Drug Screening with Genetically Encoded Fluorescent Sensors: Today and Tomorrow

 ,
,
Abstract
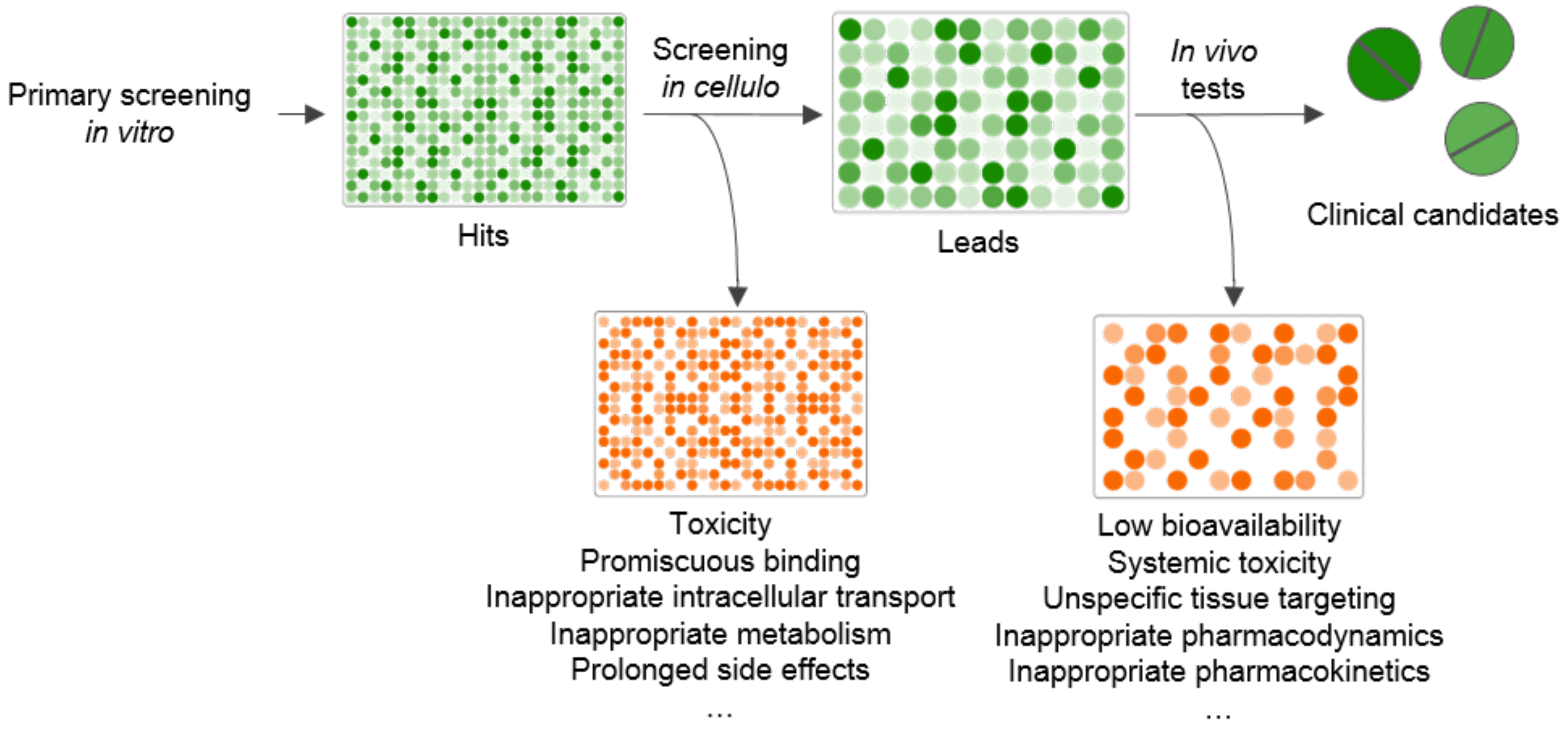
:1. Introduction
2. Anti-Cancer Compound Screening
2.1. Kinase Inhibitors Screening
2.2. Transcription Factors Regulators Screening
2.3. Cell Death Signaling Inducers Screening
2.4. Energy Metabolism Modulators Screening
3. G Protein-Coupled Receptor (GPCR) Modulator Screening
4. Membrane Potential Modulator Screening
Genetically Encoded Voltage Indicators (GEVI)
5. Fluorescent Sensors Detecting Drug Toxicity
5.1. Proteome Stress Inductors Screening
5.2. Mitochondrial Toxicants Screening
6. Anti-Parasitic Drug Screening
7. Ca2+-Signaling Modulator Screening
8. Animal Models for Drug Screening Using Genetically Encoded Sensors—Looking Towards the Future
8.1. Animal Models for Drug Screening
8.2. CRISPR Technology in Human Diseases Modeling
8.3. Fluorescent Sensors in Zebrafish Research and Drug Screening

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sensor | Sensor Specificity | Sensor Location | Ref |
|---|---|---|---|
| GCAMP3 | Ca2+ | lateral line hair cells | [185] |
| GCAMP5G | Ca2+ | retina, tectum | [186] |
| RCaMP | Ca2+ | Trigeminal Neurons | [184] |
| GCAMP6 | Ca2+ | neurons | [92] |
| Mauthner neurons | [187] | ||
| ubiquitous | [188] | ||
| neurons | [189] | ||
| sPAGCaMP6f | Ca2+ | motor neuron, Müller glia, retinal ganglion cells | [190] |
| jRGECO1a | Ca2+ | neurons | [191] |
| K-GECO1 | Ca2+ | spinal sensory neurons | [192] |
| mNG-GECO1 | Ca2+ | neurons | [193] |
| FGCaMP7 | Ca2+ | neurons | [194] |
| CaViar | Ca2+ and voltage (but dim) | miocardium | [91] |
| CaMPARI2 | activated neuronal ensembles | neurons | [195] |
| Sypher3s | pH | ubiquitous | [196] |
| pHluorin | pH | Retinal Horizontal Cell–Cone Synapse | [197] |
| syPhy | pH | lateral line hair cells | [198] |
| Bongwoorie | voltage | neurons | [199] |
| ASAP1 | voltage | neurons | [200] |
| zArchon1 | voltage | neurons | [201] |
| roGFP2-Orp1 | H2O2 | endothelial cells and cardiomyocytes | [202] |
| Hyper | H2O2 | ubiquitous | [203] |
| HyPer3 | H2O2 | ubiquitous | [204] |
| HyperRed | H2O2 | ubiquitous | [205] |
| Hyper7 | H2O2 | ubiquitous | [206] |
| Grx1-roGFP2 | Glutathione redox state | endothelial cells and cardiomyocytes | [202] |
| iGluSnFR | extracellular glutamate | optic tectum | [207] |
| glial cells throughout the nervous system | [208] | ||
| hair cell ribbon | [209] | ||
| DA1m | extracellular dophamine | neurons | [181] |
| iGABASnFR | extracellular GABA | neurons of zebrafish cerebellum | [180] |
| GRABNE1m | extracellular norepinephrine | neurons | [182] |
| iNap1 | NADPH | ubiquitous | [205] |
| SoNar | NADH/NAD+ ratio | ubiquitous | [210] |
| REX-YFP | NAD+/NADH ratio | lateral line hair cells | [211] |
| Caspase 3 FRET sensor | caspase 3 activity | ubiquitous | [212] |
| C3 | caspase 3 activity | skin cells | [213] |
| Voltron | voltage | neurons | [214] |
8.4. Perspectives on the Development of Genetically Encoded Fluorescent Biosensors Suitable for Drug HTS
9. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| AD | Alzheimer’s disease |
| AKAR | A-kinase activity reporter |
| AP | action potential |
| BRET | bioluminescence resonance energy transfer |
| CRISPR | clustered regularly interspaced short palindromic repeats |
| CML | chronic myelogenous leukemia |
| CRM | caloric restriction mimetics |
| CMs | cardiomyocytes |
| cpYFP | circularly permuted yellow fluorescent protein |
| DAG | diacylglycerol |
| EAD | early afterdepolarization |
| ERKs | extracellular signal-regulated kinases |
| ECFP | enhanced cyan fluorescent protein |
| EGFR | epidermal growth factor receptor |
| FPs | fluorescent proteins |
| FLIM | fluorescence lifetime imaging |
| FLT | fluorescence lifetime |
| FRET | Förster resonance energy transfer |
| GPCR | G protein-coupled receptor |
| GEVI | genetically encoded voltage indicators |
| GECI | genetically encoded calcium indicators |
| HTS | high-throughput screening |
| HMGB1 | high-mobility group box 1 protein |
| iPSCs | induced pluripotent stem cells |
| ICD | immunogenic cell death |
| JNKs | c-Jun N-terminal kinases |
| LDG | lactate dehydrogenase |
| MAPK | mitogen-activated protein kinase |
| NLS | nuclear localization signal |
| OXPHOS | oxidative phosphorylation |
| PIP2 | phosphatidylinositol 4,5-bisphosphate |
| PLC | phospholipase C |
| PLB | phospholamban |
| PS1 | presenilin 1 |
| PTU | 1-phenyl 2-thiourea |
| ROS | reactive oxygen species |
| RTKs | receptors of growth factors, cytokines, and hormones |
| SERCA2a | sarco/endoplasmic reticulum Ca2+ ATPase |
| SFCAI | switch-on fluorescence-based caspase-3-like protease activity indicator |
| SPB | streptavidin binding protein |
| ssODN | single-stranded DNA oligonucleotide |
| TMRM | tetramethylrhodamine, methyl ester |
| UGI | uracil DNA glycosylase inhibitor |
| VEGFR | vascular endothelial growth factor |
| VGCs | voltage-gated ion channels |
References
- Zhang, J.-H.; Chung, T.D.Y.; Oldenburg, K.R. A Simple Statistical Parameter for Use in Evaluation and Validation of High Throughput Screening Assays. J. Biomol. Screen. 1999, 4, 67–73. [Google Scholar] [CrossRef]
- Arora, A.; Scholar, E.M. Role of Tyrosine Kinase Inhibitors in Cancer Therapy. J. Pharm. Exp. Ther. 2005, 315, 971–979. [Google Scholar] [CrossRef] [Green Version]
- Zaytseva, Y.Y.; Valentino, J.D.; Gulhati, P.; Mark Evers, B. mTOR inhibitors in cancer therapy. Cancer Lett. 2012, 319, 1–7. [Google Scholar] [CrossRef]
- Caunt, C.J.; Sale, M.J.; Smith, P.D.; Cook, S.J. MEK1 and MEK2 inhibitors and cancer therapy: The long and winding road. Nat. Rev. Cancer 2015, 15, 577–592. [Google Scholar] [CrossRef]
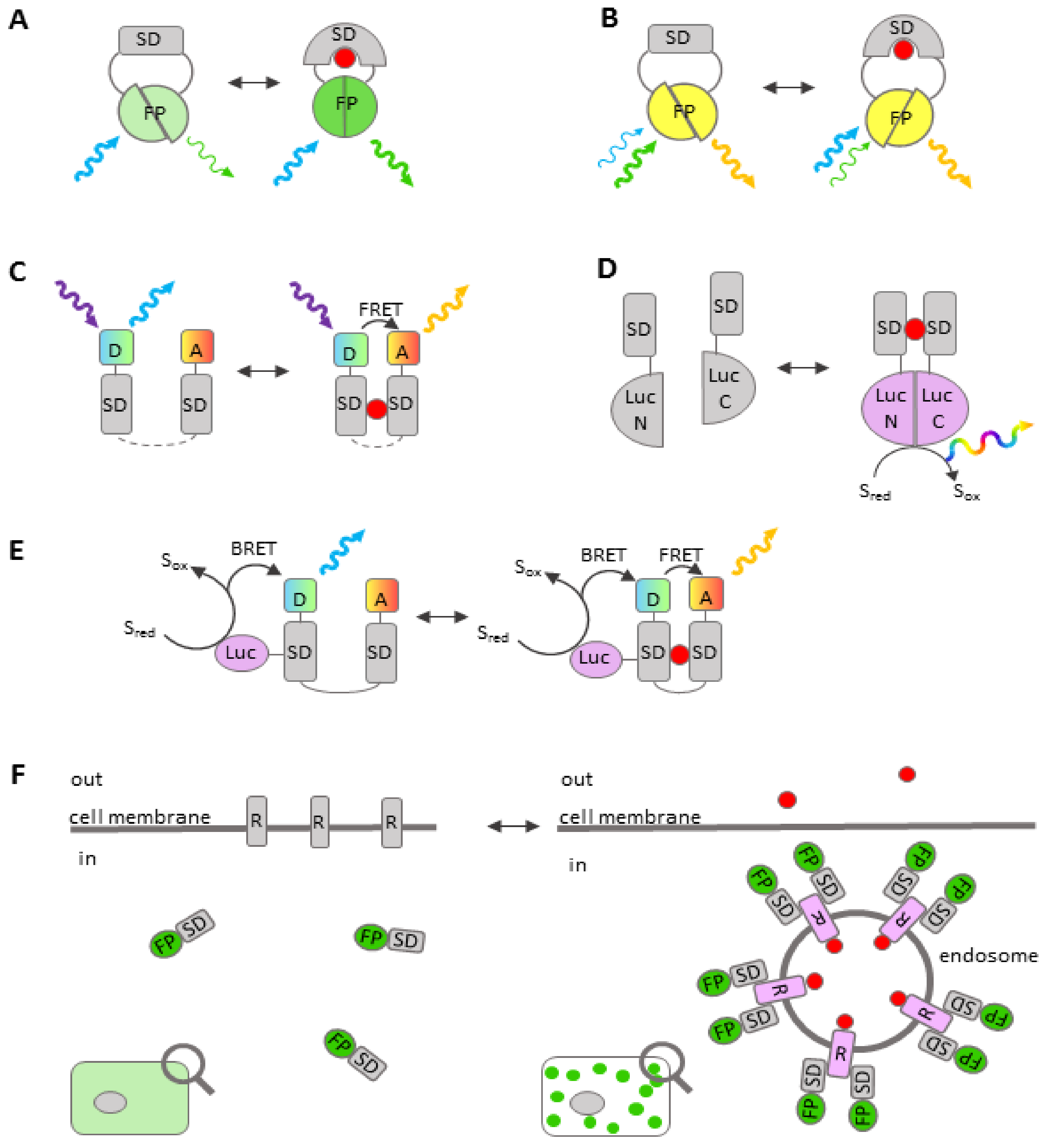
- Prével, C.; Pellerano, M.; Van, T.N.N.; Morris, M.C. Fluorescent biosensors for high throughput screening of protein kinase inhibitors. Biotechnol. J. 2014, 9, 253–265. [Google Scholar] [CrossRef] [PubMed]
- Nhu Ngoc Van, T.; Morris, M.C. Fluorescent Sensors of Protein Kinases. In Progress in Molecular Biology and Translational Science; Elsevier: Amsterdam, The Netherlands, 2013; Volume 113, pp. 217–274. ISBN 978-0-12-386932-6. [Google Scholar]
- Morris, M.C. Fluorescent biosensors—Probing protein kinase function in cancer and drug discovery. Biochim. Biophys. Acta BBA-Proteins Proteom. 2013, 1834, 1387–1395. [Google Scholar] [CrossRef]
- Gao, X.; Zhang, J. Spatiotemporal Analysis of Differential Akt Regulation in Plasma Membrane Microdomains. Mol. Biol. Cell 2008, 19, 4366–4373. [Google Scholar] [CrossRef] [Green Version]
