Overcoming Obstacles to Targeting Muscarinic Receptor Signaling in Colorectal Cancer


1
Department of Medicine, Division of Gastroenterology & Hepatology, University of Maryland School of Medicine, Baltimore, MA 21201, USA
2
Veterans Affairs Maryland Healthcare System, Baltimore, MA 21201, USA
3
Marlene and Stewart Greenebaum Comprehensive Cancer Center, University of Maryland School of Medicine, Baltimore, MA 21201, USA
4
Department of Biochemistry and Molecular Biology, University of Maryland School of Medicine, Baltimore, MA 21201, USA
*
Author to whom correspondence should be addressed.
Int. J. Mol. Sci. 2021, 22(2), 716; https://doi.org/10.3390/ijms22020716
Submission received: 18 December 2020
/
Revised: 8 January 2021
/
Accepted: 10 January 2021
/
Published: 13 January 2021
(This article belongs to the Special Issue Cholinergic Signaling in Human Health and Diseases)
{kind=link}
Abstract
:Despite great advances in our understanding of the pathobiology of colorectal cancer and the genetic and environmental factors that mitigate its onset and progression, a paucity of effective treatments persists. The five-year survival for advanced, stage IV disease remains substantially less than 20%. This review examines a relatively untapped reservoir of potential therapies to target muscarinic receptor expression, activation, and signaling in colorectal cancer. Most colorectal cancers overexpress M3 muscarinic receptors (M3R), and both in vitro and in vivo studies have shown that activating these receptors stimulates cellular programs that result in colon cancer growth, survival, and spread. In vivo studies using mouse models of intestinal neoplasia have shown that using either genetic or pharmacological approaches to block M3R expression and activation, respectively, attenuates the development and progression of colon cancer. Moreover, both in vitro and in vivo studies have shown that blocking the activity of matrix metalloproteinases (MMPs) that are induced selectively by M3R activation, i.e., MMP1 and MMP7, also impedes colon cancer growth and progression. Nonetheless, the widespread expression of muscarinic receptors and MMPs and their importance for many cellular functions raises important concerns about off-target effects and the safety of employing similar strategies in humans. As we highlight in this review, highly selective approaches can overcome these obstacles and permit clinicians to exploit the reliance of colon cancer cells on muscarinic receptors and their downstream signal transduction pathways for therapeutic purposes.
1. Introduction
Colorectal cancer (CRC) is the second and third most commonly occurring cancer in women and men, respectively; approximately 1.8 million new cases were reported worldwide in 2018 [1]. Most cases occur in industrialized countries with high consumption of so-called ‘western diets’ [2]. In the United States (U.S.), it is estimated that 147,950 persons were diagnosed with CRC in 2020, comprising 104,610 colon and 43,340 rectal cancers. Although CRC is predominantly diagnosed in persons 50 years and older, the increasing prevalence of CRC in younger persons, particularly African American men, is concerning; it is estimated that 17,930 new cases of CRC will occur in persons younger than 50 years [3]. Overall, in the U.S. in 2020 it is anticipated 52,300 deaths will be attributed to CRC [3,4]. Despite advances in screening and public health initiatives to increase CRC awareness and the importance of screening, CRC remains a leading cause of cancer morbidity and mortality. It is particularly concerning that despite decreasing rates in those older than 55 years, for currently unknown reasons, the incidence of metastatic CRC is increasing at an alarming rate in those who are younger [5,6].
The management of CRC depends on tumor stage, location, and other patient-specific characteristics. Surgical excision remains the mainstay of therapy for early-stage CRC; however, treatment options for advanced metastatic disease remain focused on adjuvant or neoadjuvant chemotherapy, depending on whether the individual meets criteria for surgery [7]. Based on evidence of improved response rates and progression-free survival, first-line chemotherapy for metastatic CRC (mCRC), e.g., capecitabine and 5-fluorouracil (5-FU), is commonly combined with other cytotoxic agents such as irinotecan or oxaliplatin [8]. Nevertheless, despite improved treatment strategies the majority of patients with advanced mCRC succumb to disease within five years as a consequence of initial or acquired drug resistance and lack of efficacy [9].
The principal mechanism underlying resistance to chemotherapy and subsequent cancer progression is altered programmed cell death (apoptosis), which is controlled by intricate signaling pathways. Chemotherapeutic regimens such as FOLFOX (5-FU/leucovorin/oxaliplatin) and FOLFOXIRI (5-FU/leucovorin/oxaliplatin/irinotecan) generally promote apoptosis by killing susceptible cells and allowing resistant cells to proliferate and repopulate tumors [7]. Thus, targeting the mechanisms that promote cell survival (i.e., resistance to apoptosis) has become a promising strategy to treat mCRC; this is largely accomplished by combining FOLFOX or FOLFOXIRI with monoclonal antibodies and kinase inhibitors targeting specific survival pathways, e.g., those downstream of epidermal and vascular endothelial growth factor receptors (EGFR and VEGF) [10]. Nonetheless, even these advances generally provide limited response rates, measured in months gained, and mCRC continues to have a dismal five-year survival in the range of 14.3 % [11].
Novel immunotherapeutics have shown great benefit for some cancers, but their efficacy for mCRC appears limited to those with sporadic or inherited mismatch repair defects. Overall, it is estimated that fewer than 10% of patients with mCRC will derive benefit from immune checkpoint inhibitors and, even then, responses in a small subset of patients are limited to only months of added life [12,13].
To address these challenges, newer approaches to target key signaling pathways that mediate cancer cell proliferation, survival, invasion, and metastasis are imperative. For example, as mutations in receptor or downstream targets such as c-MYC, EGFR, KRAS, NRAS, BRAF, PTEN, or SMAD2/4 confer resistance to therapy and shorter survival, major efforts are underway to overcome these obstacles to successful therapy [14]. In addition to genetic mutations, alterations in non-coding RNAs, e.g., microRNAs, that mediate post-transcriptional regulation of protein expression may alter tumor progression and serve as both prognostic biomarkers and therapeutic targets. While further treatments are being developed and tested for efficacy, a better understanding of disease pathobiology is likely to identify novel therapeutic targets [15].
In this context, the M3 subtype cholinergic muscarinic receptor (M3R) was identified as a promotor and regulator of colon cancer cell progression. M3R activation promotes CRC progression by both EGFR-dependent and -independent mechanisms [16]. Elucidating the molecular mechanisms underlying these pathways has already provided valuable insights into potential downstream targets. Here, we review current knowledge regarding how muscarinic agonists activate M3R, downstream signaling, and kinase activation, and how these actions result in disease progression. Importantly, we consider how these advances in knowledge may identify novel targets and consider current obstacles to leveraging these targets therapeutically.
2. Muscarinic Receptor Subtypes in Normal Physiology
The seven transmembrane domain G protein-coupled receptor (GPCR) muscarinic receptor family comprises five subtypes, designated M1R to M5R, that are expressed in a wide variety of organs and tissues [17,18]. Consequently, these receptors regulate a variety of important biological functions including ion channel transport, smooth muscle contraction, lipid turnover, and adenylyl cyclase activity [19,20,21,22]. There is an extensive sequence homology within the seven transmembrane spanning domains of these receptors [23]. Sequence variability is exhibited at the extracellular amino terminus and within the third intracellular loop [24]. The selectivity of G protein coupling depends on the individual muscarinic receptor, defined by amino acids within the second intracellular loop and further in the membrane portion of the third loop [24]. The physiological activity of muscarinic receptors depends on tissue expression, G protein coupling, and the downstream signal transduction pathway. In general, stimulation of odd-numbered muscarinic receptors, M1R, M3R, and M5R, is coupled to activation of G proteins in the Gq11 family and downstream activation of phospholipase C-β and phosphatidylinositol trisphosphate turnover that enhances calcium flux. The even-numbered muscarinic receptors, M2R and M4R, are generally coupled to G proteins within the Gi/o family, and their stimulation inhibits adenylyl cyclase activity, thereby reducing levels of intracellular cAMP [25].
Although muscarinic receptor expression may be ubiquitous, patterns of subtype distribution vary from organ to organ and receptor subtypes may have overlapping, or distinct functions. For example, both the brain and the eye express all five muscarinic receptor subtypes; however, M5R predominates in the brain, whereas M3R predominates in the eye [26,27,28,29,30,31]. M1R is widely distributed in the heart, uterus, GI tract, and central nervous system. In the heart, M1R activation increases calcium concentration and stimulates tachycardia, whereas M2R activation results in bradycardia [32,33]. Activation of M3R relaxes systemic vascular tone [34] while M5R activation relaxes cerebral vascular tone [35]. Within the gut, pulmonary airways [36], bladder [37], uterus [38], and smooth muscle, M1R, M2R, and M3R are the predominantly expressed and studied muscarinic receptor subtypes [39]. Whereas M1R and M3R appear to play a key role in gut epithelial cell function, M2R appears to be more important for subepithelial smooth muscle cell function. For example, M3R regulates acid secretion from gastric parietal cells, and a mix of M1R and M3R regulate pepsinogen release from gastric chief cells [40,41,42]. Nevertheless, even these functions may be complicated by interactions between the muscarinic receptor subtypes. Therefore, while smooth muscle cell function per se may be regulated by M2R activity, overall smooth muscle tone is regulated tightly by the autonomic and enteric nervous systems that signal via M1R and M3R [43].
3. M3R Expression in CRC
The M3R muscarinic receptor subtype, encoded by CHRM3, plays a prominent role in CRC progression. CHRM3 is a conditional oncogene whose expression stimulates cell proliferation and invasion, resistance to apoptosis, and, in general, cell functions that result in the progression of CRC and metastasis [44,45]. Most colon cancers overexpress M3R/CHRM3 [44,46,47,48]. Likewise, several human colon cancer cell lines commonly used in biomedical research, e.g., HT-29 and H508 cells, overexpress M3R/CHRM3 [44,49]. Compared to normal colon tissues, Yang et al. detected eight-fold greater CHRM3 RNA expression in colon cancer specimens [45]. Experiments inhibiting M3R activity in HT-29 cells [46] or comparing M3R expression in CRC to normal colon tissue [44] confirmed the impact of M3R/CHRM3 expression and activity on CRC progression.
