Succinic Semialdehyde Dehydrogenase Deficiency: In Vitro and In Silico Characterization of a Novel Pathogenic Missense Variant and Analysis of the Mutational Spectrum of ALDH5A1

 , , , , , and
, , , , , and
Abstract
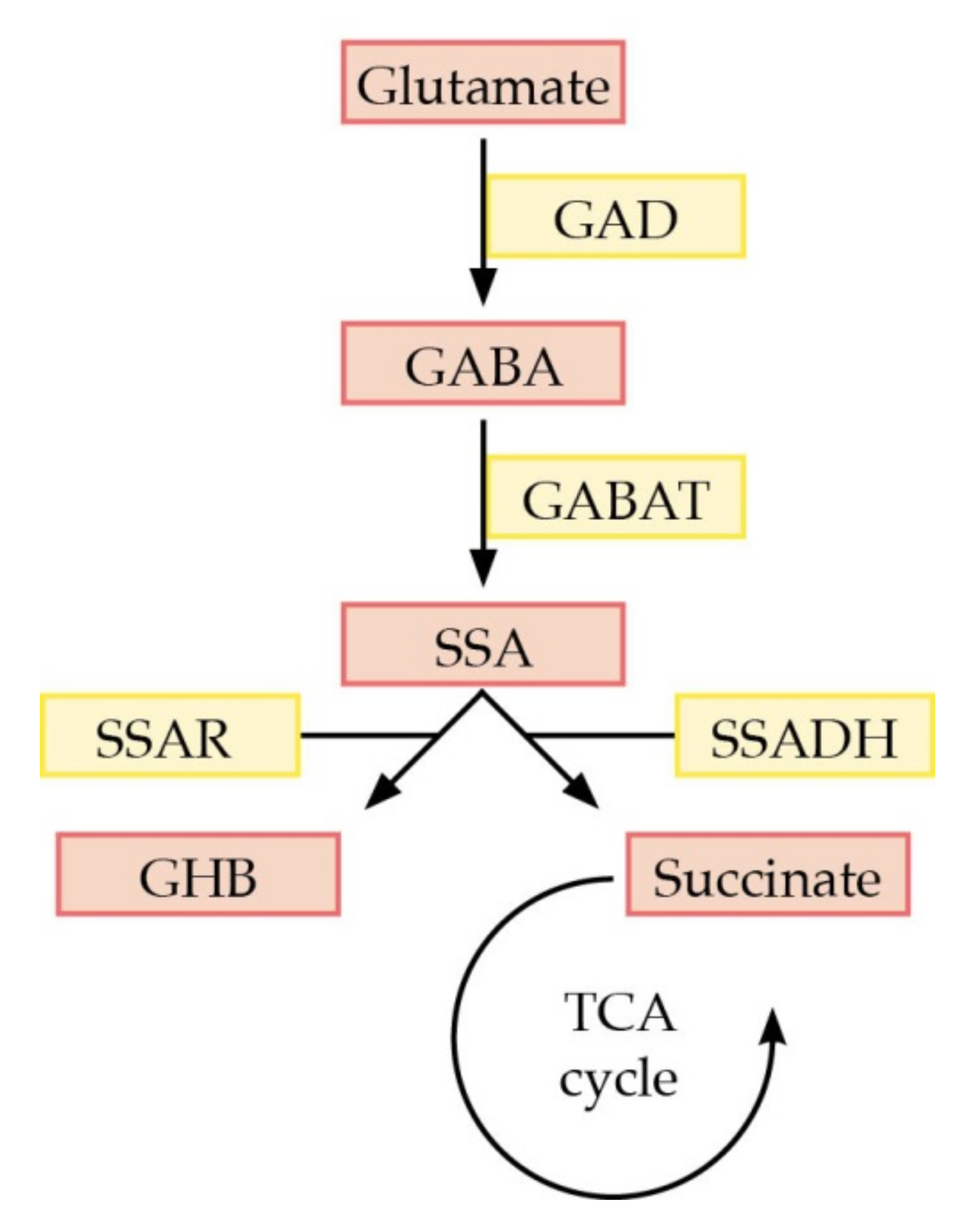
:1. Introduction
2. Results
2.1. Clinical Synopsis
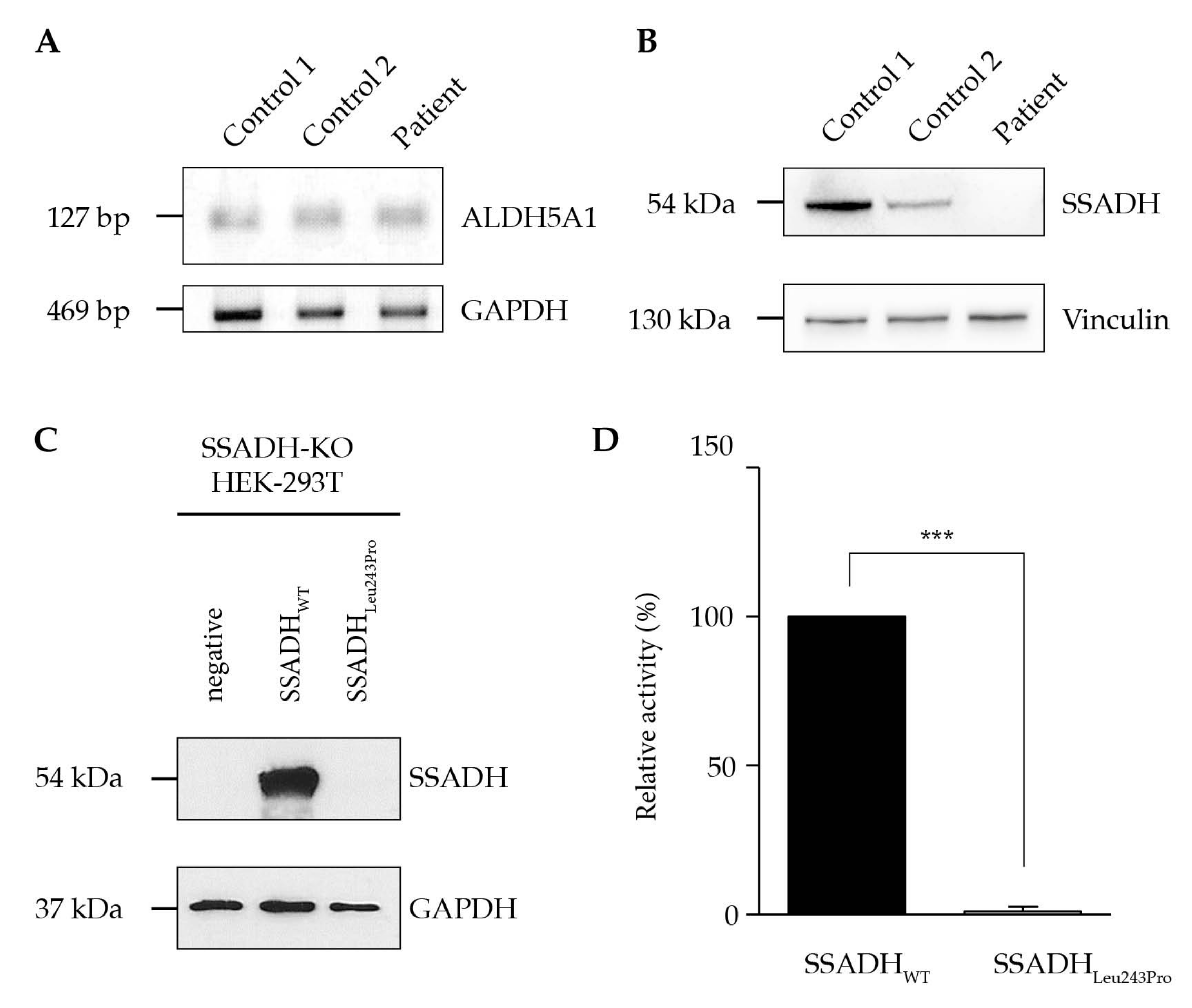
2.2. Biochemical Data and Sequence Analysis
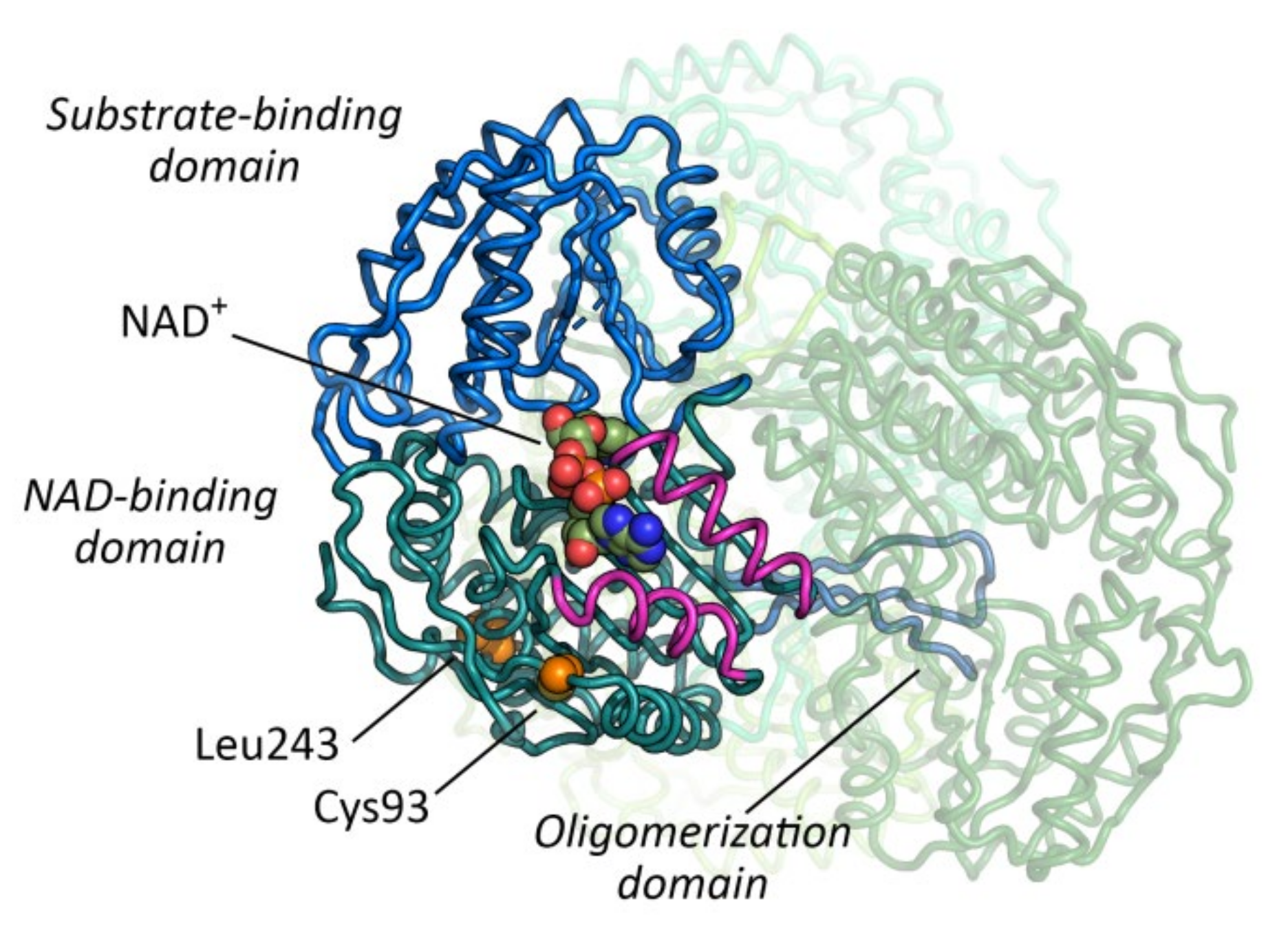
2.3. In Silico Analysis of Variant Stability
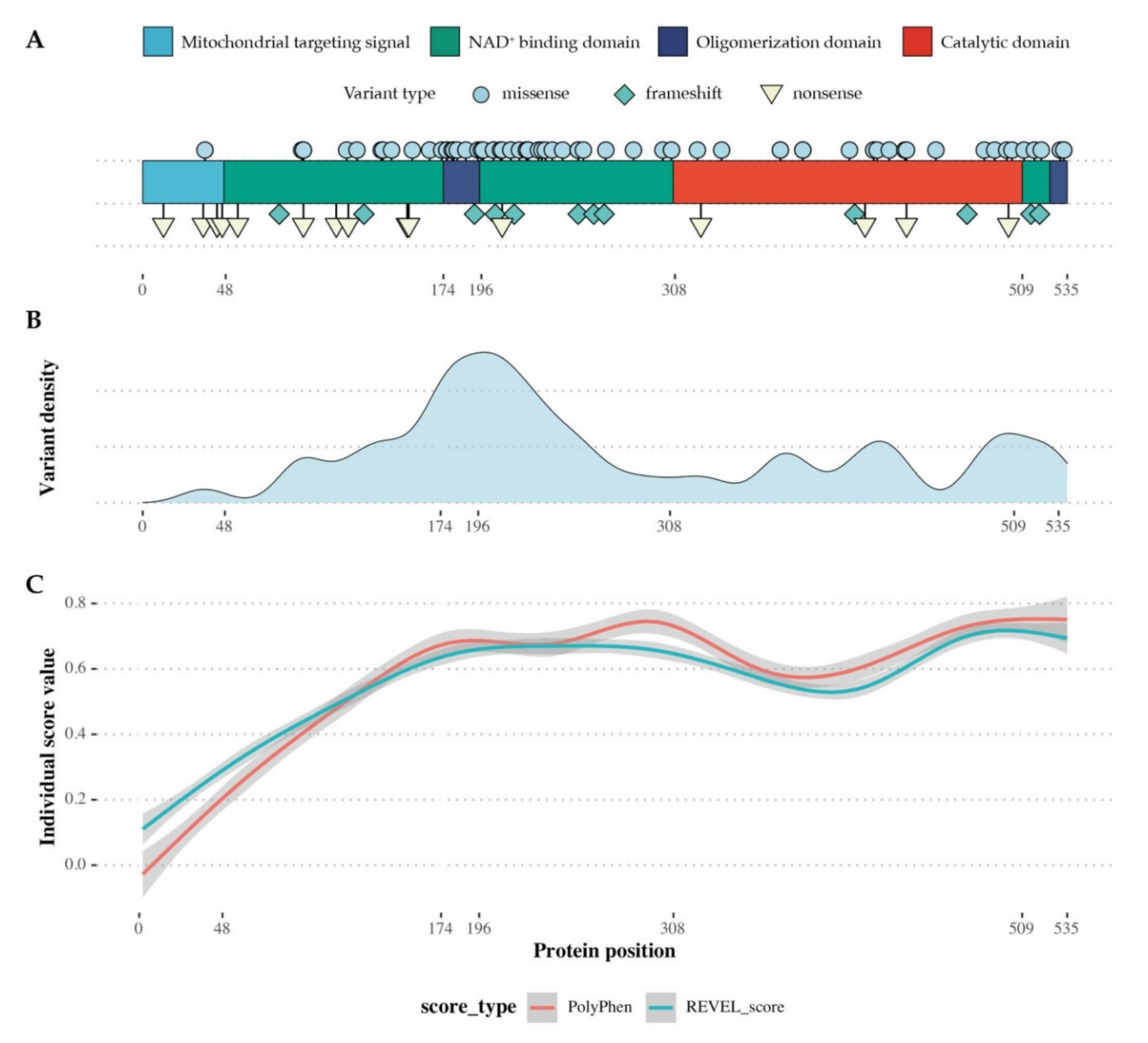
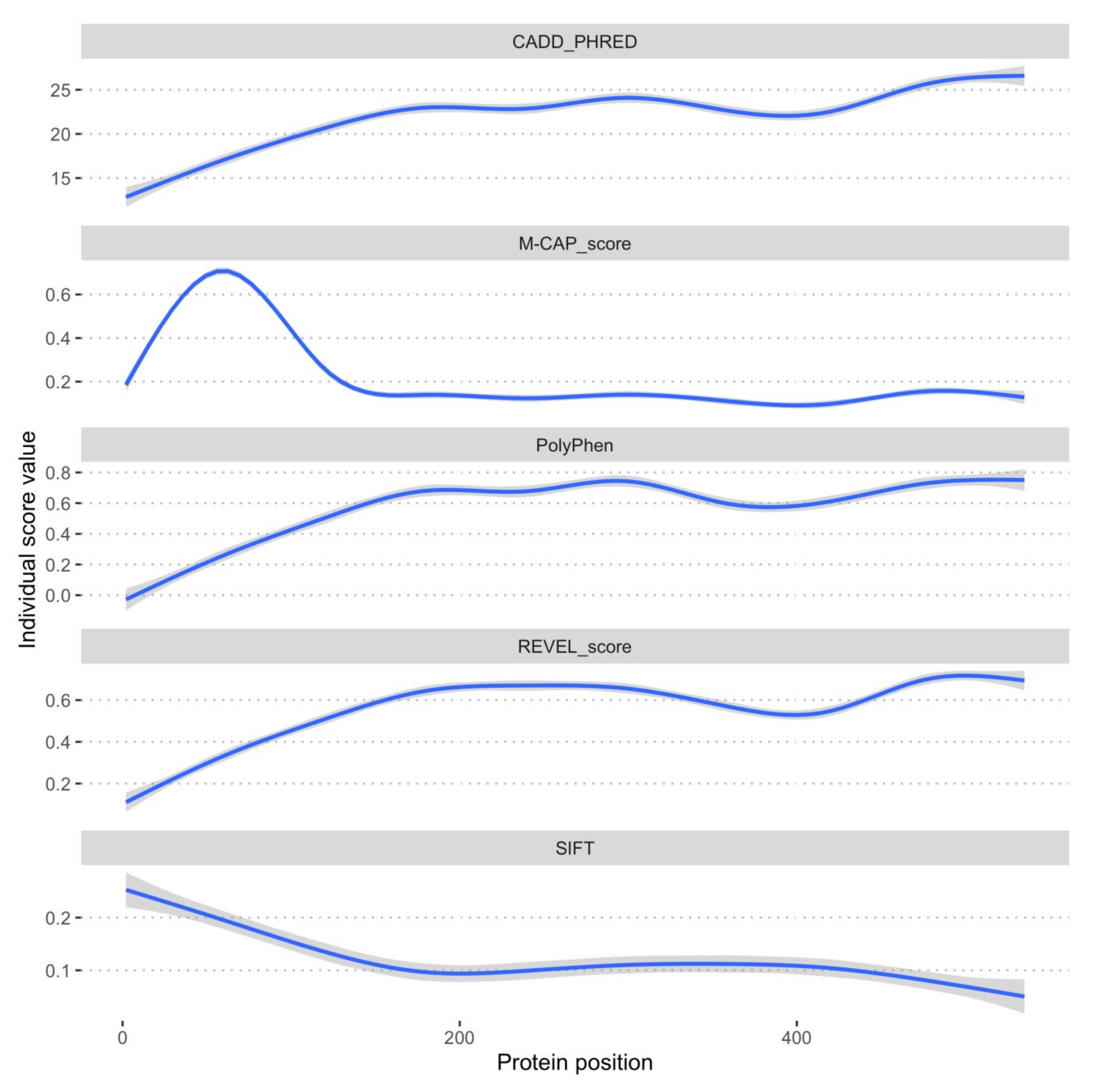
2.4. SSADH Variant Distribution
3. Discussion
4. Materials and Methods
4.1. Genetic Analysis
4.2. Ethics Committee Statement and Informed Consent
4.3. PCR Analysis
4.4. Semi-Quantitative Protein Analysis
4.5. Generation of the SSADH-Deficient HEK-293T Cell Line
4.6. Transient Expression of the Constructs
4.7. SSADH Enzyme Activity
4.8. Statistics
4.9. In Silico Analysis of Missense Variants in ALDH5A1
4.10. FoldX Calculation of Mutamer Stability and Free Energy Change
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| CSF | Cerebrospinal fluid |
| ECL | Enhanced chemiluminescence |
| GABA | γ-amino butyric acid |
| GABAT | GABA transaminase |
| GAD | Glutamate decarboxylase |
| GAPDH | Glyceraldehyde 3-phosphate dehydrogenase |
| GHB | γ-hydroxybutyrate |
| LOVD | Leiden Open Variation Database |
| NAD+ | Nicotinamide adenine dinucleotide |
| PBS | Phosphate-buffered saline |
| PCR | Polymerase chain reaction |
| PISA | Proteins, Interfaces, Structures and Assemblies |
| SSA | Succinic semialdehyde |
| SSADH | Succinic semialdehyde dehydrogenase |
| SSADHD | Succinic semialdehyde dehydrogenase deficiency |
| SSR | Succinic semialdehyde reductase |
| SUDEP | Sudden unexpected death in epilepsy |
| TBS-T | Tris-buffered saline and tween |
| TCA | Tricarboxylic acid |
| VEP | Variant Effect Prediction |
| WT | Wild-type |
Appendix A

References
- Jakobs, C.; Bojasch, M.; Mönch, E.; Rating, D.; Siemes, H.; Hanefeld, F. Urinary excretion of gamma-hydroxybutyric acid in a patient with neurological abnormalities. The probability of a new inborn error of metabolism. Clin. Chim. Acta 1981, 111, 169–178. [Google Scholar] [CrossRef]
- Gibson, K.M.; Jakobs, C.; Pearl, P.L.; Snead, O.C. Murine succinate semialdehyde dehydrogenase (SSADH) deficiency, a heritable disorder of GABA metabolism with epileptic phenotype. IUBMB Life 2005, 57, 639–644. [Google Scholar] [CrossRef] [PubMed]
