Discovery of 5-Phenoxy-2-aminopyridine Derivatives as Potent and Selective Irreversible Inhibitors of Bruton’s Tyrosine Kinase

 ,
,
Abstract
:
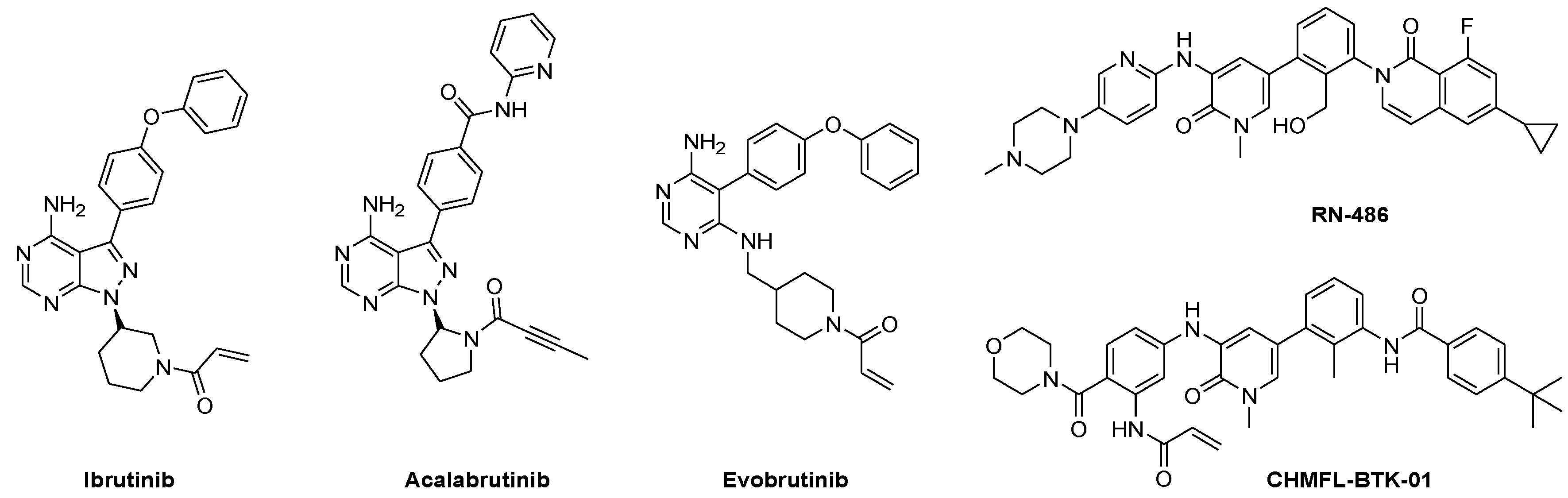
1. Introduction
2. Results and Discussion
