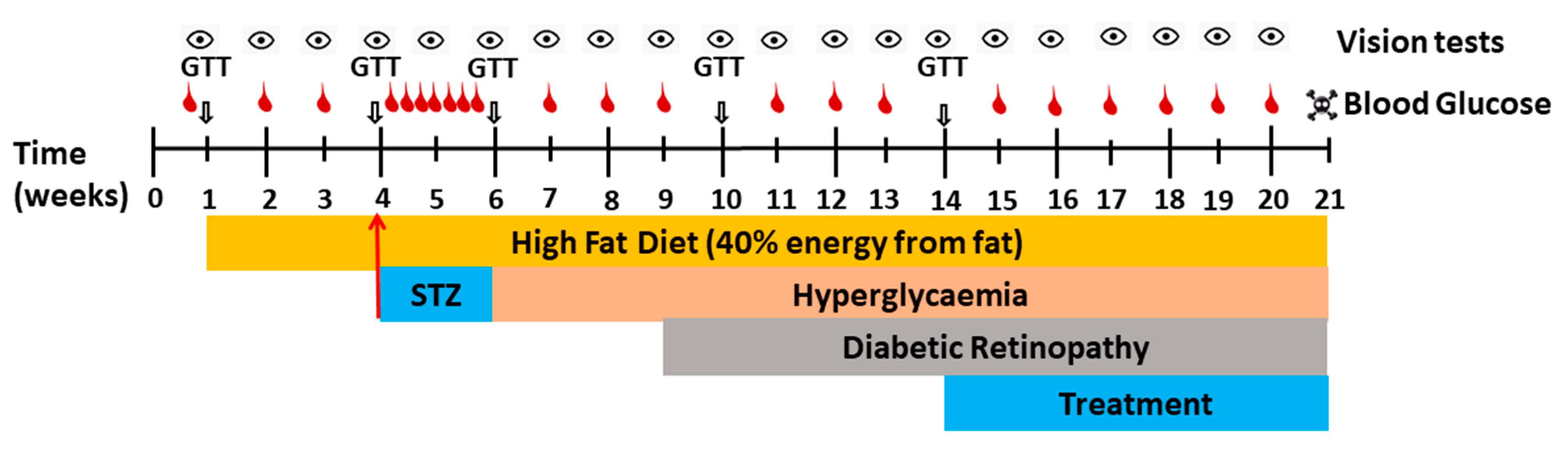
4.5. Induction of Diabetes Using STZ via Osmotic Minipump
4.6. Treatment with Test Compounds
4.7. Assessment of Visual Acuity in Rats Using Optokinetic Response
4.8. Immunohistochemistry
4.9. Image Acquisition
4.10. Exclusion Criteria
4.11. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Duh, E.J.; Sun, J.K.; Stitt, A.W. Diabetic retinopathy: Current understanding, mechanisms, and treatment strategies. JCI Insight 2017, 2. [Google Scholar] [CrossRef] [PubMed]
- Heng, L.Z.; Comyn, O.; Peto, T.; Tadros, C.; Ng, E.; Sivaprasad, S.; Hykin, P.G. Diabetic retinopathy: Pathogenesis, clinical grading, management and future developments. Diabet. Med. 2013, 30, 640–650. [Google Scholar] [CrossRef] [PubMed]
- Kirwin, S.J.; Kanaly, S.T.; Hansen, C.R.; Cairns, B.J.; Ren, M.; Edelman, J.L. Retinal Gene Expression and Visually Evoked Behavior in Diabetic Long Evans Rats. Investig. Ophthalmol. Vis. Sci. 2011, 52, 7654–7663. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rodrigues, E.B.; Urias, M.G.; Penha, F.M.; Badaró, E.; Novais, E.A.; Meirelles, R.; Farah, M. Diabetes induces changes in neuroretina before retinal vessels: A spectral-domain optical coherence tomography study. Int. J. Retin. Vitr. 2015, 1, 4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, W.; Lo, A.C.Y. Diabetic Retinopathy: Pathophysiology and Treatments. Int. J. Mol. Sci. 2018, 19, 1816. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bandello, F.; Toni, D.; Porta, M.; Varano, M. Diabetic retinopathy, diabetic macular edema, and cardiovascular risk: The importance of a long-term perspective and a multidisciplinary approach to optimal intravitreal therapy. Acta Diabetol. 2020, 57, 513–526. [Google Scholar] [CrossRef]
- Bonfiglio, V.; Platania, C.B.M.; Lazzara, F.; Conti, F.; Pizzo, C.; Reibaldi, M.; Russo, A.; Fallico, M.; Ortisi, E.; Pignatelli, F.; et al. TGF-β Serum Levels in Diabetic Retinopathy Patients and the Role of Anti-VEGF Therapy. Int. J. Mol. Sci. 2020, 21, 9558. [Google Scholar] [CrossRef]
- Alam, N.M.; Mills, W.C.; Wong, A.A.; Douglas, R.M.; Szeto, H.H.; Prusky, G.T. A mitochondrial therapeutic reverses visual decline in mouse models of diabetes. Dis. Model. Mech. 2015, 8, 701–710. [Google Scholar] [CrossRef] [Green Version]
- Arévalo, J.F.; Sanchez, J.G.; Lasave, A.F.; Wu, L.; Maia, M.; Bonafonte, S.; Brito, M.; Alezzandrini, A.A.; Restrepo, N.; Berrocal, M.H.; et al. Intravitreal Bevacizumab (Avastin) for Diabetic Retinopathy: The 2010 GLADAOF Lecture. J. Ophthalmol. 2011, 2011, 1–13. [Google Scholar] [CrossRef]
- Aung, M.H.; Kim, M.K.; Olson, D.E.; Thule, P.M.; Pardue, M.T. Early Visual Deficits in Streptozotocin-Induced Diabetic Long Evans Rats. Investig. Ophthalmol. Vis. Sci. 2013, 54, 1370–1377. [Google Scholar] [CrossRef]
- Lee, H.B.; Yu, M.-R.; Yang, Y.; Jiang, Z.; Ha, H. Reactive Oxygen Species-Regulated Signaling Pathways in Diabetic Nephropathy. J. Am. Soc. Nephrol. 2003, 14, S241–S245. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kowluru, R.A. Diabetic Retinopathy: Mitochondrial Dysfunction and Retinal Capillary Cell Death. Antioxid. Redox Signal. 2005, 7, 1581. [Google Scholar] [CrossRef] [PubMed]