- Malaspina, P.; Roullet, J.B.; Pearl, P.; Ainslie, G.; Vogel, K.; Gibson, K. Succinic semialdehyde dehydrogenase deficiency (SSADHD): Pathophysiological complexity and multifactorial trait associations in a rare monogenic disorder of GABA metabolism. Neurochem. Int. 2016, 99, 72–84. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pearl, P.L.; Gibson, K.M.; Acosta, M.T.; Vezina, L.G.; Theodore, W.H.; Rogawski, M.A.; Novotny, E.J.; Gropman, A.; Conry, J.A.; Berry, G.T.; et al. Clinical spectrum of succinic semialdehyde dehydrogenase deficiency. Neurology 2003, 60, 1413–1417. [Google Scholar] [CrossRef] [PubMed]
- DiBacco, M.L.; Roullet, J.B.; Kapur, K.; Brown, M.N.; Walters, D.C.; Gibson, K.M.; Pearl, P.L. Age-related phenotype and biomarker changes in SSADH deficiency. Ann. Clin. Transl. Neurol. 2019, 6, 114–120. [Google Scholar] [CrossRef]
- Didiasova, M.; Banning, A.; Brennenstuhl, H.; Jung-Klawitter, S.; Cinquemani, C.; Opladen, T.; Tikkanen, R. Succinic Semialdehyde Dehydrogenase Deficiency: An Update. Cells 2020, 9, 477. [Google Scholar] [CrossRef] [Green Version]
- Gibson, K.M.; Lee, C.F.; Chambliss, K.L.; Kamali, V.; Francois, B.; Jaeken, J.; Jakobs, C. 4-Hydroxybutyric aciduria: Application of a fluorometric assay to the determination of succinic semialdehyde dehydrogenase activity in extracts of cultured human lymphoblasts. Clin. Chim. Acta 1991, 196, 219–221. [Google Scholar] [CrossRef]
- Vogel, K.R.; Pearl, P.L.; Theodore, W.H.; McCarter, R.C.; Jakobs, C.; Gibson, K.M. Thirty years beyond discovery—Clinical trials in succinic semialdehyde dehydrogenase deficiency, a disorder of GABA metabolism. J. Inherit. Metab. Dis. 2013, 36, 401–410. [Google Scholar] [CrossRef] [Green Version]
- Cash, C.D.; Maitre, M.; Mandel, P. Purification from human brain and some properties of two nadph-linked aldehyde reductases which reduce succinic semialdehyde to 4-hydroxybutyrate. J. Neurochem. 1979, 33, 1169–1175. [Google Scholar] [CrossRef]
- Kim, Y.G.; Lee, S.J.; Kwon, O.S.; Park, S.Y.; Park, B.J.; Kim, K.J. Redox-switch modulation of human SSADH by dynamic catalytic loop. EMBO J. 2009, 28, 959–968. [Google Scholar] [CrossRef] [Green Version]