- Irby, R.B.; Yeatman, T.J. Role of Src expression and activation in human cancer. Oncogene 2000, 19, 5636–5642. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.; Botvinick, E.L.; Zhao, Y.; Berns, M.W.; Usami, S.; Tsien, R.Y.; Chien, S. Visualizing the mechanical activation of Src. Nature 2005, 434, 1040–1045. [Google Scholar] [CrossRef] [PubMed]
- Nobis, M.; McGhee, E.J.; Morton, J.P.; Schwarz, J.P.; Karim, S.A.; Quinn, J.; Edward, M.; Campbell, A.D.; McGarry, L.C.; Evans, T.R.J.; et al. Intravital FLIM-FRET Imaging Reveals Dasatinib-Induced Spatial Control of Src in Pancreatic Cancer. Cancer Res. 2013, 73, 4674–4686. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, W.; Mehta, S.; Zhang, J. Genetically encoded fluorescent biosensors illuminate kinase signaling in cancer. J. Biol. Chem. 2019, 294, 14814–14822. [Google Scholar] [CrossRef] [PubMed] [Green Version]
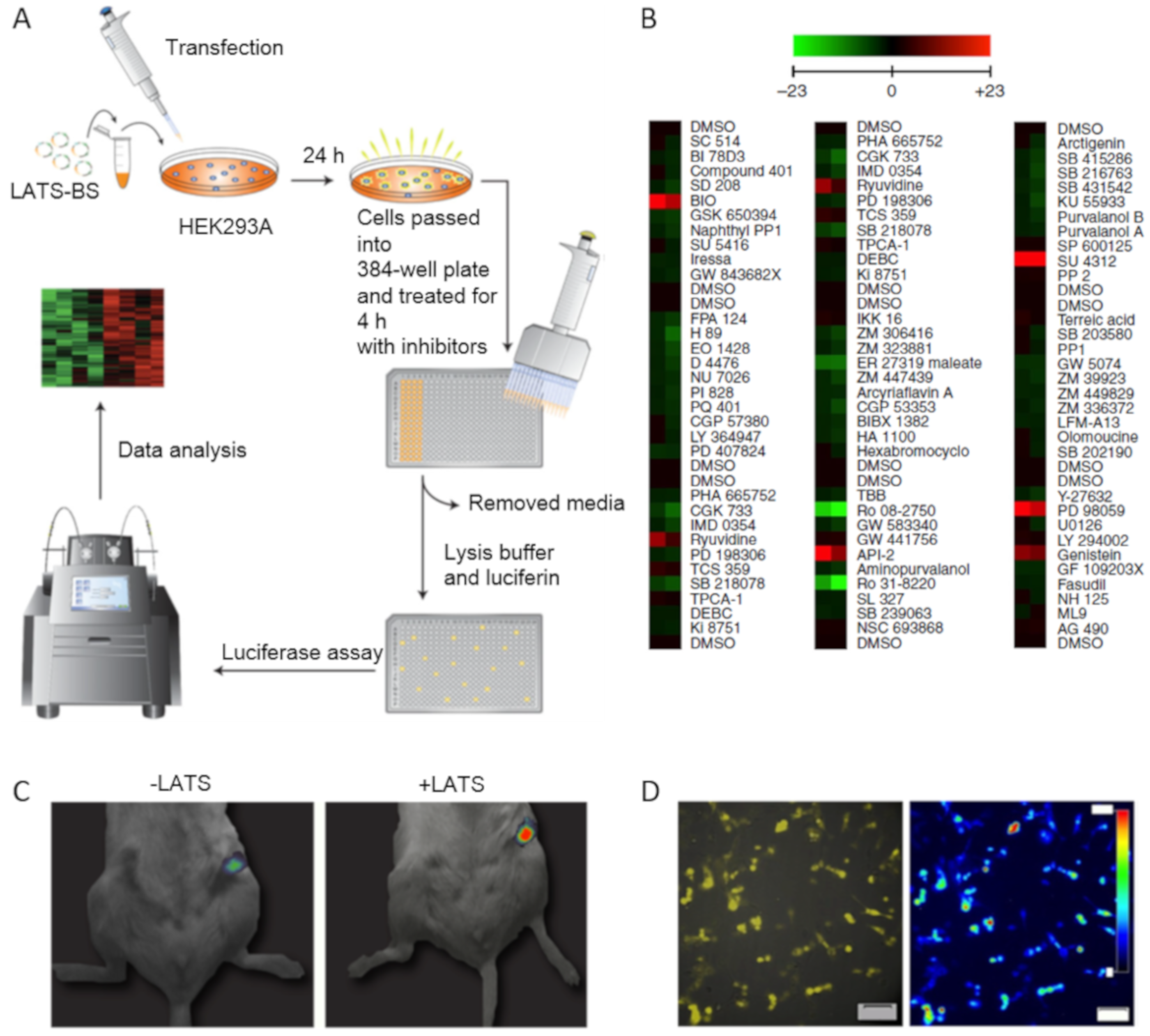
- Plouffe, S.W.; Hong, A.W.; Guan, K.-L. Disease implications of the Hippo/YAP pathway. Trends Mol. Med. 2015, 21, 212–222. [Google Scholar] [CrossRef] [PubMed]
- Azad, T.; Janse van Rensburg, H.J.; Lightbody, E.D.; Neveu, B.; Champagne, A.; Ghaffari, A.; Kay, V.R.; Hao, Y.; Shen, H.; Yeung, B.; et al. A LATS biosensor screen identifies VEGFR as a regulator of the Hippo pathway in angiogenesis. Nat. Commun. 2018, 9, 1061. [Google Scholar] [CrossRef] [PubMed]
- Harvey, C.D.; Ehrhardt, A.G.; Cellurale, C.; Zhong, H.; Yasuda, R.; Davis, R.J.; Svoboda, K. A genetically encoded fluorescent sensor of ERK activity. Proc. Natl. Acad. Sci. USA 2008, 105, 19264–19269. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bogoyevitch, M.A.; Kobe, B. Uses for JNK: The Many and Varied Substrates of the c-Jun N-Terminal Kinases. Microbiol. Mol. Biol. Rev. 2006, 70, 1061–1095. [Google Scholar] [CrossRef] [Green Version]
- Komatsu, N.; Aoki, K.; Yamada, M.; Yukinaga, H.; Fujita, Y.; Kamioka, Y.; Matsuda, M. Development of an optimized backbone of FRET biosensors for kinases and GTPases. Mol. Biol. Cell 2011, 22, 4647–4656. [Google Scholar] [CrossRef] [Green Version]
- Komatsu, N.; Terai, K.; Imanishi, A.; Kamioka, Y.; Sumiyama, K.; Jin, T.; Okada, Y.; Nagai, T.; Matsuda, M. A platform of BRET-FRET hybrid biosensors for optogenetics, chemical screening, and in vivo imaging. Sci. Rep. 2018, 8, 8984. [Google Scholar] [CrossRef]
- Bennett, B. JNK: A new therapeutic target for diabetes. Curr. Opin. Pharmacol. 2003, 3, 420–425. [Google Scholar] [CrossRef]
- Borsello, T.; Forloni, G. JNK Signalling: A Possible Target to Prevent Neurodegeneration. Curr. Pharm. Des. 2007, 13, 1875–1886. [Google Scholar] [CrossRef]
- Rose, B.A.; Force, T.; Wang, Y. Mitogen-Activated Protein Kinase Signaling in the Heart: Angels Versus Demons in a Heart-Breaking Tale. Physiol. Rev. 2010, 90, 1507–1546. [Google Scholar] [CrossRef] [Green Version]
- Bubici, C.; Papa, S. JNK signalling in cancer: In need of new, smarter therapeutic targets: JNKs in cancer. Br. J. Pharm. 2014, 171, 24–37. [Google Scholar] [CrossRef] [PubMed]
- Fosbrink, M.; Aye-Han, N.-N.; Cheong, R.; Levchenko, A.; Zhang, J. Visualization of JNK activity dynamics with a genetically encoded fluorescent biosensor. Proc. Natl. Acad. Sci. USA 2010, 107, 5459–5464. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rossari, F.; Minutolo, F.; Orciuolo, E. Past, Present, and Future of Bcr-Abl Inhibitors: From Chemical Development to Clinical Efficacy; BioMed Central Ltd.: London, UK, 2018; Volume 11. [Google Scholar]
- Kurokawa, K.; Mochizuki, N.; Ohba, Y.; Mizuno, H.; Miyawaki, A.; Matsuda, M. A Pair of Fluorescent Resonance Energy Transfer-based Probes for Tyrosine Phosphorylation of the CrkII Adaptor Protein in Vivo. J. Biol. Chem. 2001, 276, 31305–31310. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tunceroglu, A.; Matsuda, M.; Birge, R.B. Real-time Fluorescent Resonance Energy Transfer Analysis to Monitor Drug Resistance in Chronic Myelogenous Leukemia. Mol. Cancer Ther. 2010, 9, 3065–3073. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mizutani, T.; Kondo, T.; Darmanin, S.; Tsuda, M.; Tanaka, S.; Tobiume, M.; Asaka, M.; Ohba, Y. A Novel FRET-Based Biosensor for the Measurement of BCR-ABL Activity and Its Response to Drugs in Living Cells. Clin. Cancer Res. 2010, 16, 3964–3975. [Google Scholar] [CrossRef] [Green Version]
- Gschwind, A.; Fischer, O.M.; Ullrich, A. The discovery of receptor tyrosine kinases: Targets for cancer therapy. Nat. Rev. Cancer 2004, 4, 361–370. [Google Scholar] [CrossRef]
- Antczak, C.; Bermingham, A.; Calder, P.; Malkov, D.; Song, K.; Fetter, J.; Djaballah, H. Domain-Based Biosensor Assay to Screen for Epidermal Growth Factor Receptor Modulators in Live Cells. Assay Drug Dev. Technol. 2012, 10, 24–36. [Google Scholar] [CrossRef] [Green Version]
- Antczak, C.; Mahida, J.P.; Bhinder, B.; Calder, P.A.; Djaballah, H. A high-content biosensor-based screen identifies cell-permeable activators and inhibitors of EGFR function: Implications in drug discovery. J. Biomol. Screen. 2012, 17, 885–899. [Google Scholar] [CrossRef] [Green Version]
- Moll, U.M.; Petrenko, O. The MDM2-p53 Interaction. Mol. Cancer Res. 2003, 1, 1001–1008. [Google Scholar]
- Dudgeon, D.D.; Shinde, S.; Hua, Y.; Shun, T.Y.; Lazo, J.S.; Strock, C.J.; Giuliano, K.A.; Taylor, D.L.; Johnston, P.A.; Johnston, P.A. Implementation of a 220,000-Compound HCS Campaign to Identify Disruptors of the Interaction between p53 and hDM2 and Characterization of the Confirmed Hits. J. Biomol. Screen. 2010, 15, 766–782. [Google Scholar] [CrossRef] [Green Version]
- Yeh, E.; Cunningham, M.; Arnold, H.; Chasse, D.; Monteith, T.; Ivaldi, G.; Hahn, W.C.; Stukenberg, P.T.; Shenolikar, S.; Uchida, T.; et al. A signalling pathway controlling c-Myc degradation that impacts oncogenic transformation of human cells. Nat. Cell Biol. 2004, 6, 308–318. [Google Scholar] [CrossRef] [PubMed]
- Fan-Minogue, H.; Cao, Z.; Paulmurugan, R.; Chan, C.T.; Massoud, T.F.; Felsher, D.W.; Gambhir, S.S. Noninvasive molecular imaging of c-Myc activation in living mice. Proc. Natl. Acad. Sci. USA 2010, 107, 15892–15897. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fan-Minogue, H.; Bodapati, S.; Solow-Cordero, D.; Fan, A.; Paulmurugan, R.; Massoud, T.F.; Felsher, D.W.; Gambhir, S.S. A c-Myc Activation Sensor-Based High-Throughput Drug Screening Identifies an Antineoplastic Effect of Nitazoxanide. Mol. Cancer Ther. 2013, 12, 1896–1905. [Google Scholar] [CrossRef] [Green Version]
- Rello-Varona, S.; Kepp, O.; Vitale, I.; Michaud, M.; Senovilla, L.; Jemaà, M.; Joza, N.; Galluzzi, L.; Castedo, M.; Kroemer, G. An automated fluorescence videomicroscopy assay for the detection of mitotic catastrophe. Cell Death Dis. 2010, 1, e25. [Google Scholar] [CrossRef] [PubMed]
- Porter, A.G.; Jänicke, R.U. Emerging roles of caspase-3 in apoptosis. Cell Death Differ. 1999, 6, 99–104. [Google Scholar] [CrossRef] [PubMed]
