Circulating 16S RNA in Biofluids: Extracellular Vesicles as Mirrors of Human Microbiome?


 , ,
, ,  and
and
Abstract
:1. The Human Microbiome
2. The Microbiome as a Gateway to Detect the Microbiota Composition: 16S Sequencing
3. The “Healthy” Microbiome
4. Microbiota and Disease
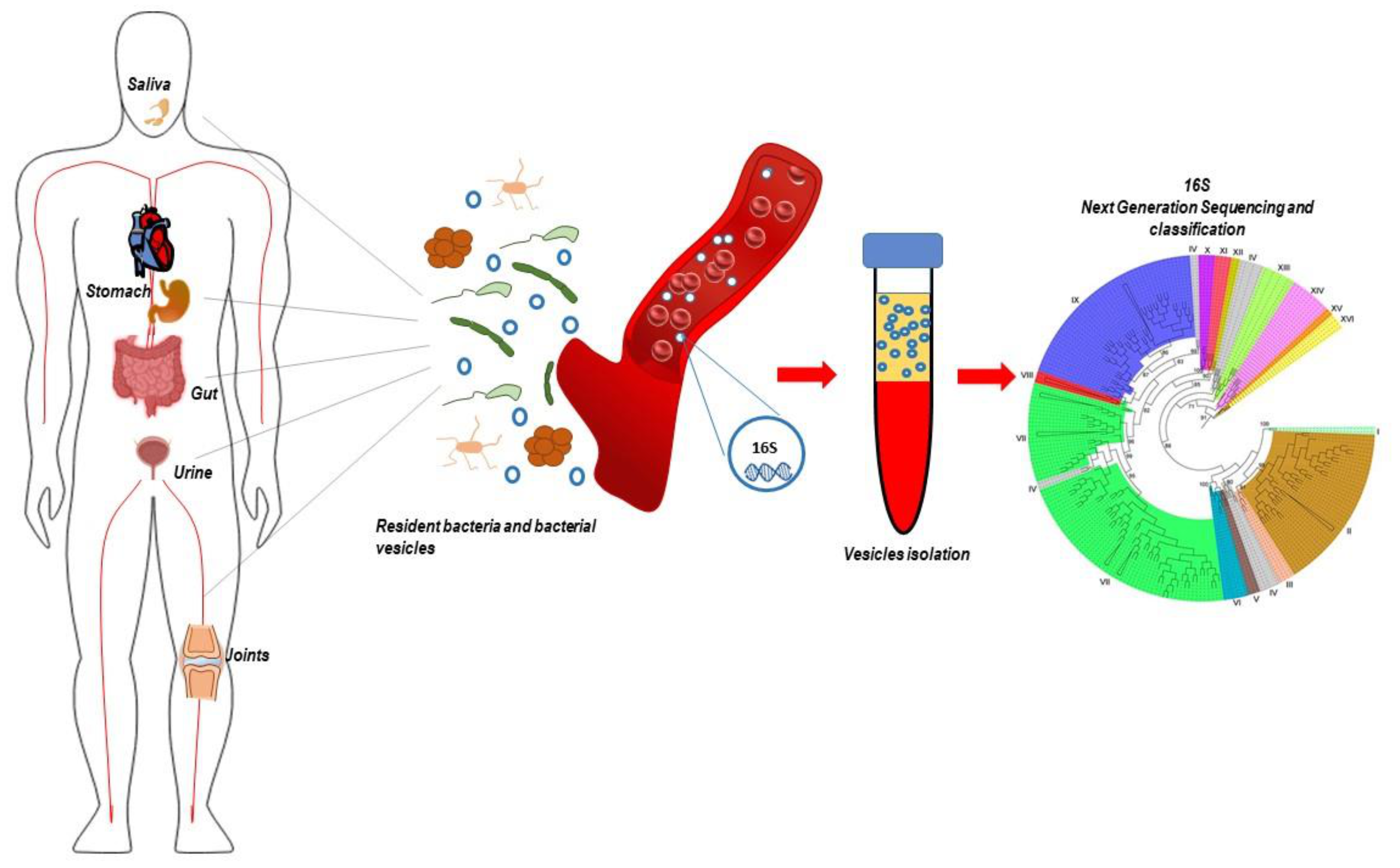
5. Bacteria in the Blood: To Be or Not to Be? Circulating 16S Detection
6. Circulating 16S in a Disease Context
7. Source of Circulating 16S RNA: Are We Really Looking at Bacteria in the Blood?
8. Bacteria-Derived Vesicles and 16s RNA
9. Limitations and Functional Perspectives
10. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Marchesi, J.R.; Ravel, J. The vocabulary of microbiome research: A proposal. Microbiome 2015, 3, 1–3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Leewenhoeck, A. Observations, communicated to the publisher by Mr. Antony van Leewenhoeck, in a Dutch letter of the 9th October 1676. here English’d: Concerning little animals by him observed in rain-well-sea-and snow water; as also in water wherein pepper had lain infused. Philos. Trans. R. Soc. Lond. 1677, 12, 821–831. [Google Scholar] [CrossRef]
- Greenhalgh, K.; Meyer, K.M.; Aagaard, K.M.; Wilmes, P. The human gut microbiome in health: Establishment and resilience of microbiota over a lifetime. Environ. Microbiol. 2016, 18, 2103–2116. [Google Scholar] [CrossRef] [PubMed]
- Guinane, C.M.; Cotter, P.D. Role of the gut microbiota in health and chronic gastrointestinal disease: Understanding a hidden metabolic organ. Ther. Adv. Gastroenterol. 2013, 6, 295–308. [Google Scholar] [CrossRef] [Green Version]
- Lloyd-Price, J.; Abu-Ali, G.; Huttenhower, C. The healthy human microbiome. Genome Med. 2016, 8, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Fraher, M.H.; O’Toole, P.W.; Quigley, E.M.M. Techniques used to characterize the gut microbiota: A guide for the clinician. Nat. Rev. Gastroenterol. Hepatol. 2012, 9, 312–322. [Google Scholar] [CrossRef]
- Srinivasan, R.; Karaoz, U.; Volegova, M.; MacKichan, J.; Kato-Maeda, M.; Miller, S.; Nadarajan, R.; Brodie, E.L.; Lynch, S.V. Use of 16S rRNA gene for identification of a broad range of clinically relevant bacterial pathogens. PLoS ONE 2015, 10, e0117617. [Google Scholar] [CrossRef]
- Hiergeist, A.; Gläsner, J.; Reischl, U.; Gessner, A. Analyses of intestinal microbiota: Culture versus sequencing: Figure 1. ILAR J. 2015, 56, 228–240. [Google Scholar] [CrossRef] [Green Version]
- Giovannoni, S.J.; Britschgi, T.B.; Moyer, C.L.; Field, K.G. Genetic diversity in Sargasso Sea bacterioplankton. Nature 1990, 345, 60–63. [Google Scholar] [CrossRef]
- Metzker, M.L. Emerging technologies in DNA sequencing. Genome Res. 2005, 15, 1767–1776. [Google Scholar] [CrossRef] [Green Version]