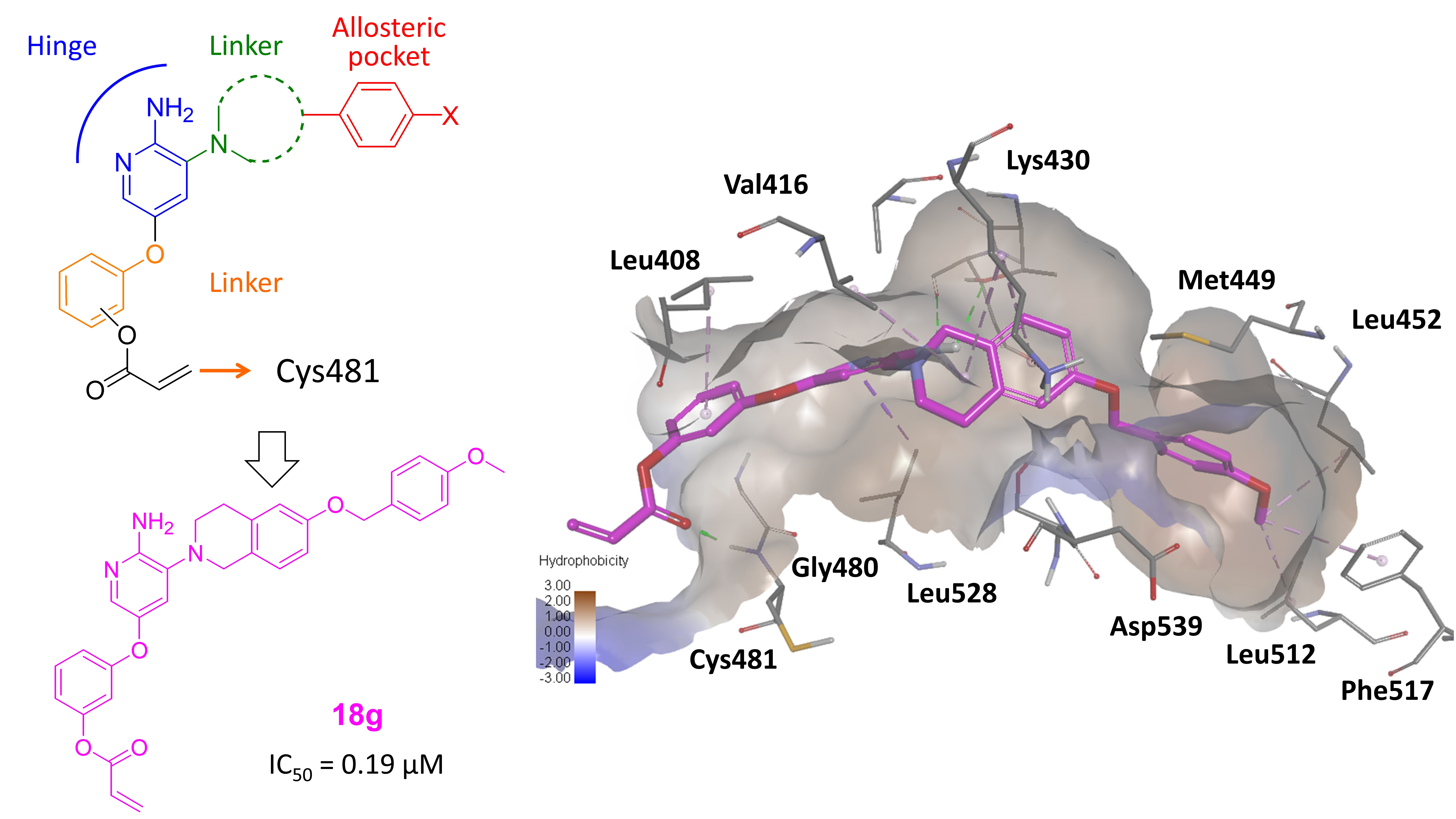
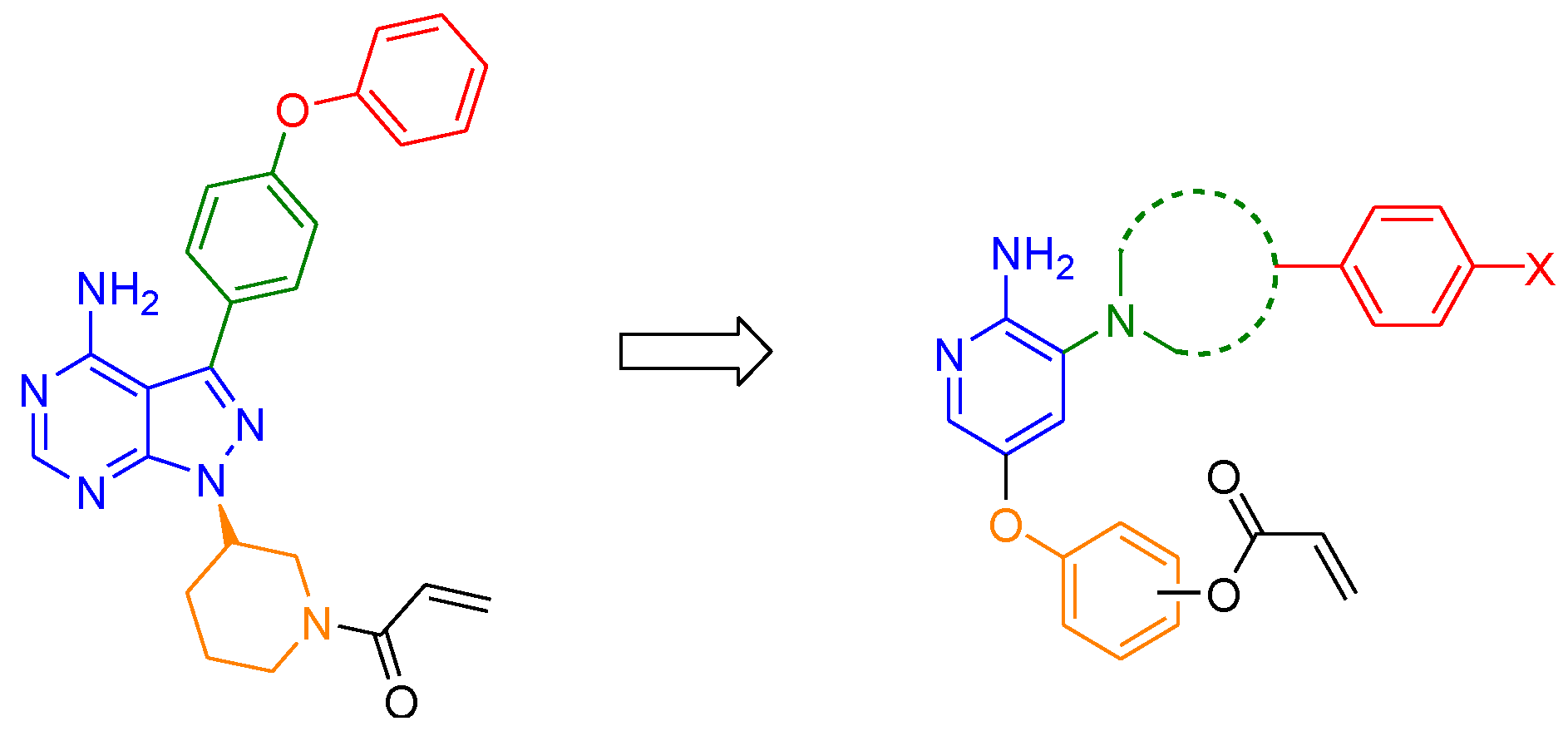
2.1. Drug Design
