Knocking down USP39 Inhibits the Growth and Metastasis of Non-Small-Cell Lung Cancer Cells through Activating the p53 Pathway

 ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
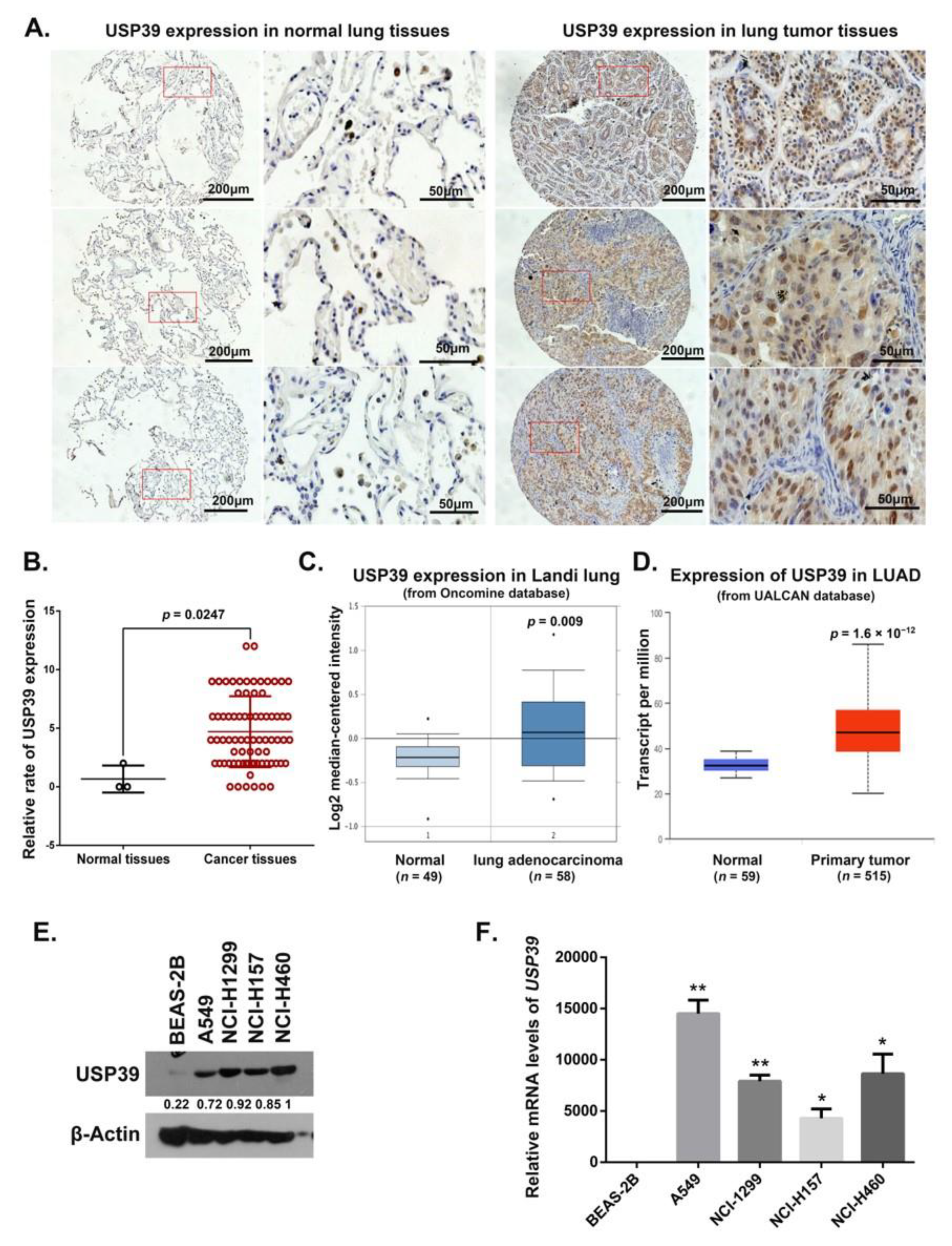
2.1. Expression of USP39 Is Significantly Increased in Human Lung Cancer Tissues and NSCLC Cell Lines
2.2. Knocking Down USP39 Inhibits A549 Cell Growth in Vivo and In Vitro
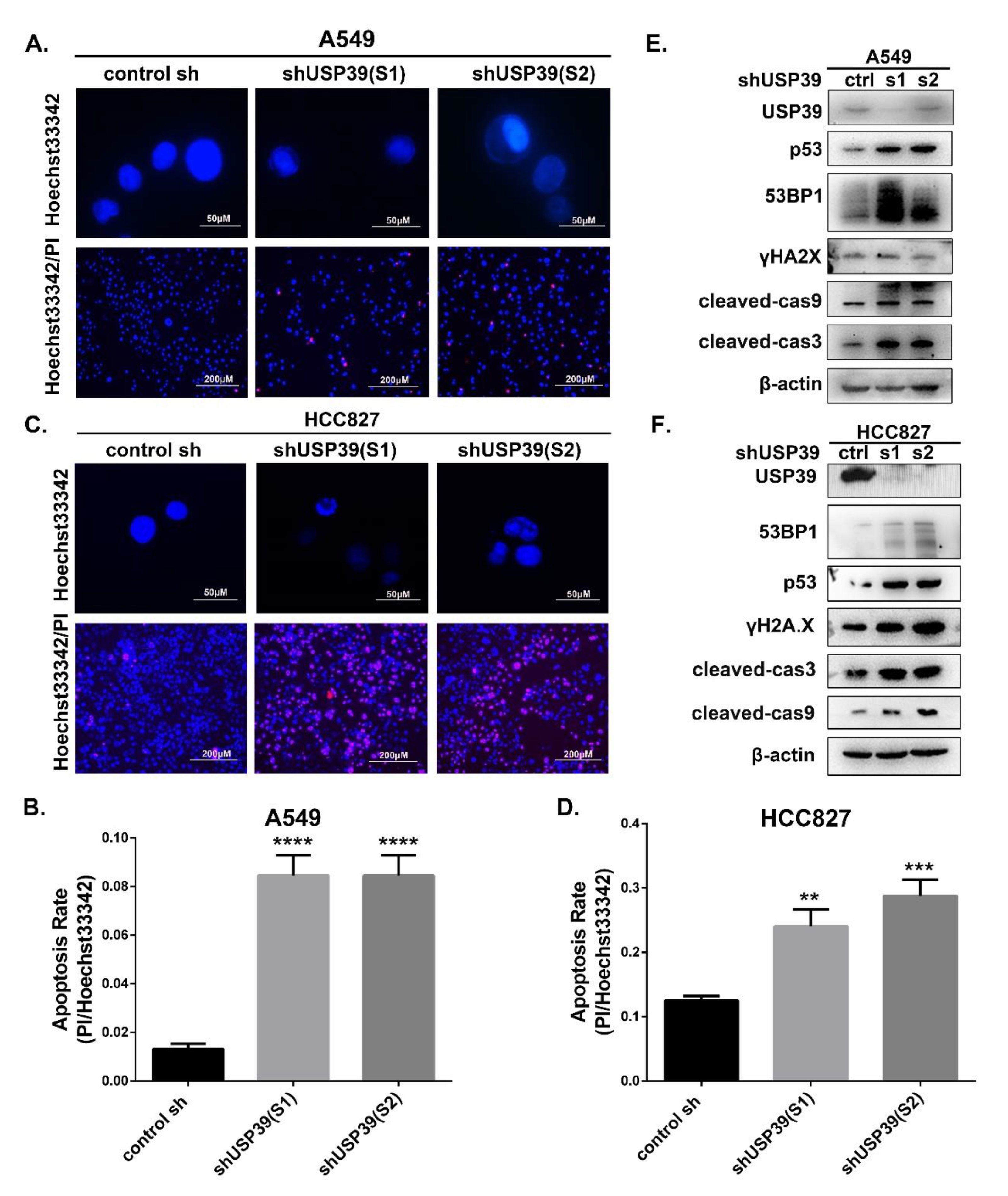
2.3. Knocking Down USP39 Inhibits the G2/M Cell Cycle Transition and Induces Apoptosis
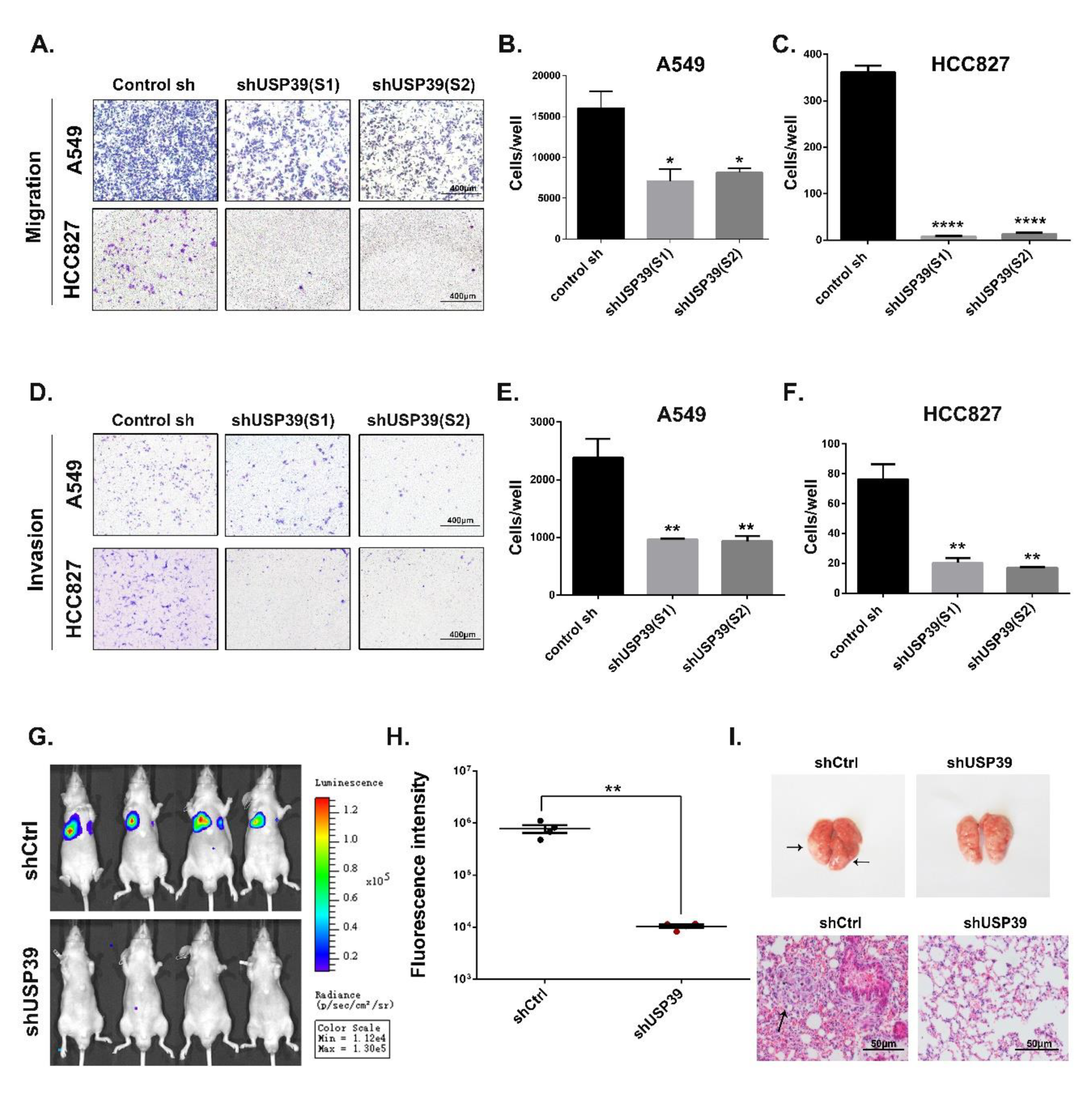
2.4. Knockdown of USP39 Inhibits Metastatic of Lung Cancer Cells In Vivo and In Vitro
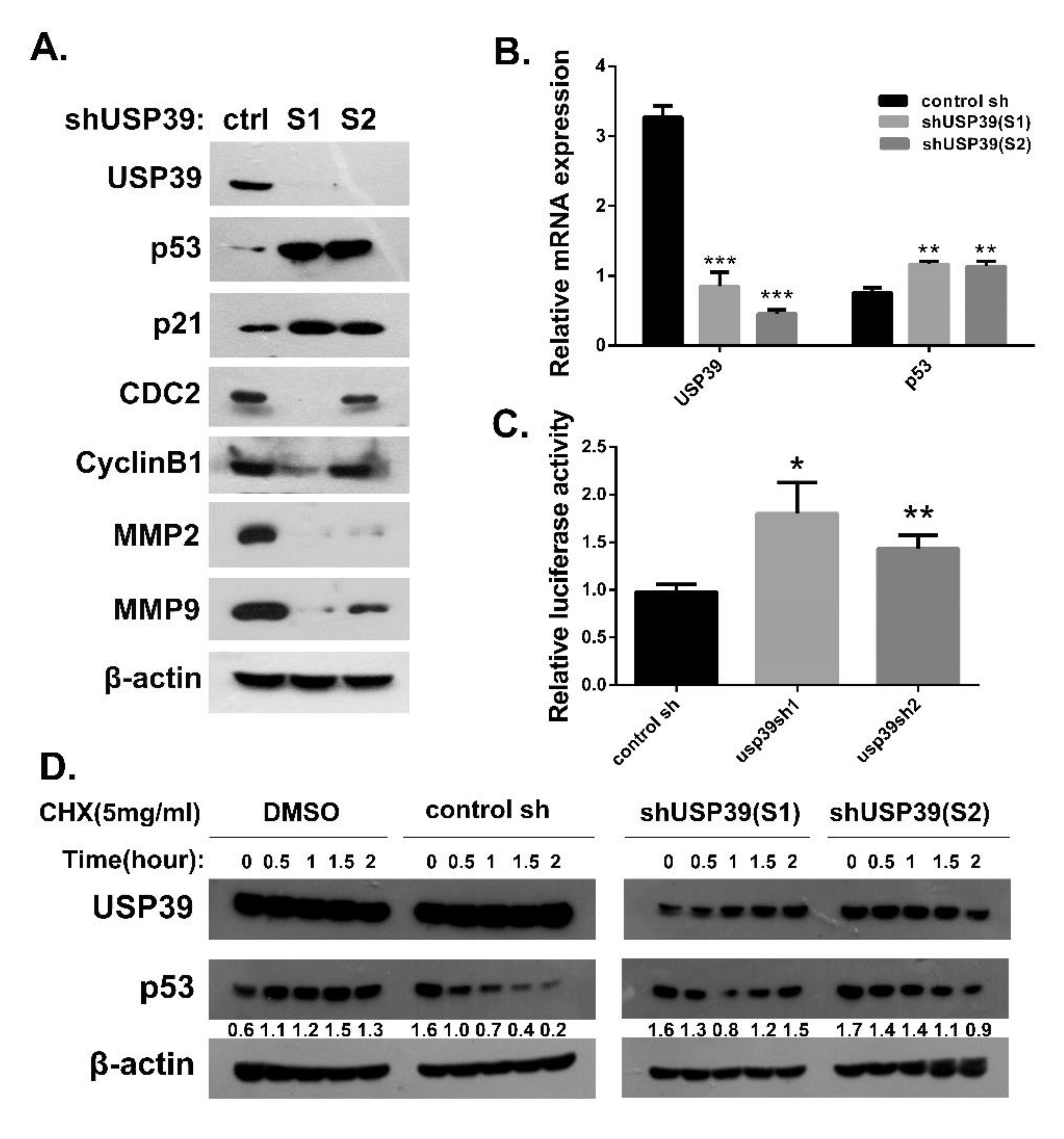
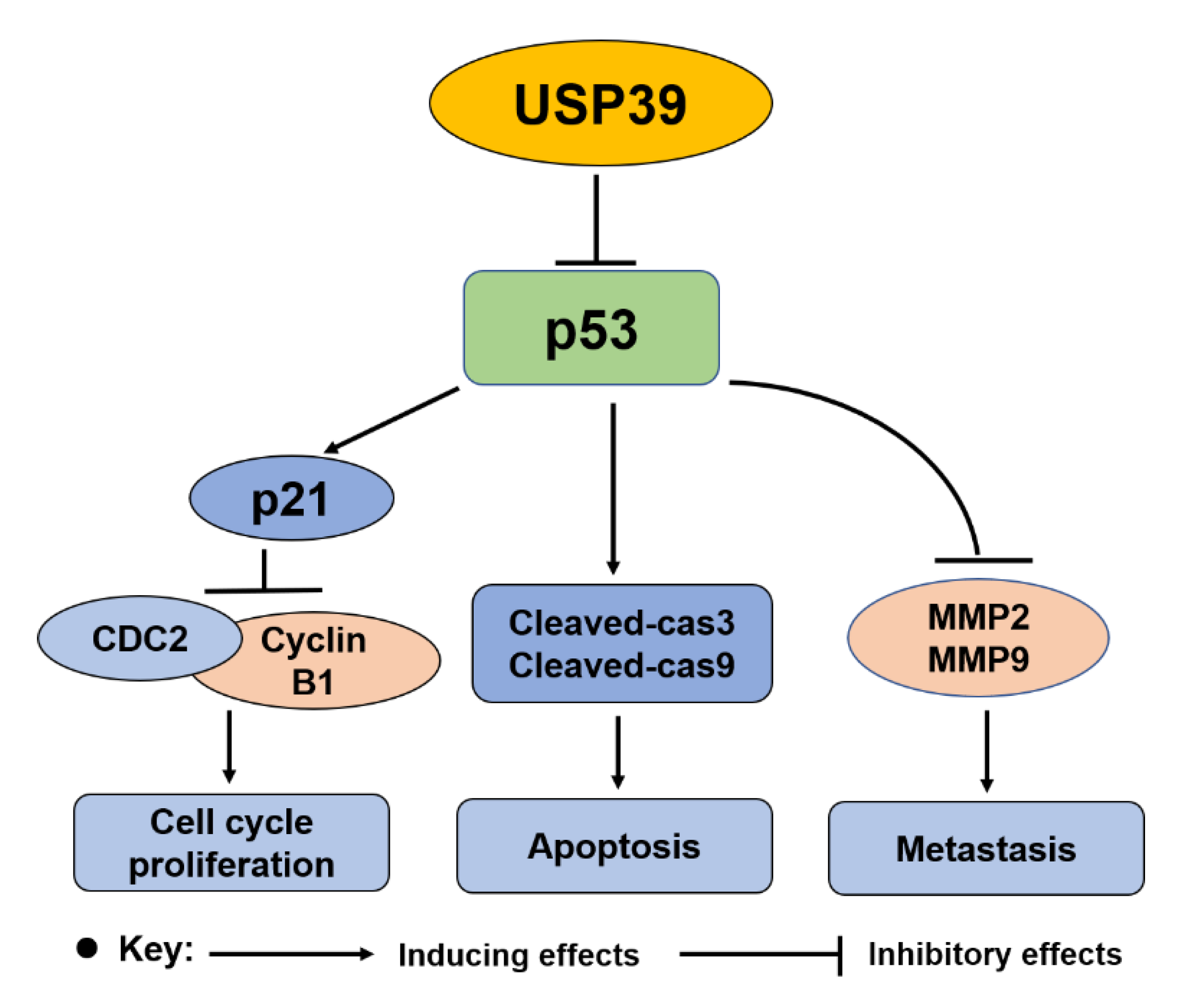
2.5. Knocking Down USP39 Promotes Activation of p53 Signaling
3. Discussion
4. Materials and Methods
4.1. Lung Cancer Microarray and Immunohistochemistry
4.2. Cell Lines and Cell Culture
4.3. Lentivirus Production and USP39 Knockdown
4.4. Protein Extraction and Western Blot
4.5. RNA Extraction and Quantitative PCR Analysis
4.6. Cell Proliferation and Colony Formation Assay
4.7. Flow Cytometric Assay
4.8. Detection of Apoptosis by Hoechst Staining and Hoechst33342/PI Double Staining
4.9. Migration and Invasion Assay
4.10. In Vivo Tumorigenesis
4.11. In Vivo Metastasis Experiment
4.12. Luciferase Reporter Assay