- Turnbaugh, P.J.; Ley, R.E.; Hamady, M.; Fraser-Liggett, C.M.; Knight, R.; Gordon, J.I. The human microbiome project. Nature 2007, 449, 804–810. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, N.-P.; Warnow, T.; Pop, M.; White, B. A perspective on 16S rRNA operational taxonomic unit clustering using sequence similarity. NPJ Biofilms Microbiomes 2016, 2, 16004. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuczynski, J.; Stombaugh, J.; Walters, W.A.; González, A.; Caporaso, J.G.; Knight, R. Using QIIME to analyze 16S rRNA gene sequences from microbial communities. Curr. Protoc. Microbiol. 2012, 27, 1E.5.1–1E.5.20. [Google Scholar]
- Chappidi, S.; Villa, E.C.; Cantarel, B.L. Using mothur to determine bacterial community composition and structure in 16S ribosomal RNA datasets. Curr. Protoc. Bioinform. 2019, 27, 1E.5.1–1E.5.20. [Google Scholar] [CrossRef] [PubMed]
- Calle, M.L. Statistical analysis of metagenomics data. Genom. Inform. 2019, 17, e6. [Google Scholar] [CrossRef] [PubMed]
- Kim, B.-R.; Shin, J.; Guevarra, R.B.; Lee, J.H.; Kim, D.W.; Seol, K.H.; Lee, J.H.; Kim, H.B.; Isaacson, R. Deciphering diversity indices for a better understanding of microbial communities. J. Microbiol. Biotechnol. 2017, 27, 2089–2093. [Google Scholar] [CrossRef] [Green Version]
- Modin, O.; Liébana, R.; Saheb-Alam, S.; Wilén, B.M.; Suarez, C.; Hermansson, M.; Persson, F. Hill-based dissimilarity indices and null models for analysis of microbial community assembly. Microbiome 2020, 8, 132. [Google Scholar] [CrossRef]
- Mueller, N.T.; Bakacs, E.; Combellick, J.; Grigoryan, Z.; Dominguez-Bello, M.G. The infant microbiome development: Mom matters. Trends Mol. Med. 2015, 21, 109–117. [Google Scholar] [CrossRef] [Green Version]
- Fouhy, F.; Ross, R.P.; Fitzgerald, G.F.; Stanton, C.; Cotter, P.D. Composition of the early intestinal microbiota: Knowledge, knowledge gaps and the use of high-throughput sequencing to address these gaps. Gut Microbes 2012, 3, 203–220. [Google Scholar] [CrossRef] [Green Version]
- Aagaard, K.M.; Riehle, K.; Ma, J.; Segata, N.; Mistretta, T.A.; Coarfa, C.; Raza, S.; Rosenbaum, S.; Van den Veyver, I.; Milosavljevic, A.; et al. A metagenomic approach to characterization of the vaginal microbiome signature in pregnancy. PLoS ONE 2012, 7, e36466. [Google Scholar] [CrossRef]
