Risperidone Exacerbates Glucose Intolerance, Nonalcoholic Fatty Liver Disease, and Renal Impairment in Obese Mice

 ,
,
Abstract
:1. Introduction
2. Results
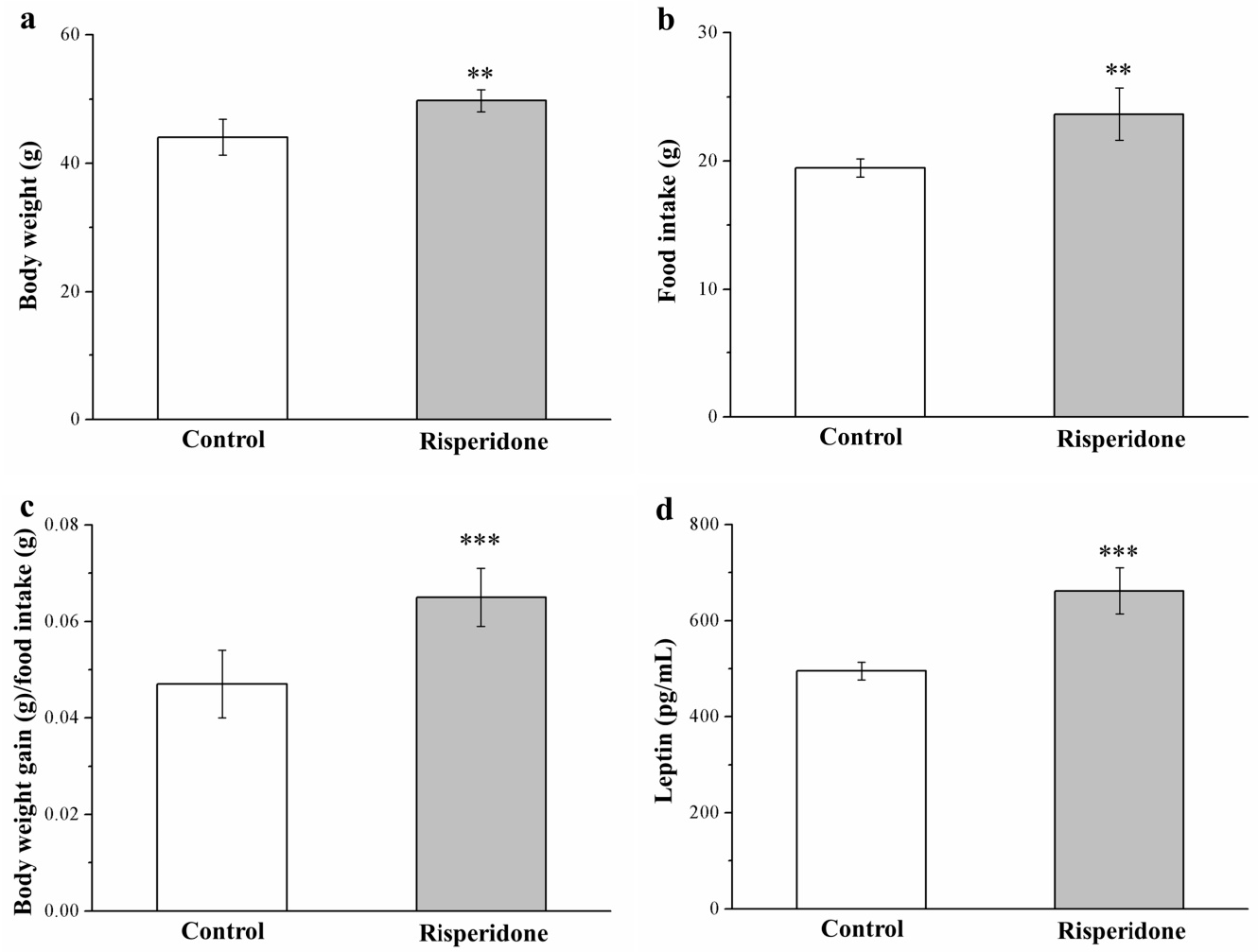
2.1. Risperidone Affects the Food Efficiency and Intake and Morphometric Parameters
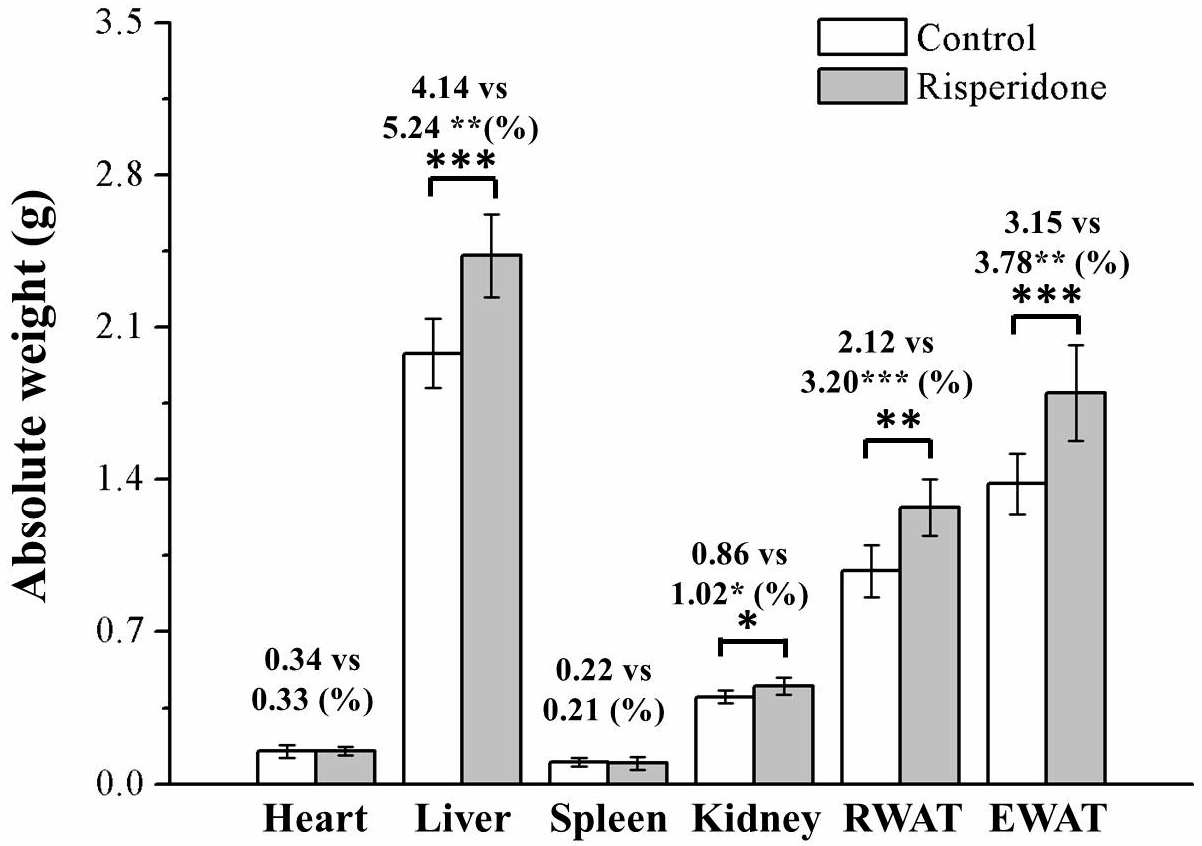
2.2. Risperidone Increases the Weight of Organs and Fat Pads
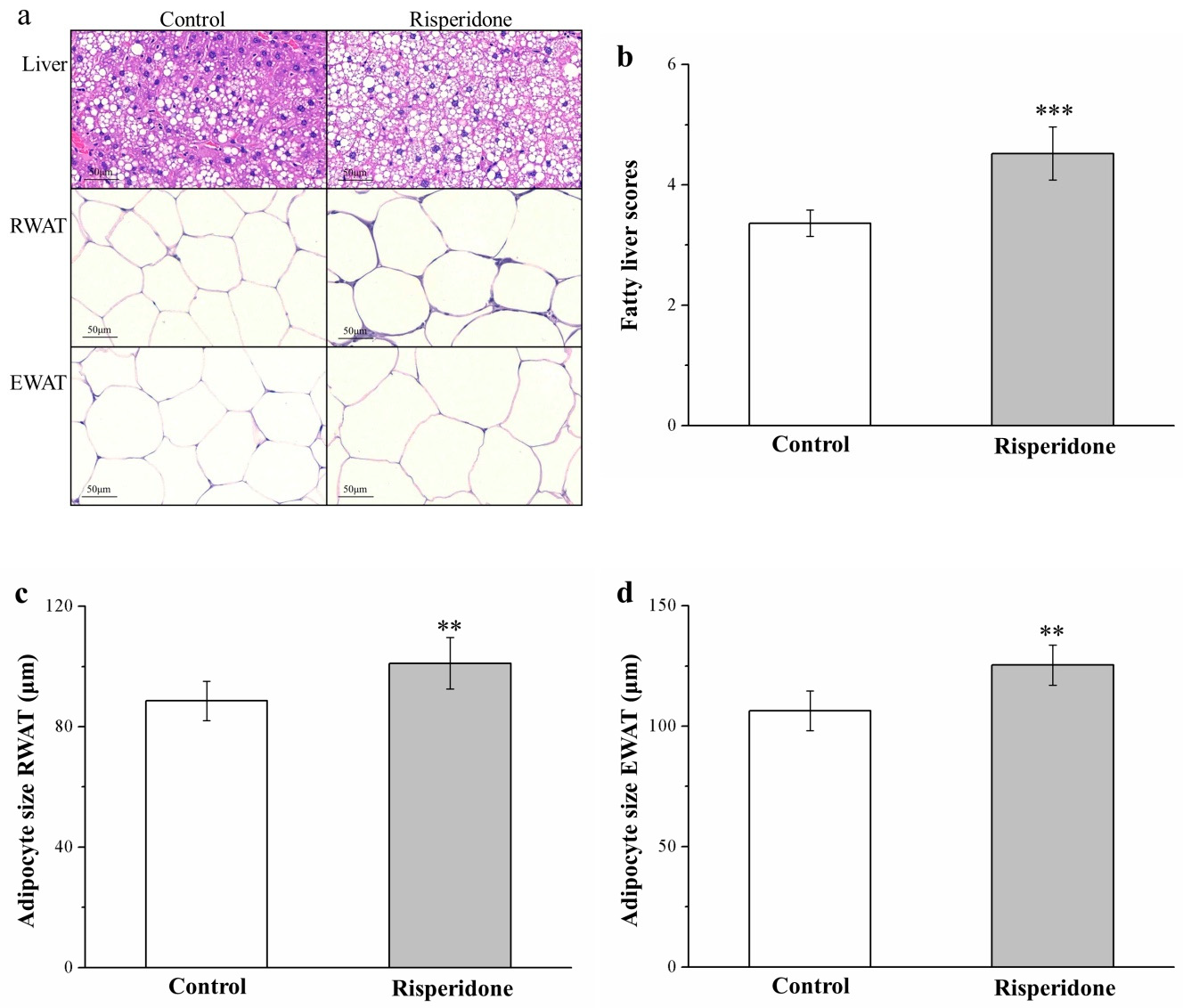
2.3. Risperidone Restricts the Accumulation of Liver Fat and Size of Adipocytes