4.13. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2020. CA Cancer J. Clin. 2020, 70, 7–30. [Google Scholar] [CrossRef] [PubMed]
- Torre, L.A.; Siegel, R.L.; Jemal, A. Lung Cancer Statistics. In Lung Cancer and Personalized Medicine; Springer International Publishing: Berlin, Germany, 2016; pp. 1–19. [Google Scholar]
- Herbst, R.S.; Heymach, J.V.; Lippman, S.M. Lung cancer. N. Engl. J. Med. 2008, 359, 1367–1380. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heist, R.S.; Engelman, J.A. SnapShot: Non-small cell lung cancer. Cancer Cell 2012, 21, 448. [Google Scholar] [CrossRef] [PubMed]
- Spira, A.; Ettinger, D.S. Multidisciplinary Management of Lung Cancer. N. Engl. J. Med. 2004, 350, 2008–2010. [Google Scholar] [CrossRef] [PubMed]
- Rosenzweig, K.E.; Gomez, J.E. Concurrent chemotherapy and radiation therapy for inoperable locally advanced non-small-cell lung cancer. J. Clin. Oncol. 2017, 35, 6–10. [Google Scholar] [CrossRef] [PubMed]
- Nijman, S.M.; Luna-Vargas, M.P.; Velds, A.; Brummelkamp, T.R.; Dirac, A.M.; Sixma, T.K.; Bernards, R. A genomic and functional inventory of deubiquitinating enzymes. Cell 2005, 123, 773–786. [Google Scholar] [CrossRef] [Green Version]
- Sowa, M.E.; Bennett, E.J.; Gygi, S.P.; Harper, J.W. Defining the Human Deubiquitinating Enzyme Interaction Landscape. Cell 2009, 138, 389–403. [Google Scholar] [CrossRef] [Green Version]