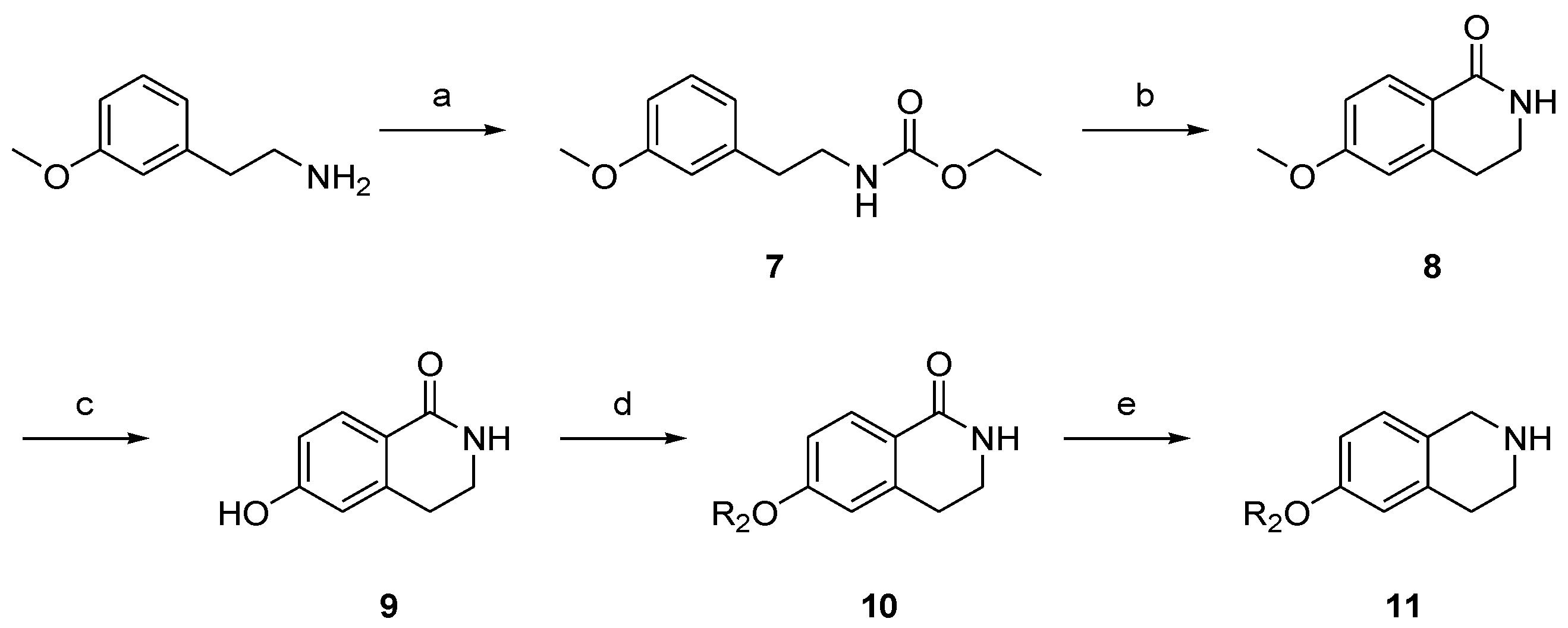
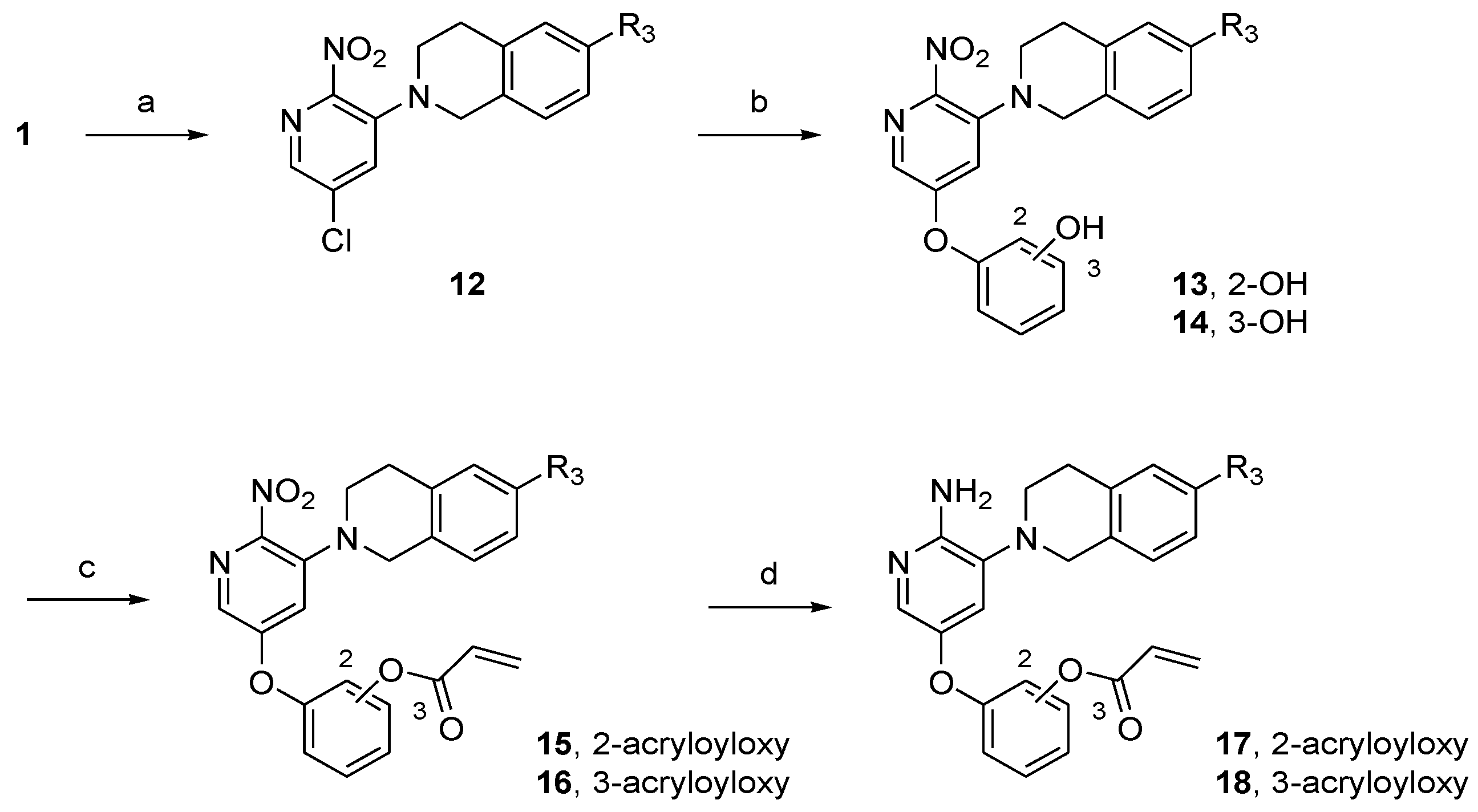
2.2. Synthesis