2.4. Risperidone Increases the Serum Levels of Alanine Aminotransferase, Aspartate Aminotransferase, Fatty Acid-Binding Protein 4 mRNA, and Sterol Regulatory Element-Binding Protein 1 mRNA
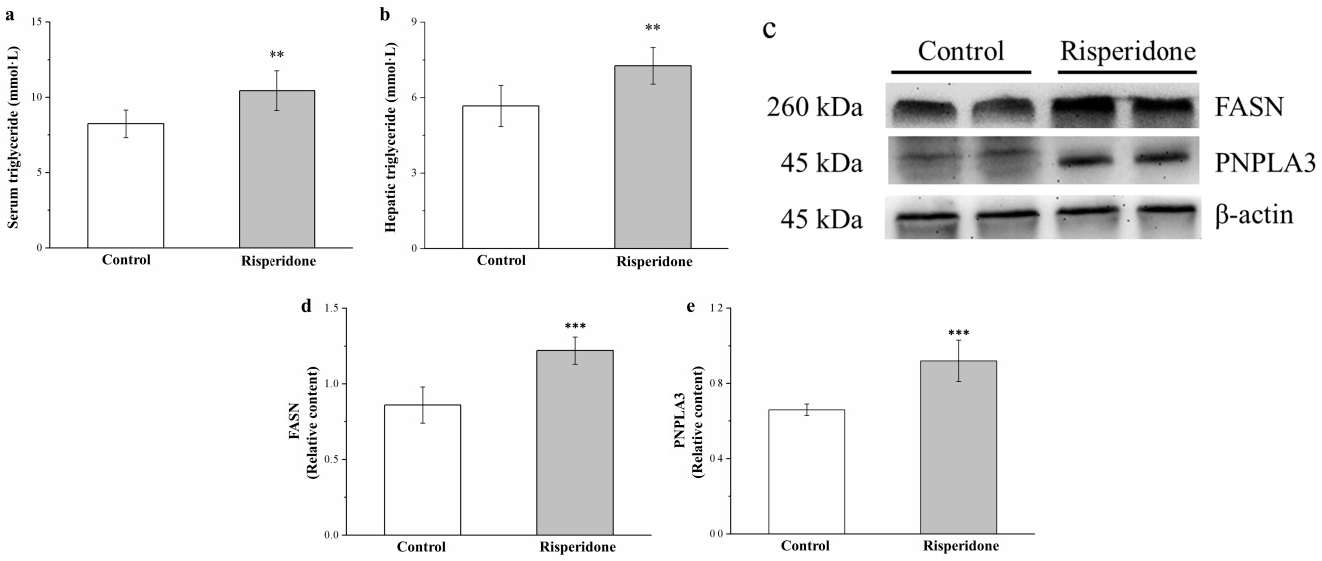
2.5. Risperidone Increases the Serum and Hepatic Levels of Triglycerides as Well as the Hepatic Expression of Patatin-Like Phospholipid Domain Containing Protein 3 and Fatty Acid Synthase but Reduces the Hepatic Expression of Adiponectin and AMPK