- Gueven, N.; Nadikudi, M.; Daniel, A.; Chhetri, J. Targeting mitochondrial function to treat optic neuropathy. Mitochondrion 2017, 36, 7–14. [Google Scholar] [CrossRef] [PubMed]
- Haefeli, R.H.; Erb, M.; Gemperli, A.C.; Robay, D.; Fruh, I.C.; Anklin, C.; Dallmann, R.; Gueven, N. NQO1-Dependent Redox Cycling of Idebenone: Effects on Cellular Redox Potential and Energy Levels. PLoS ONE 2011, 6, e17963. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Becker, C.; Bray-French, K.; Drewe, J. Pharmacokinetic evaluation of idebenone. Expert Opin. Drug Metab. Toxicol. 2010, 6, 1437–1444. [Google Scholar] [CrossRef]
- Li, J.; Chen, X.; Xiao, W.; Ma, W.; Li, T.; Huang, J.; Liu, X.; Liang, X.; Tang, S.; Luo, Y. Mitochondria-targeted antioxidant peptide SS31 attenuates high glucose-induced injury on human retinal endothelial cells. Biochem. Biophys. Res. Commun. 2011, 404, 349–356. [Google Scholar] [CrossRef]
- Premilovac, D.; Gasperini, R.J.; Sawyer, S.; West, A.; Keske, M.A.; Taylor, B.; Foa, L. A New Method for Targeted and Sustained Induction of Type 2 Diabetes in Rodents. Sci. Rep. 2017, 7, 14158. [Google Scholar] [CrossRef] [Green Version]
- Woolley, K.L.; Nadikudi, M.; Koupaei, M.N.; Corban, M.; McCartney, P.; Bissember, A.C.; Lewis, T.W.; Gueven, N.; Smith, J.A.; Gueven, N. Amide linked redox-active naphthoquinones for the treatment of mitochondrial dysfunction. MedChemComm 2019, 10, 399–412. [Google Scholar] [CrossRef]
- Feng, Z.; Smith, J.A.; Gueven, N.; Quirino, J.P. Metabolic Stability of New Mito-Protective Short-Chain Naphthoquinones. Pharmaceuticals 2020, 13, 29. [Google Scholar] [CrossRef] [Green Version]
- Nguyen, Q.D.; Brown, D.M.; Marcus, D.M.; Boyer, D.; Patel, S.; Feiner, L.; Gibson, A.; Sy, J.P.; Rundle, A.C.; Hopkins, J.J.; et al. Ranibizumab for Diabetic Macular Edema. Ophthalmology 2012, 119, 789–801. [Google Scholar] [CrossRef]
- Sohn, E.H.; Van Dijk, H.W.; Jiao, C.; Kok, P.H.B.; Jeong, W.; Demirkaya, N.; Garmager, A.; Wit, F.; Kucukevcilioglu, M.; Van Velthoven, M.E.J.; et al. Retinal neurodegeneration may precede microvascular changes characteristic of diabetic retinopathy in diabetes mellitus. Proc. Natl. Acad. Sci. USA 2016, 113, E2655–E2664. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vujosevic, S.; Muraca, A.; Alkabes, M.; Villani, E.; Cavarzeran, F.; Rossetti, L.; De Cilla, S. Early microvascular and neural changes in patients with type 1 and type 2 diabetes mellitus without clinical signs of diabetic retinopathy. Retina 2019, 39, 435–445. [Google Scholar] [CrossRef] [PubMed]
- Platania, C.B.; Maisto, R.; Trotta, M.C.; D’Amico, M.; Rossi, S.; Gesualdo, C.; D’Amico, G.; Balta, C.; Herman, H.; Hermenean, A.; et al. Retinal and circulating miRNA expression patterns in diabetic retinopathy: An in silico and in vivo approach. Br. J. Pharmacol. 2019, 176, 2179–2194. [Google Scholar] [CrossRef] [PubMed]