In the normal colon, relatively weak M3R expression is restricted primarily to basolateral membranes of surface epithelial cells. However, in CRC, M3R is expressed diffusely along cell membranes, consistent with the loss of cell polarity in neoplasia [44]. Interestingly, although there is a significant association between the level of M3R/CHRM3 expression in primary tumors and the presence of CRC metastases, M3R/CHRM3 expression within metastases is not increased, suggesting M3R/CHRM3 overexpression is less important and impactful for cancer cell function once CRC cells have metastasized [44]. It would be of great interest to uncover and possibly leverage the biological cues and signaling programs that lead to and mediate this reduction in M3R/CHRM3 expression.
4. Muscarinic Receptor Agonism
Many cellular functions are impacted by muscarinic receptor activation; however, the most impactful in CRC are likely to be those related to cell migration and invasion since the predominant cause of CRC morbidity and mortality is metastatic, stage IV disease. Hence, although M3R activation may stimulate CRC cell proliferation, the size of the primary tumor is only a concern as it may correlate with the likelihood of extraintestinal spread of disease.
M3R overexpression per se does not account for its impact on CRC; the sources, availability, and concentrations of M3R agonists within the CRC microenvironment able to interact with M3R on neoplastic cells may play an equally important role. At present, only two endogenous ligands, acetylcholine (ACh) [25] and selected bile acids (BAs) [21,50], are known to activate muscarinic receptors. Regardless of whether M3R are activated by ACh or BAs, the propagation of downstream cell transduction stimulates CRC cell proliferation, resistance to apoptosis (survival), migration, and invasion [49]. Similar actions can be achieved by treating cells or mice with “designer” ACh mimetics, e.g., bethanechol, which are more resistant to hydrolysis by acetylcholinesterases.
Although ACh, a neurotransmitter, is typically produced by neurons [51], non-neuronal ACh can promote neoplasia [52,53,54,55,56] and for some cancers may even be the predominant source of ACh. In the tumor microenvironment, ACh may be produced by and released from enteric neurons, immunocytes, and CRC cells themselves [53,57]. Choline acetyltransferase (ChAT) plays an important catalytic role in the biosynthesis of both neuronal and non-neuronal ACh and its expression is reported in several organs and cancers, and is sometimes used as a surrogate marker of non-neuronal ACh production [25]. Using quantitative-PCR, Cheng et al. demonstrated ChAT expression and ACh production and release by H508, WiDr, and Caco-2 human colon cancer cells [56]. Notably, treating CRC cells with either selective or non-selective muscarinic receptor antagonists attenuated H508 colon cancer cell proliferation by 40% supporting the impact of endogenous production of ACh and autocrine effects. Inhibiting acetylcholinesterase activity increased H508 cell proliferation by as much as 2.5-fold, providing additional evidence that ACh can function as an autocrine growth factor for CRC [56].
These biological phenomena may have clinical consequences. Pheochromocytomas, uncommon neuroendocrine tumors that secrete excess catecholamines, may also produce excess ACh [58,59]. Despite previous endoscopic resection of a small focus of rectal cancer and vigilant surveillance, an elderly man with an unresectable pheochromocytoma experienced rapid recurrence of the rectal adenocarcinoma [60]. Analysis of tissue from the rectal carcinoma and pheochromocytoma revealed overexpression of M3R and ChAT, respectively [60]. For proof-of-principle, Rosenvinge et al. demonstrated that conditioned media from pheochromocytoma cells can stimulate the proliferation of H508 colon cancer cells, an action blocked by pretreating cells with the muscarinic receptor antagonist atropine [60]. These findings are consistent with the hypothesis that ACh released from the neuroendocrine tumor stimulated swift regrowth of remnant cells after endoscopic resection of the rectal cancer [59,60].
M3R can also be activated by non-traditional muscarinic ligands, such as selected BAs and their derivatives [61,62,63,64]. The potential for physiological BA signaling by this mechanism was discovered in 1998, when an interaction between BAs and M3R was observed in secretory gastric epithelial cells [50]. Structural similarities between BAs and cholesterol may explain the ability of the former to mimic the actions of the latter [61] and interact functionally with muscarinic receptor subtypes expressed selectively on Chinese hamster ovary (CHO) cells [61]; cholesterol is reported to act as an allosteric modulator of other GPCRs [65]. Further studies elucidated similarities between ACh and BA-induced post-muscarinic receptor signaling, primarily using H508 and HT-29 colon cancer cells. A prominent feature of these pathways is transactivation of epidermal growth factor receptors (EGFR) [63,66]; Cheng et al. showed that M3R and EGFR inhibitors and antibodies independently blocked the signaling and proliferative actions of BAs [64]. Thus, BA-induced colon cancer cell proliferation is M3R-dependent and mediated, in part, by transactivation of EGFR [64].
Post-mortem analysis of cecal contents from 19 persons without colorectal neoplasia revealed the presence of BA concentrations within the range capable of promoting colon cancer cell proliferation via M3R activation in vitro [67]; it is unknown whether fecal BA concentrations vary between persons with or without colonic neoplasia although, in rodents, increased concentration of fecal bile acids promote colon neoplasia. Mice fed a diet enriched in a secondary BA, deoxycholic acid, developed both precursor lesions of colon neoplasia and frank cancers [68,69]. Likewise, in mouse models of CRC, genetic ablation of a key intestinal BA transporter, ASBT, or of FGF15, a feedback inhibitor of hepatic BA synthesis, results in both increased fecal BA levels and promotion of colon neoplasia [68,69]. Notably, these findings may help to explain why consumption of a Western diet enriched in fats, beef, and processed meats that increase BA production may also increase the risk of CRC [70,71].
The effects of M3R agonism on the promotion of colon neoplasia are supported by in vivo evidence. In mice treated with azoxymethane (AOM), a procarcinogen that selectively induces colon neoplasia in rodents and mimics sporadic CRC in humans [72], M3R-deficiency dose-dependently reduced colon neoplasia [73]; similar effects were observed in ApcMin/+ mice, a genetic model of colon neoplasia [74]. Conversely, Peng et al. showed that adding bethanechol, a non-selective muscarinic receptor agonist, to the drinking water of mice treated with azoxymethane significantly increased both the number and volume of colon tumors [75].
5. Targeting Muscarinic Receptors
As discussed above, M1 and M3 subtype muscarinic receptors are co-expressed in both normal and neoplastic intestinal epithelial cells [76]. Whereas, in azoxymethane-treated mice, ablating CHRM3 expression attenuates colon neoplasia [72,73], ablating CHRM1 did not significantly alter colon tumor number or size and, surprisingly, may have trended towards promoting colon neoplasia [76]. More importantly, concurrent ablation of both CHRM3 and CHRM1 negated the beneficial effects of CHRM3 ablation [76]. These divergent effects of M3R and M1R on colon neoplasia in this animal model highlight the concern that to be effective, muscarinic receptor antagonists must be highly selective—off-target effects on other muscarinic receptor subtypes may either negate beneficial effects or, even more concerning, aggravate disease.
Amongst other factors, developing selective muscarinic receptor antagonists is challenging due to the highly similar orthosteric binding sites among the receptor subtypes [74]. Currently, no muscarinic receptor antagonists have been approved to treat cancer. However, in vivo studies using non-selective muscarinic antagonists such as scopolamine butylbromide demonstrated they can reduce intestinal tumor number and volume, albeit not as effectively as M3R gene (CHRM3) ablation [77]. Although scopolamine butylbromide does not cross the blood–brain barrier, besides being a non-selective muscarinic receptor antagonist, it is not specifically directed against intestinal muscarinic receptors and, therefore, its use may result in a wide variety of unwanted anti-cholinergic side effects [15]. As implied by the promising anti-neoplastic effects of genetic ablation of M3R/CHRM3 [76,77], M3R targeting via highly selective antagonism is necessary to maximize efficacy and to prevent off-target effects on other muscarinic receptor subtypes, specifically M1R [76].
Given the widespread clinical use of M3R antagonists for other conditions, repurposing these medications for use as adjuvants to current CRC therapies should be considered and studied in clinical trials. In particular, the highly selective M3R antagonist, darifenacin, approved by the U.S. Food and Drug Administration (FDA) to treat genitourinary conditions, may be a viable option [78]. Notably, darifenacin, which is prescribed primarily for symptoms of an “overactive” bladder, arrested tumor progression in nude mice, further highlighting its potential for repurposed use [79]. Although muscarinic antagonists have been safely tolerated by most persons for many years, repurposing these medications warrants further investigation for potential dose-dependent toxicities at the levels and durations required to achieve anti-neoplastic effects. For example, case-control studies suggest long-term use of anti-cholinergic medications may be associated with an increased risk of dementia, particularly in those with pre-existing Parkinson Disease [80,81].
6. Targeting Matrix Metalloproteinases
In human CRC cell experimental models, muscarinic receptor agonists stimulate cancer progression via complex interacting signal transduction pathways involving both EGFR-dependent and -independent pathways (Figure 1). Downstream effectors of these pathways induce gene transcription programs resulting in cell proliferation, survival, migration, and invasion [49,66,82]. In particular, because of their critical role in degrading components of the extracellular matrix (ECM), M3R-induced matrix metalloproteinase (MMP) gene transcription is important for cell migration and invasion [83,84]; MMPs are key promoters of cancer progression [85]. MMPs are part of the metzincin family of metalloproteinases, comprised of 24 zinc-containing proteases that cleave components of the ECM in both health and disease [83,84]. Each class of MMPs, such as collagenases, gelatinases, stromelysins, and matrilysins, plays a different role [85,86]. Normally, MMP activity is closely regulated by tissue inhibitors, e.g., TIMPs [86]; in cancer, dysregulated MMP-TIMP expression may favor proteolysis [86], thereby contributing to cancer spread [87]. Giambernardi et al. reported abnormal expression of several MMPs in colon, breast, and prostate cancer cell lines [84]. MMP7 and MMP1 appear to play particularly important roles in CRC.
In transgenic mouse models, MMP7 overexpression early in colon neoplasia promotes tumorigenesis; the converse is observed in MMP7-deficient mice [88,89]. Increased MMP7 expression in human CRC correlates with advanced disease and worse outcomes [90]. As a consequence of its ability to degrade ECM, MMP1, a collagenase, is a key player in colon cancer cell migration and metastasis [83,84], and its expression in human CRC is also associated with cancer progression, metastasis, and a poor prognosis [83,91]. Shiozawa et al. found absent MMP1 expression in colorectal adenomas, but its expression in 76% of CRC [73]. Enhanced MMP1 expression in invasive versus intramucosal CRC suggests increased expression of MMP1 in the earlier stages of tumor invasion, a similar pattern to that for increased M3R expression [73]. MMP1 expression correlates with infiltrative CRC, specifically with lymph node and liver metastasis [73]. MMP9 may also be associated with metastasis in CRC; high levels of both MMP1 and MMP9 expression in tumor-free mucosa correlated with TNM-stage and lymph node involvement [92].