- Langendorf, C.G.; Key, T.L.G.; Fenalti, G.; Kan, W.T.; Buckle, A.M.; Caradoc-Davies, T.; Tuck, K.L.; Law, R.H.P.; Whisstock, J.C. The X-Ray Crystal Structure of Escherichia coli Succinic Semialdehyde Dehydrogenase; Structural Insights into NADP+/Enzyme Interactions. PLoS ONE 2010, 5, e9280. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Akaboshi, S.; Hogema, B.M.; Novelletto, A.; Malaspina, P.; Salomons, G.S.; Maropoulos, G.D.; Jakobs, C.; Grompe, M.; Gibson, K.M. Mutational spectrum of the succinate semialdehyde dehydrogenase (ALDH5A1) gene and functional analysis of 27 novel disease-causing mutations in patients with SSADH deficiency. Hum. Mutat. 2003, 22, 442–450. [Google Scholar] [CrossRef] [PubMed]
- Krissinel, E.; Henrick, K. Inference of Macromolecular Assemblies from Crystalline State. J. Mol. Biol. 2007, 372, 774–797. [Google Scholar] [CrossRef] [PubMed]
- Fokkema, I.F.A.C.; den Dunnen, J.; Taschner, P.E.M. LOVD: Easy creation of a locus-specific sequence variation database using an “LSDB-in-a-box” approach. Hum. Mutat. 2005, 26, 63–68. [Google Scholar] [CrossRef]
- Fokkema, I.F.A.C.; Taschner, P.E.; Schaafsma, G.C.P.; Celli, J.; Laros, J.F.J.; Dunnen, J.T.D. LOVD v.2.0: The next generation in gene variant databases. Hum. Mutat. 2011, 32, 557–563. [Google Scholar] [CrossRef]
- Pop, A.; Smith, D.E.; Kirby, T.; Walters, D.; Gibson, K.M.; Mahmoudi, S.; Van Dooren, S.J.; Kanhai, W.A.; Fernandez-Ojeda, M.R.; Wever, E.J.; et al. Functional analysis of thirty-four suspected pathogenic missense variants in ALDH5A1 gene associated with succinic semialdehyde dehydrogenase deficiency. Mol. Genet. Metab. 2020, 130, 172–178. [Google Scholar] [CrossRef]
- Lapalme-Remis, S.; Lewis, E.C.; De Meulemeester, C.; Chakraborty, P.; Gibson, K.M.; Torres, C.; Guberman, A.; Salomons, G.S.; Jakobs, C.; Ali-Ridha, A.; et al. Natural history of succinic semialdehyde dehydrogenase deficiency through adulthood. Neurology 2015, 85, 861–865. [Google Scholar] [CrossRef]
- DiBacco, M.L.; Pop, A.; Salomons, G.S.; Hanson, E.; Roullet, J.B.; Gibson, K.M.; Pearl, P.L. Novel ALDH5A1 variants and genotype: Phenotype correlation in SSADH deficiency. Neurology 2020, 95, e2675–e2682. [Google Scholar] [CrossRef]
- Pearl, P.L.; Parviz, M.; Vogel, K.; Schreiber, J.; Theodore, W.H.; Gibson, K.M. Inherited disorders of gamma-aminobutyric acid metabolism and advances inALDH5A1mutation identification. Dev. Med. Child Neurol. 2015, 57, 611–617. [Google Scholar] [CrossRef]