- Flowers, L.; Grice, E.A. The skin microbiota: Balancing risk and reward. Cell Host Microbe 2020, 28, 190–200. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Ryu, E.; Hathcock, M.; Ballman, K.; Chia, N.; Olson, J.E.; Nelson, H. Impact of demographics on human gut microbial diversity in a US Midwest population. PeerJ 2016, 4, e1514. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kolodziejczyk, A.A.; Zheng, D.; Elinav, E. Diet–microbiota interactions and personalized nutrition. Nat. Rev. Genet. 2019, 17, 742–753. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.; Shi, G. Smoking and microbiome in oral, airway, gut and some systemic diseases. J. Transl. Med. 2019, 17, 1–15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Benedict, C.; Vogel, H.; Jonas, W.; Woting, A.; Blaut, M.; Schürmann, A.; Cedernaes, J. Gut microbiota and glucometabolic alterations in response to recurrent partial sleep deprivation in normal-weight young individuals. Mol. Metab. 2016, 5, 1175–1186. [Google Scholar] [CrossRef]
- Shively, C.A.; Register, T.C.; Appt, S.E.; Clarkson, T.B.; Uberseder, B.; Clear, K.Y.J.; Wilson, A.S.; Chiba, A.; Tooze, J.A.; Cook, K.L. Consumption of Mediterranean versus Western diet leads to distinct mammary gland microbiome populations. Cell Rep. 2018, 25, 47–56.e3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Belizário, J.E.; Napolitano, M. Human microbiomes and their roles in dysbiosis, common diseases, and novel therapeutic approaches. Front. Microbiol. 2015, 6, 1050. [Google Scholar] [CrossRef] [Green Version]
- Rutebemberwa, A.; Stevens, M.J.; Pérez, M.J.; Smith, L.P.; Sanders, L.; Cosgrove, G.; Robertson, C.E.; Tuder, R.M.; Harris, J.K. Novosphingobium and its potential role in chronic obstructive pulmonary diseases: Insights from microbiome studies. PLoS ONE 2014, 9, e111150. [Google Scholar] [CrossRef]
- O’Dwyer, D.N.; Dickson, R.P.; Moore, B.B. The lung microbiome, immunity, and the pathogenesis of chronic lung disease. J. Immunol. 2016, 196, 4839–4847. [Google Scholar] [CrossRef] [Green Version]
- Zhao, Y.; Wang, Z. Gut microbiome and cardiovascular disease. Curr. Opin. Cardiol. 2020, 35, 207–218. [Google Scholar] [CrossRef]
- Zhu, Q.; Gao, R.; Zhang, Y.; Pan, D.; Zhu, Y.; Zhang, X.; Yang, R.; Jiang, R.; Xu, Y.; Qin, H. Dysbiosis signatures of gut microbiota in coronary artery disease. Physiol. Genom. 2018, 50, 893–903. [Google Scholar] [CrossRef] [PubMed]
- Parsonnet, J.; Friedman, G.D.; Vandersteen, D.P.; Chang, Y.; Vogelman, J.H.; Orentreich, N.; Sibley, R.K. Helicobacter pylori infection and the risk of gastric carcinoma. N. Engl. J. Med. 1991, 325, 1127–1131. [Google Scholar] [CrossRef] [PubMed]
- Raza, M.H.; Gul, K.; Arshad, A.; Riaz, N.; Waheed, U.; Rauf, A.; Aldakheel, F.; Alduraywish, S.; Rehman, M.U.; Abdullah, M. Microbiota in cancer development and treatment. J. Cancer Res. Clin. Oncol. 2019, 145, 49–63. [Google Scholar] [CrossRef] [PubMed]
