Identification and Characterization of the Intra-Articular Microbiome in the Osteoarthritic Knee

 , , ,
, , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
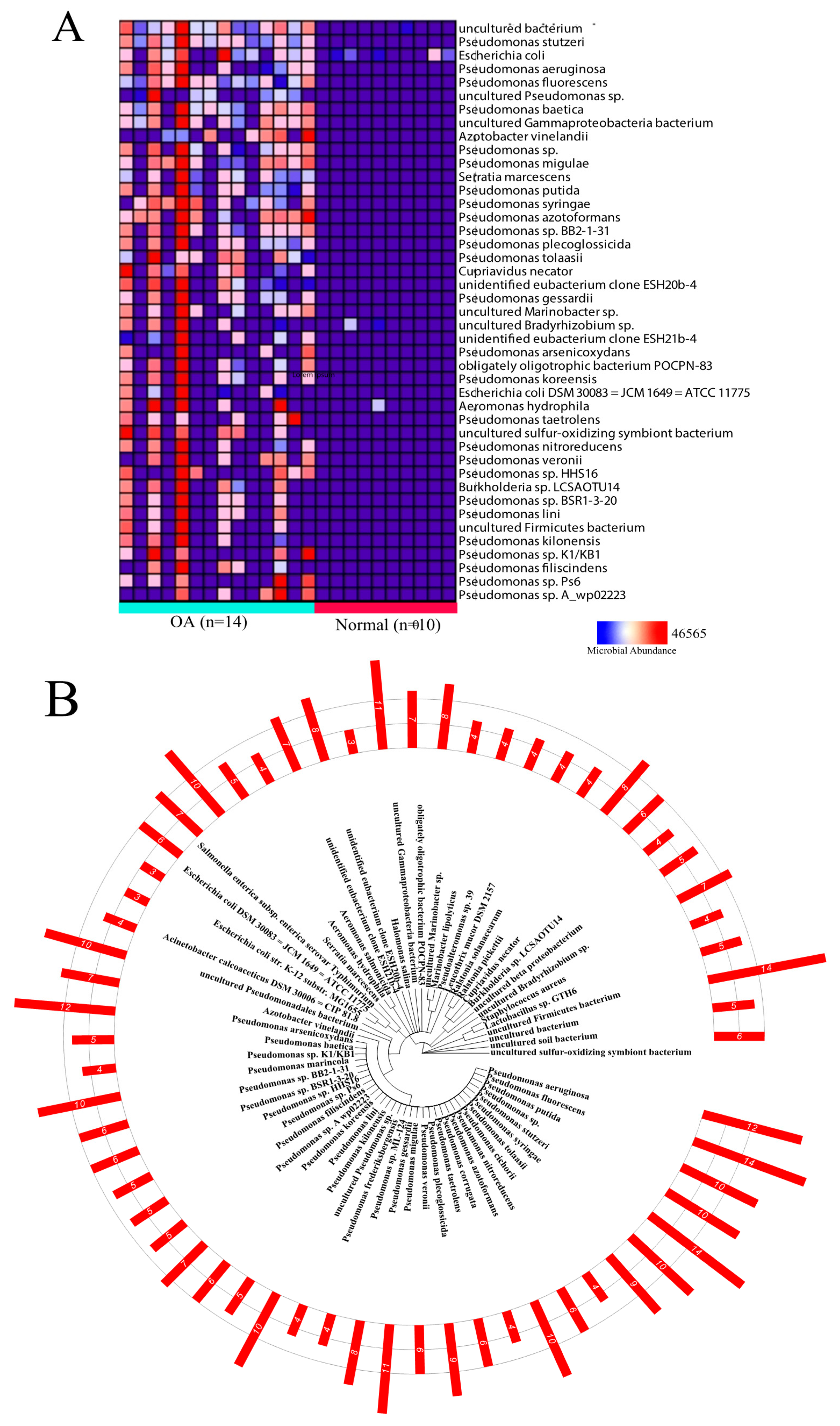
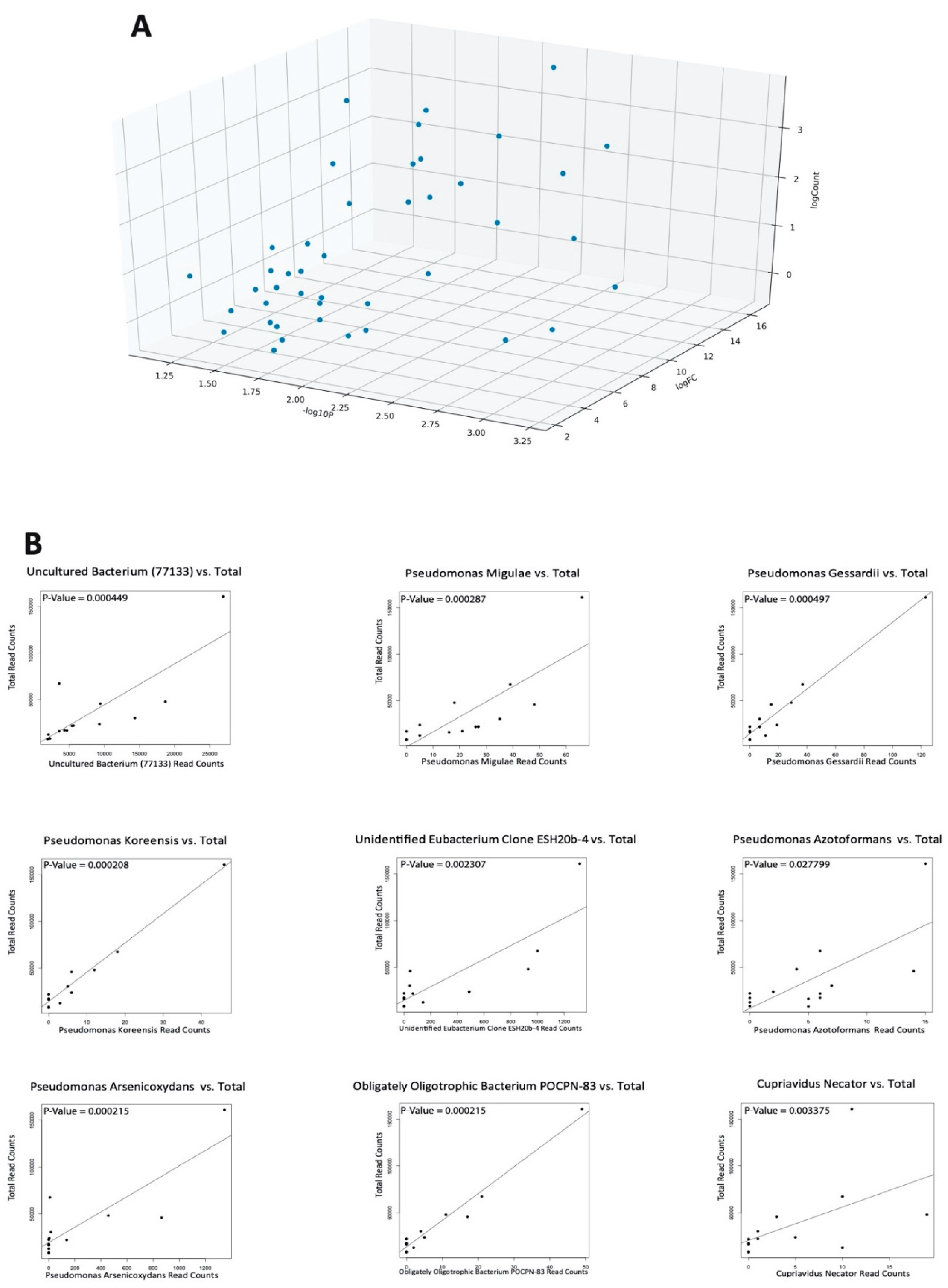
2.1. Differential Microbial Abundance in OA and Normal Tissue
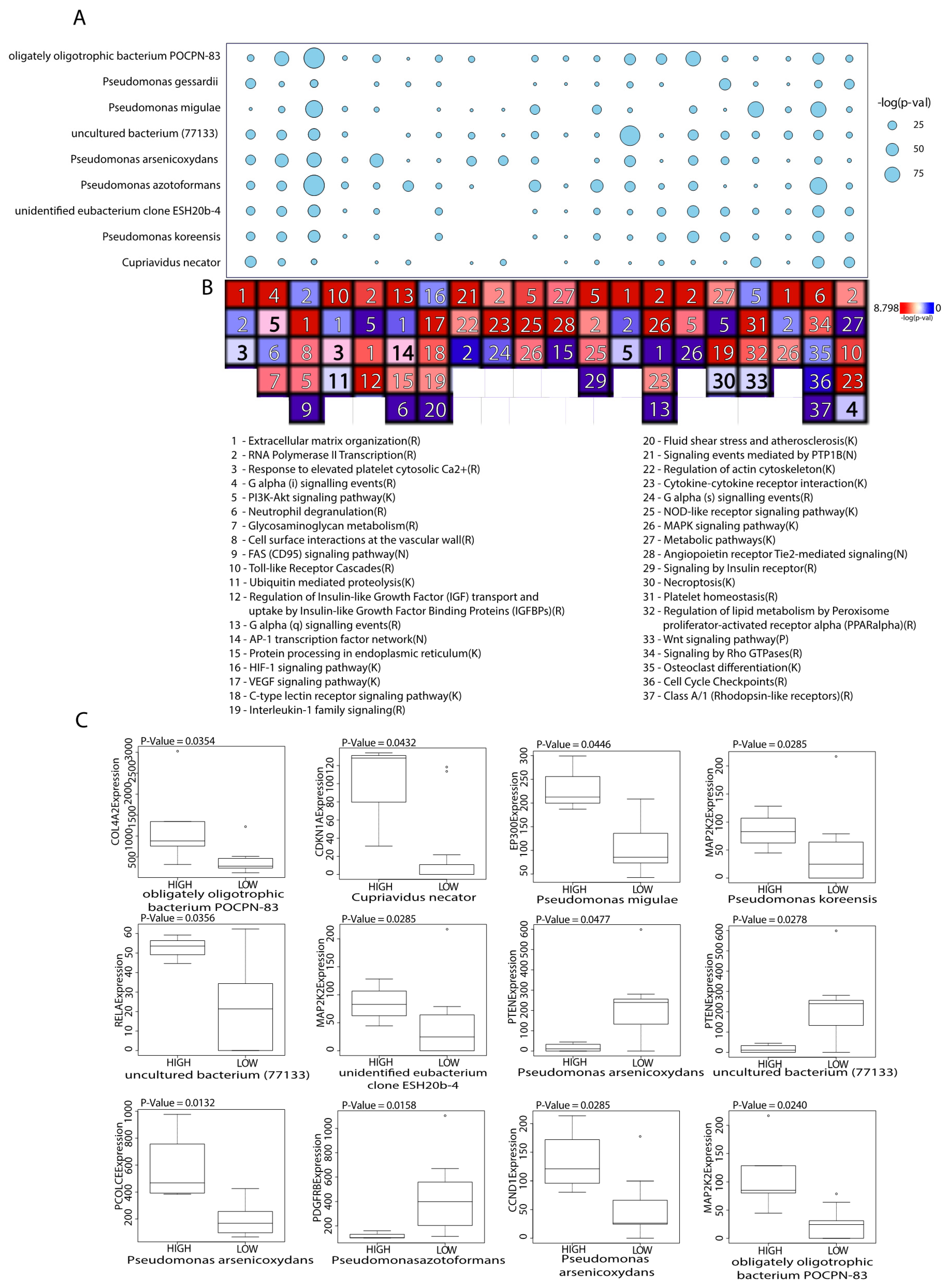
2.2. Microbes Are Significantly Correlated with Catabolic and Anabolic Pathways
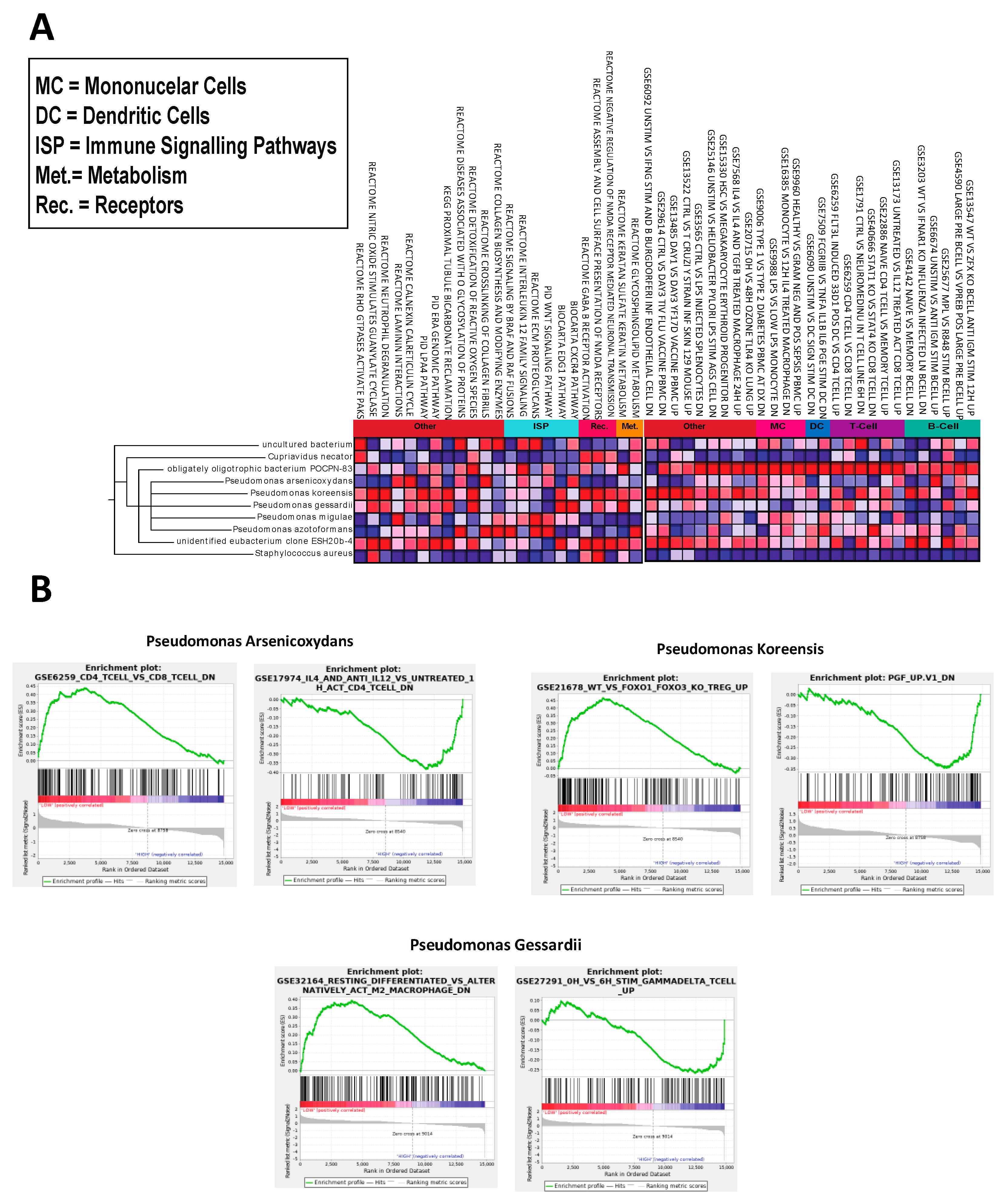
2.3. Microbes Are Significantly Correlated with OA Immunologic Gene Signatures
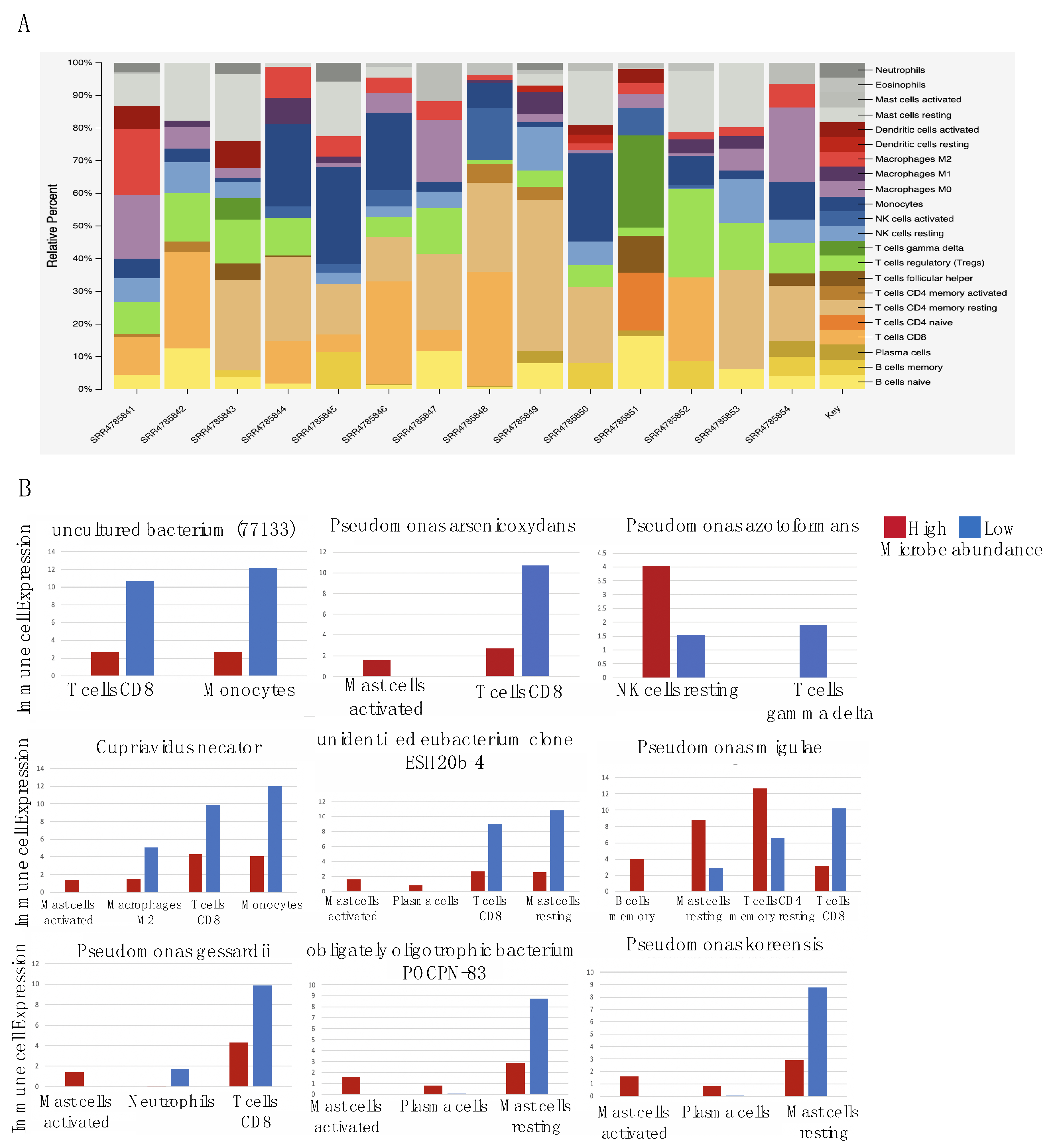
2.4. Microbes Are Significantly Correlated with Immune Cell Types
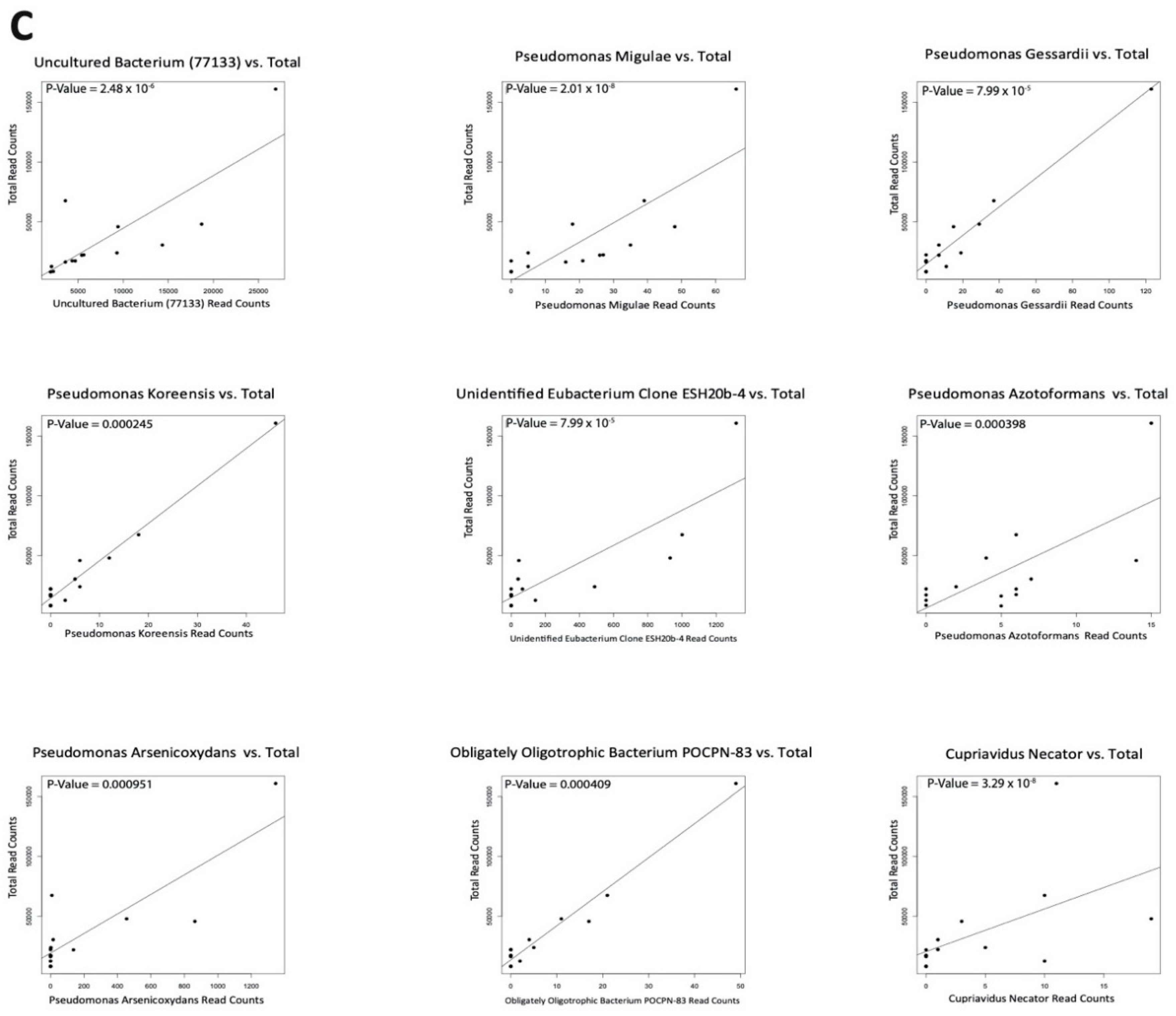
2.5. Negligible Contaminants Found in Differentially Abundant Microbes
3. Discussion
4. Materials and Methods
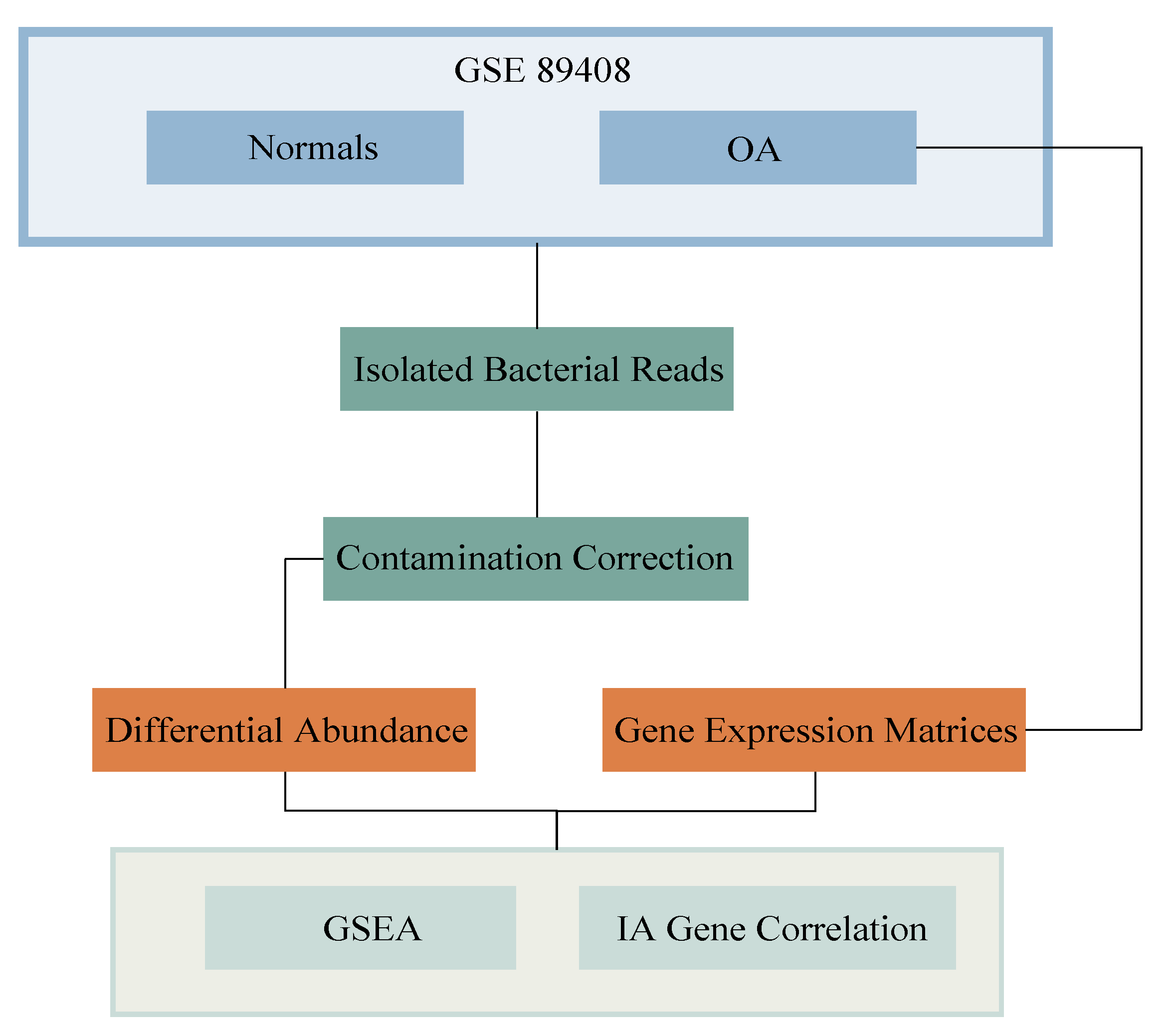
4.1. Acquisition of Data and Extraction of Isolated Bacterial Reads
4.2. Differential Abundance
4.3. Acquisition of RNA Read Counts
4.4. Gene Set Enrichment Analysis (GSEA)
4.5. Gene Co-Expression Analysis
4.6. CIBERSORTx
4.7. Contamination Correction Using Read Depth
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| OA | Osteoarthritis |
| CPM | Counts per million |
| GSEA | Gene Set Enrichment Analysis |
| KEGG | Kyoto Encyclopedia of Genes and Genotype |
| WGCNA | Weighted Gene Correlation Network Analysis |
| ECM | Extracellular Matrix |
| IA | Immune-Associated |
References
- Dequeker, J.; Luyte, F.P. The history of osteoarthritis-osteoarthrosis. Ann. Rheum. Dis. 2008, 67, 5–10. [Google Scholar] [CrossRef] [PubMed]
- Lawrence, R.C.; Felson, D.T.; Helmick, C.G.; Arnold, L.M.; Choi, H.; Deyo, R.A.; Gabriel, S.; Hirsch, R.; Hochberg, M.C.; Hunder, G.G.; et al. Estimates of the prevalence of arthritis and other rheumatic conditions in the United States. Part II. Arthritis Rheumatol. 2008, 58, 26–35. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Neogi, T. The epidemiology and impact of pain in osteoarthritis. Osteoarthr. Cartil. 2013, 21, 1145–1153. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mora, J.C.; Przkora, R.; Cruz-Almeida, Y. Knee osteoarthritis: Pathophysiology and current treatment modalities. J. Pain Res. 2008, 11, 2189–2196. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berthelot, J.M.; Sellam, J.; Maugars, Y.; Berenbaum, F. Cartilage-gut microbiome axis: A new paradigm for novel therapeutic opportunities in osteoarthritis. RMD Open 2009, 5, e001037. [Google Scholar] [CrossRef]