Xie et al. showed that activating human colon cancer cell muscarinic receptors with ACh selectively induced expression of MMP 1, 7, and 10 [93], and Raufman et al. showed that treating CRC cells with atropine to block M3R activation or with a neutralizing anti-MMP1 antibody abolished ACh-induced cell invasion [83]. Nonetheless, despite their importance for CRC progression and promising in vitro and pre-clinical studies, two decades after initial clinical trials failed to show benefit for small molecule broad spectrum MMP inhibitors, targeting MMPs for cancer treatment remains challenging [94].
Initial therapeutic efforts were directed at developing inhibitors consisting primarily of a zinc-chelating moiety intended to target the active site of the MMP catalytic domain [95]. Approximately 50 such agents were tested and showed promising results in pre-clinical animal models—nevertheless, nearly all failed in clinical trials [95,96], largely due to off-target actions, metabolic instability, poor bioavailability, or dose-limiting side effects [97]. Disease stage may also have impacted discrepancies between effectiveness in pre-clinical studies and clinical trials; compared to murine models, subjects in clinical trials had more advanced disease [85,95,97]. Additionally, MMPs share structural similarity with the active site of other members of the metzincin family, resulting in broad and unexpected adverse effects [95,98]. Batimastat and marimastat, among the first MMP inhibitors tested in clinical trials [99,100], inhibit MMP 1, 2, 7 and 9, by binding to the active zinc site [101]. Although animal models demonstrated promising antitumor effects of batimastat [100,102], the clinical performance of batimastat, marimastat, and related compounds was disappointing and adverse events resulted in termination of clinical trials [99,103,104]; side-effects were largely attributed to off-target effects on molecules involved in vital cell functions such as cell–matrix interactions, cellular adhesion, and growth factor availability [105,106]. Another obstacle to developing MMP-targeted therapies is that expression of MMP substrates differs in mice and humans. For example, MMP7 activates intestinal α-defensin in murine but not human Paneth cells [85,107].
Advances in understanding structure-function relationships [95,108] led to renewed interest in selectively targeting the cancer-promoting actions of MMPs while retaining their important beneficial effects [109]. Notably, 10 of 24 MMPs have anti-tumorigenic and anti-inflammatory effects; inhibiting or downregulating their activity is likely to have adverse effects [109]. Using this information, investigators developed highly selective small-molecule inhibitors and antibodies against MMP 12 and 14 [110,111,112], monoclonal antibodies directed at the catalytic zinc-protein complex and enzyme surface conformational epitope of MMP 2 and 9 [15,113], and a highly selective compound, JNJ0966, that inhibits MMP9 activation [15,114]. JNJ0966, which interacts with a structural pocket distinct from the catalytic domain and proximate to the cleavage site of the MMP9 zymogen, attenuated disease severity in a mouse model of autoimmune encephalomyelitis, without altering the catalytic activity of MMP 1, 2, 3, or 14 [15,114]. These innovative approaches are likely to spur similar efforts to target other MMPs selectively. Nonetheless, considering the broad range of biochemical activities of MMPs, even selective inhibition may cause harm [109]. Alternative or adjunct strategies to avoid toxic systemic levels of MMP inhibitors may involve directed and targeted administration, likely as an adjunct to current chemotherapeutic regimens [15,85,94].
7. Epidermal Growth Factor Receptors
As a consequence of MMP7-mediated release of an EGFR ligand HB-EGF, the epidermal growth factor receptor (EGFR), a receptor tyrosine kinase [66], may be transactivated after M3R activation (Figure 1) [16,115]. Consistent with these observations, inhibiting MMP activation with batimastat can block EGFR activation [116]. The prominent role of EGFR signaling in colon cancer cell [16] and its ability to modulate key hallmarks of cancer progression [117,118] has already stimulated the development of several anti-EGFR monoclonal antibodies and receptor tyrosine kinase inhibitors [118,119,120]. Nonetheless, M3R-stimulated EGFR transactivation may provide an additional therapeutic target [66]; it is likely that directly targeting MMP7, an enzyme that cleaves and releases HB-EGF from pro-HB-EGF, may augment the therapeutic benefits of other approaches (Figure 1).
8. Additional Potential Therapeutic Targets Downstream of M3R
Potential targets downstream of M3R and EGFR include RAS, BRAF, and components of mitogen activated protein kinase (MAPK) signaling. Recently, Liu et al. studied a well-known transcriptional repressor associated with several cancers, forkhead box D3 (FOXD3). Their findings revealed FOXD3 knockdown considerably enhanced the proliferation and invasiveness of human colon cancer cells [121]. FOXD3 knockdown activated a key signaling pathway in human colon cancer cells, EGFR-Ras-Raf-MEK-ERK [121]. While the exact mechanism of FOXD3 association with the EGFR-Ras-Raf-MEK-ERK signaling pathway is unclear, enhancing the expression of FOXD3 or promoting its activation may have potential as a therapeutic strategy [15,121]. Investigators have also attempted direct BRAF inhibition, especially in BRAF mutant mCRC [122]. Unfortunately, drugs such as vemurafenib, an oral single-agent BRAFV600E inhibitor, have not shown meaningful in vivo activity. The limited activity of single-agent BRAF inhibitors appears due primarily to mechanisms of resistance and feedback regulation intrinsic to the RAS/RAF/MAPK signaling pathway [122]. It is noteworthy that over 30 years following the discovery of the role of KRAS in cancer growth and development, no drugs targeting KRAS are currently in clinical trials [123,124]. These challenges are due in large part to RAS being the most frequently mutated oncogene across all malignancies, the pervasiveness of compensatory RAS-mediated signal transduction feedback loops, and signaling elicited by oncogenic gain-of-function mutations [123].
As demonstrated in human embryonic kidney cells, MAPK activation was attenuated after inhibiting Src, a non-receptor protein tyrosine kinase protooncogene whose activation supports cell survival [125]. Src regulates multiple pathways and is overexpressed in CRC where its activity promotes metastasis and may contribute to chemotherapy resistance [126]. Through its kinase activity, Src potentiates the effects of EGFR activation [127,128,129]. In H508 colon cancer cells, inhibiting Src attenuated ACh- and EGF-induced ERK1/2 phosphorylation (activation), identifying Src as a key link between M3R-induced transactivation of EGFR and the subsequent downstream activation of MAPK (ERK1/2) in CRC [66]. Moreover, warranting consideration is a related pathway involving receptors for corticotrophin-releasing factor-2 (CRF2), an important neuromodulator of stress. Crosstalk between CRF2 and M3R signaling augments colon cancer cell migration, invasion, and other attributes promoting cancer progression [130]. Pelissier-Rota et al. revealed a novel intercellular circuit whereby CRF2 agonists in conjunction with ACh-induced activation of M3R and a feedback loop resulting in additional release of a CRF2 agonist, urocortin-3, modulates activation of Src/Erk and focal adhesion kinase (FAK). Besides unveiling unique crosstalk between muscarinic receptors and CRF2, interactions between these signal transduction pathways may alter colonic mucosal barrier function, inflammation, and the risk of developing colitis-associated cancer, particularly in those with inflammatory bowel diseases [130].
Muscarinic receptor activation promotes protein biosynthesis, thereby enhancing colon cancer cell proliferation by ERK1/2-mediated pathways. In SNU-407 colon cancer cells, Park et al. explored how modulating muscarinic receptor activity affected eukaryotic translation elongation factor 2 (eEF2), the protein responsible for ribosomal translocation [131,132,133]. Treatment with muscarinic receptor agonists reduced eEF2 phosphorylation, thereby inhibiting its activity and the rate of translation, effects blocked by pre-treatment with atropine [134,135,136]. Treating cells with a potent MEK1/2 inhibitor (U0126) or protein kinase C inhibitor, GF109203X, decreased carbamylcholine-induced eEF2 dephosphorylation. These findings provide further evidence of the importance of MEK1/2-ERK1/2 and PKC signaling downstream of muscarinic receptor activation and implicate a novel role for eEF2 dephosphorylation, another potential therapeutic target.
Notably, chlorpyrifos (CPF) and other widely used organophosphate insecticides associated with exposure-dependent cancer risk activate signaling pathways similar to those following muscarinic receptor activation [137]; in H508 colon cancer cells, CPF increased EGFR phosphorylation and downstream activation of ERK1/2 [138]. These effects were attenuated by treatment with U0126, a MEK1/2 inhibitor, and AG-1478, an EGFR tyrosine kinase inhibitor [138]. As organophosphates act by inhibiting acetylcholinesterase activity, thereby increasing ACh levels, these findings suggest that interventions that block the pro-neoplastic effects of muscarinic receptor activation can also be leveraged to block the actions of this family of carcinogens that hijack components of the same signaling mechanisms.
9. Conclusions and Future Directions
The illustration depicted in Figure 1 summarizes the current state of knowledge regarding key elements of muscarinic receptor signaling and therapeutically targetable nodes in CRC. These nodes include the machinery for ACh production in the tumor microenvironment, selective inhibitors of M3R activation and MMPs, like MMP7 which mediates release of HB-EGF and activation of EGFR, and a host of key downstream signaling molecules. In addition to advances in selective drug design and development and preclinical and clinical trials of new and repurposed pharmaceuticals the conundrum underlying the apparent opposing actions of M3R and M1R in colon cancer progression must be solved as this may reveal an entirely novel, potentially exciting therapeutic strategy [76]. Given the tumor cell heterogeneity within colon cancers and redundant signaling pathways, it is likely that combinatorial therapy will be required to impede the complex interacting signal transduction pathways shown in Figure 1 and provide meaningful therapeutic gains for patients with advanced CRC.