- Liu, N.; Kong, X.D.; Kan, Q.C.; Shi, H.; Wu, Q.H.; Zhuo, Z.H.; Bai, Q.L.; Jiang, M. Mutation analysis and prenatal diagnosis in a Chinese family with succinic semialdehyde dehydrogenase and a systematic review of the literature of reported ALDH5A1 mutations. J. Périnat. Med. 2016, 44, 441–451. [Google Scholar] [CrossRef]
- Jiang, S.Z.; Shu, J.B.; Zhang, Y.Q.; Fan, W.X.; Meng, Y.T.; Song, L. Analysis of ALDH5A1 gene mutation in a Chinese Han family with succinic semialdehyde dehydrogenase deficiency. Zhonghua Yi Xue Yi Chuan Xue Za Zhi 2013, 30, 389–393. [Google Scholar] [CrossRef]
- Blasi, P.; Boyl, P.P.; Ledda, M.; Novelletto, A.; Gibson, K.; Jakobs, C.; Hogema, B.; Akaboshi, S.; Loreni, F.; Malaspina, P. Structure of human succinic semialdehyde dehydrogenase gene: Identification of promoter region and alternatively processed isoforms. Mol. Genet. Metab. 2002, 76, 348–362. [Google Scholar] [CrossRef]
- Jung, R.; Rauch, A.; Salomons, G.S.; Verhoeven, N.M.; Jakobs, C.; Gibson, K.M.; Lachmann, E.; Sass, J.O.; Trautmann, U.; Zweier, C.; et al. Clinical, cytogenetic and molecular characterization of a patient with combined succinic semialdehyde dehydrogenase deficiency and incomplete WAGR syndrome with obesity. Mol. Genet. Metab. 2006, 88, 256–260. [Google Scholar] [CrossRef] [PubMed]
- Shu, J.; Cai, F.; Fan, W.; Meng, Y.; Zhang, C.; Cai, C.; Zhang, Y.; Lin, S. Identification of ALDH5A1 gene mutations in a Chinese family affected with succinic semialdehyde dehydrogenase deficiency. Zhonghua Yi Xue Yi Chuan Xue Za Zhi 2017, 34, 6–9. [Google Scholar] [CrossRef] [PubMed]
- Di Rosa, G.; Malaspina, P.; Blasi, P.; Dionisi-Vici, C.; Rizzo, C.; Tortorella, G.; Crutchfield, S.R.; Gibson, K.M. Visual evoked potentials in succinate semialdehyde dehydrogenase (SSADH) deficiency. J. Inherit. Metab. Dis. 2009, 32 (Suppl. 1), 201–205. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, X.; Ding, Y.; Liu, Y.; Zhang, Y.; Song, J.; Wang, Q.; Li, M.; Qin, Y.; Huang, S.; Yang, Y. Succinic semialdehyde dehydrogenase deficiency of four Chinese patients and prenatal diagnosis for three fetuses. Gene 2015, 574, 41–47. [Google Scholar] [CrossRef]
- Puttmann, L.; Stehr, H.; Garshasbi, M.; Hu, H.; Kahrizi, K.; Lipkowitz, B.; Jamali, P.; Tzschach, A.; Najmabadi, H.; Ropers, H.H.; et al. A novel ALDH5A1 mutation is associated with succinic semialdehyde dehydrogenase deficiency and severe intellectual disability in an Iranian family. Am. J. Med Genet. Part A 2013, 161, 1915–1922. [Google Scholar] [CrossRef]