- Hunter, D.J.; Beavers, D.P.; Eckstein, F.; Guermazi, A.; Loeser, R.F.; Nicklas, B.J.; Mihalko, S.L.; Miller, G.D.; Lyles, M.; DeVita, P.; et al. The Intensive Diet and Exercise for Arthritis (IDEA) trial: 18-Month radiographic and MRI outcomes. Osteoarthr. Cartil. 2015, 23, 1090–1098. [Google Scholar] [CrossRef] [Green Version]
- Yu, S.P.; Hunter, D.J. Emerging drugs for the treatment of knee osteoarthritis. Expert Opin. Emerg. Drugs 2015, 20, 361–378. [Google Scholar] [CrossRef]
- Crimmins, E.M. Lifespan and Healthspan: Past, Present, and Promise. Gerontologist 2015, 55, 901–911. [Google Scholar] [CrossRef] [Green Version]
- Friedrich, M. Global Obesity Epidemic Worsening. JAMA 2017, 318, 603. [Google Scholar] [CrossRef]
- Orlowsky, E.W.; Kraus, V.B. The role of innate immunity in osteoarthritis: When our first line of defense goes on the offensive. J. Rheumatol. 2005, 42, 363–371. [Google Scholar] [CrossRef] [Green Version]
- Shreiner, A.B.; Kao, J.Y.; Young, V.B. The gut microbiome in health and in disease. Curr. Opin. Gastroenterol. 2015, 31, 69–75. [Google Scholar] [CrossRef]
- Trompette, A.; Gollwitzer, E.S.; Yadava, K.; Sichelstiel, A.K.; Sprenger, N.; Ngom-Bru, C.; Blanchard, C.; Junt, T.; Nicod, L.P.; Harris, N.L.; et al. Gut microbiota metabolism of dietary fiber influences allergic airway disease and hematopoiesis. Nat. Med. 2014, 20, 159–166. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Zhang, D.; Jia, H.; Feng, Q.; Wang, D.; Liang, D.; Wu, X.; Li, J.; Tang, L.; Li, Y.; et al. The oral and gut microbiomes are perturbed in rheumatoid arthritis and partly normalized after treatment. Nat. Med. 2015, 21, 895–905. [Google Scholar] [CrossRef] [PubMed]
- Li, J.Y.; Chassaing, B.; Tyagi, A.M.; Vaccaro, C.; Luo, T.; Adams, J.; Darby, T.M.; Weitzmann, M.N.; Mulle, J.G.; Gewirtz, A.T.; et al. Sex steroid deficiency-associated bone loss is microbiota dependent and prevented by probiotics. J. Clin. Investig. 2016, 126, 2049–2063. [Google Scholar] [CrossRef] [Green Version]