- Fraile, J.M.; Quesada, V.; Rodríguez, D.; Freije, J.M.P.; Otín, C.L. Deubiquitinases in cancer: New functions and therapeutic options. Oncogene 2011, 31, 2373–2388. [Google Scholar] [CrossRef] [Green Version]
- Van Leuken, R.J.; Luna-Vargas, M.P.; Sixma, T.K.; Wolthuis, R.M.; Medema, R. Usp39 is essential for mitotic spindle checkpoint integrity and controls mRNA-levels of aurora B. Cell Cycle 2008, 7, 2710–2719. [Google Scholar] [CrossRef]
- Hadjivassiliou, H.; Rosenberg, O.S.; Guthrie, C. The crystal structure of S. cerevisiae Sad1, a catalytically inactive deubiquitinase that is broadly required for pre-mRNA splicing. RNA 2014, 20, 656–669. [Google Scholar] [CrossRef] [Green Version]
- Chen, Z.; Gui, B.; Zhang, Y.; Xie, G.; Li, W.; Liu, S.; Xu, B.; Wu, C.; He, L.; Yang, J.; et al. Identification of a 35S U4/U6.U5 tri-small nuclear ribonucleoprotein (tri-snRNP) complex intermediate in spliceosome assembly. J. Biol. Chem. 2017, 292, 18113–18128. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, Y.; Pan, X.W.; Li, L.; Chen, L.; Liu, X.; Lu, J.L.; Zhu, X.M.; Huang, H.; Yang, Q.W.; Ye, J.Q.; et al. Overexpression of USP39 predicts poor prognosis and promotes tumorigenesis of prostate cancer via promoting EGFR mRNA maturation and transcription elongation. Oncotarget 2016, 7, 22016–22030. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ríos, Y.; Melmed, S.; Lin, S.; Liu, N.A. Zebrafish usp39 Mutation Leads to rb1 mRNA Splicing Defect and Pituitary Lineage Expansion. PLoS Genet. 2011, 7, e1001271. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dong, X.; Su, H.; Jiang, F.; Li, H.; Shi, G.; Fan, L. miR-133a, directly targeted USP39, suppresses cell proliferation and predicts prognosis of gastric cancer. Oncol. Lett. 2018, 15, 8311–8318. [Google Scholar] [CrossRef]
- Jing, C.; Liu, T.; Peng, H.; Yan, W.; Guo, C.; Xiong, L.; Liu, A. USP39, a direct target of microRNA-133a, promotes progression of pancreatic cancer via the AKT pathway. Biochem. Biophys. Res. Commun. 2017, 486, 184–190. [Google Scholar]
- Xing, Z.; Sun, F.; He, W.; Wang, Z.; Song, X.; Song, X. Downregulation of ubiquitin-specific peptidase 39 suppresses the proliferation and induces the apoptosis of human colorectal cancer cells. Oncol. Lett. 2018, 15, 5443–5450. [Google Scholar]
- Xu, Y.; Zhu, M.R.; Zhang, J.Y.; Si, G.M.; Lv, J.J. Knockdown of ubiquitin-specific peptidase 39 inhibits the malignant progression of human renal cell carcinoma. Mol. Med. Rep. 2018, 17, 4729–4735. [Google Scholar] [CrossRef]
- Lin, Z.; Xiong, L.; Lin, Q. Ubiquitin-specific protease 39 is overexpressed in human lung cancer and promotes tumor cell proliferation in vitro. Mol. Cell Biochem. 2016, 422, 97–107. [Google Scholar] [CrossRef]