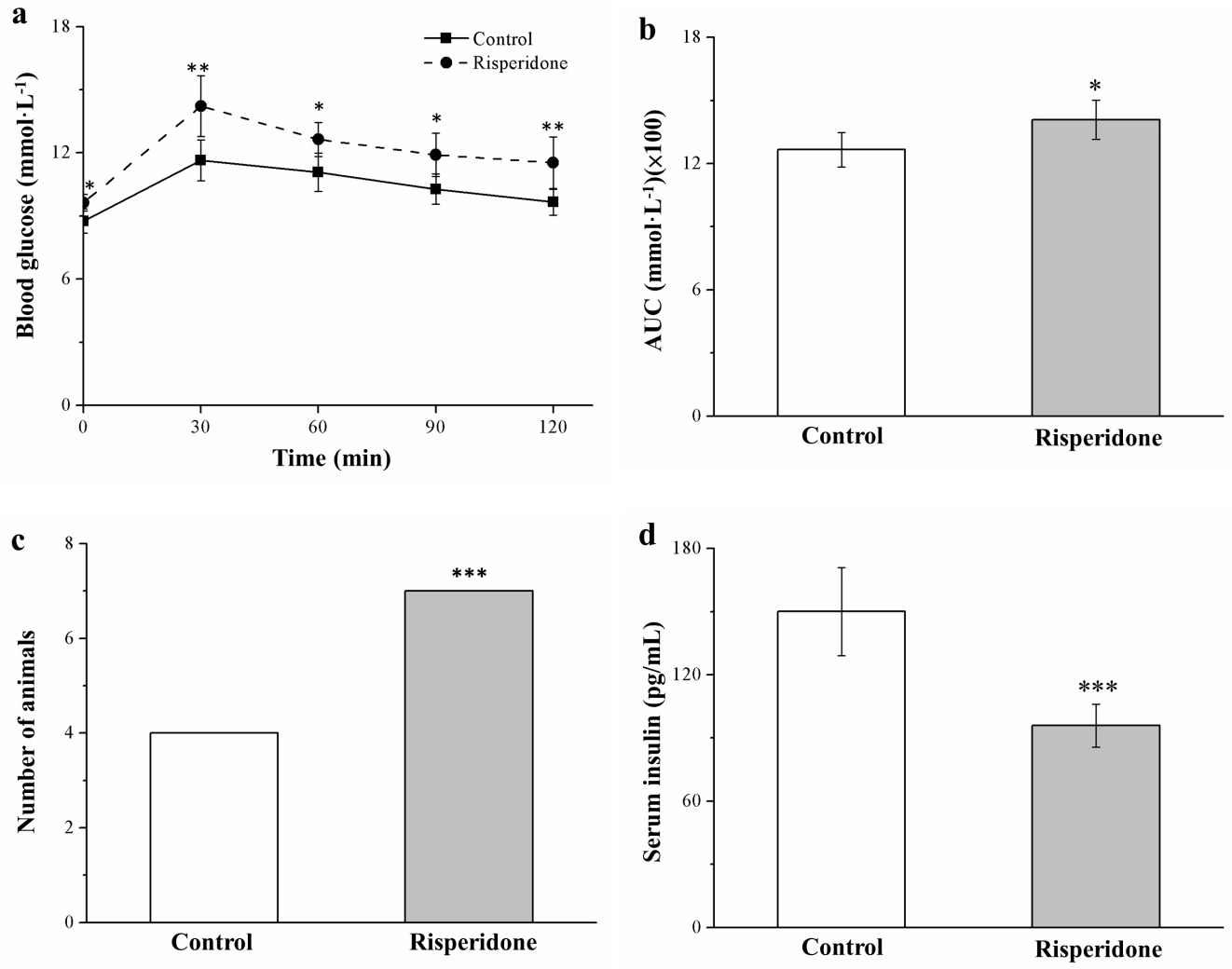
2.6. Risperidone Reduces Insulin Levels and Worsens Glucose Intolerance
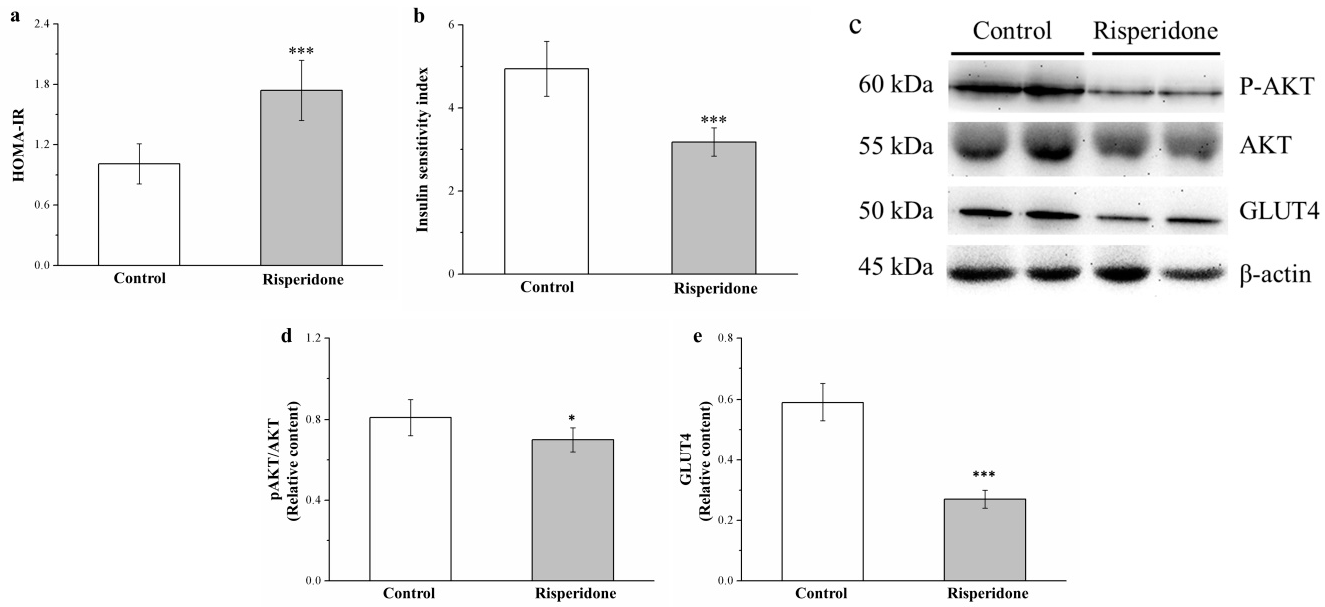
2.7. Risperidone Reduces IS by Attenuating Phosphorylated Akt and Glucose Transporter 4 Expression
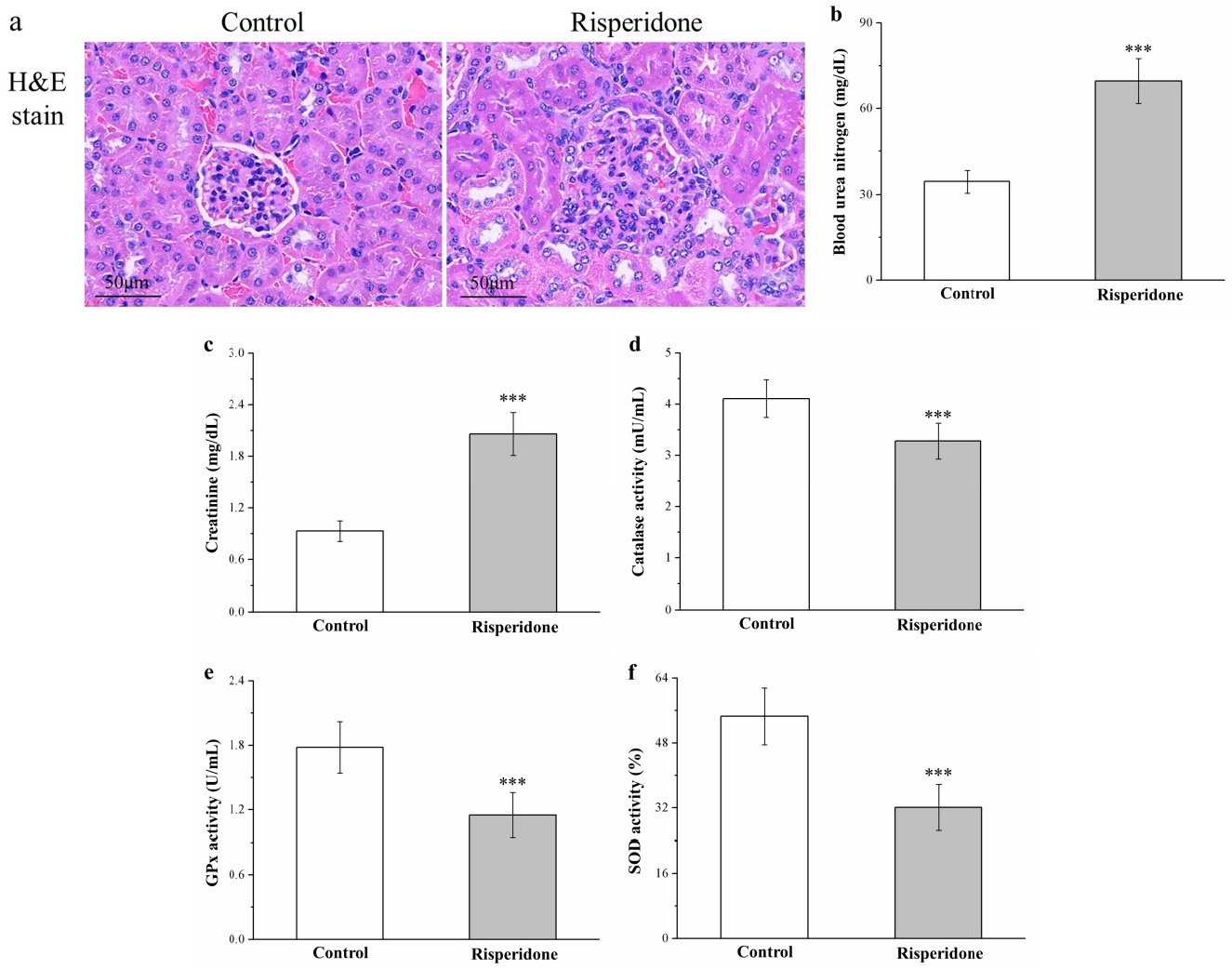
2.8. Risperidone Induces Renal Injury and Increases Serum Blood Urea Nitrogen and Creatinine Levels but Reduces Antioxidant Enzymes in the Kidneys
3. Discussion
4. Materials and Methods
4.1. Animals, Diet-Based Obesity Induction, and Risperidone Therapy
4.2. Food Intake, Body Weight, Leptin, and Insulin Level Measurement
4.3. Measurement of Serum and Hepatic Levels of Triglycerides and Serum Levels of Creatinine, Bun, ALT, and Ast
4.4. IPGTT
4.5. IR and IS Indices
- -
- HOMA-IR index = [fasting insulin (in mU/L) × fasting glucose (in mmol/L)]/22.5,
- -
- IS index = [1/fasting insulin (in mU/L) × fasting glucose (in mmol/L)] × 1000.