- Claudio, F.; Judith, C. Chronic Inflammation (Inflammaging) and Its Potential Contribution to Age-Associated Diseases. J. Gerontol. Ser. A Biomed. Sci. Med Sci. 2014, 69, S4–S9. [Google Scholar] [CrossRef]
- Thevaranjan, N.; Puchta, A.; Schulz, C.; Naidoo, A.; Szamosi, J.C.; Verschoor, C.P.; Loukov, D.; Schenck, L.P.; Jury, J.; Foley, K.P.; et al. Age-Associated Microbial Dysbiosis Promotes Intestinal Permeability, Systemic Inflammation, and Macrophage Dysfunction. Cell Host Microbe 2017, 21, 455–466.e4. [Google Scholar] [CrossRef]
- Qi, Y.; Goel, R.; Kim, S.; Richards, E.M.; Carter, C.S.; Pepine, C.J.; Raizada, M.K.; Buford, T.W. Intestinal Permeability Biomarker Zonulin is Elevated in Healthy Aging. J. Am. Med. Dir. Assoc. 2017, 18, 810.e1–810.e4. [Google Scholar] [CrossRef] [PubMed]
- Biagi, E.; Candela, M.; Franceschi, C.; Brigidi, P. The aging gut microbiota: New perspectives. Ageing Res. Rev. 2011, 10, 428–429. [Google Scholar] [CrossRef]
- Chase, J.; Fouquier, J.; Zare, M.; Sonderegger, D.L.; Knight, R.; Kelley, S.T.; Siegel, J.; Caporaso, J.G. Geography and location are the primary drivers of office microbiome composition. mSystems 2016, 1, e00022-16. [Google Scholar] [CrossRef] [Green Version]
- Collins, K.H.; Paul, H.A.; Reimer, R.A.; Seerattan, R.A.; Hart, D.A.; Herzog, W. Relationship between inflammation, the gut microbiota, and metabolic osteoarthritis development: Studies in a rat model. Osteoarthr. Cartil. 2015, 23, 1989–1998. [Google Scholar] [CrossRef] [Green Version]
- Ulici, V.; Kelley, K.L.; Azcarate-Peril, M.A.; Cleveland, R.J.; Sartor, R.B.; Schwartz, T.A.; Loeser, R.F. Osteoarthritis induced by destabilization of the medial meniscus is reduced in germ-free mice. Osteoarthr. Cartil. 2018, 26, 1098–1109. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hammad, D.; Liyanapathirana, V.; Tonge, D. Molecular characterisation of the synovial fluid microbiome in rheumatoid arthritis patients and healthy control subjects. PLoS ONE 2019, 14, e0225110. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, Y.; Chen, B.; Li, S.; Yang, L.; Zhu, D.; Wang, Y.; Wang, H.; Wang, T.; Shi, B.; Gai, Z.; et al. Detection and characterization of bacterial nucleic acids in culture-negative synovial tissue and fluid samples from rheumatoid arthritis or osteoarthritis patients. Sci. Rep. 2018, 8, 14305. [Google Scholar] [CrossRef] [PubMed]