- Wu, J.; Chen, Y.; Geng, G.; Li, L.; Yin, P.; Nowsheen, S.; Li, Y.; Wu, C.; Liu, J.; Zhao, F.; et al. USP39 regulates DNA damage response and chemo-radiation resistance by deubiquitinating and stabilizing CHK2. Cancer Lett. 2019, 449, 114–124. [Google Scholar] [CrossRef]
- Fraile, J.M.; Manchado, E.; Lujambio, A.; Quesada, V.; Campos-Iglesias, D.; Webb, T.R.; Lowe, S.W.; López-Otín, C.; Freije, J.M.P. USP39 Deubiquitinase Is Essential for KRAS Oncogene-driven Cancer. J. Biol. Chem. 2017, 292, 4164–4175. [Google Scholar] [CrossRef] [Green Version]
- Allende-Vega, N.; Dayal, S.; Agarwala, U.; Sparks, A.; Bourdon, J.C.; Saville, M.K. p53 is activated in response to disruption of the pre-mRNA splicing machinery. Oncogene 2013, 32, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Yuan, X.; Sun, X.; Shi, X.; Jiang, C.; Yu, D.; Zhang, W.; Ding, Y. USP39 regulates the growth of SMMC-7721 cells via FoxM1. Exp. Ther. Med. 2017, 13, 1506–1513. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, K.Y.; Zhang, J.; Jiang, L.C.; Jiang, C.; Yu, D.; Zhang, W.; Ding, Y. Knockdown of USP39 by lentivirus-mediated RNA interference suppresses the growth of oral squamous cell carcinoma. Cancer Biomark. 2016, 16, 137–144. [Google Scholar] [CrossRef] [PubMed]