4.6. Histological and Morphometric Analyses of Tissues
4.7. RNA Extraction and Real-Time Quantitative Polymerase Chain Reaction
4.8. Western Blotting
4.9. Measurement of Renal Catalase, GPx, and SOD Levels
4.10. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| HFD | High-fat diet |
| SD | Standard diet |
| RWAT | Retroperitoneal white adipose tissue |
| EWAT | Epididymal white adipose tissue |
| H&E | Hematoxylin and eosin |
| ALT | Alanine aminotransferase |
| AST | Aspartate aminotransferase |
| FABP4 | Fatty acid-binding protein 4 |
| SREBP1 | Sterol regulatory element-binding protein 1 |
| FASN | Fatty acid synthase |
| PNPLA3 | Patatin-like phospholipid domain containing protein 3 |
| AUC | Area under the curves |
| IR | Insulin resistance |
| IS | Insulin sensitivity |
| HOMA | Homeostatic model assessment |
| GLUT | Glucose transport |
| BUN | Blood urea nitrogen |
| GPx | Glutathione peroxidase |
| SOD | Superoxide dismutase |
| ERK | Extracellular signal-related kinase |
| SOCS | Suppressor of cytokine signaling |
| FGF-21 | Fibroblast growth factor-21 |
| NLRP3 | Leucine-rich repeat domain containing 3 |
| IL-1β | Interleukin-1β |
| IL-18 | Interleukin-18 |
| IPGTT | Intraperitoneal glucose tolerance test |
| TNF-β | Tumor necrosis factor-β |
| IHC | Immunohistochemical |
References
- Llerena, A.; Berecz, R.; Peñas-Lledó, E.; Süveges, A.; Fariñas, H. Pharmacogenetics of clinical response to risperidone. Pharmacogenomics 2013, 14, 177–194. [Google Scholar] [CrossRef] [PubMed]
- Ricci, L.A.; Connor, D.F.; Morrison, R.; Melloni, R.H.J. Risperidone exerts potent anti-aggressive effects in a developmentally immature animal model of escalated aggression. Biol. Psychiatry 2007, 62, 218–225. [Google Scholar] [CrossRef] [PubMed]
- Shen, S.P.; Liu, Y.; Qiu, H.; Tsai, K.Y.; Wu, H.C.; Liang, W.M.; Shu, M.; Chou, F.H. The risk of bone fracture after long-term risperidone exposure is not increased compared to other atypical antipsychotics: A retrospective cohort study. PLoS ONE 2019, 14, e0221948. [Google Scholar] [CrossRef] [PubMed]
- McDougle, C.J.; Epperson, C.N.; Pelton, G.H.; Wasylink, S.; Price, L.H. A double-blind, placebo-controlled study of risperidone addition in serotonin reuptake inhibitor-refractory obsessive-compulsive disorder. Arch. Gen. Psychiatry 2000, 57, 794–801. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Muñoz-Negro, J.E.; Cuadrado, L.; Cervilla, J.A. Current evidences on psychopharmacology of schizoaffective disorder. Actas Esp. Psiquiatr. 2019, 47, 190–201. [Google Scholar] [PubMed]
- Luo, C.; Liu, J.; Wang, X.; Mao, X.; Zhou, H.; Liu, Z. Pharmacogenetic correlates of antipsychotic-induced weight gain in the Chinese population. Neurosci. Bull. 2019, 35, 561–580. [Google Scholar] [CrossRef] [PubMed]
- Krogmann, A.; Peters, L.; von Hardenberg, L.; Bödeker, K.; Nöhles, V.B.; Correll, C.U. Keeping up with the therapeutic advances in schizophrenia: A review of novel and emerging pharmacological entities. CNS Spectr. 2019, 24, 38–69. [Google Scholar] [CrossRef]
- Kaushal, J.; Bhutani, G.; Gupta, R. Comparison of fasting blood sugar and serum lipid profile changes after treatment with atypical antipsychotics olanzapine and risperidone. Singapore Med. J. 2012, 53, 488–492. [Google Scholar]
- Kaur, K.K.; Gautam Allahbadia, G.; Singh, M. Importance of simultaneous treatment of obesity and diabetes mellitus: A sequelae to the understanding of diabesity-a review. Obes. Res. Open J. 2018, 6, 1–10. [Google Scholar] [CrossRef]
- Raeder, M.B.; Fernø, J.; Vik-Mo, A.O.; Steen, V.M. SREBP activation by antipsychotic- and antidepressant-drugs in cultured human liver cells: Relevance for metabolic side-effects? Mol. Cell. Biochem. 2006, 289, 167–173. [Google Scholar] [CrossRef]
- Khoza, S.; Barner, J.C. Glucose dysregulation associated with antidepressant agents: An analysis of 17 published case reports. Int. J. Clin. Pharm. 2011, 33, 484–492. [Google Scholar] [CrossRef] [PubMed]
- Nagamine, T. Severe hypoglycemia associated with risperidone. Psychiatry Clin. Neurosci. 2016, 70, 421. [Google Scholar] [CrossRef] [PubMed]
- Omi, T.; Riku, K.; Fukumoto, M.; Kanai, K.; Omura, Y.; Takada, H.; Matunaga, H. Paliperidone induced hypoglycemia by increasing insulin secretion. Case Rep. Psychiatry 2016, 2016, 1805414. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, P.L.; Lane, H.Y.; Su, K.P. Risperidone alternative for a schizophrenic patient with olanzapine-exacerbated diabetic mellitus. Psychiatry Clin. Neurosci. 2006, 60, 115–116. [Google Scholar] [CrossRef]
- Gupta, S.; Steinmeyer, C.; Frank, B.; Madhusoodanan, S.; Lockwood, K.; Lentz, B.; Keller, P. Hyperglycemia and hypertriglyceridemia in real world patients on antipsychotic therapy. Am. J. Ther. 2003, 10, 348–355. [Google Scholar] [CrossRef]
- Koller, E.A.; Cross, J.T.; Doraiswamy, P.M.; Schneider, B.S. Risperidone-associated diabetes mellitus: A pharmacovigilance study. Pharmacotherapy 2003, 23, 735–744. [Google Scholar] [CrossRef]
- Konishi, H.; Shirakawa, J.; Arai, M.; Terauchi, Y. Drug-induced hyperglycemia in the Japanese Adverse Drug Event Report database: Association of evelolimus use with diabetes. Endocr. J. 2019, 66, 571–574. [Google Scholar] [CrossRef] [Green Version]
- Auger, F.; Duriez, P.; Martin-Nizard, F.; Durieux, N.; Bordet, R.; Pétrault, O. Long-term risperidone treatment induces visceral adiposity associated with hepatic steatosis in mice: A magnetic resonance approach. Schizophr. Res. Treat. 2014, 2014, 429291. [Google Scholar] [CrossRef]