- Tian, H.; Ip, L.; Luo, H.; Chang, D.C.; Luo, K.Q. A high throughput drug screen based on fluorescence resonance energy transfer (FRET) for anticancer activity of compounds from herbal medicine. Br. J. Pharmacol. 2007, 150, 321–334. [Google Scholar] [CrossRef]
- Takemoto, K.; Nagai, T.; Miyawaki, A.; Miura, M. Spatio-Temporal Activation of Caspase Revealed by Indicator That Is Insensitive to Environmental Effects. J. Cell Biol. 2003, 160, 235–243. [Google Scholar] [CrossRef]
- Zhang, J.; Wang, X.; Cui, W.; Wang, W.; Zhang, H.; Liu, L.; Zhang, Z.; Li, Z.; Ying, G.; Zhang, N.; et al. Visualization of caspase-3-like activity in cells using a genetically encoded fluorescent biosensor activated by protein cleavage. Nat. Commun. 2013, 4, 2157. [Google Scholar] [CrossRef] [PubMed]
- Silva, M.T. Secondary necrosis: The natural outcome of the complete apoptotic program. FEBS Lett. 2010, 584, 4491–4499. [Google Scholar] [CrossRef] [Green Version]
- Lekshmi, A.; Varadarajan, S.N.; Lupitha, S.S.; Nair, M.; Chandrasekharan, A.; Santhoshkumar, T.R. A Real-Time Image-Based Approach to Distinguish and Discriminate Apoptosis from Necrosis. Curr. Protoc. Toxicol. 2018, 75, 2.27.1–2.27.16. [Google Scholar] [CrossRef]
- Kroemer, G.; Galluzzi, L.; Kepp, O.; Zitvogel, L. Immunogenic Cell Death in Cancer Therapy. Annu. Rev. Immunol. 2013, 31, 51–72. [Google Scholar] [CrossRef] [PubMed]
- Tanida, I.; Ueno, T.; Kominami, E. LC3 and autophagy. In Methods in Molecular Biology; Humana Press: Totowa, NJ, USA, 2008; Volume 445, pp. 77–88. ISBN 978-1-58829-853-9. [Google Scholar]
- Liu, P.; Zhao, L.; Pol, J.; Levesque, S.; Petrazzuolo, A.; Pfirschke, C.; Engblom, C.; Rickelt, S.; Yamazaki, T.; Iribarren, K.; et al. Crizotinib-induced immunogenic cell death in non-small cell lung cancer. Nat. Commun. 2019, 10, 1486. [Google Scholar] [CrossRef] [PubMed]
- Zhao, L.; Liu, P.; Kepp, O.; Kroemer, G. Methods for Measuring HMGB1 Release during Immunogenic Cell Death, 1st ed.; Elsevier Inc.: Amsterdam, The Netherlands, 2019; Volume 629, ISBN 978-0-12-818671-8. [Google Scholar]
- Zhao, L.; Liu, P.; Boncompain, G.; Loos, F.; Lachkar, S.; Bezu, L.; Chen, G.; Zhou, H.; Perez, F.; Kepp, O.; et al. Identification of pharmacological inhibitors of conventional protein secretion. Sci. Rep. 2018, 8, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Chen, G.; Xie, W.; Nah, J.; Sauvat, A.; Liu, P.; Pietrocola, F.; Sica, V.; Carmona-Gutierrez, D.; Zimmermann, A.; Pendl, T.; et al. 3,4-Dimethoxychalcone induces autophagy through activation of the transcription factors TFE 3 and TFEB. EMBO Mol. Med. 2019, 11, e10469. [Google Scholar] [CrossRef]
- Zheng, J. Energy metabolism of cancer: Glycolysis versus oxidative phosphorylation (review). Oncol. Lett. 2012, 4, 1151–1157. [Google Scholar] [CrossRef] [Green Version]
- Imamura, H.; Nhat, K.P.H.; Togawa, H.; Saito, K.; Iino, R.; Kato-Yamada, Y.; Nagai, T.; Noji, H. Visualization of ATP levels inside single living cells with fluorescence resonance energy transfer-based genetically encoded indicators. Proc. Natl. Acad Sci. USA 2009, 106, 15651–15656. [Google Scholar] [CrossRef] [Green Version]
- Zhao, Z.; Rajagopalan, R.; Zweifach, A. A Novel Multiple-Read Screen for Metabolically Active Compounds Based on a Genetically Encoded FRET Sensor for ATP. SLAS Discov. 2018, 23, 907–918. [Google Scholar] [CrossRef]
- Zhao, Y.; Hu, Q.; Cheng, F.; Su, N.; Wang, A.; Zou, Y.; Hu, H.; Chen, X.; Zhou, H.-M.; Huang, X.; et al. SoNar, a Highly Responsive NAD+/NADH Sensor, Allows High-Throughput Metabolic Screening of Anti-tumor Agents. Cell Metab. 2015, 777–789. [Google Scholar] [CrossRef] [Green Version]
- Williams, C. cAMP detection methods in HTS: Selecting the best from the rest. Nat. Rev. Drug Discov. 2004, 3, 125–135. [Google Scholar] [CrossRef]
- Allen, M.D.; DiPilato, L.M.; Rahdar, M.; Ren, Y.R.; Chong, C.; Liu, J.O.; Zhang, J. Reading Dynamic Kinase Activity in Living Cells for High-Throughput Screening. ACS Chem. Biol. 2006, 1, 371–376. [Google Scholar] [CrossRef]
- Nikolaev, V.O.; Bünemann, M.; Hein, L.; Hannawacker, A.; Lohse, M.J. Novel Single Chain cAMP Sensors for Receptor-induced Signal Propagation. J. Biol. Chem. 2004, 279, 37215–37218. [Google Scholar] [CrossRef] [Green Version]
- Mazina, O.; Reinart-Okugbeni, R.; Kopanchuk, S.; Rinken, A. BacMam System for FRET-Based cAMP Sensor Expression in Studies of Melanocortin MC1 Receptor Activation. J. Biomol. Screen. 2012, 17, 1096–1101. [Google Scholar] [CrossRef] [Green Version]
- Klarenbeek, J.B.; Goedhart, J.; Hink, M.A.; Gadella, T.W.J.; Jalink, K. A mTurquoise-Based cAMP Sensor for Both FLIM and Ratiometric Read-Out Has Improved Dynamic Range. PLoS ONE 2011, 6, e19170. [Google Scholar] [CrossRef]
- Mazina, O.; Allikalt, A.; Heinloo, A.; Reinart-Okugbeni, R.; Kopanchuk, S.; Rinken, A. cAMP Assay for GPCR Ligand Characterization: Application of BacMam Expression System. In G Protein-Coupled Receptor Screening Assays; Prazeres, D.M.F., Martins, S.A.M., Eds.; Methods in Molecular Biology; Springer: New York, NY, USA, 2015; Volume 1272, pp. 65–77. ISBN 978-1-4939-2335-9. [Google Scholar]
- Tewson, P.H.; Quinn, A.M.; Hughes, T.E. A multiplexed fluorescent assay for independent second-messenger systems: Decoding GPCR activation in living cells. J. Biomol. Screen. 2012, 18, 797–806. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- O’Dowd, B.F.; Ji, X.; Alijaniaram, M.; Rajaram, R.D.; Kong, M.M.C.; Rashid, A.; Nguyen, T.; George, S.R. Dopamine Receptor Oligomerization Visualized in Living Cells. J. Biol. Chem. 2005, 280, 37225–37235. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eglen, R.M. Enzyme Fragment Complementation: A Flexible High Throughput Screening Assay Technology. Assay Drug Dev. Technol. 2002, 1, 97–105. [Google Scholar] [CrossRef] [PubMed]
- O’Dowd, B.F.; Alijaniaram, M.; Ji, X.; Nguyen, T.; Eglen, R.M.; George, S.R. Using Ligand-Induced Conformational Change to Screen for Compounds Targeting G-Protein-Coupled Receptors. J. Biomol. Screen. 2007, 12, 175–185. [Google Scholar] [CrossRef] [Green Version]
- Bourque, K.; Jones-Tabah, J.; Mnasri, N.; Martin, R.; Hébert, T. Combining Optical Approaches with Human Inducible Pluripotent Stem Cells in G Protein-Coupled Receptor Drug Screening and Development. Biomolecules 2018, 8, 180. [Google Scholar] [CrossRef] [Green Version]
- Tanner, M.R.; Beeton, C. Differences in ion channel phenotype and function between humans and animal models. Front. Biosci. 2018, 23, 43–64. [Google Scholar]
- Ion Channels|HUGO Gene Nomenclature Committee. Available online: https://www.genenames.org/data/genegroup/#!/group/177 (accessed on 5 July 2020).