- Menduti, G.; Biamino, E.; Vittorini, R.; Vesco, S.; Puccinelli, M.P.; Porta, F.; Capo, C.; Leo, S.; Ciminelli, B.M.; Iacovelli, F.; et al. Succinic semialdehyde dehydrogenase deficiency: The combination of a novel ALDH5A1 gene mutation and a missense SNP strongly affects SSADH enzyme activity and stability. Mol. Genet. Metab. 2018, 124, 210–215. [Google Scholar] [CrossRef]
- Yamakawa, Y.; Nakazawa, T.; Ishida, A.; Saito, N.; Komatsu, M.; Matsubara, T.; Obinata, K.; Hirose, S.; Okumura, A.; Shimizu, T. A boy with a severe phenotype of succinic semialdehyde dehydrogenase deficiency. Brain Dev. 2012, 34, 107–112. [Google Scholar] [CrossRef]
- Aoshima, T.; Kajita, M.; Sekido, Y.; Ishiguro, Y.; Tsuge, I.; Kimura, M.; Yamaguchi, S.; Watanabe, K.; Shimokata, K.; Niwa, T. Mutation Analysis in a Patient with Succinic Semialdehyde Dehydrogenase Deficiency: A Compound Heterozygote with 103-121del and 1460T > A of the ALDH5A1 Gene. Hum. Hered. 2002, 53, 42–44. [Google Scholar] [CrossRef]
- Leo, S.; Capo, C.; Ciminelli, B.M.; Iacovelli, F.; Menduti, G.; Funghini, S.; Donati, M.A.; Falconi, M.; Rossi, L.; Malaspina, P. SSADH deficiency in an Italian family: A novel ALDH5A1 gene mutation affecting the succinic semialdehyde substrate binding site. Metab. Brain Dis. 2017, 32, 1383–1388. [Google Scholar] [CrossRef] [PubMed]
- Wang, P.; Cai, F.; Cao, L.; Wang, Y.; Zou, Q.; Zhao, P.; Wang, C.; Zhang, Y.; Cai, C.; Shu, J. Clinical diagnosis and mutation analysis of four Chinese families with succinic semialdehyde dehydrogenase deficiency. BMC Med. Genet. 2019, 20, 88. [Google Scholar] [CrossRef] [PubMed]
- Akiyama, T.; Osaka, H.; Shimbo, H.; Kuhara, T.; Shibata, T.; Kobayashi, K.; Kurosawa, K.; Yoshinaga, H. SSADH deficiency possibly associated with enzyme activity-reducing SNPs. Brain Dev. 2016, 38, 871–874. [Google Scholar] [CrossRef] [PubMed]
- Ran, F.A.; Hsu, P.D.; Wright, J.; Agarwala, V.; Scott, D.A.; Zhang, F. Genome engineering using the CRISPR-Cas9 system. Nat. Protoc. 2013, 8, 2281–2308. [Google Scholar] [CrossRef] [Green Version]
- Guerois, R.; Nielsen, J.E.; Serrano, L. Predicting Changes in the Stability of Proteins and Protein Complexes: A Study of More Than 1000 Mutations. J. Mol. Biol. 2002, 320, 369–387. [Google Scholar] [CrossRef]