- Azirak, S.; Bilgic, S.; Korkmaz, D.T.; Guvenc, A.N.; Kocaman, N.; Ozer, M.K. The protective effect of resveratrol against risperidone-induced liver damage through an action on FAS gene expression. Gen. Physiol. Biophys. 2019, 38, 215–225. [Google Scholar] [CrossRef]
- Jiang, Y.; McCombs, J.S.; Park, S.H. A Retrospective Cohort Study of Acute Kidney Injury Risk Associated with Antipsychotics. CNS Drugs 2017, 31, 319–326. [Google Scholar] [CrossRef]
- Højlund, M.; Lund, L.C.; Herping, J.L.E.; Haastrup, M.B.; Damkier, P.; Henriksen, D.P. Second-generation antipsychotics and the risk of chronic kidney disease: A population-based case-control study. BMJ Open 2020, 10, e038247. [Google Scholar] [CrossRef] [PubMed]
- Chang, G.R.; Chiu, Y.S.; Wu, Y.Y.; Chen, W.Y.; Liao, J.W.; Chao, T.H.; Mao, F.C. Rapamycin protects against high fat diet-induced obesity in C57BL/6J mice. J. Pharmacol. Sci. 2009, 109, 496–503. [Google Scholar] [CrossRef] [PubMed]
- Hou, P.H.; Chang, G.R.; Chen, C.P.; Lin, Y.L.; Chao, I.S.; Shen, T.T.; Mao, F.C. Long-term administration of olanzapine induces adiposity and increases hepatic fatty acid desaturation protein in female C57BL/6J mice. Iran. J. Basic Med. Sci. 2018, 21, 495–501. [Google Scholar] [PubMed]
- Chang, G.R.; Wu, Y.Y.; Chiu, Y.S.; Chen, W.Y.; Liao, J.W.; Hsu, H.M.; Chao, T.H.; Hung, S.W.; Mao, F.C. Long-term administration of rapamycin reduces adiposity, but impairs glucose tolerance in high-fat diet-fed KK/HlJ mice. Basic Clin. Pharmacol. Toxicol. 2009, 105, 188–198. [Google Scholar] [CrossRef]
- Wu, C.F.; Hou, P.H.; Mao, F.C.; Su, Y.C.; Wu, C.Y.; Yang, W.C.; Lin, C.S.; Tsai, H.P.; Liao, H.Y.; Chang, G.R. Mirtazapine reduces adipocyte hypertrophy and increases glucose transporter expression in obese mice. Animals 2020, 10, 1423. [Google Scholar] [CrossRef]
- Kim, J.W.; Lee, Y.S.; Park, Y.S.; Kim, B.H.; Lee, S.Y.; Yeon, J.E.; Lee, C.H. Multiparametric MR index for the diagnosis of non-alcoholic steatohepatitis in patients with non-alcoholic fatty liver disease. Sci. Rep. 2020, 10, 2671. [Google Scholar] [CrossRef] [Green Version]
- Scaini, G.; Quevedo, J.; Velligan, D.; Roberts, D.L.; Raventos, H.; Walss-Bass, C. Second generation antipsychotic-induced mitochondrial alterations: Implications for increased risk of metabolic syndrome in patients with schizophrenia. Eur. Neuropsychopharmacol. 2018, 28, 369–380. [Google Scholar] [CrossRef]
- Rojo, L.E.; Gaspar, P.A.; Silva, H.; Risco, L.; Arena, P.; Cubillos-Robles, K.; Jara, B. Metabolic syndrome and obesity among users of second generation antipsychotics: A global challenge for modern psychopharmacology. Pharmacol. Res. 2015, 101, 74–85. [Google Scholar] [CrossRef]
- Carney, C.P.; Jones, L.; Woolson, R.F. Medical comorbidity in women and men with schizophrenia: A population-based controlled study. J. Gen. Intern. Med. 2006, 21, 1133–1137. [Google Scholar] [CrossRef]
- Hsu, J.H.; Chien, I.C.; Lin, C.H.; Chou, Y.J.; Chou, P. Increased risk of chronic liver disease in patients with schizophrenia: A population based cohort study. Psychosomatics 2014, 55, 163–171. [Google Scholar] [CrossRef]
- Wang, H.Y.; Huang, C.L.; Feng, I.J.; Tsuang, H.C. Second-generation antipsychotic medications and risk of chronic kidney disease in schizophrenia: Population-based nested case-control study. BMJ Open 2018, 8, e019868. [Google Scholar] [CrossRef] [PubMed]
- Chang, G.R.; Chiu, Y.S.; Wu, Y.Y.; Lin, Y.C.; Hou, P.H.; Mao, F.C. Rapamycin impairs HPD-induced beneficial effects on glucose homeostasis. Br. J. Pharmacol. 2015, 172, 3793–3804. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ipsen, D.H.; Lykkesfeldt, J.; Tveden-Nyborg, P. Molecular mechanisms of hepatic lipid accumulation in non-alcoholic fatty liver disease. Cell Mol Life Sci 2018, 75, 3313–3327. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, A.; Wang, Y.; Keshaw, H.; Xu, L.Y.; Lam, K.S.; Cooper, G.J. The fat-derived hormone adiponectin alleviates alcoholic and nonalcoholic fatty liver diseases in mice. J. Clin. Investig. 2003, 112, 91–100. [Google Scholar] [CrossRef] [Green Version]
- Chang, G.R.; Chen, W.K.; Hou, P.H.; Mao, F.C. Isoproterenol exacerbates hyperglycemia and modulates chromium distribution in mice fed with a high fat diet. J. Trace Elem. Med. Biol. 2017, 44, 315–321. [Google Scholar] [CrossRef]
- Chang, G.R.; Hou, P.H.; Chen, W.K.; Lin, C.T.; Tsai, H.P.; Mao, F.C. Exercise affects blood glucose levels and tissue chromium distribution in high-fat diet-fed C57BL6 mice. Molecules 2020, 25, 1658. [Google Scholar] [CrossRef] [Green Version]
- Coimbra, T.M.; Janssen, U.; Gröne, H.J.; Ostendorf, T.; Kunter, U.; Schmidt, H.; Brabant, G.; Floege, J. Early events leading to renal injury in obese Zucker (fatty) rats with type II diabetes. Kidney Int. 2000, 57, 167–182. [Google Scholar] [CrossRef] [Green Version]
- Abd El-Twab, S.M.; Hozayen, W.G.; Hussein, O.E.; Mahmoud, A.M. 18β-Glycyrrhetinic acid protects against methotrexate-induced kidney injury by up-regulating the Nrf2/ARE/HO-1 pathway and endogenous antioxidants. Ren. Fail. 2016, 38, 1516–1527. [Google Scholar] [CrossRef]
- Bahr, S.M.; Weidemann, B.J.; Castro, A.N.; Walsh, J.W.; deLeon, O.; Burnett, C.M.; Pearson, N.A.; Murry, D.J.; Grobe, J.L.; Kirby, J.R. Risperidone-induced weight gain is mediated through shifts in the gut microbiome and suppression of energy expenditure. EBioMedicine 2015, 2, 1725–1734. [Google Scholar] [CrossRef] [Green Version]