- Voltage-Gated Ion Channels|HUGO Gene Nomenclature Committee. Available online: https://www.genenames.org/data/genegroup/#!/group/178 (accessed on 5 July 2020).
- Streit, J.; Kleinlogel, S. Dynamic all-optical drug screening on cardiac voltage-gated ion channels. Sci. Rep. 2018, 8, 1153. [Google Scholar] [CrossRef] [Green Version]
- Annecchino, L.A.; Schultz, S.R. Progress in automating patch clamp cellular physiology. Brain Neurosci. Adv. 2018, 2, 1–16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Braubach, O.; Cohen, L.B.; Choi, Y. Historical Overview and General Methods of Membrane Potential Imaging. In Membrane Potential Imaging in the Nervous System and Heart; Canepari, M., Zecevic, D., Bernus, O., Eds.; Advances in Experimental Medicine and Biology; Springer International Publishing: Cham, Switzerland, 2015; Volume 859, pp. 3–26. ISBN 978-3-319-17640-6. [Google Scholar]
- Miller, E.W. Small molecule fluorescent voltage indicators for studying membrane potential. Curr. Opin. Chem. Biol. 2016, 33, 74–80. [Google Scholar] [CrossRef] [Green Version]
- Xu, Y.; Zou, P.; Cohen, A.E. Voltage imaging with genetically encoded indicators. Curr. Opin. Chem. Biol. 2017, 39, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Knöpfel, T.; Song, C. Optical voltage imaging in neurons: Moving from technology development to practical tool. Nat. Rev. Neurosci. 2019, 20, 719–727. [Google Scholar] [CrossRef] [PubMed]
- Joshi, J.; Rubart, M.; Zhu, W. Optogenetics: Background, Methodological Advances and Potential Applications for Cardiovascular Research and Medicine. Front. Bioeng. Biotechnol. 2020, 7, 466. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hochbaum, D.R.; Zhao, Y.; Farhi, S.L.; Klapoetke, N.; Werley, C.A.; Kapoor, V.; Zou, P.; Kralj, J.M.; Maclaurin, D.; Smedemark-Margulies, N.; et al. All-optical electrophysiology in mammalian neurons using engineered microbial rhodopsins. Nat. Methods 2014, 11, 825–833. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bando, Y.; Sakamoto, M.; Kim, S.; Ayzenshtat, I.; Yuste, R. Comparative Evaluation of Genetically Encoded Voltage Indicators. Cell Rep. 2019, 26, 802–813.e4. [Google Scholar] [CrossRef] [Green Version]
- Zhang, H.; Reichert, E.; Cohen, A.E. Optical electrophysiology for probing function and pharmacology of voltage-gated ion channels. eLife 2016, 5, e15202. [Google Scholar] [CrossRef]
- Hsu, H.; Huang, E.; Yang, X.C.; Karschin, A.; Labarca, C.; Figl, A.; Ho, B.; Davidson, N.; Lester, H.A. Slow and incomplete inactivations of voltage-gated channels dominate encoding in synthetic neurons. Biophys. J. 1993, 65, 1196–1206. [Google Scholar] [CrossRef] [Green Version]
- Kirkton, R.D.; Bursac, N. Engineering biosynthetic excitable tissues from unexcitable cells for electrophysiological and cell therapy studies. Nat. Commun. 2011, 2, 300. [Google Scholar] [CrossRef] [Green Version]
- Park, J.; Werley, C.A.; Venkatachalam, V.; Kralj, J.M.; Dib-Hajj, S.D.; Waxman, S.G.; Cohen, A.E. Screening Fluorescent Voltage Indicators with Spontaneously Spiking HEK Cells. PLoS ONE 2013, 8, e85221. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, H.; Zou, B.; Du, F.; Xu, K.; Li, M. Reporting Sodium Channel Activity Using Calcium Flux: Pharmacological Promiscuity of Cardiac Nav1.5. Mol. Pharm. 2015, 87, 207–217. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nagel, G.; Szellas, T.; Huhn, W.; Kateriya, S.; Adeishvili, N.; Berthold, P.; Ollig, D.; Hegemann, P.; Bamberg, E. Channelrhodopsin-2, a directly light-gated cation-selective membrane channel. Proc. Natl. Acad. Sci. USA 2003, 100, 13940–13945. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Han, X.; Chow, B.Y.; Zhou, H.; Klapoetke, N.C.; Chuong, A.; Rajimehr, R.; Yang, A.; Baratta, M.V.; Winkle, J.; Desimone, R.; et al. A High-Light Sensitivity Optical Neural Silencer: Development and Application to Optogenetic Control of Non-Human Primate Cortex. Front. Syst. Neurosci. 2011, 5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, H.H.; St-Pierre, F. Genetically Encoded Voltage Indicators: Opportunities and Challenges. J. Neurosci. 2016, 36, 9977–9989. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Moyer, B.D.; Yu, V.; McGivern, J.G.; Jarosh, M.; Werley, C.A.; Hecht, V.C.; Babcock, R.J.; Dong, K.; Dempsey, G.T.; et al. Correlation of Optical and Automated Patch Clamp Electrophysiology for Identification of Na V 1.7 Inhibitors. SLAS Discov. Adv. Sci. Drug Discov. 2020, 25, 434–446. [Google Scholar] [CrossRef]
- Zou, P.; Zhao, Y.; Douglass, A.D.; Hochbaum, D.R.; Brinks, D.; Werley, C.A.; Harrison, D.J.; Campbell, R.E.; Cohen, A.E. Bright and fast multicoloured voltage reporters via electrochromic FRET. Nat. Commun. 2014, 5, 4625. [Google Scholar] [CrossRef] [Green Version]
- Preziosi, P. Science, pharmacoeconomics and ethics in drug R&D: A sustainable future scenario? Nat. Rev. Drug Discov. 2004, 3, 521–526. [Google Scholar] [CrossRef]
- Piccini, J.P.; Whellan, D.J.; Berridge, B.R.; Finkle, J.K.; Pettit, S.D.; Stockbridge, N.; Valentin, J.-P.; Vargas, H.M.; Krucoff, M.W. Current challenges in the evaluation of cardiac safety during drug development: Translational medicine meets the Critical Path Initiative. Am. Heart J. 2009, 158, 317–326. [Google Scholar] [CrossRef]
- Redfern, W.; Carlsson, L.; Davis, A.; Lynch, W.; Mackenzie, I.; Palethorpe, S.; Siegl, P.; Strang, I.; Sullivan, A.; Wallis, R. Relationships between preclinical cardiac electrophysiology, clinical QT interval prolongation and torsade de pointes for a broad range of drugs: Evidence for a provisional safety margin in drug development. Cardiovasc. Res. 2003, 58, 32–45. [Google Scholar] [CrossRef]
- Lu, H.R.; Vlaminckx, E.; Hermans, A.N.; Rohrbacher, J.; Van Ammel, K.; Towart, R.; Pugsley, M.; Gallacher, D.J. Predicting drug-induced changes in QT interval and arrhythmias: QT-shortening drugs point to gaps in the ICHS7B Guidelines. Br. J. Pharmacol. 2008, 154, 1427–1438. [Google Scholar] [CrossRef] [PubMed]
- Dempsey, G.T.; Chaudhary, K.W.; Atwater, N.; Nguyen, C.; Brown, B.S.; McNeish, J.D.; Cohen, A.E.; Kralj, J.M. Cardiotoxicity screening with simultaneous optogenetic pacing, voltage imaging and calcium imaging. J. Pharmacol. Toxicol. Methods 2016, 81, 240–250. [Google Scholar] [CrossRef] [PubMed]
- Hou, J.H.; Kralj, J.M.; Douglass, A.D.; Engert, F.; Cohen, A.E. Simultaneous mapping of membrane voltage and calcium in zebrafish heart in vivo reveals chamber-specific developmental transitions in ionic currents. Front. Physiol. 2014, 5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, T.-W.; Wardill, T.J.; Sun, Y.; Pulver, S.R.; Renninger, S.L.; Baohan, A.; Schreiter, E.R.; Kerr, R.A.; Orger, M.B.; Jayaraman, V.; et al. Ultrasensitive fluorescent proteins for imaging neuronal activity. Nature 2013, 499, 295–300. [Google Scholar] [CrossRef] [Green Version]
- Sakai, R.; Repunte-Canonigo, V.; Raj, C.D.; Knöpfel, T. Design and characterization of a DNA-encoded, voltage-sensitive fluorescent protein: A DNA-encoded, voltage-sensitive fluorescent protein. Eur. J. Neurosci. 2001, 13, 2314–2318. [Google Scholar] [CrossRef]
- Lam, A.J.; St-Pierre, F.; Gong, Y.; Marshall, J.D.; Cranfill, P.J.; Baird, M.A.; McKeown, M.R.; Wiedenmann, J.; Davidson, M.W.; Schnitzer, M.J.; et al. Improving FRET dynamic range with bright green and red fluorescent proteins. Nat. Methods 2012, 9, 1005–1012. [Google Scholar] [CrossRef] [Green Version]
- Chen, Z.; Xian, W.; Bellin, M.; Dorn, T.; Tian, Q.; Goedel, A.; Dreizehnter, L.; Schneider, C.M.; Ward-van Oostwaard, D.; Ng, J.K.M.; et al. Subtype-specific promoter-driven action potential imaging for precise disease modelling and drug testing in hiPSC-derived cardiomyocytes. Eur. Heart J. 2016, ehw189. [Google Scholar] [CrossRef] [Green Version]