- An, Y.; Yang, S.; Guo, K.; Ma, B.; Wang, Y. Reduced USP39 expression inhibits malignant proliferation of medullary thyroid carcinoma in vitro. World J. Surg. Oncol. 2015, 13, 255. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Giaccia, A.J.; Kastan, M.B. The complexity of p53 modulation: Emerging patterns from divergent signals. Genes Dev. 1998, 12, 2973–2983. [Google Scholar] [CrossRef] [Green Version]
- Rufini, A.; Tucci, P.J.F.; Celardo, I.; Melino, G. Senescence and aging: The critical roles of p53. Oncogene 2013, 32, 5129–5143. [Google Scholar] [CrossRef]
- Reed, S.M.; Quelle, D.E. p53 Acetylation: Regulation and Consequences. Cancers 2014, 7, 30–69. [Google Scholar] [CrossRef]
- Fragkos, M.; Jurvansuu, J.; Beard, P. H2AX is required for cell cycle arrest via the p53/p21 pathway. Mol. Cell. Biol. 2009, 29, 2828–2840. [Google Scholar] [CrossRef] [Green Version]
- Lu, Z.; Miao, Y.; Muhammad, I.; Tian, E.; Hu, W.; Wang, J.; Wang, B.; Wang, B.; Li, J. Colistin-induced autophagy and apoptosis involves the JNK-Bcl2-Bax signaling pathway and JNK-p53-ROS positive feedback loop in PC-12 cells. Chem. Biol. Interact. 2017, 277, 62–73. [Google Scholar] [CrossRef]
- Yan, C.; Wang, H.; Boyd, D.D. ATF3 represses 72-kDa type IV collagenase (MMP-2) expression by antagonizing p53-dependent trans-activation of the collagenase promoter. J. Biol. Chem. 2002, 277, 10804–10812. [Google Scholar] [CrossRef] [Green Version]