- Zheng, L.; Du, Y.; Miller, C.; Gubitosi-Klug, R.A.; Kern, T.S.; Ball, S.; Berkowitz, B.A. Critical role of inducible nitric oxide synthase in degeneration of retinal capillaries in mice with streptozotocin-induced diabetes. Diabetologia 2007, 50, 1987–1996. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hajdú, R.I.; Laurik, L.K.; Szabó, K.; Dékány, B.; Almási, Z.; Énzsöly, A.; Szabó, A.; Radovits, T.; Mátyás, C.; Oláh, A.; et al. Detailed Evaluation of Possible Ganglion Cell Loss in the Retina of Zucker Diabetic Fatty (ZDF) Rats. Sci. Rep. 2019, 9, 1–16. [Google Scholar] [CrossRef] [Green Version]
- Howell, S.J.; Mekhail, M.N.; Azem, R.; Ward, N.L.; Kern, T.S. Degeneration of retinal ganglion cells in diabetic dogs and mice: Relationship to glycemic control and retinal capillary degeneration. Mol. Vis. 2013, 19, 1413–1421. [Google Scholar]
- Énzsöly, A.; Szabo, A.; Szabó, K.; Szél, Á; Németh, J.; Lukáts, Á. Novel features of neurodegeneration in the inner retina of early diabetic rats. Histol. Histopathol. 2015, 30, 971–985. [Google Scholar] [CrossRef]
- Howell, N. Leber hereditary optic neuropathy: Respiratory chain dysfunction and degeneration of the optic nerve. Vis. Res. 1998, 38, 1495–1504. [Google Scholar] [CrossRef] [Green Version]
- Romano, G.L.; Amato, R.; Lazzara, F.; Porciatti, V.; Chou, T.-H.; Drago, F.; Bucolo, C. P2X7 receptor antagonism preserves retinal ganglion cells in glaucomatous mice. Biochem. Pharmacol. 2020, 180, 114199. [Google Scholar] [CrossRef]
- Gueven, N. Optic Neurodegeneration: Time to Act. Biol. Med. 2014, 1. [Google Scholar] [CrossRef] [Green Version]
- Pemp, B.; Kircher, K.; Reitner, A. Visual function in chronic Leber’s hereditary optic neuropathy during idebenone treatment initiated 5 to 50 years after onset. Graefe’s Arch. Clin. Exp. Ophthalmol. 2019, 257, 2751–2757. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heitz, F.D.; Erb, M.; Anklin, C.; Robay, D.; Pernet, V.; Gueven, N. Idebenone Protects against Retinal Damage and Loss of Vision in a Mouse Model of Leber’s Hereditary Optic Neuropathy. PLoS ONE 2012, 7, e45182. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weerasekera, L.Y.; Balmer, L.A.; Ram, R.; Morahan, G. Characterization of Retinal Vascular and Neural Damage in a Novel Model of Diabetic Retinopathy. Investig. Ophthalmol. Vis. Sci. 2015, 56, 3721–3730. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abu El-Asrar, A.M.; Desmet, S.; Meersschaert, A.; Dralands, L.; Missotten, L.; Geboes, K. Expression of the inducible isoform of nitric oxide synthase in the retinas of human subjects with diabetes mellitus. Am. J. Ophthalmol. 2001, 132, 551–556. [Google Scholar] [CrossRef]
- Bringmann, A.; Iandiev, I.; Pannicke, T.; Wurm, A.; Hollborn, M.; Wiedemann, P.; Osborne, N.N.; Reichenbach, A. Cellular signaling and factors involved in Müller cell gliosis: Neuroprotective and detrimental effects. Prog. Retin. Eye Res. 2009, 28, 423–451. [Google Scholar] [CrossRef]