- Jin, L.; Han, Z.; Platisa, J.; Wooltorton, J.R.A.; Cohen, L.B.; Pieribone, V.A. Single Action Potentials and Subthreshold Electrical Events Imaged in Neurons with a Fluorescent Protein Voltage Probe. Neuron 2012, 75, 779–785. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shinnawi, R.; Huber, I.; Maizels, L.; Shaheen, N.; Gepstein, A.; Arbel, G.; Tijsen, A.J.; Gepstein, L. Monitoring Human-Induced Pluripotent Stem Cell-Derived Cardiomyocytes with Genetically Encoded Calcium and Voltage Fluorescent Reporters. Stem Cell Rep. 2015, 5, 582–596. [Google Scholar] [CrossRef] [Green Version]
- Shaheen, N.; Shiti, A.; Huber, I.; Shinnawi, R.; Arbel, G.; Gepstein, A.; Setter, N.; Goldfracht, I.; Gruber, A.; Chorna, S.V.; et al. Human Induced Pluripotent Stem Cell-Derived Cardiac Cell Sheets Expressing Genetically Encoded Voltage Indicator for Pharmacological and Arrhythmia Studies. Stem Cell Rep. 2018, 10, 1879–1894. [Google Scholar] [CrossRef]
- Sun, Y.-H.; Kao, H.K.J.; Chang, C.-W.; Merleev, A.; Overton, J.L.; Pretto, D.; Yechikov, S.; Maverakis, E.; Chiamvimonvat, N.; Chan, J.W.; et al. Human induced pluripotent stem cell line with genetically encoded fluorescent voltage indicator generated via CRISPR for action potential assessment post-cardiogenesis. Stem Cells 2020, 38, 90–101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Los, G.V.; Encell, L.P.; McDougall, M.G.; Hartzell, D.D.; Karassina, N.; Zimprich, C.; Wood, M.G.; Learish, R.; Ohana, R.F.; Urh, M.; et al. HaloTag: A Novel Protein Labeling Technology for Cell Imaging and Protein Analysis. ACS Chem. Biol. 2008, 3, 373–382. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Fares, M.; Dunham, N.P.; Gao, Z.; Miao, K.; Jiang, X.; Bollinger, S.S.; Boal, A.K.; Zhang, X. AgHalo: A Facile Fluorogenic Sensor to Detect Drug-Induced Proteome Stress. Angew. Chem. 2017, 129, 8798–8802. [Google Scholar] [CrossRef]
- Contreras-Baeza, Y.; Ceballo, S.; Arce-Molina, R.; Sandoval, P.Y.; Alegría, K.; Barros, L.F.; San Martín, A. MitoToxy assay: A novel cell-based method for the assessment of metabolic toxicity in a multiwell plate format using a lactate FRET nanosensor, Laconic. PLoS ONE 2019, 14, e0224527. [Google Scholar] [CrossRef] [Green Version]
- San Martín, A.; Ceballo, S.; Ruminot, I.; Lerchundi, R.; Frommer, W.B.; Barros, L.F. A Genetically Encoded FRET Lactate Sensor and Its Use to Detect the Warburg Effect in Single Cancer Cells. PLoS ONE 2013, 8, e57712. [Google Scholar] [CrossRef]
- Baenke, F.; Dubuis, S.; Brault, C.; Weigelt, B.; Dankworth, B.; Griffiths, B.; Jiang, M.; Mackay, A.; Saunders, B.; Spencer-Dene, B.; et al. Functional screening identifies MCT4 as a key regulator of breast cancer cell metabolism and survival. J. Pathol. 2015, 237, 152–165. [Google Scholar] [CrossRef]
- Chandrasekharan, A.; Varadarajan, S.N.; Lekshmi, A.; Lupitha, S.S.; Darvin, P.; Chandrasekhar, L.; Pillai, P.R.; Santhoshkumar, T.R.; Pillai, M.R. A high-throughput real-time in vitro assay using mitochondrial targeted roGFP for screening of drugs targeting mitochondria. Redox Biol. 2019, 20, 379–389. [Google Scholar] [CrossRef]
- Hanson, G.T.; Aggeler, R.; Oglesbee, D.; Cannon, M.; Capaldi, R.A.; Tsien, R.Y.; Remington, S.J. Investigating mitochondrial redox potential with redox-sensitive green fluorescent protein indicators. J. Biol. Chem. 2004, 279, 13044–13053. [Google Scholar] [CrossRef] [Green Version]
- World Malaria Report 2019; World Health Organization: Geneva, Switzerland, 2019; ISBN 978-92-4-156572-1.
- Joet, T.; Eckstein-Ludwig, U.; Morin, C.; Krishna, S. Validation of the hexose transporter of Plasmodium falciparum as a novel drug target. Proc. Natl. Acad. Sci. USA 2003, 100, 7476–7479. [Google Scholar] [CrossRef] [Green Version]
- Slavic, K.; Straschil, U.; Reininger, L.; Doerig, C.; Morin, C.; Tewari, R.; Krishna, S. Life cycle studies of the hexose transporter of Plasmodium species and genetic validation of their essentiality. Mol. Microbiol. 2010, 75, 1402–1413. [Google Scholar] [CrossRef] [Green Version]
- Bermejo, C.; Haerizadeh, F.; Takanaga, H.; Chermak, D.; Frommer, W.B. Optical sensors for measuring dynamic changes of cytosolic metabolite levels in yeast. Nat. Protoc. 2011, 6, 1806–1817. [Google Scholar] [CrossRef] [PubMed]
- Kraft, T.E.; Heitmeier, M.R.; Putanko, M.; Edwards, R.L.; Ilagan, M.X.G.; Payne, M.A.; Autry, J.M.; Thomas, D.D.; Odom, A.R.; Hruz, P.W. A Novel Fluorescence Resonance Energy Transfer-Based Screen in High-Throughput Format to Identify Inhibitors of Malarial and Human Glucose Transporters. Antimicrob. Agents Chemother. 2016, 60, 7407–7414. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heitmeier, M.R.; Hresko, R.C.; Edwards, R.L.; Prinsen, M.J.; Ilagan, M.X.G.; Odom John, A.R.; Hruz, P.W. Identification of druggable small molecule antagonists of the Plasmodium falciparum hexose transporter PfHT and assessment of ligand access to the glucose permeation pathway via FLAG-mediated protein engineering. PLoS ONE 2019, 14, e0216457. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brun, R.; Blum, J.; Chappuis, F.; Burri, C. Human African trypanosomiasis. Lancet 2010, 375, 148–159. [Google Scholar] [CrossRef] [Green Version]
- Voyton, C.M.; Morris, M.T.; Ackroyd, P.C.; Morris, J.C.; Christensen, K.A. A FRET Flow Cytometry-Based High Throughput Screening Assay to Identify Disrupters of Glucose Levels in Trypanosoma brucei. ACS Infect. Dis. 2018, 4, 1058–1066. [Google Scholar] [CrossRef] [PubMed]
- Deuschle, K.; Okumoto, S.; Fehr, M.; Looger, L.L.; Kozhukh, L.; Frommer, W.B. Construction and optimization of a family of genetically encoded metabolite sensors by semirational protein engineering. Protein Sci. 2005, 14, 2304–2314. [Google Scholar] [CrossRef]
- Monteith, G.R.; Bird, G.S.J. Techniques: High-throughput measurement of intracellular Ca2+–back to basics. Trends Pharmacol. Sci. 2005, 26, 218–223. [Google Scholar] [CrossRef]
- Bassett, J.J.; Monteith, G.R. Chapter Five-Genetically Encoded Calcium Indicators as Probes to Assess the Role of Calcium Channels in Disease and for High-Throughput Drug Discovery. In Advances in Pharmacology; Geraghty, D.P., Rash, L.D., Eds.; Ion Channels DownUnder; Academic Press: Cambridge, MA, USA, 2017; Volume 79, pp. 141–171. [Google Scholar]
- Zhao, Y.; Araki, S.; Wu, J.; Teramoto, T.; Chang, Y.-F.; Nakano, M.; Abdelfattah, A.S.; Fujiwara, M.; Ishihara, T.; Nagai, T.; et al. An Expanded Palette of Genetically Encoded Ca2+ Indicators. Science 2011, 333, 1888–1891. [Google Scholar] [CrossRef] [Green Version]
- Subach, O.M.; Sotskov, V.P.; Plusnin, V.V.; Gruzdeva, A.M.; Barykina, N.V.; Ivashkina, O.I.; Anokhin, K.V.; Nikolaeva, A.Y.; Korzhenevskiy, D.A.; Vlaskina, A.V.; et al. Novel Genetically Encoded Bright Positive Calcium Indicator NCaMP7 Based on the mNeonGreen Fluorescent Protein. Int. J. Mol. Sci. 2020, 21, 1644. [Google Scholar] [CrossRef] [Green Version]
- Qian, Y.; Piatkevich, K.D.; McLarney, B.; Abdelfattah, A.S.; Mehta, S.; Murdock, M.H.; Gottschalk, S.; Molina, R.S.; Zhang, W.; Chen, Y.; et al. A genetically encoded near-infrared fluorescent calcium ion indicator. Nat. Methods 2019, 16, 171–174. [Google Scholar] [CrossRef]
- Akerboom, J.; Tian, L.; Marvin, J.S.; Looger, L.L. Engineering and Application of Genetically Encoded Calcium Indicators. In Genetically Encoded Functional Indicators; Martin, J.-R., Ed.; Neuromethods; Humana Press: Totowa, NJ, USA, 2012; pp. 125–147. ISBN 978-1-62703-014-4. [Google Scholar]
- Pérez Koldenkova, V.; Nagai, T. Genetically encoded Ca2+ indicators: Properties and evaluation. Biochim. Biophys. Acta BBA-Mol. Cell Res. 2013, 1833, 1787–1797. [Google Scholar] [CrossRef] [Green Version]
- Mank, M.; Griesbeck, O. Genetically Encoded Calcium Indicators. Chem. Rev. 2008, 108, 1550–1564. [Google Scholar] [CrossRef] [PubMed]
- Kotlikoff, M.I. Genetically encoded Ca2+ indicators: Using genetics and molecular design to understand complex physiology. J. Physiol. 2007, 578, 55–67. [Google Scholar] [CrossRef] [PubMed]