- Schymkowitz, J.; Borg, J.; Stricher, F.; Nys, R.; Rousseau, F.; Serrano, L. The FoldX web server: An online force field. Nucleic Acids Res. 2005, 33, W382–W388. [Google Scholar] [CrossRef] [Green Version]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Configuration | Combination | ∆∆G (kcal/mol) | St.Dev. ∆∆G |
|---|---|---|---|
| Monomers | F | 7.20 | ±1.69 |
| P | 7.63 | ±0.62 | |
| Tetramers | FFFF | 28.09 | ±3.32 |
| PPPP | 29.36 | ±1.11 | |
| FPPP | 29.09 | ±1.94 | |
| FFPP | 27.32 | ±3.21 | |
| FFFP | 26.41 | ±3.36 | |
| FPFP | 26.74 | ±2.16 | |
| PFFP | 28.16 | ±3.32 |
| No. | Genetic Variant | Protein Change | Domain | SIFT | Poly-Phen | REVEL | Source |
|---|---|---|---|---|---|---|---|
| 1 | c.106G > C | p.Gly36Arg | Mitochondrial targeting signal | 0.82 | 0 | 0.103 | LOVD [14,15] |
| 2 | c.275A > G | p.Asp92Gly | NAD+ binding domain | 0.03 | 0.964 | 0.477 | [16] |
| 3 | c.277T > C | p.Cys93Arg | NAD+ binding domain | 0.05 | 0.979 | 0.677 | [16] |
| 4 | c.278G > T | p.Cys93Phe | NAD+ binding domain | 0.16 | 0.972 | 0.638 | [12] |
| 5 | c.354G > C | p.Lys118Arn | NAD+ binding domain | 0.01 | 0.514 | 0.550 | [16] |
| 6 | c.371T > G | p.Leu124Arg | NAD+ binding domain | 0 | 0.986 | 0.938 | [16] |
| 7 | c.412C > T | p.Leu138Pro | NAD+ binding domain | 0 | 1 | 0.948 | [17] |
| 8 | c.416C > A | p.Ala139Asp | NAD+ binding domain | 0 | 0.999 | 0.886 | [18] |
| 9 | c.431C > A | p.Ala144Asp | NAD+ binding domain | 0.08 | 0.854 | 0.522 | [16] |
| 10 | c.466G > A | p.Glu156Lys | NAD+ binding domain | 0 | 1 | 0.924 | [19] |
| 11 | c.496T > C | p.Trp166Arg | NAD+ binding domain | 0 | 0.998 | 0.970 | [20] |
| 12 | c.517C > T | p.Arg173Cys | NAD+ binding domain | 0 | 1 | 0.836 | [19] |
| 13 | c.526G > A | p.Gly176Arg | Oligomerization domain | 0 | 1 | 0.953 | [12] |
| 14 | c.527G > A | p.Gly176Glu | Oligomerization domain | 0 | 1 | 0.924 | [21] |
| 15 | c.536T > A | p.Ile179Asn | Oligomerization domain | 0 | 0.984 | 0.765 | [16] |
| 16 | c.538C > T | p.His180Tyr | Oligomerization domain | 0.1 | 0 | 0.125 | [22] |
| 17 | c.545C > T | p.Pro182Leu | Oligomerization domain | 0.05 | 0.735 | 0.304 | [22] |
| 18 | c.559C > G | p.Arg187Gly | Oligomerization domain | 0 | 0.942 | 0.552 | [16] |
| 19 | c.581C > T | p.Pro194Leu | Oligomerization domain | 0 | 1 | 0.938 | [16] |
| 20 | c.587G > A | p.Gly196Asp | Oligomerization domain | 0 | 1 | 0.958 | [23] |
| 21 | c.589G > A | p.Val197Met | NAD+ binding domain | 0 | 1 | 0.940 | [20] |