- Boyda, H.N.; Tse, L.; Procyshyn, R.M.; Honer, W.G.; Barr, A.M. Preclinical models of antipsychotic drug-induced metabolic side effects. Trends Pharmacol. Sci. 2010, 31, 484–497. [Google Scholar] [CrossRef]
- Pavan, C.; Vindigni, V.; Michelotto, L.; Rimessi, A.; Abatangelo, G.; Cortivo, R.; Pinton, P.; Zavan, B. Weight gain related to treatment with atypical antipsychotics is due to activation of PKC-β. Pharm. J. 2010, 10, 408–417. [Google Scholar] [CrossRef] [PubMed]
- Piao, L.; Park, J.; Li, Y.; Shin, S.; Shin, S.; Kong, G.; Shrestha, R.; Tran, Q.; Hur, G.M. SOCS3 and SOCS6 are required for the risperidone-mediated inhibition of insulin and leptin signaling in neuroblastoma cells. Int. J. Mol. Med. 2014, 33, 1364–1370. [Google Scholar] [CrossRef] [PubMed]
- Oh, C.M.; Namkung, J.; Go, Y.; Shong, K.E.; Kim, K.; Kim, H.; Park, B.Y.; Lee, H.W.; Jeon, Y.H.; Song, J.; et al. Regulation of systemic energy homeostasis by serotonin in adipose tissues. Nat. Commun. 2015, 6, 6794. [Google Scholar] [CrossRef] [PubMed]
- Shong, K.E.; Oh, C.M.; Namkung, J.; Park, S.; Kim, H. Serotonin regulates de novo lipogenesis in adipose tissues through serotonin receptor 2A. Endocrinol. Metab. 2020, 35, 470–479. [Google Scholar] [CrossRef] [PubMed]
- Serretti, A.; Mandelli, L. Antidepressants and body weight: A comprehensive review and meta-analysis. J. Clin. Psychiatry 2010, 71, 1259–1272. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Choi, W.; Namkung, J.; Hwang, I.; Kim, H.; Lim, A.; Park, H.J.; Lee, H.W.; Han, K.H.; Park, S.; Jeong, J.S.; et al. Serotonin signals through a gut-liver axis to regulate hepatic steatosis. Nat. Commun. 2018, 9, 4824. [Google Scholar] [CrossRef]
- Radetti, G.; Kleon, W.; Stuefer, J.; Pittschieler, K. Non-alcoholic fatty liver disease in obese children evaluated by magnetic resonance imaging. Acta Paediatr. 2006, 95, 833–837. [Google Scholar] [CrossRef]
- Zhao, X.Y.; Xiong, X.; Liu, T.; Lin, M.; Peng, X.; Rui, C.; Guo, L.; Li, S.; Li, X.; Lin, J.D. Long noncoding RNA licensing of obesity-linked hepatic lipogenesis and NAFLD pathogenesis. Nat. Commun. 2018, 9, 2986. [Google Scholar] [CrossRef] [Green Version]
- Krebs, S.; Dormann, H.; Muth-Selbach, U.; Hahn, E.G.; Brune, K.; Schneider, H.T. Risperidone-induced cholestatic hepatitis. Eur. J. Gastroenterol. Hepatol. 2001, 13, 67–69. [Google Scholar] [CrossRef]
- Dasarathy, S.; Yang, Y.; McCullough, A.J.; Marczewski, S.; Bennett, C.; Kalhan, S.C. Elevated hepatic fatty acid oxidation, high plasma fibroblast growth factor 21, and fasting bile acids in nonalcoholic steatohepatitis. Eur. J. Gastroenterol. Hepatol. 2011, 23, 382–388. [Google Scholar] [CrossRef] [Green Version]
- Xu, J.; Lloyd, D.J.; Hale, C.; Stanislaus, S.; Chen, M.; Sivits, G.; Vonderfecht, S.; Hecht, R.; Li, Y.S.; Lindberg, R.A.; et al. Fibroblast growth factor 21 reverses hepatic steatosis, increases energy expenditure, and improves insulin sensitivity in diet-induced obese mice. Diabetes 2009, 58, 250–259. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Holtmann, M.; Kopf, D.; Mayer, M.; Bechtinger, E.; Schmidt, M.H. Risperidone-associated steatohepatitis and excessive weight-gain. Pharmacopsychiatry 2003, 36, 206–207. [Google Scholar] [CrossRef] [PubMed]
- Moon, J.S.; Lee, S.; Park, M.A.; Siempos, I.I.; Haslip, M.; Lee, P.J.; Yun, M.; Kim, C.K.; Howrylak, J.; Ryter, S.W.; et al. UCP2-induced fatty acid synthase promotes NLRP3 inflammasome activation during sepsis. J Clin Investig. 2015, 125, 665–680. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Långberg, E.C.; Seed Ahmed, M.; Efendic, S.; Gu, H.F.; Östenson, C.G. Genetic association of adrenergic receptor alpha 2A with obesity and type 2 diabetes. Obesity 2013, 21, 1720–1725. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumar, K.E.; Rani, P.D.J.; Ram, K.R.; Swathi, P.; Cupta, M.N. Pharmacodynamic and pharmacokinetic drug interaction of gliclazide and risperidone in animal models. Int. J. Pharm Pharm 2012, 4, 659–662. [Google Scholar]
- Shoelson, S.E.; Herrero, L.; Naaz, A. Obesity, inflammation, and insulin resistance. Gastroenterology 2007, 132, 2169–2180. [Google Scholar] [CrossRef]
- Sharma, A.K.; Bharti, S.; Kumar, R.; Krishnamurthy, B.; Bhatia, J.; Kumari, S.; Arya, D.S. Syzygium cumini ameliorates insulin resistance and β-cell dysfunction via modulation of PPAR, dyslipidemia, oxidative stress, and TNF-α in type 2 diabetic rats. J. Pharmacol. Sci. 2012, 119, 205–213. [Google Scholar] [CrossRef] [Green Version]
- Sharma, S.; Pathak, S.; Gupta, G.; Sharma, S.K.; Singh, L.; Sharma, R.K.; Mishra, A.; Dua, K. Pharmacological evaluation of aqueous extract of syzigium cumini for its antihyperglycemic and antidyslipidemic properties in diabetic rats fed a high cholesterol diet-role of PPARγ and PPARα. Biomed. Pharm. 2017, 89, 447–453. [Google Scholar] [CrossRef]
- Baynes, J.W. Role of oxidative stress in development of complications in diabetes. Diabetes 1991, 40, 405–412. [Google Scholar] [CrossRef]
- Dastych, M.; Synek, O.; Gottwaldová, J. Impact of long-term lithium treatment on renal function in patients with bipolar disorder based on novel biomarkers. J. Clin. Psychopharmacol. 2019, 39, 238–242. [Google Scholar] [CrossRef]
- Yeh, C.Y.; Chen, C.K.; Hsu, H.J.; Wu, I.W.; Sun, C.Y.; Chou, C.C.; Lee, C.C.; Wang, L.J. Prescription of psychotropic drugs in patients with chronic renal failure on hemodialysis. Ren. Fail. 2014, 36, 1545–1549. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Olatunji, O.J.; Chen, H.; Zhou, Y. Lycium chinense leaves extract ameliorates diabetic nephropathy by suppressing hyperglycemia mediated renal oxidative stress and inflammation. Biomed. Pharm. 2018, 102, 1145–1151. [Google Scholar] [CrossRef] [PubMed]