Author Contributions
Writing–original draft preparation, O.A., M.T., S.H., G.X. and J.-P.R.; writing—review and editing, O.A., M.T., S.H., G.X. and J.-P.R.; supervision, J.-P.R.; funding acquisition, G.X. and J.-P.R. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by the United States (U.S.) Department of Veterans Affairs Biomedical Laboratory Research and Development Program, VA Merit Award grant numbers BX002129 and BX002777. Osman Ali and Mazen Tolaymat were supported by the U.S. National Institutes of Health, grant number T32 DK067872. The contents do not represent the views of the U.S. Department of Veterans Affairs or the United States Government.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Ferlay, J.; Colombet, M.; Soerjomataram, I.; Mathers, C.; Parkin, D.M.; Piñeros, M.; Znaor, A.; Bray, F. Estimating the global cancer incidence and mortality in 2018: GLOBOCAN sources and methods. Int. J. Cancer 2019, 144, 1941–1953. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mármol, I.; Sánchez-de-Diego, C.; Pradilla Dieste, A.; Cerrada, E.; Rodriguez Yoldi, M. Colorectal Carcinoma: A General Overview and Future Perspectives in Colorectal Cancer. Int. J. Mol. Sci. 2017, 18, 197. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Siegel, R.L.; Miller, K.D.; Goding Sauer, A.; Fedewa, S.A.; Butterly, L.F.; Anderson, J.C.; Cercek, A.; Smith, R.A.; Jemal, A. Colorectal cancer statistics, 2020. CA Cancer J. Clin. 2020, 70, 145–164. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Colorectal Cancer Statistics|How Common Is Colorectal Cancer? Available online: https://www.cancer.org/cancer/colon-rectal-cancer/about/key-statistics.html#references (accessed on 10 September 2020).
- Siegel, R.L.; Fedewa, S.A.; Anderson, W.F.; Miller, K.D.; Ma, J.; Rosenberg, P.S.; Jemal, A. Colorectal Cancer Incidence Patterns in the United States, 1974–2013. JNCI J. Natl. Cancer Inst. 2017, 109. [Google Scholar] [CrossRef] [Green Version]
- Yeo, H.; Betel, D.; Abelson, J.S.; Zheng, X.E.; Yantiss, R.; Shah, M.A. Early-onset Colorectal Cancer is Distinct from Traditional Colorectal Cancer. Clin. Colorectal Cancer 2017, 16, 293–299.e6. [Google Scholar] [CrossRef]
- Benson, A.B.; Venook, A.P.; Cederquist, L.; Chan, E.; Chen, Y.-J.; Cooper, H.S.; Deming, D.; Engstrom, P.F.; Enzinger, P.C.; Fichera, A.; et al. Colon Cancer, Version 1.2017, NCCN Clinical Practice Guidelines in Oncology. J. Natl. Compr. Cancer Netw. 2017, 15, 370–398. [Google Scholar] [CrossRef]
- Labianca, R.; Nordlinger, B.; Beretta, G.D.; Mosconi, S.; Mandalà, M.; Cervantes, A.; Arnold, D. Early colon cancer: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann. Oncol. 2013, 24, vi64–vi72. [Google Scholar] [CrossRef]
- Hammond, W.A.; Swaika, A.; Mody, K. Pharmacologic resistance in colorectal cancer: A review. Ther. Adv. Med. Oncol. 2016, 8, 57–84. [Google Scholar] [CrossRef] [Green Version]
- Fong, W.; To, K.K.W. Drug repurposing to overcome resistance to various therapies for colorectal cancer. Cell. Mol. Life Sci. 2019, 76, 3383–3406. [Google Scholar] [CrossRef]
- National Cancer Institute Colorectal Cancer—Cancer Stat Facts. Available online: https://seer.cancer.gov/statfacts/html/colorect.html (accessed on 10 September 2020).
- Ganesh, K.; Stadler, Z.K.; Cercek, A.; Mendelsohn, R.B.; Shia, J.; Segal, N.H.; Diaz, L.A. Immunotherapy in colorectal cancer: Rationale, challenges and potential. Nat. Rev. Gastroenterol. Hepatol. 2019, 16, 361–375. [Google Scholar] [CrossRef]
- André, T.; Shiu, K.-K.; Kim, T.W.; Jensen, B.V.; Jensen, L.H.; Punt, C.; Smith, D.; Garcia-Carbonero, R.; Benavides, M.; Gibbs, P.; et al. Pembrolizumab in Microsatellite-Instability–High Advanced Colorectal Cancer. N. Engl. J. Med. 2020, 383, 2207–2218. [Google Scholar] [CrossRef] [PubMed]
- Peeters, M.; Price, T. Biologic therapies in the metastatic colorectal cancer treatment continuum—Applying current evidence to clinical practice. Cancer Treat. Rev. 2012, 38, 397–406. [Google Scholar] [CrossRef] [PubMed]
- Felton, J.; Hu, S.; Raufman, J.-P. Targeting M3 Muscarinic Receptors for Colon Cancer Therapy. Curr. Mol. Pharmacol. 2018, 11, 184–190. [Google Scholar] [CrossRef] [PubMed]
- Prenzel, N.; Zwick, E.; Daub, H.; Leserer, M.; Abraham, R.; Wallasch, C.; Ullrich, A. EGF receptor transactivation by G-protein-coupled receptors requires metalloproteinase cleavage of proHB-EGF. Nature 1999, 402, 884–888. [Google Scholar] [CrossRef] [PubMed]
- Wess, J. Muscarinic acetylcholine receptor knockout mice: Novel phenotypes and clinical implications. Annu. Rev. Pharmacol. Toxicol. 2004, 44, 423–450. [Google Scholar] [CrossRef]
- Eglen, R.M. Overview of Muscarinic Receptor Subtypes. In Handbook of Experimental Pharmacology; Springer: Berlin/Heidelberg, Germany, 2012; Volume 208, pp. 3–28. ISBN 9783642232732. [Google Scholar]
- Gilman, A.G. G proteins and dual control of adenylate cyclase. Cell 1984, 36, 577–579. [Google Scholar] [CrossRef]
- Malbon, C.C. G proteins in development. Nat. Rev. Mol. Cell Biol. 2005, 6, 689–701. [Google Scholar] [CrossRef]
- Von Rosenvinge, E.C.; Raufman, J.-P. Muscarinic Receptor Signaling in Colon Cancer. Cancers 2011, 3, 971–981. [Google Scholar] [CrossRef] [Green Version]
- Spindel, E.R. Muscarinic receptor agonists and antagonists: Effects on cancer. Handb. Exp. Pharmacol. 2012, 451–468. [Google Scholar] [CrossRef] [Green Version]
- Kruse, A.C.; Hu, J.; Pan, A.C.; Arlow, D.H.; Rosenbaum, D.M.; Rosemond, E.; Green, H.F.; Liu, T.; Chae, P.S.; Dror, R.O.; et al. Structure and dynamics of the M3 muscarinic acetylcholine receptor. Nature 2012, 482, 552–556. [Google Scholar] [CrossRef] [Green Version]
- Wess, J. Molecular Biology of Muscarinic Acetylcholine Receptors. Crit. Rev. Neurobiol. 1996, 10, 69–99. [Google Scholar] [CrossRef] [PubMed]
- Shah, N.; Khurana, S.; Cheng, K.; Raufman, J.-P. Muscarinic receptors and ligands in cancer. Am. J. Physiol. Physiol. 2009, 296, C221–C232. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abrams, P.; Andersson, K.-E.; Buccafusco, J.J.; Chapple, C.; de Groat, W.C.; Fryer, A.D.; Kay, G.; Laties, A.; Nathanson, N.M.; Pasricha, P.J.; et al. Muscarinic receptors: Their distribution and function in body systems, and the implications for treating overactive bladder. Br. J. Pharmacol. 2006, 148, 565–578. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Powell, H.C. The War of the Soups and the Sparks: The Discovery of Neurotransmitters and the Dispute over How Nerves Communicate (review). Bull. Hist. Med. 2006, 80, 598–599. [Google Scholar] [CrossRef]
- Gil, D.W.; Krauss, H.A.; Bogardus, A.M.; WoldeMussie, E. Muscarinic receptor subtypes in human iris-ciliary body measured by immunoprecipitation. Investig. Ophthalmol. Vis. Sci. 1997, 38, 1434–1442. [Google Scholar]
- Bognar, I.T.; Altes, U.; Beinhauer, C.; Kessler, I.; Fuder, H. A muscarinic receptor different from the M1, M2, M3 and M4 subtypes mediates the contraction of the rabbit iris sphincter. Naunyn. Schmiedebergs. Arch. Pharmacol. 1992, 345, 611–618. [Google Scholar] [CrossRef]
- Bymaster, F.P.; Carter, P.A.; Yamada, M.; Gomeza, J.; Wess, J.; Hamilton, S.E.; Nathanson, N.M.; McKinzie, D.L.; Felder, C.C. Role of specific muscarinic receptor subtypes in cholinergic parasympathomimetic responses, in vivo phosphoinositide hydrolysis, and pilocarpine-induced seizure activity. Eur. J. Neurosci. 2003, 17, 1403–1410. [Google Scholar] [CrossRef]
- Matsui, M.; Motomura, D.; Karasawa, H.; Fujikawa, T.; Jiang, J.; Komiya, Y.; Takahashi, S.-I.; Taketo, M.M. Multiple functional defects in peripheral autonomic organs in mice lacking muscarinic acetylcholine receptor gene for the M3 subtype. Proc. Natl. Acad. Sci. USA 2000, 97, 9579–9584. [Google Scholar] [CrossRef] [Green Version]