- Zitvogel, L.; Galluzzi, L.; Viaud, S.; Vétizou, M.; Daillère, R.; Merad, M.; Kroemer, G. Cancer and the gut microbiota: An unexpected link. Sci. Transl. Med. 2015, 7, 271ps1. [Google Scholar] [CrossRef] [Green Version]
- Tremlett, H.; Bauer, K.C.; Appel-Cresswell, S.; Finlay, B.B.; Waubant, E. The gut microbiome in human neurological disease: A review. Ann. Neurol. 2017, 81, 369–382. [Google Scholar] [CrossRef]
- Xu, M.; Xu, X.; Li, J.; Li, F. Association between gut microbiota and autism spectrum disorder: A systematic review and meta-analysis. Front. Psychiatry 2019, 10, 473. [Google Scholar] [CrossRef]
- Nikkari, S.; McLaughlin, I.J.; Bi, W.; Dodge, D.E.; Relman, D.A. Does blood of healthy subjects contain bacterial ribosomal DNA? J. Clin. Microbiol. 2001, 39, 1956–1959. [Google Scholar] [CrossRef] [Green Version]
- Castillo, D.J.; Rifkin, R.F.; Cowan, D.A.; Potgieter, M. The healthy human blood microbiome: Fact or fiction? Front. Cell. Infect. Microbiol. 2019, 9, 148. [Google Scholar] [CrossRef] [Green Version]
- Païssé, S.; Valle, C.; Servant, F.; Courtney, M.; Burcelin, R.; Amar, J.; Lelouvier, B. Comprehensive description of blood microbiome from healthy donors assessed by 16S targeted metagenomic sequencing. Transfusion 2016, 56, 1138–1147. [Google Scholar] [CrossRef]
- Turnbaugh, P.J.; Ley, R.E.; Mahowald, M.A.; Magrini, V.; Mardis, E.R.; Gordon, J.I. An obesity-associated gut microbiome with increased capacity for energy harvest. Nature 2006, 444, 1027–1031. [Google Scholar] [CrossRef]
- Amar, J.; Serino, M.; Lange, C.; Chabo, C.; Iacovoni, J.; Mondot, S.; Lepage, P.; Klopp, C.; Mariette, J.; Bouchez, O. Involvement of tissue bacteria in the onset of diabetes in humans: Evidence for a concept. Diabetologia 2011, 54, 3055–3061. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Amar, J.; Lange, C.; Payros, G.; Garret, C.; Chabo, C.; Lantieri, O.; Courtney, M.; Marre, M.; Charles, M.A.; Balkau, B. Blood microbiota dysbiosis is associated with the onset of cardiovascular events in a large general population: The D.E.S.I.R. Study. PLoS ONE 2013, 8, e54461. [Google Scholar] [CrossRef] [PubMed]
- Dinakaran, V.; Rathinavel, A.; Pushpanathan, M.; Sivakumar, R.; Gunasekaran, P.; Rajendhran, J. Elevated levels of circulating DNA in cardiovascular disease patients: Metagenomic profiling of microbiome in the circulation. PLoS ONE 2014, 9, e105221. [Google Scholar] [CrossRef] [PubMed]
- Lelouvier, B.; Servant, F.; Païssé, S.; Brunet, A.C.; Benyahya, S.; Serino, M.; Valle, C.; Ortiz, M.R.; Puig, J.; Courtney, M.; et al. Changes in blood microbiota profiles associated with liver fibrosis in obese patients: A pilot analysis. Hepatology 2016, 64, 2015–2027. [Google Scholar] [CrossRef] [PubMed]