- Meyer, A.; Laverny, G.; Bernardi, L.; Charles, A.L.; Alsaleh, G.; Pottecher, J.; Sibilia, J.; Geny, B. Mitochondria: An Organelle of Bacterial Origin Controlling Inflammation. Front. Immunol. 2018, 9, 536. [Google Scholar] [CrossRef] [PubMed]
- West, A.P. Mitochondrial dysfunction as a trigger of innate immune responses and inflammation. Toxicology 2017, 391, 54–63. [Google Scholar] [CrossRef]
- Barber, A.J.; Antonetti, D.A.; Gardner, T.W.; The Penn State Retina Research Group. Altered Expression of Retinal Occludin and Glial Fibrillary Acidic Protein in Experimental Diabetes. Investig. Ophthalmol. Vis. Sci. 2000, 41, 3561–3568. [Google Scholar]
- Sadik, N.A.H.; El-Boghdady, N.A.; Omar, N.; Al-Hamid, H.A. Esculetin and idebenone ameliorate galactose-induced cataract in a rat model. J. Food Biochem. 2020, 44, e13230. [Google Scholar] [CrossRef] [PubMed]
- Santos, J.M.; Kowluru, R.A. Retinal Mitochondrial Transport Machinery and Decreased Mitochondria Biogenesis In The Development Of Diabetic Retinopathy. Investig. Ophthalmol. Vis. Sci. 2012, 53, 5417. [Google Scholar]
- Tomilov, A.; Allen, S.A.; Hui, C.; Bettaieb, A.; Cortopassi, G. Idebenone is a cytoprotective insulin sensitizer whose mechanism is Shc inhibition. Pharmacol. Res. 2018, 137, 89–103. [Google Scholar] [CrossRef] [PubMed]
- Mishra, M.; Duraisamy, A.J.; Bhattacharjee, S.; Kowluru, R.A. Adaptor Protein p66Shc: A Link Between Cytosolic and Mitochondrial Dysfunction in the Development of Diabetic Retinopathy. Antioxid. Redox Signal. 2019, 30, 1621–1634. [Google Scholar] [CrossRef] [PubMed]
- Kowluru, R.A.; Kowluru, A.; Veluthakal, R.; Mohammad, G.; Syed, I.; Santos, J.M.; Mishra, M. TIAM1–RAC1 signalling axis-mediated activation of NADPH oxidase-2 initiates mitochondrial damage in the development of diabetic retinopathy. Diabetololia 2014, 57, 1047–1056. [Google Scholar] [CrossRef] [PubMed]
- Tiefenbach, J.; Magomedova, L.; Liu, J.; Reunov, A.A.; Tsai, R.; Eappen, N.S.; Jockusch, R.A.; Nislow, C.; Cummins, C.L.; Krause, H.M. Idebenone and coenzyme Q10are novel PPARα/γ ligands, with potential for treatment of fatty liver diseases. Dis. Model. Mech. 2018, 11, dmm034801. [Google Scholar] [CrossRef] [Green Version]
- Jacot, J.G.; Sherris, D. Potential Therapeutic Roles for Inhibition of the PI3K/Akt/mTOR Pathway in the Pathophysiology of Diabetic Retinopathy. J. Ophthalmol. 2011, 2011, 1–19. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.-W.; Li, Q.; Liu, C.-M.; Hall, P.A.; Jiang, J.-J.; Katchis, C.D.; Kang, S.; Dong, B.C.; Li, S.; Zhou, F.-Q. Lin28 Signaling Supports Mammalian PNS and CNS Axon Regeneration. Cell Rep. 2018, 24, 2540–2552. [Google Scholar] [CrossRef] [Green Version]
- Lei, D.; Shao, Z.; Zhou, X.; Yuan, H. Synergistic neuroprotective effect of rasagiline and idebenone against retinal ischemia-reperfusion injury via the Lin28-let-7-Dicer pathway. Oncotarget 2018, 9, 12137–12153. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- National Health and Medical Research Council. Australian Code for the Care and Use of Animals for Scientific Purposes, 8th ed.; National Health and Medical Research Council: Canberra, ACT, Australia, 2013. [Google Scholar]