- Wu, N.; Nishioka, W.K.; Derecki, N.C.; Maher, M.P. High-throughput-compatible assays using a genetically-encoded calcium indicator. Sci. Rep. 2019, 9, 12692. [Google Scholar] [CrossRef] [Green Version]
- Cai, B.; Chen, X.; Liu, F.; Li, J.; Gu, L.; Liu, J.R.; Liu, J. A Cell-Based Functional Assay Using a Green Fluorescent Protein-Based Calcium Indicator dCys-GCaMP. Assay Drug Dev. Technol. 2014, 12, 342–351. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Shi, Y.; Wei, H. Calcium Dysregulation in Alzheimer’s Disease: A Target for New Drug Development. J. Alzheimers Dis. Parkinsonism 2017, 7. [Google Scholar] [CrossRef]
- Honarnejad, K.; Daschner, A.; Giese, A.; Zall, A.; Schmidt, B.; Szybinska, A.; Kuznicki, J.; Herms, J. Development and Implementation of a High-Throughput Compound Screening Assay for Targeting Disrupted ER Calcium Homeostasis in Alzheimer’s Disease. PLoS ONE 2013, 8, e80645. [Google Scholar] [CrossRef]
- Honarnejad, K.; Daschner, A.; Gehring, A.P.; Szybinska, A.; Giese, A.; Kuznicki, J.; Bracher, F.; Herms, J. Identification of tetrahydrocarbazoles as novel multifactorial drug candidates for treatment of Alzheimer’s disease. Transl. Psychiatry 2014, 4, e489. [Google Scholar] [CrossRef] [Green Version]
- Fantuzzo, J.A.; Robles, D.A.; Mirabella, V.R.; Hart, R.P.; Pang, Z.P.; Zahn, J.D. Development of a high-throughput arrayed neural circuitry platform using human induced neurons for drug screening applications. Lab Chip 2020, 20, 1140–1152. [Google Scholar] [CrossRef]
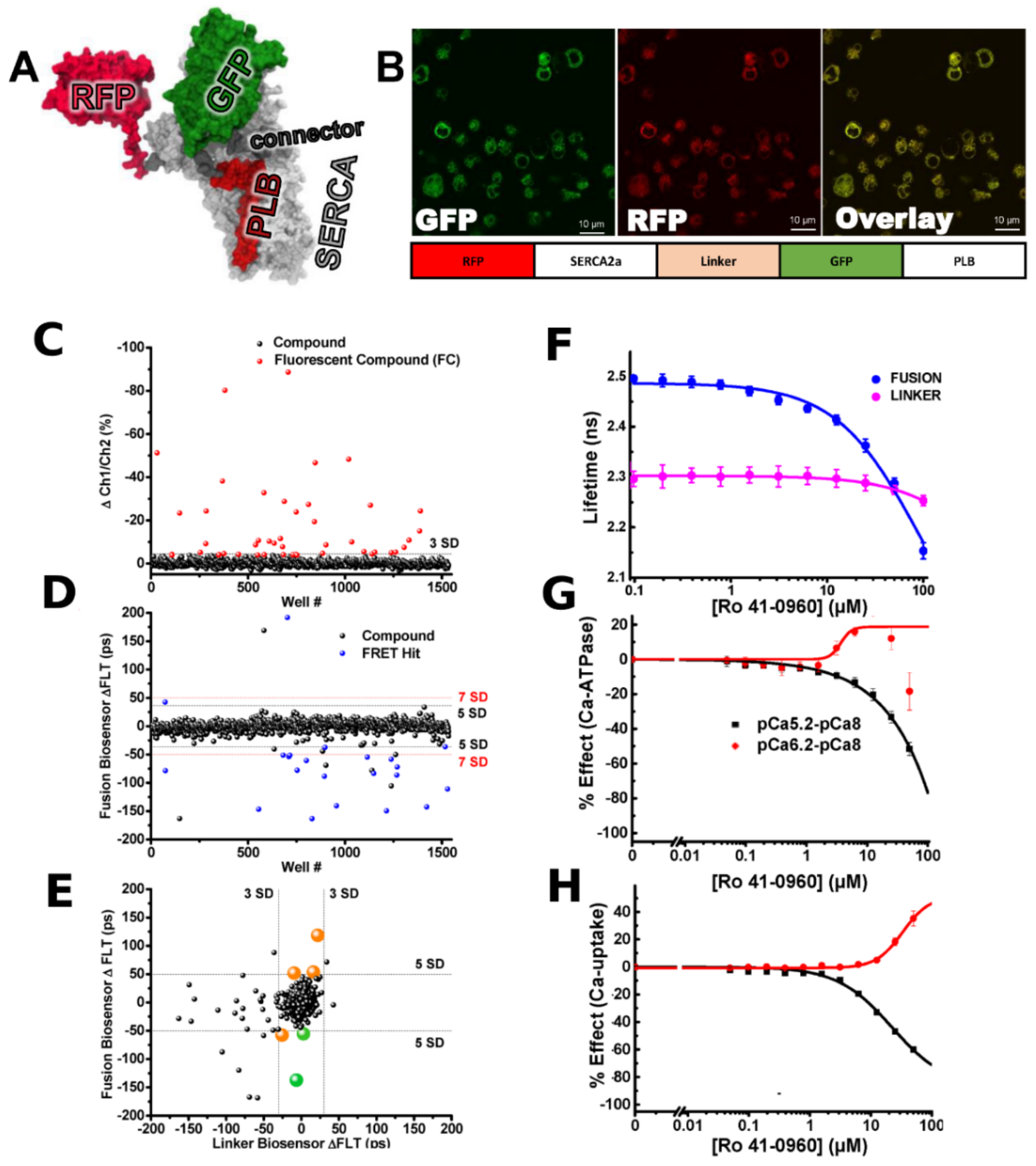
- Schaaf, T.M.; Peterson, K.C.; Grant, B.D.; Bawaskar, P.; Yuen, S.; Li, J.; Muretta, J.M.; Gillispie, G.D.; Thomas, D.D. High-throughput spectral and lifetime-based FRET screening in living cells to identify small-molecule effectors of SERCA. SLAS Discov. 2017, 22, 262–273. [Google Scholar] [CrossRef] [Green Version]
- Schaaf, T.M.; Li, A.; Grant, B.D.; Peterson, K.; Yuen, S.; Bawaskar, P.; Kleinboehl, E.; Li, J.; Thomas, D.D.; Gillispie, G.D. Red-Shifted FRET Biosensors for High-Throughput Fluorescence Lifetime Screening. Biosensors 2018, 8, 99. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schaaf, T.M.; Kleinboehl, E.; Yuen, S.L.; Roelike, L.N.; Svensson, B.; Thompson, A.R.; Cornea, R.L.; Thomas, D.D. Live-Cell Cardiac-Specific High-Throughput Screening Platform for Drug-Like Molecules That Enhance Ca2+ Transport. Cells 2020, 9, 1170. [Google Scholar] [CrossRef] [PubMed]
- Dowden, H.; Munro, J. Trends in clinical success rates and therapeutic focus. Nat. Rev. Drug Discov. 2019, 18, 495–496. [Google Scholar] [CrossRef]
- DiMasi, J.A.; Grabowski, H.G.; Hansen, R.W. Innovation in the pharmaceutical industry: New estimates of R&D costs. J. Health Econ. 2016, 47, 20–33. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Elitt, M.S.; Barbar, L.; Tesar, P.J. Drug screening for human genetic diseases using iPSC models. Hum. Mol. Genet. 2018, 27, R89–R98. [Google Scholar] [CrossRef] [Green Version]
- Mittal, R.; Woo, F.W.; Castro, C.S.; Cohen, M.A.; Karanxha, J.; Mittal, J.; Chhibber, T.; Jhaveri, V.M. Organ-on-chip models: Implications in drug discovery and clinical applications. J. Cell. Physiol. 2019, 234, 8352–8380. [Google Scholar] [CrossRef] [Green Version]
- Zhang, T.; Peterson, R.T. Zebrafish as a Platform for Drug Screening; Elsevier: Amsterdam, The Netherlands, 2020; ISBN 9780128124314. [Google Scholar]
- Strange, K. Drug discovery in fish, flies, and worms. ILAR J. 2016, 57, 133–143. [Google Scholar] [CrossRef]
- White, R.M.; Sessa, A.; Burke, C.; Bowman, T.; LeBlanc, J.; Ceol, C.; Bourque, C.; Dovey, M.; Goessling, W.; Burns, C.E.; et al. Transparent Adult Zebrafish as a Tool for In Vivo Transplantation Analysis. Cell Stem Cell 2008, 2, 183–189. [Google Scholar] [CrossRef] [Green Version]
- Antinucci, P.; Hindges, R. A crystal-clear zebrafish for in vivo imaging. Sci. Rep. 2016, 6, 29490. [Google Scholar] [CrossRef] [Green Version]
- Karlsson, J.; Von Hofsten, J.; Olsson, P.E. Generating transparent zebrafish: A refined method to improve detection of gene expression during embryonic development. Mar. Biotechnol. 2001, 3, 522–527. [Google Scholar] [CrossRef]
- Howe, K.; Clark, M.D.; Torroja, C.F.; Torrance, J.; Berthelot, C.; Muffato, M.; Collins, J.E.; Humphray, S.; McLaren, K.; Matthews, L.; et al. The zebrafish reference genome sequence and its relationship to the human genome. Nature 2013, 496, 498–503. [Google Scholar] [CrossRef] [Green Version]
- Hwang, W.Y.; Fu, Y.; Reyon, D.; Maeder, M.L.; Tsai, S.Q.; Sander, J.D.; Peterson, R.T.; Yeh, J.-R.J.; Joung, J.K. Efficient genome editing in zebrafish using a CRISPR-Cas system. Nat. Biotechnol. 2013, 31, 227–229. [Google Scholar] [CrossRef]
- Cong, L.; Ran, F.A.; Cox, D.; Lin, S.; Barretto, R.; Habib, N.; Hsu, P.D.; Wu, X.; Jiang, W.; Marraffini, L.A.; et al. Multiplex Genome Engineering Using CRISPR/Cas Systems. Science 2013, 339, 819. [Google Scholar] [CrossRef] [Green Version]
- Cho, S.W.; Kim, S.; Kim, J.M.; Kim, J.-S. Targeted genome engineering in human cells with the Cas9 RNA-guided endonuclease. Nat. Biotechnol. 2013, 31, 230–232. [Google Scholar] [CrossRef]
- Jinek, M.; East, A.; Cheng, A.; Lin, S.; Ma, E.; Doudna, J. RNA-programmed genome editing in human cells. eLife 2013, 2, e00471. [Google Scholar] [CrossRef] [Green Version]
- Chang, N.; Sun, C.; Gao, L.; Zhu, D.; Xu, X.; Zhu, X.; Xiong, J.W.; Xi, J.J. Genome editing with RNA-guided Cas9 nuclease in Zebrafish embryos. Cell Res. 2013, 23, 465–472. [Google Scholar] [CrossRef] [Green Version]
- Jao, L.E.; Wente, S.R.; Chen, W. Efficient multiplex biallelic zebrafish genome editing using a CRISPR nuclease system. Proc. Natl. Acad. Sci. USA 2013, 110, 13904–13909. [Google Scholar] [CrossRef] [Green Version]
- Burger, A.; Lindsay, H.; Felker, A.; Hess, C.; Anders, C.; Chiavacci, E.; Zaugg, J.; Weber, L.M.; Catena, R.; Jinek, M.; et al. Maximizing mutagenesis with solubilized CRISPR-Cas9 ribonucleoprotein complexes. Development 2016, 143, 2025–2037. [Google Scholar] [CrossRef] [Green Version]
- Zischewski, J.; Fischer, R.; Bortesi, L. Detection of on-target and off-target mutations generated by CRISPR/Cas9 and other sequence-specific nucleases. Biotechnol. Adv. 2017, 35, 95–104. [Google Scholar] [CrossRef]
- Sentmanat, M.F.; Peters, S.T.; Florian, C.P.; Connelly, J.P.; Pruett-Miller, S.M. A Survey of Validation Strategies for CRISPR-Cas9 Editing. Sci. Rep. 2018, 8, 1–8. [Google Scholar] [CrossRef] [Green Version]
- Liu, K.; Petree, C.; Requena, T.; Varshney, P.; Varshney, G.K. Expanding the CRISPR toolbox in zebrafish for studying development and disease. Front. Cell Dev. Biol. 2019, 7, 1–15. [Google Scholar] [CrossRef] [Green Version]
- Wu, R.S.; Lam, I.I.; Clay, H.; Duong, D.N.; Deo, R.C.; Coughlin, S.R. A Rapid Method for Directed Gene Knockout for Screening in G0 Zebrafish. Dev. Cell 2018, 46, 112–125.e4. [Google Scholar] [CrossRef] [Green Version]
- Jobst-Schwan, T.; Schmidt, J.M.; Schneider, R.; Hoogstraten, C.A.; Ullmann, J.F.P.; Schapiro, D.; Majmundar, A.J.; Kolb, A.; Eddy, K.; Shril, S.; et al. Acute multi-sgRNA knockdown of KEOPS complex genes reproduces the microcephaly phenotype of the stable knockout zebrafish model. PLoS ONE 2018, 13, e0191503. [Google Scholar] [CrossRef] [Green Version]
- Boel, A.; De Saffel, H.; Steyaert, W.; Callewaert, B.; De Paepe, A.; Coucke, P.J.; Willaert, A. CRISPR/Cas9-mediated homology-directed repair by ssODNs in zebrafish induces complex mutational patterns resulting from genomic integration of repair-template fragments. Dis. Models Mech. 2018, 11, dmm035352. [Google Scholar] [CrossRef] [Green Version]
- Prykhozhij, S.V.; Fuller, C.; Steele, S.L.; Veinotte, C.J.; Razaghi, B.; Robitaille, J.M.; McMaster, C.R.; Shlien, A.; Malkin, D.; Berman, J.N. Optimized knock-in of point mutations in zebrafish using CRISPR/Cas9. Nucleic Acids Res. 2018, 46, 1–15. [Google Scholar] [CrossRef]