- Qian, Y.; Yang, X.; Xu, S.; Wu, C.; Qin, N.; Chen, S.D.; Xiao, Q. Detection of microbial 16S rRNA gene in the blood of patients with Parkinson’s disease. Front. Aging Neurosci. 2018, 10, 156. [Google Scholar] [CrossRef] [Green Version]
- Hammad, D.B.M.; Hider, S.L.; Liyanapathirana, V.C.; Tonge, D.P. Molecular characterization of circulating microbiome signatures in rheumatoid arthritis. Front. Cell. Infect. Microbiol. 2020, 9, 440. [Google Scholar] [CrossRef]
- Whittle, E.; Leonard, M.O.; Harrison, R.; Gant, T.W.; Tonge, D.P. Multi-method characterization of the human circulating microbiome. Front. Microbiol. 2018, 9, 3266. [Google Scholar] [CrossRef] [Green Version]
- Santiago, A.; Pozuelo, M.; Poca, M.; Gely, C.; Nieto, J.C.; Torras, X.; Román, E.; Campos, D.; Sarrabayrouse, G.; Vidal, S. Alteration of the serum microbiome composition in cirrhotic patients with ascites. Sci. Rep. 2016, 6, 25001. [Google Scholar] [CrossRef] [Green Version]
- Domingues, S.; Nielsen, K.M. Membrane vesicles and horizontal gene transfer in prokaryotes. Curr. Opin. Microbiol. 2017, 38, 16–21. [Google Scholar] [CrossRef]
- Zhang, G.; Meredith, T.C.; Kahne, D. On the essentiality of lipopolysaccharide to Gram-negative bacteria. Curr. Opin. Microbiol. 2013, 16, 779–785. [Google Scholar] [CrossRef] [Green Version]
- Silhavy, T.J.; Kahne, D.; Walker, S. The bacterial cell envelope. Cold Spring Harb. Perspect. Biol. 2010, 2, a000414. [Google Scholar] [CrossRef] [PubMed]
- Schwechheimer, C.; Kuehn, M.J. Outer-membrane vesicles from Gram-negative bacteria: Biogenesis and functions. Nat. Rev. Genet. 2015, 13, 605–619. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pathirana, R.D.; Kaparakis-Liaskos, M. Bacterial membrane vesicles: Biogenesis, immune regulation and pathogenesis. Cell. Microbiol. 2016, 18, 1518–1524. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, E.-Y.; Choi, D.-Y.; Kim, D.-K.; Kim, J.W.; Park, J.O.; Kim, S.; Kim, S.H.; Desiderio, D.M.; Kim, Y.K.; Kim, K.P. Gram-positive bacteria produce membrane vesicles: Proteomics-based characterization of Staphylococcus aureus-derived membrane vesicles. Proteomics 2009, 9, 5425–5436. [Google Scholar] [CrossRef]
- Brown, L.; Wolf, J.M.; Prados-Rosales, R.; Casadevall, A. Through the wall: Extracellular vesicles in Gram-positive bacteria, mycobacteria and fungi. Nat. Rev. Genet. 2015, 13, 620–630. [Google Scholar] [CrossRef] [Green Version]
- Kulp, A.; Kuehn, M.J. Biological functions and biogenesis of secreted bacterial outer membrane vesicles. Annu. Rev. Microbiol. 2010, 64, 163–184. [Google Scholar] [CrossRef] [Green Version]
- Kang, C.-S.; Ban, M.; Choi, E.-J.; Moon, H.G.; Jeon, J.S.; Kim, D.K.; Park, S.K.; Jeon, S.G.; Roh, T.Y.; Myung, S.J. Extracellular vesicles derived from gut microbiota, especially Akkermansia muciniphila, protect the progression of dextran sulfate sodium-induced colitis. PLoS ONE 2013, 8, e76520. [Google Scholar] [CrossRef] [Green Version]