- Jiang, J.; Ma, B.; Li, X.; Jin, W.; Han, C.; Wang, L.; Wang, H. MiR-1281, a p53-responsive microRNA, impairs the survival of human osteosarcoma cells upon ER stress via targeting USP39. Am. J. Cancer Res. 2018, 8, 1764–1774. [Google Scholar] [PubMed]
- Kusinska, R.U.; Kordek, R.; Pluciennik, E.; Bednarek, A.K.; Piekarski, J.H.; Potemski, P. Does vimentin help to delineate the so-called ‘basal type breast cancer’? J. Exp. Clin. Cancer Res. 2009, 28, 118. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sakoda, T.; Kasahara, N.; Hamamori, Y.; Kedes, L. A high-titer lentiviral production system mediates efficient transduction of differentiated cells including beating cardiac myocytes. J. Mol. Cell. Cardiol. 1999, 31, 2037–2047. [Google Scholar] [CrossRef] [PubMed]
- Ma, Y.; Zhao, M.; Zhong, J.; Shi, L.; Luo, Q.; Liu, J.; Wang, J.; Yuan, X.; Huang, C. Proteomic profiling of proteins associated with lymph node metastasis in colorectal cancer. J. Cell. Biochem. 2010, 110, 1512–1519. [Google Scholar] [CrossRef] [PubMed]
- Zhong, J.; Zhao, M.; Ma, Y.; Luo, Q.; Liu, J.; Wang, J.; Wang, J.; Yuan, X.; Sang, J.; Huang, C. UCHL1 acts as a colorectal cancer oncogene via activation of the β-catenin/TCF pathway through its deubiquitinating activity. Int. J. Mol. Med. 2012, 30, 430–436. [Google Scholar] [CrossRef]