- Hong, C.; Manimaran, S.; Shen, Y.; Perez-Rogers, J.F.; Byrd, A.L.; Castro-Nallar, E.; Crandall, K.A.; Johnson, W.E. PathoScope 2.0: A complete computational framework for strain identification in environmental or clinical sequencing samples. Microbiome 2014, 2, 33. [Google Scholar] [CrossRef] [PubMed]
- Sofia, V.; Nasrul, E.; Manjas, M.; Revilla, G. Analysis of the Relationship between RELA Gene Expression and MMP-13 Gene Expression in Synoviocyte Cells after Mesenchymal Stem Cell Wharton Jelly. Open Access Maced. J. Med. Sci. 2019, 7, 543–548. [Google Scholar] [CrossRef] [Green Version]
- Pietrocola, F.; Lachkar, S.; Enot, D.P.; Niso-Santano, M.; Bravo-San Pedro, J.M.; Sica, V.; Izzo, V.; Maiuri, M.C.; Madeo, F.; Mariño, G.; et al. Spermidine induces autophagy by inhibiting the acetyltransferase EP300. Cell Death Differ. 2015, 22, 509–516. [Google Scholar] [CrossRef] [PubMed]
- Caramés, B.; Olmer, M.; Kiosses, W.B.; Lotz, M.K. The relationship of autophagy defects to cartilage damage during joint aging in a mouse model. Arthritis Rheumatol. 2015, 67, 1568–1576. [Google Scholar] [CrossRef]
- Jeon, O.H.; Kim, C.; Laberge, R.M.; Demaria, M.; Rathod, S.; Vasserot, A.P.; Chung, J.W.; Kim, D.H.; Poon, Y.; David, N.; et al. Local clearance of senescent cells attenuates the development of post-traumatic osteoarthritis and creates a pro-regenerative environment. Nat. Med. 2017, 23, 775–781. [Google Scholar] [CrossRef]
- Aki, T.; Hashimoto, K.; Ogasawara, M.; Itoi, E. A whole-genome transcriptome analysis of articular chondrocytes in secondary osteoarthritis of the hip. PLoS ONE 2018, 13, e0199734. [Google Scholar] [CrossRef]
- Cao, L.; Wang, Y.; Wang, Q.; Huang, J. LncRNA FOXD2-AS1 regulates chondrocyte proliferation in osteoarthritis by acting as a sponge of miR-206 to modulate CCND1 expression. Biomed. Pharmacother. 2018, 106, 1220–1226. [Google Scholar] [CrossRef]
- Xie, J.; Lin, J.; Wei, M.; Teng, Y.; He, Q.; Yang, G.; Yang, X. Sustained Akt signaling in articular chondrocytes causes osteoarthritis via oxidative stress-induced senescence in mice. Bone Res. 2019, 7, 23. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matsumura, T.; Saito, Y.; Suzuki, T.; Teramoto, A.; Ozasa, Y.; Yamashita, T.; Fujimiya, M.; Saito-Chikenji, T. Phosphorylated Platelet-Derived Growth Factor Receptor-Positive Cells with Anti-Apoptotic Properties Accumulate in the Synovium of Patients with Rheumatoid Arthritis. Front. Immunol. 