- Yoo, J.Y.; Rho, M.; You, Y.-A.; Kwon, E.J.; Kim, M.H.; Kym, S.; Jee, Y.K.; Kim, Y.K.; Kim, Y.J. 16S rRNA gene-based metagenomic analysis reveals differences in bacteria-derived extracellular vesicles in the urine of pregnant and non-pregnant women. Exp. Mol. Med. 2016, 48, e208. [Google Scholar] [CrossRef] [Green Version]
- Choi, H.-I.; Choi, J.-P.; Seo, J.; Kim, B.J.; Rho, M.; Han, J.K.; Kim, J.G. Helicobacter pylori-derived extracellular vesicles increased in the gastric juices of gastric adenocarcinoma patients and induced inflammation mainly via specific targeting of gastric epithelial cells. Exp. Mol. Med. 2017, 49, e330. [Google Scholar] [CrossRef]
- Park, J.-Y.; Choi, J.; Lee, Y.; Lee, J.E.; Lee, E.H.; Kwon, H.J.; Yang, J.; Jeong, B.R.; Kim, Y.K.; Han, P.L. Metagenome analysis of bodily microbiota in a mouse model of Alzheimer disease using bacteria-derived membrane vesicles in blood. Exp. Neurobiol. 2017, 26, 369–379. [Google Scholar] [CrossRef]
- Samra, M.; Nam, S.K.; Lim, D.H.; Kim, D.H.; Yang, J.; Kim, Y.K.; Kim, J.H. Urine bacteria-derived extracellular vesicles and allergic airway diseases in children. Int. Arch. Allergy Immunol. 2018, 178, 150–158. [Google Scholar] [CrossRef] [PubMed]
- Philley, J.V.; Kannan, A.; Olusola, P.; McGaha, P.; Singh, K.P.; Samten, B.; Griffith, D.E.; Dasgupta, S. Microbiome diversity in sputum of nontuberculous mycobacteria infected women with a history of breast cancer. Cell. Physiol. Biochem. 2019, 52, 263–279. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.; Lee, H.K.; Min, S.K.; Lee, W.H. 16S rDNA microbiome composition pattern analysis as a diagnostic biomarker for biliary tract cancer. World J. Surg. Oncol. 2020, 18, 1–10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rhee, S.J.; Kim, H.; Lee, Y.; Lee, H.J.; Park, C.H.K.; Yang, J.; Kim, Y.K.; Kym, S.; Ahn, Y.M. Comparison of serum microbiome composition in bipolar and major depressive disorders. J. Psychiatr. Res. 2020, 123, 31–38. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.I.; Kang, N.; Leem, S.; Yang, J.; Jo, H.; Lee, M.; Kim, H.S.; Dhanasekaran, D.N.; Kim, Y.K.; Park, T. Metagenomic analysis of serum microbe-derived extracellular vesicles and diagnostic models to differentiate ovarian cancer and benign ovarian tumor. Cancers 2020, 12, 1309. [Google Scholar] [CrossRef] [PubMed]