| 22 | c.608C > G | p.Pro203Arg | NAD+ binding domain | 0 | 1 | 0.941 | [16] |
| 23 | c.608C > T | p.Pro203Leu | NAD+ binding domain | 0 | 1 | 0.936 | LOVD [14,15] |
| 24 | c.620C > T | p.Pro207Leu | NAD+ binding domain | 0 | 0.998 | 0.952 | [16] |
| 25 | c.622A > C | p.Ser208Arg | NAD+ binding domain | 0.2 | 0.993 | 0.686 | [16] |
| 26 | c.637C > G | p.Arg213Gly | NAD+ binding domain | 0 | 0.985 | 0.699 | [16] |
| 27 | c.638G > T | p.Arg213Leu | NAD+ binding domain | 0 | 0.308 | 0.859 | [24] |
| 28 | c.653C > A | p.Ala218Asp | NAD+ binding domain | 0 | 0.952 | 0.941 | [16] |
| 29 | c.667T > C | p.Cys223Arg | NAD+ binding domain | 0 | 0.936 | 0.867 | [25] |
| 30 | c.668G > A | p.Cys223Tyr | NAD+ binding domain | 0 | 0.983 | 0.806 | [12] |
| 31 | c.685C > T | p.Pro229Ser | NAD+ binding domain | 0 | 0.961 | 0.766 | [16] |
| 32 | c.691G > A | p.Glu231Lys | NAD+ binding domain | 0.02 | 0.092 | 0.693 | [21] |
| 33 | c.698C > T | p.Thr233Met | NAD+ binding domain | 0 | 0.999 | 0.845 | [12] |
| 34 | c.709G > A | p.Ala237Thr | NAD+ binding domain | 0.01 | 0.293 | 0.798 | [16] |
| 35 | c.709G > T | p.Ala237Ser | NAD+ binding domain | 0 | 0.492 | 0.691 | [22] |
| 36 | c.728T > C | p.Leu243Pro | NAD+ binding domain | 0 | 0.999 | 0.931 | This report |
| 37 | c.754G > T | p.Gly252Cys | NAD+ binding domain | 0 | 1 | 0.845 | [16] |
| 38 | c.755G > T | p.Gly252Val | NAD+ binding domain | 0 | 1 | 0.825 | [16] |
| 39 | c.764A > G | p.Asn255Ser | NAD+ binding domain | 0.21 | 0.998 | 0.673 | [12] |
| 40 | c.800T > G | p.Val267Gly | NAD+ binding domain | 0.02 | 0.998 | 0.893 | [26] |
| 41 | c.803G > A | p.Gly268Glu | NAD+ binding domain | 0 | 1 | 0.865 | [12] |
| 42 | c.851G > A | p.Gly284Asp | NAD+ binding domain | 0 | 1 | 0.989 | [16] |
| 43 | c.901A > G | p.Lys301Glu | NAD+ binding domain | 0 | 1 | 0.931 | [27] |
| 44 | c.961G > A | p.Val321Met | Catalytic domain | 0.01 | 0.602 | 0.421 | Faruq et al. 2020, submitted |
| 45 | c.1005C > A | p.Asn335Lys | Catalytic domain | 0 | 0.999 | 0.856 | [12] |
| 46 | c.1106G > A | p.Arg369His | Catalytic domain | 0.16 | 0.003 | 0.304 | LOVD [14,15] |
| 47 | c.1145C > T | p.Pro382Leu | Catalytic domain | 0 | 1 | 0.805 | [12] |
| 48 | c.1145C > A | p.Pro382Gln | Catalytic domain | 0 | 1 | 0.819 | [12] |
| 49 | c.1226G > A | p.Gly409Asp | Catalytic domain | 0 | 1 | 0.936 | [12] |