2019, 10, 241. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, S.; Liu, H.; Wu, Y.; Heng, B.C.; Chen, P.; Liu, H.; Ouyang, H.W. Wnt and Rho GTPase signaling in osteoarthritis development and intervention: Implications for diagnosis and therapy. Arthritis Res. Ther. 2013, 15, 217. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lacy, P. Mechanisms of degranulation in neutrophils. Allergy Asthma Clin. Immunol. 2006, 2, 98–108. [Google Scholar] [CrossRef] [Green Version]
- Eisenhofer, R.; Minich, J.J.; Marotz, C.; Cooper, A.; Knight, R.; Weyrich, L.S. Contamination in Low Microbial Biomass Microbiome Studies: Issues and Recommendations. Trends Microbiol. 2018, 27, 105–117. [Google Scholar] [CrossRef]
- Leggett, R.M.; Ramirez-Gonzalez, R.H.; Clavijo, B.J.; Waite, D.; Davey, R.P. Sequencing quality assessment tools to enable data-driven informatics for high throughput genomics. Front. Genet. 2013, 4, 288. [Google Scholar] [CrossRef]
- Guo, Y.; Walsh, A.M.; Fearon, U.; Smith, M.D.; Wechalekar, M.D.; Yin, X.; Cole, S.; Orr, C.; McGarry, T.; Canavan, M.; et al. CD40L-Dependent Pathway Is Active at Various Stages of Rheumatoid Arthritis Disease Progression. J. Immunol. 2017, 198, 4490–4501. [Google Scholar] [CrossRef] [Green Version]
- Schott, E.M.; Farnsworth, C.W.; Grier, A.; Lillis, J.A.; Soniwala, S.; Dadourian, G.H.; Bell, R.D.; Doolittle, M.L.; Villani, D.A.; Awad, H.; et al. Targeting the gut microbiome to treat the osteoarthritis of obesity. JCI Insight 2018, 3, e95997. [Google Scholar] [CrossRef]
- Boer, C.G.; Radjabzadeh, D.; Medina-Gomez, C.; Garmaeva, S.; Schiphof, D.; Arp, P.; Koet, T.; Kurilshikov, A.; Fu, J.; Ikram, M.A.; et al. Intestinal microbiome composition and its relation to joint pain and inflammation. Nat. Commun. 2019, 10, 4881. [Google Scholar] [CrossRef] [Green Version]
- Favazzo, L.J.; Hendesi, H.; Villani, D.A.; Soniwala, S.; Dar, Q.A.; Schott, E.M.; Gill, S.R.; Zuscik, M.J. The gut microbiome-joint connection: Implications in osteoarthritis. Curr. Opin. Rheumatol. 2020, 32, 92–101. [Google Scholar] [CrossRef]