- Yu, J.; Cheng, Y.Y.; Tao, Q.; Cheung, K.F.; Lam, C.N.; Geng, H.; Tian, L.; Wong, Y.P.; Tong, J.H.M.; Ying, J.; et al. Methylation of protocadherin 10, a novel tumor suppressor, is associated with poor prognosis in patients with gastric cancer. Gastroenterology 2009, 136, 640–651. [Google Scholar] [CrossRef]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yuan, J.; Zhang, G.; Li, X.; Ma, Q.; Cheng, W.; Wang, W.; Zhang, B.; Hu, T.; Song, G. Knocking down USP39 Inhibits the Growth and Metastasis of Non-Small-Cell Lung Cancer Cells through Activating the p53 Pathway. Int. J. Mol. Sci. 2020, 21, 8949. https://doi.org/10.3390/ijms21238949
Yuan J, Zhang G, Li X, Ma Q, Cheng W, Wang W, Zhang B, Hu T, Song G. Knocking down USP39 Inhibits the Growth and Metastasis of Non-Small-Cell Lung Cancer Cells through Activating the p53 Pathway. International Journal of Molecular Sciences. 2020; 21(23):8949. https://doi.org/10.3390/ijms21238949
Chicago/Turabian StyleYuan, Jiahui, Gongye Zhang, Xiaomei Li, Qiujuan Ma, Weipeng Cheng, Weiwei Wang, Bing Zhang, Tianhui Hu, and Gang Song. 2020. "Knocking down USP39 Inhibits the Growth and Metastasis of Non-Small-Cell Lung Cancer Cells through Activating the p53 Pathway" International Journal of Molecular Sciences 21, no. 23: 8949. https://doi.org/10.3390/ijms21238949