- Hoshijima, K.; Jurynec, M.J.; Grunwald, D.J. Precise Editing of the Zebrafish Genome Made Simple and Efficient. Dev. Cell 2016, 36, 654–667. [Google Scholar] [CrossRef] [Green Version]
- Irion, U.; Krauss, J.; Nüsslein-Volhard, C. Precise and efficient genome editing in zebrafish using the CRISPR/Cas9 system. Development 2014, 141, 4827–4830. [Google Scholar] [CrossRef] [Green Version]
- Ablain, J.; Durand, E.M.; Yang, S.; Zhou, Y.; Zon, L.I. A CRISPR/Cas9 vector system for tissue-specific gene disruption in zebrafish. Dev. Cell 2015, 32, 756–764. [Google Scholar] [CrossRef] [Green Version]
- Yin, L.; Maddison, L.A.; Li, M.; Kara, N.; Lafave, M.C.; Varshney, G.K.; Burgess, S.M.; Patton, J.G.; Chen, W. Multiplex conditional mutagenesis using transgenic expression of Cas9 and sgRNAs. Genetics 2015, 200, 431–441. [Google Scholar] [CrossRef] [Green Version]
- Di Donato, V.; De Santis, F.; Auer, T.O.; Testa, N.; Sánchez-Iranzo, H.; Mercader, N.; Concordet, J.P.; Bene, F. Del 2C-Cas9: A versatile tool for clonal analysis of gene function. Genome Res. 2016, 26, 681–692. [Google Scholar] [CrossRef] [Green Version]
- Zhou, W.; Brown, W.; Bardhan, A.; Delaney, M.; Ilk, A.S.; Rauen, R.R.; Kahn, S.I.; Tsang, M.; Deiters, A. Spatiotemporal Control of CRISPR/Cas9 Function in Cells and Zebrafish using Light-Activated Guide RNA. Angew. Chem. Int. Ed. 2020, 59, 8998–9003. [Google Scholar] [CrossRef]
- Burg, L.; Palmer, N.; Kikhi, K.; Miroshnik, E.S.; Rueckert, H.; Gaddy, E.; MacPherson Cunningham, C.; Mattonet, K.; Lai, S.L.; Marín-Juez, R.; et al. Conditional mutagenesis by oligonucleotide-mediated integration of loxP sites in zebrafish. PLoS Genet. 2018, 14, e1007754. [Google Scholar] [CrossRef]
- Komor, A.C.; Kim, Y.B.; Packer, M.S.; Zuris, J.A.; Liu, D.R. Programmable editing of a target base in genomic DNA without double-stranded DNA cleavage. Nature 2016, 533, 420–424. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Qin, W.; Lu, X.; Xu, J.; Huang, H.; Bai, H.; Li, S.; Lin, S. Programmable base editing of zebrafish genome using a modified CRISPR-Cas9 system. Nat. Commun. 2017, 8, 6–10. [Google Scholar] [CrossRef]
- Rees, H.A.; Komor, A.C.; Yeh, W.H.; Caetano-Lopes, J.; Warman, M.; Edge, A.S.B.; Liu, D.R. Improving the DNA specificity and applicability of base editing through protein engineering and protein delivery. Nat. Commun. 2017, 8, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Gaudelli, N.M.; Komor, A.C.; Rees, H.A.; Packer, M.S.; Badran, A.H.; Bryson, D.I.; Liu, D.R. Programmable base editing of A•T to G•C in genomic DNA without DNA cleavage. Nature 2017, 551, 464–471. [Google Scholar] [CrossRef]
- Qin, W.; Lu, X.; Liu, Y.; Bai, H.; Li, S.; Lin, S. Precise A•T to G•C base editing in the zebrafish genome. BMC Biol. 2018, 16, 139. [Google Scholar] [CrossRef]
- Rees, H.A.; Liu, D.R. Base editing: Precision chemistry on the genome and transcriptome of living cells. Nat. Rev. Genet. 2018, 19, 770–788. [Google Scholar] [CrossRef]
- Carrington, B.; Weinstein, R.N.; Sood, R. BE4max and AncBE4max Are Efficient in Germline Conversion of C:G to T:A Base Pairs in Zebrafish. Cells 2020, 9, 1690. [Google Scholar] [CrossRef]
- Anzalone, A.V.; Randolph, P.B.; Davis, J.R.; Sousa, A.A.; Koblan, L.W.; Levy, J.M.; Chen, P.J.; Wilson, C.; Newby, G.A.; Raguram, A.; et al. Search-and-replace genome editing without double-strand breaks or donor DNA. Nature 2019, 576, 149–157. [Google Scholar] [CrossRef]
- Naert, T.; Vleminckx, K. CRISPR/Cas9 disease models in zebrafish and Xenopus: The genetic renaissance of fish and frogs. Drug Discov. Today Technol. 2018, 28, 41–52. [Google Scholar] [CrossRef]
- Sakai, C.; Ijaz, S.; Hoffman, E.J. Zebrafish Models of Neurodevelopmental Disorders: Past, Present, and Future. Front. Mol. Neurosci. 2018, 11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- North, T.E.; Goessling, W.; Walkley, C.R.; Lengerke, C.; Kopani, K.R.; Lord, A.M.; Weber, G.J.; Bowman, T.V.; Jang, I.; Grosser, T.; et al. Prostaglandin E2 regulates vertebrate haematopoietic stem cell homeostasis. Nature 2007, 447, 1007–1011. [Google Scholar] [CrossRef] [PubMed]
- Goessling, W.; Allen, R.S.; Guan, X.; Jin, P.; Uchida, N.; Dovey, M.; Harris, J.M.; Metzger, M.E.; Bonifacino, A.C.; Stroncek, D.; et al. Prostaglandin E2 Enhances Human Cord Blood Stem Cell Xenotransplants and Shows Long-Term Safety in Preclinical Nonhuman Primate Transplant Models. Cell Stem Cell 2011, 8, 445–458. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- White, R.M.; Cech, J.; Ratanasirintrawoot, S.; Lin, C.Y.; Rahl, P.B.; Burke, C.J.; Langdon, E.; Tomlinson, M.L.; Mosher, J.; Kaufman, C.; et al. DHODH modulates transcriptional elongation in the neural crest and melanoma. Nature 2011, 471, 518–522. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Griffin, A.; Hamling, K.R.; Knupp, K.; Hong, S.G.; Lee, L.P.; Baraban, S.C. Clemizole and modulators of serotonin signalling suppress seizures in Dravet syndrome. Brain 2017, 140, 669–683. [Google Scholar] [CrossRef]
- Lin, X.; Duan, X.; Jacobs, C.; Ullmann, J.; Chan, C.Y.; Chen, S.; Cheng, S.H.; Zhao, W.N.; Poduri, A.; Wang, X.; et al. High-throughput brain activity mapping and machine learning as a foundation for systems neuropharmacology. Nat. Commun. 2018, 9, 1–12. [Google Scholar] [CrossRef]
- Marvin, J.S.; Shimoda, Y.; Magloire, V.; Leite, M.; Kawashima, T.; Jensen, T.P.; Kolb, I.; Knott, E.L.; Novak, O.; Podgorski, K.; et al. A genetically encoded fluorescent sensor for in vivo imaging of GABA. Nat. Methods 2019, 16, 763–770. [Google Scholar] [CrossRef] [Green Version]
- Sun, F.; Zeng, J.; Jing, M.; Zhou, J.; Feng, J.; Owen, S.F.; Luo, Y.; Li, F.; Wang, H.; Yamaguchi, T.; et al. A Genetically Encoded Fluorescent Sensor Enables Rapid and Specific Detection of Dopamine in Flies, Fish, and Mice. Cell, 2018; 174, 481–496.e19. [Google Scholar] [CrossRef]
- Feng, J.; Zhang, C.; Lischinsky, J.E.; Jing, M.; Zhou, J.; Wang, H.; Zhang, Y.; Dong, A.; Wu, Z.; Wu, H.; et al. A Genetically Encoded Fluorescent Sensor for Rapid and Specific In Vivo Detection of Norepinephrine. Neuron 2019, 102, 745–761.e8. [Google Scholar] [CrossRef]
- Caballero, M.V.; Candiracci, M. Zebrafish as Toxicological model for screening and recapitulate human diseases. J. Unexplored Med. Data 2018, 3, 4. [Google Scholar] [CrossRef] [Green Version]
- Akerboom, J.; Calderón, N.C.; Tian, L.; Wabnig, S.; Prigge, M.; Tolö, J.; Gordus, A.; Orger, M.B.; Severi, K.E.; Macklin, J.J.; et al. Genetically encoded calcium indicators for multi-color neural activity imaging and combination with optogenetics. Front. Mol. Neurosci. 2013, 6, 1–29. [Google Scholar] [CrossRef] [Green Version]
- Esterberg, R.; Hailey, D.W.; Rubel, E.W.; Raible, D.W. ER–Mitochondrial Calcium Flow Underlies Vulnerability of Mechanosensory Hair Cells to Damage. J. Neurosci. 2014, 34, 9703–9719. [Google Scholar] [CrossRef] [PubMed]
- Akerboom, J.; Chen, T.-W.; Wardill, T.J.; Tian, L.; Marvin, J.S.; Mutlu, S.; Calderón, N.C.; Esposti, F.; Borghuis, B.G.; Sun, X.R.; et al. Optimization of a GCaMP Calcium Indicator for Neural Activity Imaging. J. Neurosci. 2012, 32, 13819–13840. [Google Scholar] [CrossRef] [PubMed]
- Marsden, K.C.; Granato, M. In Vivo Ca2+ Imaging Reveals that Decreased Dendritic Excitability Drives Startle Habituation. Cell Rep. 2015, 13, 1733–1740. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, J.; Xia, L.; Bruchas, M.R.; Solnica-Krezel, L. Imaging early embryonic calcium activity with GCaMP6s transgenic zebrafish. Dev. Biol. 2017, 430, 385–396. [Google Scholar] [CrossRef]
- Turrini, L.; Fornetto, C.; Marchetto, G.; Müllenbroich, M.C.; Tiso, N.; Vettori, A.; Resta, F.; Masi, A.; Mannaioni, G.; Pavone, F.S.; et al. Optical mapping of neuronal activity during seizures in zebrafish. Sci. Rep. 2017, 7, 3025. [Google Scholar] [CrossRef] [Green Version]