- De Santis, S.; Cavalcanti, E.; Mastronardi, M.; Jirillo, E.; Chieppa, M. Nutritional Keys for Intestinal Barrier Modulation. Front. Immunol. 2015, 6, 612. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Wang, P.; Su, Q.; Wang, S.; Wang, F. Myosin light chain kinase mediates intestinal barrier disruption following burn injury. PLoS ONE 2012, 7, e34946. [Google Scholar] [CrossRef] [PubMed]
- Earley, Z.M.; Akhtar, S.; Green, S.J.; Naqib, A.; Khan, O.; Cannon, A.R.; Hammer, A.M.; Morris, N.L.; Li, X.; Eberhardt, J.M.; et al. Burn Injury Alters the Intestinal Microbiome and Increases Gut Permeability and Bacterial Translocation. PLoS ONE 2015, 10, e0129996. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, P.; Stärkel, P.; Turner, J.R.; Ho, S.B.; Schnabl, B. Dysbiosis-induced intestinal inflammation activates tumor necrosis factor receptor I and mediates alcoholic liver disease in mice. Hepatology 2015, 61, 883–894. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sinusas, K. Osteoarthritis: Diagnosis and treatment. Am. Fam. Physician 2012, 85, 49–56. [Google Scholar]
- Yu, S.P.; Hunter, D.J. Managing osteoarthritis. Aust. Prescr. 2015, 38, 115–119. [Google Scholar] [CrossRef] [Green Version]
- Rogers, M.; Aronoff, D. The influence of non-steroidal anti-inflammatory drugs on the gut microbiome. Clin. Microbiol. Infect. 2016, 22, 178.e1–178.e9. [Google Scholar] [CrossRef] [Green Version]
- Wang, Q.; Lepus, C.M.; Raghu, H.; Reber, L.L.; Tsai, M.M.; Wong, H.H.; von Kaeppler, E.; Lingampalli, N.; Bloom, M.S.; Hu, N.; et al. IgE-mediated mast cell activation promotes inflammation and cartilage destruction in osteoarthritis. Elife 2019, 8, e39905. [Google Scholar] [CrossRef]
- Hsieh, J.L.; Shiau, A.L.; Lee, C.H.; Yang, S.J.; Lee, B.O.; Jou, I.; Wu, C.L.; Chen, S.H.; Shen, P.C. CD8+ T Cell-Induced Expression of Tissue Inhibitor of Metalloproteinses-1 Exacerbated Osteoarthritis. Int. J. Mol. Sci. 2013, 14, 19951–19970. [Google Scholar] [CrossRef] [Green Version]
- Saito, I.; Koshino, T.; Nakashima, K.; Uesugi, M.; Saito, T. Increased cellular infiltrate in inflammatory synovia of osteoarthritic knees. Osteoarthr. Cartil. 2002, 10, 156–162. [Google Scholar] [CrossRef] [Green Version]