- Hornung, B.V.H.; Zwittink, R.D.; Kuijper, E.J. Issues and current standards of controls in microbiome research. FEMS Microbiol. Ecol. 2019, 95. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eisenhofer, R.; Minich, J.J.; Marotz, C.; Cooper, A.; Knight, R.; Weyrich, L.S. Contamination in low microbial biomass microbiome studies: Issues and recommendations. Trends Microbiol. 2019, 27, 105–117. [Google Scholar] [CrossRef]
- Turner, P.; Turner, C.; Jankhot, A.; Helen, N.; Lee, S.J.; Day, N.P.; White, N.J.; Nosten, F.; Goldblatt, D. A longitudinal study of Streptococcus pneumoniae carriage in a cohort of infants and their mothers on the Thailand-Myanmar border. PLoS ONE 2012, 7, e38271. [Google Scholar] [CrossRef] [Green Version]
- Bentley, S.D.; Cox, M.J.; Turek, E.M.; Calus, S.T.; Cookson, W.O.; Moffatt, M.F.; Turner, P.; Parkhill, J.; Loman, N.J.; Walker, A.W. Reagent and laboratory contamination can critically impact sequence-based microbiome analyses. BMC Biol. 2014, 12, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Korpela, K.; Helve, O.; Kolho, K.-L.; Saisto, T.; Skogberg, K.; Dikareva, E.; Stefanovic, V.; Salonen, A.; Andersson, S.; de Vos, W.M. Maternal fecal microbiota transplantation in cesarean-born infants rapidly restores normal gut microbial development: A proof-of-concept study. Cell 2020, 183, 324–334.e5. [Google Scholar] [CrossRef]
- Andersen, V.; Möller, S.; Jensen, P.B.; Møller, F.T.; Green, A. Caesarean delivery and risk of chronic inflammatory diseases (inflammatory bowel disease, rheumatoid arthritis, coeliac disease, and diabetes mellitus): A population based registry study of 2,699,479 births in denmark during 1973–2016. Clin. Epidemiol. 2020, 12, 287–293. Available online: https://www.dovepress.com/caesarean-delivery-and-risk-of-chronic-inflammatory-diseases-inflammat-peer-reviewed-article-CLEP (accessed on 17 November 2020). [CrossRef] [PubMed] [Green Version]
- Witjes, J.J.; Smits, L.P.; Pekmez, C.T.; Prodan, A.; Meijnikman, A.S.; Troelstra, M.A.; Bouter, K.E.C.; Herrema, H.; Levin, E.; Holleboom, A.G. Donor fecal microbiota transplantation alters gut microbiota and metabolites in obese individuals with steatohepatitis. Hepatol. Commun. 2020, 4, 1578–1590. [Google Scholar] [CrossRef] [PubMed]
- Choi, Y.; Kwon, Y.; Kim, D.-K.; Jeon, J.; Jang, S.C.; Wang, T.; Ban, M.; Kim, M.H.; Jeon, S.G.; Kim, M.S. Gut microbe-derived extracellular vesicles induce insulin resistance, thereby impairing glucose metabolism in skeletal muscle. Sci. Rep. 2015, 5, 15878. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mohammadzadeh, R.; Ghazvini, K.; Farsiani, H.; Soleimanpour, S. Mycobacterium Tuberculosis extracellular vesicles: Exploitation for vaccine technology and diagnostic methods. Crit. Rev. Microbiol. 2020, 12, 1–21. [Google Scholar] [CrossRef] [PubMed]