- Colecraft, H.M.; Egamino, J.P.; Sharma, V.K.; Sheu, S.-S. Signaling Mechanisms Underlying Muscarinic Receptor-mediated Increase in Contraction Rate in Cultured Heart Cells. J. Biol. Chem. 1998, 273, 32158–32166. [Google Scholar] [CrossRef] [Green Version]
- Dhein, S.; van Koppen, C.J.; Brodde, O.-E. Muscarinic receptors in the Mammalian Heart. Pharmacol. Res. 2001, 44, 161–182. [Google Scholar] [CrossRef]
- Khurana, S.; Chacon, I.; Xie, G.; Yamada, M.; Wess, J.; Raufman, J.-P.; Kennedy, R.H. Vasodilatory effects of cholinergic agonists are greatly diminished in aorta from M3R−/− mice. Eur. J. Pharmacol. 2004, 493, 127–132. [Google Scholar] [CrossRef] [PubMed]
- Yamada, M.; Lamping, K.G.; Duttaroy, A.; Zhang, W.; Cui, Y.; Bymaster, F.P.; McKinzie, D.L.; Felder, C.C.; Deng, C.-X.; Faraci, F.M.; et al. Cholinergic dilation of cerebral blood vessels is abolished in M5 muscarinic acetylcholine receptor knockout mice. Proc. Natl. Acad. Sci. USA 2001, 98, 14096–14101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Donfack, J.; Kogut, P.; Forsythe, S.; Solway, J.; Ober, C. Sequence variation in the promoter region of the cholinergic receptor muscarinic 3 gene and asthma and atopy. J. Allergy Clin. Immunol. 2003, 111, 527–532. [Google Scholar] [CrossRef] [PubMed]
- Chess-Williams, R. Muscarinic receptors of the urinary bladder: Detrusor, urothelial and prejunctional. Auton. Autacoid Pharmacol. 2002, 22, 133–145. [Google Scholar] [CrossRef] [PubMed]
- Kitazawa, T.; Hirama, R.; Masunaga, K.; Nakamura, T.; Asakawa, K.; Cao, J.; Teraoka, H.; Unno, T.; Komori, S.; Yamada, M.; et al. Muscarinic receptor subtypes involved in carbachol-induced contraction of mouse uterine smooth muscle. Naunyn. Schmiedebergs. Arch. Pharmacol. 2008, 377, 503–513. [Google Scholar] [CrossRef]
- Chiba, T.; Bharucha, A.E.; Thomforde, G.M.; Kost, L.J.; Phillips, S.F. Model of rapid gastrointestinal transit in dogs: Effects of muscarinic antagonists and a nitric oxide synthase inhibitor. Neurogastroenterol. Motil. 2002, 14, 535–541. [Google Scholar] [CrossRef] [PubMed]
- Rosenfeld, G.C. Isolated parietal cells: Adrenergic response and pharmacology. J. Pharmacol. Exp. Ther. 1984, 229, 763–767. [Google Scholar]
- Raufman, J.P.; Sutliff, V.E.; Kasbekar, D.K.; Jensen, R.T.; Gardner, J.D. Pepsinogen secretion from dispersed chief cells from guinea pig stomach. Am. J. Physiol. Liver Physiol. 1984, 247, G95–G104. [Google Scholar] [CrossRef]
- Xie, G.; Drachenberg, C.; Yamada, M.; Wess, J.; Raufman, J.-P. Cholinergic agonist-induced pepsinogen secretion from murine gastric chief cells is mediated by M 1 and M 3 muscarinic receptors. Am. J. Physiol. Liver Physiol. 2005, 289, G521–G529. [Google Scholar] [CrossRef]
- Moro, E.; Crema, F.; Dandolo, C.; Ponti, F.; Frigo, G. Effect of muscarinic receptor blockade on canine gastric tone and compliance in vivo. Pharmacol. Res. 2005, 51, 289–296. [Google Scholar] [CrossRef]
- Cheng, K.; Shang, A.C.; Drachenberg, C.B.; Zhan, M.; Raufman, J.P. Differential expression of M3 muscarinic receptors in progressive colon neoplasia and metastasis. Oncotarget 2017, 8, 21106–21114. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, W.-L. Cholinergic receptor up-regulates COX-2 expression and prostaglandin E2 production in colon cancer cells. Carcinogenesis 2000, 21, 1789–1793. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kopp, R.; Lambrecht, G.; Mutschler, E.; Moser, U.; Tacke, R.; Pfeiffer, A. Human HT-29 colon carcinoma cells contain muscarinic M3 receptors coupled to phosphoinositide metabolism. Eur. J. Pharmacol. Mol. Pharmacol. 1989, 172, 397–405. [Google Scholar] [CrossRef]
- Zhang, W.; Roomans, G.M. Evidence for Muscarinic 3 Receptor Mediated Ion Transport in HT29 Cells Studied by X-ray Microanalysis. Cell Struct. Funct. 1997, 22, 379–385. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frucht, H.; Jensen, R.T.; Dexter, D.; Yang, W.L.; Xiao, Y. Human colon cancer cell proliferation mediated by the M3 muscarinic cholinergic receptor. Clin. Cancer Res. 1999, 5, 2532–2539. [Google Scholar] [PubMed]
- Belo, A.; Cheng, K.; Chahdi, A.; Shant, J.; Xie, G.; Khurana, S.; Raufman, J.-P. Muscarinic receptor agonists stimulate human colon cancer cell migration and invasion. Am. J. Physiol. Liver Physiol. 2011, 300, G749–G760. [Google Scholar] [CrossRef] [PubMed]
- Raufman, J.-P.; Zimniak, P.; Bartoszko-Malik, A. Lithocholyltaurine interacts with cholinergic receptors on dispersed chief cells from guinea pig stomach. Am. J. Physiol. Liver Physiol. 1998, 274, G997–G1004. [Google Scholar] [CrossRef] [PubMed]
- Amenta, F.; Tayebati, S. Pathways of Acetylcholine Synthesis, Transport and Release as Targets for Treatment of Adult-Onset Cognitive Dysfunction. Curr. Med. Chem. 2008, 15, 488–498. [Google Scholar] [CrossRef]
- Schuller, H.M. Is cancer triggered by altered signalling of nicotinic acetylcholine receptors? Nat. Rev. Cancer 2009, 9, 195–205. [Google Scholar] [CrossRef]
- Bayer, G.; Wense, T. Über den Nachweis von Hormonen in einzelligen Tieren. Pflug. Arch. Gesamte Physiol. Menschen Tiere 1936, 237, 417–422. [Google Scholar] [CrossRef]
- Grando, S.A. Biological Functions of Keratinocyte Cholinergic Receptors. J. Investig. Dermatol. Symp. Proc. 1997, 2, 41–48. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Song, P.; Sekhon, H.; Proskocil, B.; Blusztajn, J.; Mark, G.; Spindel, E. Synthesis of acetylcholine by lung cancer. Life Sci. 2003, 72, 2159–2168. [Google Scholar] [CrossRef]
- Cheng, K.; Samimi, R.; Xie, G.; Shant, J.; Drachenberg, C.; Wade, M.; Davis, R.J.; Nomikos, G.; Raufman, J.-P. Acetylcholine release by human colon cancer cells mediates autocrine stimulation of cell proliferation. Am. J. Physiol. Liver Physiol. 2008, 295, G591–G597. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klapproth, H.; Reinheimer, T.; Metzen, J.; Münch, M.; Bittinger, F.; Kirkpatrick, C.J.; Höhle, K.-D.; Schemann, M.; Racké, K.; Wessler, I. Non-neuronal acetylcholine, a signalling molecule synthezised by surface cells of rat and man. Naunyn. Schmiedebergs. Arch. Pharmacol. 1997, 355, 515–523. [Google Scholar] [CrossRef] [PubMed]
- Greene, L.A.; Rein, G. Synthesis, storage and release of acetylcholine by a noradrenergic pheochromocytoma cell line. Nature 1977, 268, 349–351. [Google Scholar] [CrossRef] [PubMed]
- Tolaymat, M.; Larabee, S.; Hu, S.; Xie, G.; Raufman, J.-P. The Role of M3 Muscarinic Receptor Ligand-Induced Kinase Signaling in Colon Cancer Progression. Cancers 2019, 11, 308. [Google Scholar] [CrossRef] [Green Version]
- Von Rosenvinge, E.C.; Cheng, K.; Drachenberg, C.B.; Fowler, C.B.; Evers, D.L.; Xie, G.; Raufman, J.P. Bedside to bench: Role of muscarinic receptor activation in ultrarapid growth of colorectal cancer in a patient with pheochromocytoma. Mayo Clin. Proc. 2013, 88, 1340–1346. [Google Scholar] [CrossRef] [Green Version]
- Raufman, J.P.; Chen, Y.; Cheng, K.; Compadre, C.; Compadre, L.; Zimniak, P. Selective interaction of bile acids with muscarinic receptors: A case of molecular mimicry. Eur. J. Pharmacol. 2002, 457, 77–84. [Google Scholar] [CrossRef]
- Farhana, L.; Nangia-Makker, P.; Arbit, E.; Shango, K.; Sarkar, S.; Mahmud, H.; Hadden, T.; Yu, Y.; Majumdar, A.P.N. Bile acid: A potential inducer of colon cancer stem cells. Stem Cell Res. Ther. 2016, 7. [Google Scholar] [CrossRef] [Green Version]
- Cheng, K.; Chen, Y.; Zimniak, P.; Raufman, J.P.; Xiao, Y.; Frucht, H. Functional interaction of lithocholic acid conjugates with M3 muscarinic receptors on a human colon cancer cell line. Biochim. Biophys. Acta 2002, 1588, 48–55. [Google Scholar] [CrossRef] [Green Version]
- Cheng, K.; Raufman, J.-P.P. Bile acid-induced proliferation of a human colon cancer cell line is mediated by transactivation of epidermal growth factor receptors. Biochem. Pharmacol. 2005, 70, 1035–1047. [Google Scholar] [CrossRef] [PubMed]