- Hsu, C.W.; Lee, Y.; Lee, C.Y.; Lin, P.Y. Neurotoxicity and nephrotoxicity caused by combined use of lithium and risperidone: A case report and literature review. BMC Pharmacol. Toxicol. 2016, 17, 59. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vanwong, N.; Srisawasdi, P.; Ngamsamut, N.; Nuntamool, N.; Puangpetch, A.; Chamkrachangpada, B.; Hongkaew, Y.; Limsila, P.; Kittitharaphan, W.; Sukasem, C. Hyperuricemia in children and adolescents with autism spectrum disorder treated with risperidone: The risk factors for metabolic adverse effects. Front. Pharmacol. 2017, 7, 527. [Google Scholar] [CrossRef] [Green Version]
- Al Hroob, A.M.; Abukhalil, M.H.; Alghonmeen, R.D.; Mahmoud, A.M. Ginger alleviates hyperglycemia-induced oxidative stress, inflammation and apoptosis and protects rats against diabetic nephropathy. Biomed Pharm. 2018, 106, 381–389. [Google Scholar] [CrossRef]
- Nikolić-Kokić, A.; Mijušković, A.; Tatalović, N.; Nestorov, J.; Miler, M.; Oreščanin-Dušić, Z.; Nikolić, M.; Milošević, V.; Blagojević, D.; Spasić, M.; et al. Effects of antipsychotic drug administration on antioxidative defense enzymes in male rat kidney. J. Toxicol. Environ. Health A 2016, 79, 905–911. [Google Scholar]
- Noto, C.; Ota, V.K.; Gadelha, A.; Noto, M.N.; Barbosa, D.S.; Bonifácio, K.L.; Nunes, S.O.; Cordeiro, Q.; Belangero, S.I.; Bressan, R.A.; et al. Oxidative stress in drug naïve first episode psychosis and antioxidant effects of risperidone. Psychiatr. Res. 2015, 68, 210–216. [Google Scholar] [CrossRef]
- Yoon, S.; Noh, J.S.; Choi, S.Y.; Baik, J.H. Effects of atypical antipsychotic drugs on body weight and food intake in dopamine D2 receptor knockout mice. Biochem. Biophys. Res. Commun. 2010, 393, 235–241. [Google Scholar] [CrossRef]
- May, M.; Beauchemin, M.; Vary, C.; Barlow, D.; Houseknecht, K.L. The antipsychotic medication, risperidone, causes global immunosuppression in healthy mice. PLoS ONE 2019, 14, e0218937. [Google Scholar] [CrossRef]
- Beauchemin, M.; Geguchadze, R.; Guntur, A.R.; Nevola, K.; Le, P.T.; Barlow, D.; Rue, M.; Vary, C.P.H.; Lary, C.W.; Motyl, K.J.; et al. Exploring mechanisms of increased cardiovascular disease risk with antipsychotic medications: Risperidone alters the cardiac proteomic signature in mice. Pharmacol. Res. 2020, 152, 104589. [Google Scholar] [CrossRef]
- London, E.; Castonguay, T.W. High fructose diets increase 11β-hydroxysteroid dehydrogenase type 1 in liver and visceral adipose in rats within 24-h exposure. Obesity 2011, 19, 925–932. [Google Scholar] [CrossRef] [PubMed]
- Hallman, T.M.; Peng, M.; Meade, R.; Hancock, W.W.; Madaio, M.P.; Gasser, D.L. The mitochondrial and kidney disease phenotypes of kd/kd mice under germfree conditions. J. Autoimmun. 2006, 26, 1–6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, R.Z.; Li, X.; Godbout, R. A novel fatty acid-binding protein (FABP) gene resulting from tandem gene duplication in mammals: Transcription in rat retina and testis. Genomics 2008, 92, 436–445. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miyazaki, T.; Shirakami, Y.; Kubota, M.; Ideta, T.; Kochi, T.; Sakai, H.; Shimizu, M. Sodium alginate prevents progression of non-alcoholic steatohepatitis and liver carcinogenesis in obese and diabetic mice. Oncotarget 2016, 7, 10448. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mkhwanazi, B.N.; Serumula, M.R.; Myburg, R.B.; Van Heerden, F.R.; Musabayane, C.T. Antioxidant effects of maslinic acid in livers, hearts and kidneys of streptozotocin-induced diabetic rats: Effects on kidney function. Ren. Fail. 2014, 36, 419–431. [Google Scholar] [CrossRef] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Variable | Control | Risperidone |
|---|---|---|
| RWAT | ||
| Adipocyte diameter | ||
| 0–50 μm (%) | 21.67 ± 2.90 | 12.67 ± 2.88 *** |
| 50–100 μm (%) | 48.33 ± 3.85 | 27.67 ± 3.57 *** |
| 100–150 μm (%) | 28.33 ± 2.18 | 42.99 ± 3.22 *** |
| >150 μm (%) | 1.67 ± 0.25 | 16.67 ± 3.05 *** |
| EWAT | ||
| Adipocyte diameter | ||
| 0–50 μm (%) | 8.33 ± 1.48 | 0 ± 0 *** |
| 50–100 μm (%) | 30.01 ± 2.97 | 3.33 ± 0.37 *** |
| 100–150 μm (%) | 33.33 ± 3.25 | 40.00 ± 3.57 ** |
| >150 μm (%) | 28.33 ± 2.59 | 56.67 ± 4.94 *** |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tsai, H.-P.; Hou, P.-H.; Mao, F.-C.; Chang, C.-C.; Yang, W.-C.; Wu, C.-F.; Liao, H.-J.; Lin, T.-C.; Chou, L.-S.; Hsiao, L.-W.; et al. Risperidone Exacerbates Glucose Intolerance, Nonalcoholic Fatty Liver Disease, and Renal Impairment in Obese Mice. Int. J. Mol. Sci. 2021, 22, 409. https://doi.org/10.3390/ijms22010409
Tsai H-P, Hou P-H, Mao F-C, Chang C-C, Yang W-C, Wu C-F, Liao H-J, Lin T-C, Chou L-S, Hsiao L-W, et al. Risperidone Exacerbates Glucose Intolerance, Nonalcoholic Fatty Liver Disease, and Renal Impairment in Obese Mice. International Journal of Molecular Sciences. 2021; 22(1):409. https://doi.org/10.3390/ijms22010409
Chicago/Turabian StyleTsai, Hsiao-Pei, Po-Hsun Hou, Frank-Chiahung Mao, Chia-Chia Chang, Wei-Cheng Yang, Ching-Feng Wu, Huei-Jyuan Liao, Tzu-Chun Lin, Lan-Szu Chou, Li-Wei Hsiao, and et al. 2021. "Risperidone Exacerbates Glucose Intolerance, Nonalcoholic Fatty Liver Disease, and Renal Impairment in Obese Mice" International Journal of Molecular Sciences 22, no. 1: 409. https://doi.org/10.3390/ijms22010409