- Kempsell, K.E.; Cox, C.J.; Hurle, M.; Wong, A.; Wilkie, S.; Zanders, E.D.; Gaston, J.H.; Crowe, J.S. Reverse transcriptase-PCR analysis of bacterial rRNA for detection and characterization of bacterial species in arthritis synovial tissue. Infect. Immun. 2000, 68, 6012–6026. [Google Scholar] [CrossRef] [Green Version]
- Olsen-Bergem, H.; Kristoffersen, A.K.; Bjørnland, T.; Reseland, J.E.; Aas, J.A. Juvenile idiopathic arthritis and rheumatoid arthritis: Bacterial diversity in temporomandibular joint synovial fluid in comparison with immunological and clinical findings. Int. J. Oral Maxillofac. Surg. 2016, 45, 318–322. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Siala, M.; Gdoura, R.; Fourati, H.; Rihl, M.; Jaulhac, B.; Younes, M.; Sibilia, J.; Baklouti, S.; Bargaoui, N.; Sellami, S.; et al. Broad-range PCR, cloning and sequencing of the full 16S rRNA gene for detection of bacterial DNA in synovial fluid samples of Tunisian patients with reactive and undifferentiated arthritis. Arthritis Res. Ther. 2009, 11, R102. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- He, Y.; Wu, W.; Zheng, H.M.; Li, P.; McDonald, D.; Sheng, H.F.; Chen, M.X.; Chen, Z.H.; Ji, G.Y.; Mujagond, P.; et al. Regional variation limits applications of healthy gut microbiome reference ranges and disease models. Nat. Med. 2018, 24, 1532–1535. [Google Scholar] [CrossRef]
- Ozkan, J.; Coroneo, M.; Willcox, M.; Wemheuer, B.; Thomas, T. Identification and Visualization of a Distinct Microbiome in Ocular Surface Conjunctival Tissue. Investig. Ophthalmol. Vis. Sci. 2018, 59, 4268–4276. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smith, D.J.; Badrick, A.C.; Zakrzewski, M.; Krause, L.; Bell, S.C.; Anderson, G.J.; Reid, D.W. Pyrosequencing reveals transient cystic fibrosis lung microbiome changes with intravenous antibiotics. Eur. Respir. J. 2014, 44, 922–930. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dinsdale, N.; Castano-Rodriquez, N.; Quinllivan, J.; Mendz, G.L. The genital microbiome of pregnant Australian women. In Proceedings of the 28th ECCMID European Congress of Clinical Microbiology and Infectious Diseases, Madrid, Spain, 21–24 April 2018; p. P0776. [Google Scholar]
- Dunn, C.M.; Velasco, C.; Rivas, A.; Andrews, M.; Garman, C.; Jacob, P.B.; Jeffries, M.A. Identification of Cartilage Microbial DNA Signatures and Associations with Knee and Hip Osteoarthritis. Arthritis Rheumatol. 2020, 72, 1111–1122. [Google Scholar] [CrossRef]