- Guixà-González, R.; Albasanz, J.L.; Rodriguez-Espigares, I.; Pastor, M.; Sanz, F.; Martí-Solano, M.; Manna, M.; Martinez-Seara, H.; Hildebrand, P.W.; Martín, M.; et al. Membrane cholesterol access into a G-protein-coupled receptor. Nat. Commun. 2017, 8, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Cheng, K.; Zimniak, P.; Raufman, J.-P. Transactivation of the Epidermal Growth Factor Receptor Mediates Cholinergic Agonist-Induced Proliferation of H508 Human Colon Cancer Cells. Cancer Res. 2003, 63, 6744–6750. [Google Scholar] [CrossRef]
- Hamilton, J.P.; Xie, G.; Raufman, J.-P.; Hogan, S.; Griffin, T.L.; Packard, C.A.; Chatfield, D.A.; Hagey, L.R.; Steinbach, J.H.; Hofmann, A.F. Human cecal bile acids: Concentration and spectrum. Am. J. Physiol. Liver Physiol. 2007, 293, G256–G263. [Google Scholar] [CrossRef] [Green Version]
- Raufman, J.-P.; Dawson, P.A.; Rao, A.; Drachenberg, C.B.; Heath, J.; Shang, A.C.; Hu, S.; Zhan, M.; Polli, J.E.; Cheng, K. Slc10a2 -null mice uncover colon cancer-promoting actions of endogenous fecal bile acids. Carcinogenesis 2015, 36, 1193–1200. [Google Scholar] [CrossRef] [Green Version]
- Cheng, K.; Metry, M.; Felton, J.; Shang, A.C.; Drachenberg, C.B.; Xu, S.; Zhan, M.; Schumacher, J.; Guo, G.L.; Polli, J.E.; et al. Diminished gallbladder filling, increased fecal bile acids, and promotion of colon epithelial cell proliferation and neoplasia in fibroblast growth factor 15-deficient mice. Oncotarget 2018, 9, 25572–25585. [Google Scholar] [CrossRef] [Green Version]
- Mehta, R.S.; Song, M.; Nishihara, R.; Drew, D.A.; Wu, K.; Qian, Z.R.; Fung, T.T.; Hamada, T.; Masugi, Y.; da Silva, A.; et al. Dietary Patterns and Risk of Colorectal Cancer: Analysis by Tumor Location and Molecular Subtypes. Gastroenterology 2017, 152, 1944–1953.e1. [Google Scholar] [CrossRef] [Green Version]
- Bianchini, F.; Caderni, G.; Dolara, P.; Fantetti, L.; Kriebel, D. Effect of Dietary Fat, Starch and Cellulose on Fecal Bile Acids in Mice. J. Nutr. 1989, 119, 1617–1624. [Google Scholar] [CrossRef]
- Boivin, G.P.; Washington, K.; Yang, K.; Ward, J.M.; Pretlow, T.P.; Russell, R.; Besselsen, D.G.; Godfrey, V.L.; Doetschman, T.; Dove, W.F.; et al. Pathology of mouse models of intestinal cancer: Consensus report and recommendations. Gastroenterology 2003, 124, 762–777. [Google Scholar] [CrossRef] [Green Version]
- Raufman, J.-P.P.; Samimi, R.; Shah, N.; Khurana, S.; Shant, J.; Drachenberg, C.; Xie, G.; Wess, J.; Cheng, K. Genetic ablation of M3 muscarinic receptors attenuates murine colon epithelial cell proliferation and neoplasia. Cancer Res. 2008, 68, 3573–3578. [Google Scholar] [CrossRef] [Green Version]
- Liu, H.; Hofmann, J.; Fish, I.; Schaake, B.; Eitel, K.; Bartuschat, A.; Kaindl, J.; Rampp, H.; Banerjee, A.; Hübner, H.; et al. Structure-guided development of selective M3 muscarinic acetylcholine receptor antagonists. Proc. Natl. Acad. Sci. USA 2018, 115, 12045–12050. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peng, Z.; Heath, J.; Drachenberg, C.; Raufman, J.P.; Xie, G. Cholinergic muscarinic receptor activation augments murine intestinal epithelial cell proliferation and tumorigenesis. BMC Cancer 2013, 13, 204. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheng, K.; Xie, G.; Khurana, S.; Heath, J.; Drachenberg, C.B.; Timmons, J.; Shah, N.; Raufman, J.-P. Divergent effects of muscarinic receptor subtype gene ablation on murine colon tumorigenesis reveals association of M3R and zinc finger protein 277 expression in colon neoplasia. Mol. Cancer 2014, 13, 77. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Raufman, J.P.; Shant, J.; Xie, G.; Cheng, K.; Gao, X.M.; Shiu, B.; Shah, N.; Drachenberg, C.B.; Heath, J.; Wess, J.; et al. Muscarinic receptor subtype-3 gene ablation and scopolamine butylbromide treatment attenuate small intestinal neoplasia in Apcmin/+ mice. Carcinogenesis 2011, 32, 1396–1402. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Drugs@FDA: FDA-Approved Drugs. Available online: https://www.accessdata.fda.gov/scripts/cder/daf/index.cfm?event=overview.process&ApplNo=207302 (accessed on 7 January 2021).
- Tata, A. Muscarinic Acetylcholine Receptors: New Potential Therapeutic Targets in Antinociception and in Cancer Therapy. Recent Pat. CNS Drug Discov. 2008, 3, 94–103. [Google Scholar] [CrossRef] [PubMed]
- Richardson, K.; Fox, C.; Maidment, I.; Steel, N.; Loke, Y.K.; Arthur, A.; Myint, P.K.; Grossi, C.M.; Mattishent, K.; Bennett, K.; et al. Anticholinergic drugs and risk of dementia: Case-control study. BMJ 2018, 361, k1315. [Google Scholar] [CrossRef] [Green Version]
- Coupland, C.A.C.; Hill, T.; Dening, T.; Morriss, R.; Moore, M.; Hippisley-Cox, J. Anticholinergic Drug Exposure and the Risk of Dementia. JAMA Intern. Med. 2019, 179, 1084. [Google Scholar] [CrossRef] [PubMed]
- Roberts, R.B.; Min, L.; Washington, M.K.; Olsen, S.J.; Settle, S.H.; Coffey, R.J.; Threadgill, D.W. Importance of epidermal growth factor receptor signaling in establishment of adenomas and maintenance of carcinomas during intestinal tumorigenesis. Proc. Natl. Acad. Sci. USA 2002, 99, 1521–1526. [Google Scholar] [CrossRef] [Green Version]
- Raufman, J.P.; Cheng, K.; Saxena, N.; Chahdi, A.; Belo, A.; Khurana, S.; Xie, G. Muscarinic receptor agonists stimulate matrix metalloproteinase 1-dependent invasion of human colon cancer cells. Biochem. Biophys. Res. Commun. 2011, 415, 319–324. [Google Scholar] [CrossRef] [Green Version]
- Giambernardi, T.A.; Grant, G.M.; Taylor, G.P.; Hay, R.J.; Maher, V.M.; Mccormick, J.J.; Klebe, R.J. Overview of matrix metalloproteinase expression in cultured human cells. Matrix Biol. 1998, 16, 483–496. [Google Scholar] [CrossRef]
- Vandenbroucke, R.E.; Libert, C. Is there new hope for therapeutic matrix metalloproteinase inhibition? Nat. Rev. Drug Discov. 2014, 13, 904–927. [Google Scholar] [CrossRef] [PubMed]
- Baker, E.A.; Bergin, F.G.; Leaper, D.J. Matrix metalloproteinases, their tissue inhibitors and colorectal cancer staging. Br. J. Surg. 2000, 87, 1215–1221. [Google Scholar] [CrossRef] [PubMed]
- Shapiro, S.D. Matrix metalloproteinase degradation of extracellular matrix: Biological consequences. Curr. Opin. Cell Biol. 1998, 10, 602–608. [Google Scholar] [CrossRef]
- McCawley, L.J.; Matrisian, L.M. Matrix metalloproteinases: Multifunctional contributors to tumor progression. Mol. Med. Today 2000, 6, 149–156. [Google Scholar] [CrossRef]
- Coussens, L.M.; Fingleton, B.; Matrisian, L.M. Matrix metalloproteinase inhibitors and cancer: Trials and tribulations. Science 2002, 295, 2387–2392. [Google Scholar] [CrossRef]
- Kirkegaard, H.; Johnsen, N.F.; Christensen, J.; Frederiksen, K.; Overvad, K.; Tjonneland, A. Association of adherence to lifestyle recommendations and risk of colorectal cancer: A prospective Danish cohort study. BMJ 2010, 341, c5504. [Google Scholar] [CrossRef] [Green Version]
- Murray, G.I.; Duncan, M.E.; O’Neil, P.; Melvin, W.T.; Fothergill, J.E. Matrix metalloproteinase–1 is associated with poor prognosis in colorectal cancer. Nat. Med. 1996, 2, 461–462. [Google Scholar] [CrossRef]
- Langenskiöld, M.; Ivarsson, M.L.; Holmdahl, L.; Falk, P.; Kåbjörn-Gustafsson, C.; Angenete, E. Intestinal mucosal MMP-1-a prognostic factor in colon cancer. Scand. J. Gastroenterol. 2013, 48, 563–569. [Google Scholar] [CrossRef]
- Xie, G.; Cheng, K.; Shant, J.; Raufman, J.P. Acetylcholine-induced activation of M3 muscarinic receptors stimulates robust matrix metalloproteinase gene expression in human colon cancer cells. Am. J. Physiol. Gastrointest. Liver Physiol. 2009, 296, G755–G763. [Google Scholar] [CrossRef] [Green Version]
- Shay, G.; Lynch, C.C.; Fingleton, B. Moving targets: Emerging roles for MMPs in cancer progression and metastasis. Matrix Biol. 2015, 44–46, 200–206. [Google Scholar] [CrossRef]
- Murphy, G. Riding the metalloproteinase roller coaster. J. Biol. Chem. 2017, 292, 7708–7718. [Google Scholar] [CrossRef] [Green Version]