- Samanta, D.; Mulye, M.; Clemente, T.M.; Justis, A.V.; Gilk, S.D. Manipulation of host cholesterol by obligate intracellular bacteria. Front. Cell. Infect. Microbiol. 2017, 7, 165. [Google Scholar] [CrossRef] [Green Version]
- Babatunde, K.A.; Subramanian, B.Y.; Ahouidi, A.D.; Murillo, P.M.; Walch, M.; Mantel, P.-Y. Role of extracellular vesicles in cellular cross talk in malaria. Front. Immunol. 2020, 11, 22. [Google Scholar] [CrossRef] [Green Version]



{kind=link}
{kind=link}
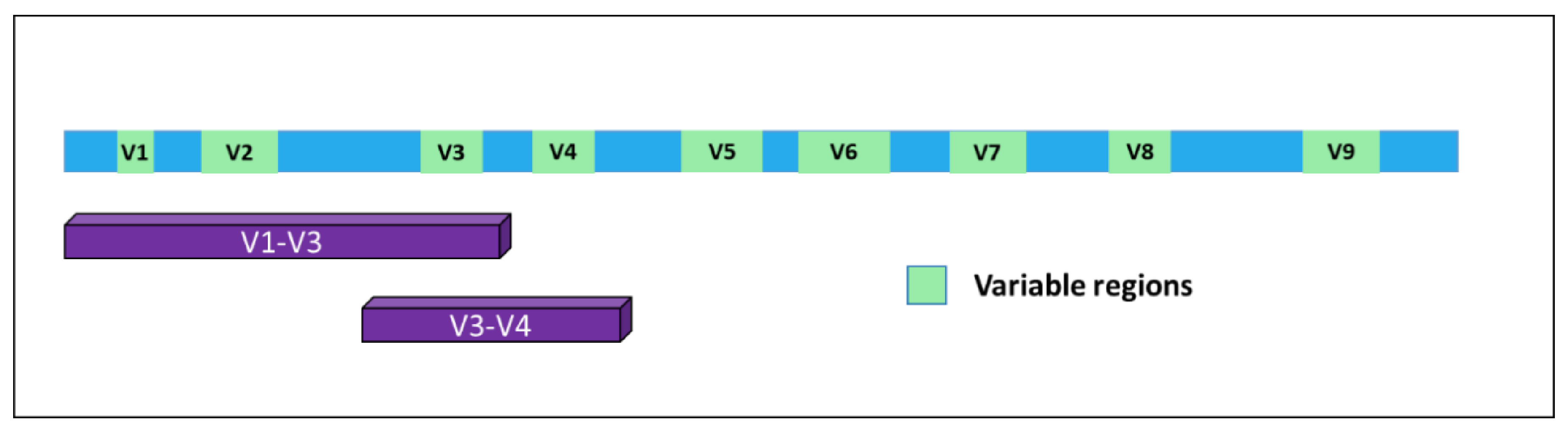
| Manuscript Ref. | Context of the Study | Extracellular Vesicles (EV) Source | Vesicles Isolation Method | 16S Regions | Sequencing Tool |
|---|---|---|---|---|---|
| [57] | Acute colitis mouse model | Small intestinal fluids, stools, and culture media | Ultracentrifugation, 200.000× g for 2 h at 4 °C | Unspecified | Roche 454 GS FLX Titanium |
| [58] | Pregnant vs. non-pregnant | Human urine | Differential centrifugation method | V1—V3 | Roche 454 GS FLX |
| [59] | Gastric cancer vs. gastric ulcers vs. duodenal ulcers. | Human gastric juices | Differential centrifugation method | V1—V3 | Roche 454 GS FLX |
| [60] | Alzheimer disease mouse model | Mouse blood | Differential centrifugation method | V3—V4 | Illumina MySeq |
| [61] | Chronic rhinitis vs. allergic rhinitis vs. atopic asthma. | Human urine | Differential centrifugation method | V3—V4 | Illumina MySeq |
| [62] | Non-tuberculous mycobacterial lung disease (NTM) and NTM + breast cancer | Human sputum | Commercial Exosome Isolation Kit | V4 | Illumina MySeq |
| [63] | Biliary tract cancer | Human blood samples | Differential centrifugation method | V3—V4 | Illumina MySeq |
| [64] | Bipolar disorder and major depressive disorder. | Human serum | Differential centrifugation method | V3—V4 | Illumina MySeq |
| [65] | Ovarian cancer and benign ovarian tumors. | Human serum | Differential centrifugation method | V3—V4 | Illumina MySeq |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ricci, V.; Carcione, D.; Messina, S.; Colombo, G.I.; D’Alessandra, Y. Circulating 16S RNA in Biofluids: Extracellular Vesicles as Mirrors of Human Microbiome? Int. J. Mol. Sci. 2020, 21, 8959. https://doi.org/10.3390/ijms21238959
Ricci V, Carcione D, Messina S, Colombo GI, D’Alessandra Y. Circulating 16S RNA in Biofluids: Extracellular Vesicles as Mirrors of Human Microbiome? International Journal of Molecular Sciences. 2020; 21(23):8959. https://doi.org/10.3390/ijms21238959
Chicago/Turabian StyleRicci, Veronica, Davide Carcione, Simone Messina, Gualtiero I. Colombo, and Yuri D’Alessandra. 2020. "Circulating 16S RNA in Biofluids: Extracellular Vesicles as Mirrors of Human Microbiome?" International Journal of Molecular Sciences 21, no. 23: 8959. https://doi.org/10.3390/ijms21238959