- Premilovac, D.; Bradley, E.A.; Ng, H.L.; Richards, S.M.; Rattigan, S.; Keske, M.A. Muscle insulin resistance resulting from impaired microvascular insulin sensitivity in Sprague Dawley rats. Cardiovasc. Res. 2013, 98, 28–36. [Google Scholar] [CrossRef] [Green Version]
- Prusky, G.T.; West, P.W.; Douglas, R.M. Behavioral assessment of visual acuity in mice and rats. Vis. Res. 2000, 40, 2201–2209. [Google Scholar] [CrossRef] [Green Version]
- Lund, R.D.; Adamson, P.; Sauve, Y.; Keegan, D.J.; Girman, S.V.; Wang, S.; Winton, H.; Kanuga, N.; Kwan, A.S.L.; Beauchène, L.; et al. Subretinal transplantation of genetically modified human cell lines attenuates loss of visual function in dystrophic rats. Proc. Natl. Acad. Sci. USA 2001, 98, 9942–9947. [Google Scholar] [CrossRef] [Green Version]
- Coffey, P.J.; Girman, S.; Wang, S.M.; Hetherington, L.; Keegan, D.J.; Adamson, P.; Greenwood, J.; Lund, R.D. Long-term preservation of cortically dependent visual function in RCS rats by transplantation. Nat. Neurosci. 2001, 5, 53–56. [Google Scholar] [CrossRef] [PubMed]
- Mead, B.; Thompson, A.; Scheven, B.A.; Logan, A.; Berry, M.; Leadbeater, W. Comparative Evaluation of Methods for Estimating Retinal Ganglion Cell Loss in Retinal Sections and Wholemounts. PLoS ONE 2014, 9, e110612. [Google Scholar] [CrossRef] [PubMed]
- Hernández, C.; Bogdanov, P.; Solà-Adell, C.; Sampedro, J.; Valeri, M.; Genís, X.; Simó-Servat, O.; García-Ramírez, M.; Simó, R. Topical administration of DPP-IV inhibitors prevents retinal neurodegeneration in experimental diabetes. Diabetologia 2017, 60, 2285–2298. [Google Scholar] [CrossRef] [PubMed]
- Anderson, P.J.; Watts, H.; Hille, C.; Philpott, K.; Clark, P.; Gentleman, M.C.S.; Jen, L.-S. Glial and endothelial blood-retinal barrier responses to amyloid-β in the neural retina of the rat. Clin. Ophthalmol. 2008, 2, 801–816. [Google Scholar] [CrossRef] [PubMed]








Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Daniel, A.; Premilovac, D.; Foa, L.; Feng, Z.; Shah, K.; Zhang, Q.; Woolley, K.L.; Bye, N.; Smith, J.A.; Gueven, N. Novel Short-Chain Quinones to Treat Vision Loss in a Rat Model of Diabetic Retinopathy. Int. J. Mol. Sci. 2021, 22, 1016. https://doi.org/10.3390/ijms22031016
Daniel A, Premilovac D, Foa L, Feng Z, Shah K, Zhang Q, Woolley KL, Bye N, Smith JA, Gueven N. Novel Short-Chain Quinones to Treat Vision Loss in a Rat Model of Diabetic Retinopathy. International Journal of Molecular Sciences. 2021; 22(3):1016. https://doi.org/10.3390/ijms22031016
Chicago/Turabian StyleDaniel, Abraham, Dino Premilovac, Lisa Foa, Zikai Feng, Krupali Shah, Qianyi Zhang, Krystel L. Woolley, Nicole Bye, Jason A. Smith, and Nuri Gueven. 2021. "Novel Short-Chain Quinones to Treat Vision Loss in a Rat Model of Diabetic Retinopathy" International Journal of Molecular Sciences 22, no. 3: 1016. https://doi.org/10.3390/ijms22031016