- Pavlaki, M.; Zucker, S. Matrix metalloproteinase inhibitors (MMPIs): The beginning of phase I or the termination of phase III clinical trials. Cancer Metastasis Rev. 2003, 22, 177–203. [Google Scholar] [CrossRef]
- Zucker, S.; Cao, J.; Chen, W.-T. Critical appraisal of the use of matrix metalloproteinase inhibitors in cancer treatment. Oncogene 2000, 19, 6642–6650. [Google Scholar] [CrossRef] [PubMed]
- Rivera, S.; Khrestchatisky, M.; Kaczmarek, L.; Rosenberg, G.A.; Jaworski, D.M. Metzincin Proteases and Their Inhibitors: Foes or Friends in Nervous System Physiology? J. Neurosci. 2010, 30, 15337–15357. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goffin, J.R.; Anderson, I.C.; Supko, J.G.; Eder, J.P.; Shapiro, G.I.; Lynch, T.J.; Shipp, M.; Johnson, B.E.; Skarin, A.T. Phase I Trial of the Matrix Metalloproteinase Inhibitor Marimastat Combined with Carboplatin and Paclitaxel in Patients with Advanced Non–Small Cell Lung Cancer. Clin. Cancer Res. 2005, 11, 3417–3424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Macaulay, V.M.; O’Byrne, K.J.; Saunders, M.P.; Braybrooke, J.P.; Long, L.; Gleeson, F.; Mason, C.S.; Harris, A.L.; Brown, P.; Talbot, D.C. Phase I study of intrapleural batimastat (BB-94), a matrix metalloproteinase inhibitor, in the treatment of malignant pleural effusions. Clin. Cancer Res. 1999, 5, 513–520. [Google Scholar]
- Acharya, M.R.; Venitz, J.; Figg, W.D.; Sparreboom, A. Chemically modified tetracyclines as inhibitors of matrix metalloproteinases. Drug Resist. Updates 2004, 7, 195–208. [Google Scholar] [CrossRef]
- Giavazzi, R.; Garofalo, A.; Ferri, C.; Lucchini, V.; Bone, E.A.; Chiari, S.; Brown, P.D.; Nicoletti, M.I.; Taraboletti, G. Batimastat, a synthetic inhibitor of matrix metahoproteinases, potentiates the antitumor activity of cisplatin in ovarian carcinoma xenografts. Clin. Cancer Res. 1998, 4, 985–992. [Google Scholar]
- Zucker, S.; Vacirca, J. Role of matrix metalloproteinases (MMPs) in colorectal cancer. Cancer Metastasis Rev. 2004, 23, 101–117. [Google Scholar] [CrossRef]
- Sparano, J.A.; Bernardo, P.; Stephenson, P.; Gradishar, W.J.; Ingle, J.N.; Zucker, S.; Davidson, N.E. Randomized phase III trial of marimastat versus placebo in patients with metastatic breast cancer who have responding or stable disease after first-line chemotherapy: Eastern Cooperative Oncology Group Trial E2196. J. Clin. Oncol. 2004, 22, 4631–4638. [Google Scholar] [CrossRef]
- Edwards, D.; Handsley, M.; Pennington, C. The ADAM metalloproteinases. Mol. Aspects Med. 2008, 29, 258–289. [Google Scholar] [CrossRef] [PubMed]
- De Arao Tan, I.; Ricciardelli, C.; Russell, D.L. The metalloproteinase ADAMTS1: A comprehensive review of its role in tumorigenic and metastatic pathways. Int. J. Cancer 2013, 133, 2263–2276. [Google Scholar] [CrossRef] [PubMed]
- Vandenbroucke, R.E.; Vanlaere, I.; Van Hauwermeiren, F.; Van Wonterghem, E.; Wilson, C.; Libert, C. Pro-inflammatory effects of matrix metalloproteinase 7 in acute inflammation. Mucosal Immunol. 2014, 7, 579–588. [Google Scholar] [CrossRef] [PubMed]
- Apte, S.S.; Parks, W.C. Metalloproteinases: A parade of functions in matrix biology and an outlook for the future. Matrix Biol. 2015, 44–46, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Dufour, A.; Overall, C.M. Missing the target: Matrix metalloproteinase antitargets in inflammation and cancer. Trends Pharmacol. Sci. 2013, 34, 233–242. [Google Scholar] [CrossRef]
- Johnson, J.L.; Devel, L.; Czarny, B.; George, S.J.; Jackson, C.L.; Rogakos, V.; Beau, F.; Yiotakis, A.; Newby, A.C.; Dive, V. A Selective Matrix Metalloproteinase-12 Inhibitor Retards Atherosclerotic Plaque Development in Apolipoprotein E–Knockout Mice. Arterioscler. Thromb. Vasc. Biol. 2011, 31, 528–535. [Google Scholar] [CrossRef] [Green Version]
- Devel, L.; Rogakos, V.; David, A.; Makaritis, A.; Beau, F.; Cuniasse, P.; Yiotakis, A.; Dive, V. Development of selective inhibitors and substrate of matrix metalloproteinase-12. J. Biol. Chem. 2006, 281, 11152–11160. [Google Scholar] [CrossRef] [Green Version]
- Devy, L.; Huang, L.; Naa, L.; Yanamandra, N.; Pieters, H.; Frans, N.; Chang, E.; Tao, Q.; Vanhove, M.; Lejeune, A.; et al. Selective Inhibition of Matrix Metalloproteinase-14 Blocks Tumor Growth, Invasion, and Angiogenesis. Cancer Res. 2009, 69, 1517–1526. [Google Scholar] [CrossRef] [Green Version]
- Sela-Passwell, N.; Kikkeri, R.; Dym, O.; Rozenberg, H.; Margalit, R.; Arad-Yellin, R.; Eisenstein, M.; Brenner, O.; Shoham, T.; Danon, T.; et al. Antibodies targeting the catalytic zinc complex of activated matrix metalloproteinases show therapeutic potential. Nat. Med. 2012, 18, 143–147. [Google Scholar] [CrossRef]
- Scannevin, R.H.; Alexander, R.; Haarlander, T.M.; Burke, S.L.; Singer, M.; Huo, C.; Zhang, Y.-M.; Maguire, D.; Spurlino, J.; Deckman, I.; et al. Discovery of a highly selective chemical inhibitor of matrix metalloproteinase-9 (MMP-9) that allosterically inhibits zymogen activation. J. Biol. Chem. 2017, 292, 17963–17974. [Google Scholar] [CrossRef] [Green Version]
- Daub, H.; Ulrich Weiss, F.; Wallasch, C.; Ullrich, A. Role of transactivation of the EGF receptor in signalling by G-protein-coupled receptors. Nature 1996, 379, 557–560. [Google Scholar] [CrossRef]
- Mitamura, T.; Higashiyama, S.; Taniguchi, N.; Klagsbrun, M.; Mekada, E. Diphtheria toxin binds to the epidermal growth factor (EGF)-like domain of human heparin-binding EGF-like growth factor/diphtheria toxin receptor and inhibits specifically its mitogenic activity. J. Biol. Chem. 1995. [Google Scholar] [CrossRef] [Green Version]
- Cohen, S.; Carpenter, G. Human epidermal growth factor: Isolation and chemical and biological properties. Proc. Natl. Acad. Sci. USA 1975, 72, 1317–1321. [Google Scholar] [CrossRef] [Green Version]
- Seeber, A.; Gastl, G. Targeted Therapy of Colorectal Cancer. Oncol. Res. Treat. 2016, 39, 796–802. [Google Scholar] [CrossRef]
- Pai, S.G. Novel therapeutic agents in the treatment of metastatic colorectal cancer. World J. Gastrointest. Oncol. 2016, 8, 99. [Google Scholar] [CrossRef] [Green Version]
- Ciardiello, F.; Tortora, G. A novel approach in the treatment of cancer: Targeting the epidermal growth factor receptor. Clin. Cancer Res. 2001, 7, 2958–2970. [Google Scholar]
- Li, K.; Guo, Q.; Yang, J.; Chen, H.; Hu, K.; Zhao, J.; Zheng, S.; Pang, X.; Zhou, S.; Dang, Y.; et al. FOXD3 is a tumor suppressor of colon cancer by inhibiting EGFRRas- Raf-MEK-ERK signal pathway. Oncotarget 2017, 8, 5048–5056. [Google Scholar] [CrossRef] [Green Version]
- Gong, J.; Cho, M.; Fakih, M. RAS and BRAF in metastatic colorectal cancer management. J. Gastrointest. Oncol. 2016, 7, 687–704. [Google Scholar] [CrossRef] [Green Version]
- Dienstmann, R.; Connor, K.; Byrne, A.T.; Fridman, W.H.; Lambrechts, D.; Sadanandam, A.; Trusolino, L.; Prehn, J.H.M.; Tabernero, J.; Kolch, W. Precision Therapy in RAS Mutant Colorectal Cancer. Gastroenterology 2020, 158, 806–811. [Google Scholar] [CrossRef]
- Porru, M.; Pompili, L.; Caruso, C.; Biroccio, A.; Leonetti, C. Targeting KRAS in metastatic colorectal cancer: Current strategies and emerging opportunities. J. Exp. Clin. Cancer Res. 2018, 37, 57. [Google Scholar] [CrossRef] [Green Version]
- Slack, B.E. The m3 muscarinic acetylcholine receptor is coupled to mitogen-activated protein kinase via protein kinase C and epidermal growth factor receptor kinase. Biochem. J. 2000, 348, 381–387. [Google Scholar] [CrossRef]
- Chen, J.; Elfiky, A.; Han, M.; Chen, C.; Saif, M.W. The Role of Src in Colon Cancer and Its Therapeutic Implications. Clin. Colorectal Cancer 2014, 13, 5–13. [Google Scholar] [CrossRef] [Green Version]
- Leu, T.H.; Maa, M.C. Functional implication of the interaction between EGF receptor and c-Src. Front. Biosci. 2003, 8, s28–s38. [Google Scholar] [CrossRef] [Green Version]