| 50 | c.1267A > T | p.Thr423Ser | Catalytic domain | 0.01 | 0.998 | 0.908 | [28] |
| 51 | c.1274T > C | p.Leu425Pro | Catalytic domain | 0 | 1 | 0.928 | [26] |
| 52 | c.1294A > C | p.Met432Leu | Catalytic domain | 0 | 0.827 | 0.877 | [29] |
| 53 | c.1321G > A | p.Gly441Arg | Catalytic domain | 0 | 1 | 0.860 | [18] |
| 54 | c.1324C > T | p.Pro442Ser | Catalytic domain | 0 | 1 | 0.961 | [16] |
| 55 | c.1460T > A | p.Val487Glu | Catalytic domain | 0.03 | 0.989 | 0.776 | [30] |
| 56 | c.1478A > G | p.Asn493Ser | Catalytic domain | 0 | 0.997 | 0.871 | [16] |
| 57 | c.1498G > C | p.Val500Leu | Catalytic domain | 0.08 | 0.113 | 0.456 | [31] |
| 58 | c.1508C > G | p.Pro503Arg | Catalytic domain | 0 | 0.933 | 0.829 | [16] |
| 59 | c.1529C > T | p.Ser510Phe | NAD+ binding domain | 0 | 1 | 0.908 | [32] |
| 60 | c.1547G > A | p.Gly516Glu | NAD+ binding domain | 0 | 1 | 0.951 | [33] |
| 61 | c.1592G > A | p.Cys531Tyr | Oligomerization domain | 0 | 0.971 | 0.869 | [16] |
| 62 | c.1597G > A | p.Gly533Arg | Oligomerization domain | 0 | 1 | 0.890 | [12] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Brennenstuhl, H.; Didiasova, M.; Assmann, B.; Bertoldi, M.; Molla, G.; Jung-Klawitter, S.; Kuseyri Hübschmann, O.; Schröter, J.; Opladen, T.; Tikkanen, R. Succinic Semialdehyde Dehydrogenase Deficiency: In Vitro and In Silico Characterization of a Novel Pathogenic Missense Variant and Analysis of the Mutational Spectrum of ALDH5A1. Int. J. Mol. Sci. 2020, 21, 8578. https://doi.org/10.3390/ijms21228578
Brennenstuhl H, Didiasova M, Assmann B, Bertoldi M, Molla G, Jung-Klawitter S, Kuseyri Hübschmann O, Schröter J, Opladen T, Tikkanen R. Succinic Semialdehyde Dehydrogenase Deficiency: In Vitro and In Silico Characterization of a Novel Pathogenic Missense Variant and Analysis of the Mutational Spectrum of ALDH5A1. International Journal of Molecular Sciences. 2020; 21(22):8578. https://doi.org/10.3390/ijms21228578
Chicago/Turabian StyleBrennenstuhl, Heiko, Miroslava Didiasova, Birgit Assmann, Mariarita Bertoldi, Gianluca Molla, Sabine Jung-Klawitter, Oya Kuseyri Hübschmann, Julian Schröter, Thomas Opladen, and Ritva Tikkanen. 2020. "Succinic Semialdehyde Dehydrogenase Deficiency: In Vitro and In Silico Characterization of a Novel Pathogenic Missense Variant and Analysis of the Mutational Spectrum of ALDH5A1" International Journal of Molecular Sciences 21, no. 22: 8578. https://doi.org/10.3390/ijms21228578