- CDC. Osteoarthritis (OA). Available online: https://www.cdc.gov/arthritis/basics/osteoarthritis.htm (accessed on 30 October 2020).
- Anders, S.; Pyl, P.T.; Huber, W. HTSeq—A Python framework to work with highthroughput sequencing data. Bioinformatics 2015, 31, 166–169. [Google Scholar] [CrossRef]
- Dobin, A.; Davis, C.A.; Schlesinger, F.; Drenkow, J.; Zaleski, C.; Jha, S.; Batut, P.; Chaisson, M.; Gingeras, T.R. STAR: Ultrafast universal RNA-seq aligner. Bioinformatics 2013, 29, 15–21. [Google Scholar] [CrossRef] [PubMed]
- Subramanian, A.; Tamayo, P.; Mootha, V.K.; Mukherjee, S.; Ebert, B.L.; Gillette, M.A.; Paulovich, A.; Pomeroy, S.L.; Golub, T.R.; Lander, E.S.; et al. Gene Set Enrichment Analysis: A Knowledge-Based Approach for Interpreting Genome-Wide Expression Profiles. Proc. Natl. Acad. Sci. USA 2005, 102, 15545–15550. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Langfelder, P.; Horvath, S. WGCNA: An R package for weighted correlation network analysis. BMC Bioinform. 2008, 9, 559. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, B.; Khodadoust, M.S.; Liu, C.L.; Newman, A.M.; Alizadeh, A.A. Profiling tumor infiltrating immune cells with CIBERSORT. Methods Mol. Biol. 2018, 1711, 243–259. [Google Scholar] [CrossRef] [PubMed]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tsai, J.C.; Casteneda, G.; Lee, A.; Dereschuk, K.; Li, W.T.; Chakladar, J.; Lombardi, A.F.; Ongkeko, W.M.; Chang, E.Y. Identification and Characterization of the Intra-Articular Microbiome in the Osteoarthritic Knee. Int. J. Mol. Sci. 2020, 21, 8618. https://doi.org/10.3390/ijms21228618
Tsai JC, Casteneda G, Lee A, Dereschuk K, Li WT, Chakladar J, Lombardi AF, Ongkeko WM, Chang EY. Identification and Characterization of the Intra-Articular Microbiome in the Osteoarthritic Knee. International Journal of Molecular Sciences. 2020; 21(22):8618. https://doi.org/10.3390/ijms21228618
Chicago/Turabian StyleTsai, Joseph C., Grant Casteneda, Abby Lee, Kypros Dereschuk, Wei Tse Li, Jaideep Chakladar, Alecio F. Lombardi, Weg M. Ongkeko, and Eric Y. Chang. 2020. "Identification and Characterization of the Intra-Articular Microbiome in the Osteoarthritic Knee" International Journal of Molecular Sciences 21, no. 22: 8618. https://doi.org/10.3390/ijms21228618