- Sato, K.I.; Sato, A.; Aoto, M.; Fukami, Y. c-Src Phosphorylates Epidermal Growth Factor Receptor on Tyrosine 845. Biochem. Biophys. Res. Commun. 1995, 215, 1078–1087. [Google Scholar] [CrossRef]
- Biscardi, J.S.; Maa, M.C.; Tice, D.A.; Cox, M.E.; Leu, T.H.; Parsons, S.J. c-Src-mediated phosphorylation of the epidermal growth factor receptor on Tyr845 and Tyr1101 is associated with modulation of receptor function. J. Biol. Chem. 1999, 274, 8335–8343. [Google Scholar] [CrossRef] [Green Version]
- Pelissier-Rota, M.; Chartier, N.T.; Bonaz, B.; Jacquier-Sarlin, M.R. A crosstalk between muscarinic and CRF2 receptors regulates cellular adhesion properties of human colon cancer cells. Biochim. Biophys. Acta Mol. Cell Res. 2017, 1864, 1246–1259. [Google Scholar] [CrossRef]
- Park, Y.S.; Liu, Z.; Vasamsetti, B.M.K.; Cho, N.J. The ERK1/2 and mTORC1 Signaling Pathways Are Involved in the Muscarinic Acetylcholine Receptor-Mediated Proliferation of SNU-407 Colon Cancer Cells. J. Cell. Biochem. 2016, 2854–2863. [Google Scholar] [CrossRef]
- Park, Y.-S.; Cho, N.-J. Enhanced proliferation of SNU-407 human colon cancer cells by muscarinic acetylcholine receptors. BMB Rep. 2008, 41, 803–807. [Google Scholar] [CrossRef] [Green Version]
- Park, Y.S.; Cho, N.J. EGFR and PKC are involved in the activation of ERK1/2 and p90 RSK and the subsequent proliferation of SNU-407 colon cancer cells by muscarinic acetylcholine receptors. Mol. Cell. Biochem. 2012, 370, 191–198. [Google Scholar] [CrossRef]
- Ryazanov, A.G.; Shestakova, E.A.; Natapov, P.G. Phosphorylation of elongation factor 2 by EF-2 kinase affects rate of translation. Nature 1988, 334, 170–173. [Google Scholar] [CrossRef]
- Vasamsetti, B.M.K.; Liu, Z.; Park, Y.S.; Cho, N.J. Muscarinic acetylcholine receptors regulate the dephosphorylation of eukaryotic translation elongation factor 2 in SNU-407 colon cancer cells. Biochem. Biophys. Res. Commun. 2019, 516, 424–429. [Google Scholar] [CrossRef] [PubMed]
- Nairn, A.C.; Palfrey, H.C. Identification of the major Mr 100,000 substrate for calmodulin-dependent protein kinase III in mammalian cells as elongation factor-2. J. Biol. Chem. 1987, 262, 17299–17303. [Google Scholar] [CrossRef]
- Lee, W.J.; Blair, A.; Hoppin, J.A.; Lubin, J.H.; Rusiecki, J.A.; Sandler, D.P.; Dosemeci, M.; Alavanja, M.C.R. Cancer Incidence Among Pesticide Applicators Exposed to Chlorpyrifos in the Agricultural Health Study. JNCI J. Natl. Cancer Inst. 2004, 96, 1781–1789. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suriyo, T.; Tachachartvanich, P.; Visitnonthachai, D.; Watcharasit, P.; Satayavivad, J. Chlorpyrifos promotes colorectal adenocarcinoma H508 cell growth through the activation of EGFR/ERK1/2 signaling pathway but not cholinergic pathway. Toxicology 2015, 338, 117–129. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
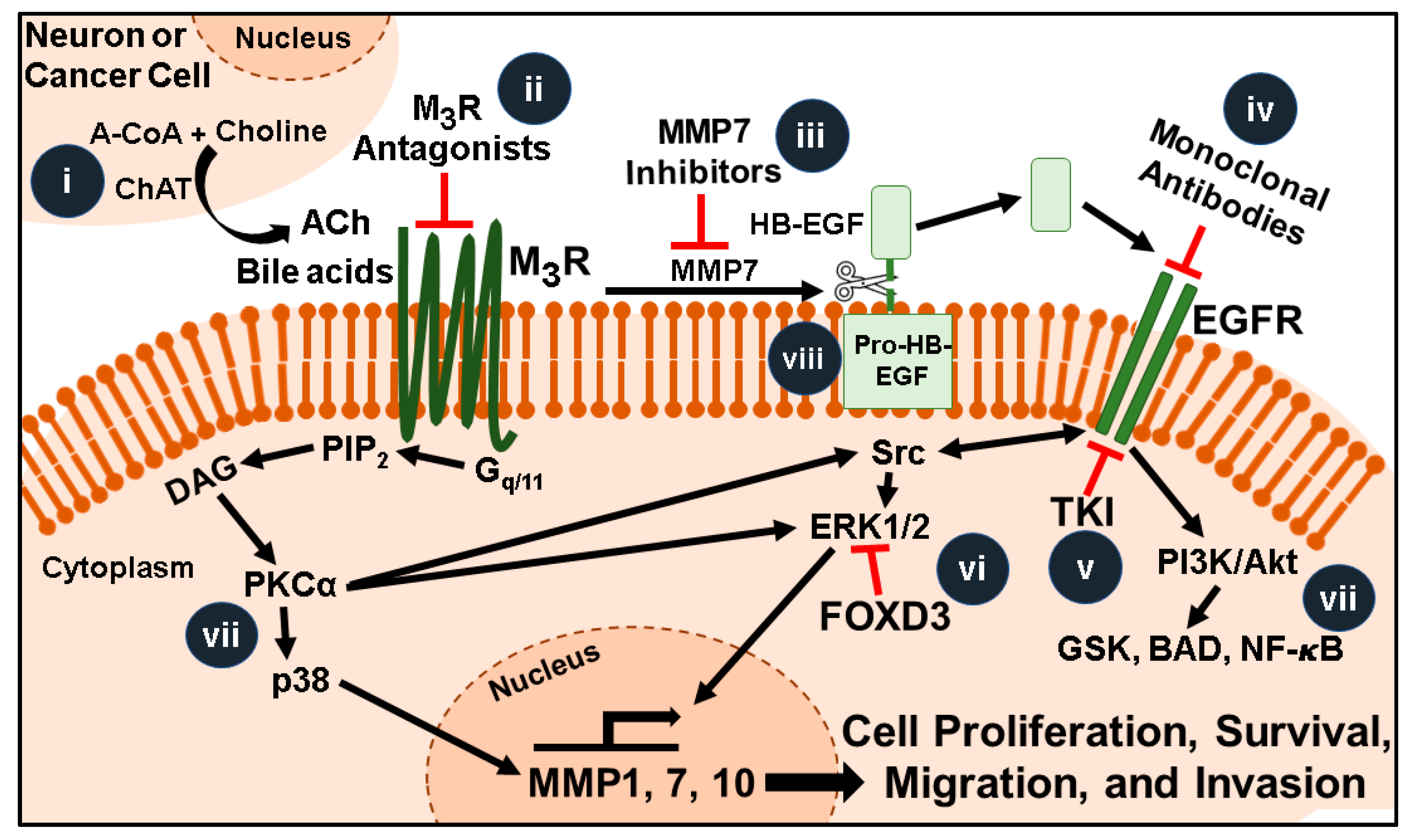
Muscarinic receptor signaling pathways and promising therapeutic targets for colorectal cancer. Acetylcholine (ACh, released from neurons, immunocytes, or adjacent cancer cells) and bile acids (BAs, in the fecal stream) activate M3 muscarinic receptors (M3R) overexpressed in colon cancer. Post M3R-signaling involves activation of protein kinase C-α (PKCα) and matrix metalloproteinase 7 (MMP7)-catalyzed release of heparin binding epidermal growth factor (EGF)-like growth factor (HB-EGF) which activates EGFR (EGFR-independent and -dependent signaling, respectively). Ultimately interactions between these complex signaling pathways induce transcription of genes that stimulate cell proliferation, survival, migration, and invasion. Amongst these genes, induction of matrix metalloproteinase-7 (MMP7) provides a “feed forward mechanism” to increase HB-EGF release. This illustration highlights features that are current or potentially future therapeutic targets: (i) Colon cancer cells can produce and release ACh, thus the molecules necessary for this process (e.g., choline acetyltransferase, ChAT) are potential therapeutic targets. (ii) Selective inhibitors of M3R can be repurposed to block the effects of ACh and bile acids. (iii) Inhibitors that selectively target MMP1 or MMP7 could block post-M3R signaling and degradation of the extracellular matrix. (iv) Monoclonal antibodies that block the EGFR binding site and (v) tyrosine kinase inhibitors that inhibit EGFR activation could be used in conjunction with selective M3R inhibitors to potentiate their inhibitory actions. (vi) FOXD3 overexpression or small molecular mimics block M3R- and EGFR-induced ERK activation. (vii) Highly selective inhibitors of the plethora of signaling molecules downstream of M3R and EGFR are in various stages of development. These include agents targeting Ras, ERK, PKC-α, and molecules comprising the apoptosis program (GSK, BAD, NF-ĸB). (viii) Pro-HB-EGF, a substrate for MMP7-activated release of HB-EGF and EGFR activation, is an unexploited therapeutic target.
Figure 1.
Muscarinic receptor signaling pathways and promising therapeutic targets for colorectal cancer. Acetylcholine (ACh, released from neurons, immunocytes, or adjacent cancer cells) and bile acids (BAs, in the fecal stream) activate M3 muscarinic receptors (M3R) overexpressed in colon cancer. Post M3R-signaling involves activation of protein kinase C-α (PKCα) and matrix metalloproteinase 7 (MMP7)-catalyzed release of heparin binding epidermal growth factor (EGF)-like growth factor (HB-EGF) which activates EGFR (EGFR-independent and -dependent signaling, respectively). Ultimately interactions between these complex signaling pathways induce transcription of genes that stimulate cell proliferation, survival, migration, and invasion. Amongst these genes, induction of matrix metalloproteinase-7 (MMP7) provides a “feed forward mechanism” to increase HB-EGF release. This illustration highlights features that are current or potentially future therapeutic targets: (i) Colon cancer cells can produce and release ACh, thus the molecules necessary for this process (e.g., choline acetyltransferase, ChAT) are potential therapeutic targets. (ii) Selective inhibitors of M3R can be repurposed to block the effects of ACh and bile acids. (iii) Inhibitors that selectively target MMP1 or MMP7 could block post-M3R signaling and degradation of the extracellular matrix. (iv) Monoclonal antibodies that block the EGFR binding site and (v) tyrosine kinase inhibitors that inhibit EGFR activation could be used in conjunction with selective M3R inhibitors to potentiate their inhibitory actions. (vi) FOXD3 overexpression or small molecular mimics block M3R- and EGFR-induced ERK activation. (vii) Highly selective inhibitors of the plethora of signaling molecules downstream of M3R and EGFR are in various stages of development. These include agents targeting Ras, ERK, PKC-α, and molecules comprising the apoptosis program (GSK, BAD, NF-ĸB). (viii) Pro-HB-EGF, a substrate for MMP7-activated release of HB-EGF and EGFR activation, is an unexploited therapeutic target.

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Ali, O.; Tolaymat, M.; Hu, S.; Xie, G.; Raufman, J.-P. Overcoming Obstacles to Targeting Muscarinic Receptor Signaling in Colorectal Cancer. Int. J. Mol. Sci. 2021, 22, 716. https://doi.org/10.3390/ijms22020716
AMA Style
Ali O, Tolaymat M, Hu S, Xie G, Raufman J-P. Overcoming Obstacles to Targeting Muscarinic Receptor Signaling in Colorectal Cancer. International Journal of Molecular Sciences. 2021; 22(2):716. https://doi.org/10.3390/ijms22020716
Chicago/Turabian StyleAli, Osman, Mazen Tolaymat, Shien Hu, Guofeng Xie, and Jean-Pierre Raufman. 2021. "Overcoming Obstacles to Targeting Muscarinic Receptor Signaling in Colorectal Cancer" International Journal of Molecular Sciences 22, no. 2: 716. https://doi.org/10.3390/ijms22020716
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.