- Berlin, S.; Carroll, E.C.; Newman, Z.L.; Okada, H.O.; Quinn, C.M.; Kallman, B.; Rockwell, N.C.; Martin, S.S.; Lagarias, J.C.; Isacoff, E.Y. Photoactivatable genetically encoded calcium indicators for targeted neuronal imaging. Nat. Methods 2015, 12, 852–858. [Google Scholar] [CrossRef] [Green Version]
- Dana, H.; Mohar, B.; Sun, Y.; Narayan, S.; Gordus, A.; Hasseman, J.P.; Tsegaye, G.; Holt, G.T.; Hu, A.; Walpita, D.; et al. Sensitive red protein calcium indicators for imaging neural activity. eLife 2016, 5, e12727. [Google Scholar] [CrossRef]
- Shen, Y.; Dana, H.; Abdelfattah, A.S.; Patel, R.; Shea, J.; Molina, R.S.; Rawal, B.; Rancic, V.; Chang, Y.-F.; Wu, L.; et al. A genetically encoded Ca2+ indicator based on circularly permutated sea anemone red fluorescent protein eqFP578. BMC Biol. 2018, 16, 9. [Google Scholar] [CrossRef] [Green Version]
- Zarowny, L.; Aggarwal, A.; Rutten, V.M.S.; Kolb, I.; Patel, R.; Huang, H.-Y.; Chang, Y.-F.; Phan, T.; Kanyo, R.; Ahrens, M.B.; et al. Bright and High-Performance Genetically Encoded Ca2+ Indicator Based on mNeonGreen Fluorescent Protein. ACS Sens. 2020, 5, 1959–1968. [Google Scholar] [CrossRef]
- Barykina, N.V.; Sotskov, V.P.; Gruzdeva, A.M.; Wu, Y.K.; Portugues, R.; Subach, O.M.; Chefanova, E.S.; Plusnin, V.V.; Ivashkina, O.I.; Anokhin, K.V.; et al. FGCaMP7, an Improved Version of Fungi-Based Ratiometric Calcium Indicator for In Vivo Visualization of Neuronal Activity. Int. J. Mol. Sci. 2020, 21, 3012. [Google Scholar] [CrossRef] [PubMed]
- Moeyaert, B.; Holt, G.; Madangopal, R.; Perez-Alvarez, A.; Fearey, B.C.; Trojanowski, N.F.; Ledderose, J.; Zolnik, T.A.; Das, A.; Patel, D.; et al. Improved methods for marking active neuron populations. Nat. Commun. 2018, 9, 4440. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ermakova, Y.G.; Pak, V.V.; Bogdanova, Y.A.; Kotlobay, A.A.; Yampolsky, I.V.; Shokhina, A.G.; Panova, A.S.; Marygin, R.A.; Staroverov, D.B.; Bilan, D.S.; et al. SypHer3s: A genetically encoded fluorescent ratiometric probe with enhanced brightness and an improved dynamic range. Chem. Commun. 2018, 54, 2898–2901. [Google Scholar] [CrossRef] [PubMed]
- Beckwith-Cohen, B.; Holzhausen, L.C.; Wang, T.-M.; Rajappa, R.; Kramer, R.H. Localizing Proton-Mediated Inhibitory Feedback at the Retinal Horizontal Cell–Cone Synapse with Genetically-Encoded pH Probes. J. Neurosci. 2019, 39, 651–662. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Li, S.; Wong, H.-T.C.; He, X.J.; Beirl, A.; Petralia, R.S.; Wang, Y.-X.; Kindt, K.S. Synaptically silent sensory hair cells in zebrafish are recruited after damage. Nat. Commun. 2018, 9, 1388. [Google Scholar] [CrossRef] [PubMed]
- Kibat, C.; Krishnan, S.; Ramaswamy, M.; Baker, B.J.; Jesuthasan, S. Imaging voltage in zebrafish as a route to characterizing a vertebrate functional connectome: Promises and pitfalls of genetically encoded indicators. J. Neurogenet. 2016, 30, 80–88. [Google Scholar] [CrossRef]
- Miyazawa, H.; Okumura, K.; Hiyoshi, K.; Maruyama, K.; Kakinuma, H.; Amo, R.; Okamoto, H.; Yamasu, K.; Tsuda, S. Optical interrogation of neuronal circuitry in zebrafish using genetically encoded voltage indicators. Sci. Rep. 2018, 8, 6048. [Google Scholar] [CrossRef] [Green Version]
- Piatkevich, K.D.; Jung, E.E.; Straub, C.; Linghu, C.; Park, D.; Suk, H.-J.; Hochbaum, D.R.; Goodwin, D.; Pnevmatikakis, E.; Pak, N.; et al. A robotic multidimensional directed evolution approach applied to fluorescent voltage reporters. Nat. Chem. Biol. 2018, 14, 352–360. [Google Scholar] [CrossRef]
- Panieri, E.; Millia, C.; Santoro, M.M. Real-time quantification of subcellular H2O2 and glutathione redox potential in living cardiovascular tissues. Free Radic. Biol. Med. 2017, 109, 189–200. [Google Scholar] [CrossRef]
- Niethammer, P.; Grabher, C.; Look, A.T.; Mitchison, T.J. A tissue-scale gradient of hydrogen peroxide mediates rapid wound detection in zebrafish. Nature 2009, 459, 996–999. [Google Scholar] [CrossRef]
- Bilan, D.S.; Pase, L.; Joosen, L.; Gorokhovatsky, A.Y.; Ermakova, Y.G.; Gadella, T.W.J.; Grabher, C.; Schultz, C.; Lukyanov, S.; Belousov, V.V. HyPer-3: A Genetically Encoded H2O2 Probe with Improved Performance for Ratiometric and Fluorescence Lifetime Imaging. ACS Chem. Biol. 2013, 8, 535–542. [Google Scholar] [CrossRef] [Green Version]
- Tao, R.; Zhao, Y.; Chu, H.; Wang, A.; Zhu, J.; Chen, X.; Zou, Y.; Shi, M.; Liu, R.; Su, N.; et al. Genetically encoded fluorescent sensors reveal dynamic regulation of NADPH metabolism. Nat. Methods 2017, 14, 720–728. [Google Scholar] [CrossRef] [PubMed]
- Pak, V.V.; Ezeriņa, D.; Lyublinskaya, O.G.; Pedre, B.; Tyurin-Kuzmin, P.A.; Mishina, N.M.; Thauvin, M.; Young, D.; Wahni, K.; Gache, S.A.M.; et al. Ultrasensitive Genetically Encoded Indicator for Hydrogen Peroxide Identifies Roles for the Oxidant in Cell Migration and Mitochondrial Function. Cell Metab. 2020, 31, 642–653.e6. [Google Scholar] [CrossRef] [PubMed]
- Marvin, J.S.; Borghuis, B.G.; Tian, L.; Cichon, J.; Harnett, M.T.; Akerboom, J.; Gordus, A.; Renninger, S.L.; Chen, T.-W.; Bargmann, C.I.; et al. An optimized fluorescent probe for visualizing glutamate neurotransmission. Nat. Methods 2013, 10, 162–170. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- MacDonald, R.B.; Kashikar, N.D.; Lagnado, L.; Harris, W.A. A Novel Tool to Measure Extracellular Glutamate in the Zebrafish Nervous System In Vivo. Zebrafish 2016, 14, 284–286. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pichler, P.; Lagnado, L. The Transfer Characteristics of Hair Cells Encoding Mechanical Stimuli in the Lateral Line of Zebrafish. J. Neurosci. 2019, 39, 112–124. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zou, Y.; Wang, A.; Shi, M.; Chen, X.; Liu, R.; Li, T.; Zhang, C.; Zhang, Z.; Zhu, L.; Ju, Z.; et al. Analysis of redox landscapes and dynamics in living cells and in vivo using genetically encoded fluorescent sensors. Nat. Protoc. 2018, 13, 2362–2386. [Google Scholar] [CrossRef] [PubMed]
- Wong, H.C.; Zhang, Q.; Beirl, A.J.; Petralia, R.S.; Wang, Y.-X.; Kindt, K. Synaptic mitochondria regulate hair-cell synapse size and function. eLife 2019, 8, e48914. [Google Scholar] [CrossRef]
- Andrews, N.; Ramel, M.-C.; Kumar, S.; Alexandrov, Y.; Kelly, D.J.; Warren, S.C.; Kerry, L.; Lockwood, N.; Frolov, A.; Frankel, P.; et al. Visualising apoptosis in live zebrafish using fluorescence lifetime imaging with optical projection tomography to map FRET biosensor activity in space and time. J. Biophotonics 2016, 9, 414–424. [Google Scholar] [CrossRef] [Green Version]
- Jia, H.; Song, Y.; Huang, B.; Ge, W.; Luo, K.Q. Engineered Sensor Zebrafish for Fast Detection and Real-Time Tracking of Apoptosis at Single-Cell Resolution in Live Animals. ACS Sens. 2020, 5, 823–830. [Google Scholar] [CrossRef]
- Abdelfattah, A.S.; Kawashima, T.; Singh, A.; Novak, O.; Liu, H.; Shuai, Y.; Huang, Y.-C.; Campagnola, L.; Seeman, S.C.; Yu, J.; et al. Bright and photostable chemigenetic indicators for extended in vivo voltage imaging. Science 2019, 365, 699–704. [Google Scholar] [CrossRef] [PubMed]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Potekhina, E.S.; Bass, D.Y.; Kelmanson, I.V.; Fetisova, E.S.; Ivanenko, A.V.; Belousov, V.V.; Bilan, D.S. Drug Screening with Genetically Encoded Fluorescent Sensors: Today and Tomorrow. Int. J. Mol. Sci. 2021, 22, 148. https://doi.org/10.3390/ijms22010148
Potekhina ES, Bass DY, Kelmanson IV, Fetisova ES, Ivanenko AV, Belousov VV, Bilan DS. Drug Screening with Genetically Encoded Fluorescent Sensors: Today and Tomorrow. International Journal of Molecular Sciences. 2021; 22(1):148. https://doi.org/10.3390/ijms22010148
Chicago/Turabian StylePotekhina, Ekaterina S., Dina Y. Bass, Ilya V. Kelmanson, Elena S. Fetisova, Alexander V. Ivanenko, Vsevolod V. Belousov, and Dmitry S. Bilan. 2021. "Drug Screening with Genetically Encoded Fluorescent Sensors: Today and Tomorrow" International Journal of Molecular Sciences 22, no. 1: 148. https://doi.org/10.3390/ijms22010148