2.3. Structure–Activity Relationship Analysis
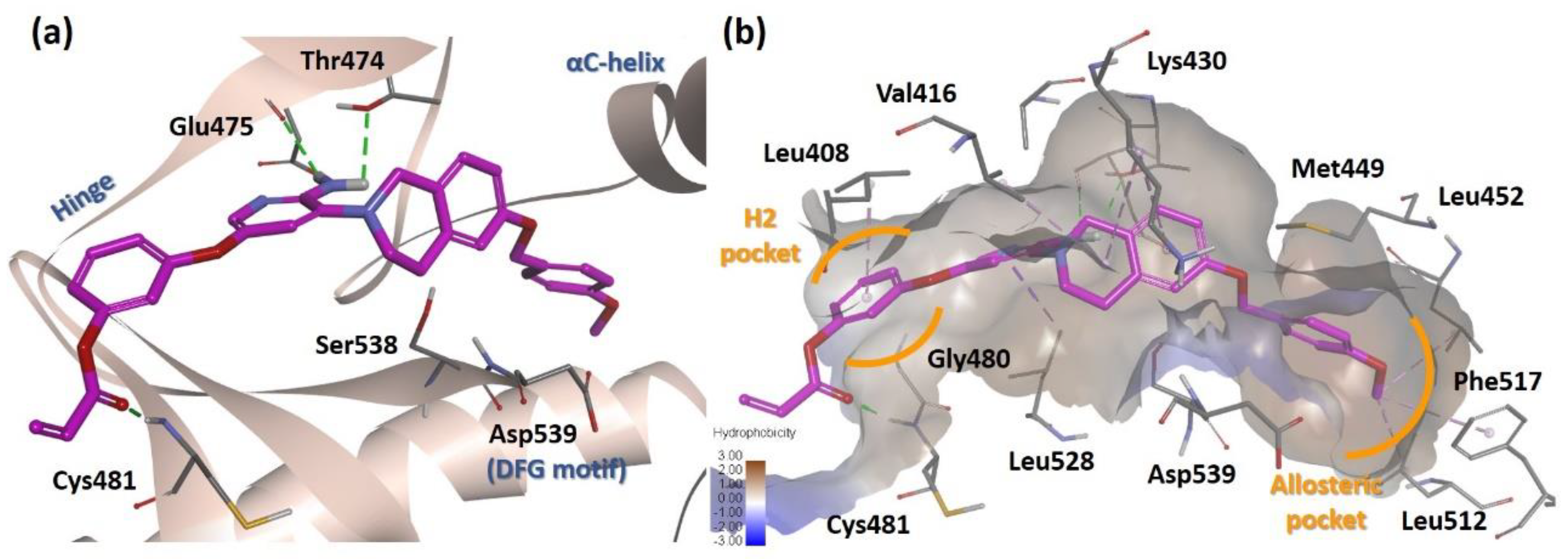
2.4. Molecular Docking Analysis
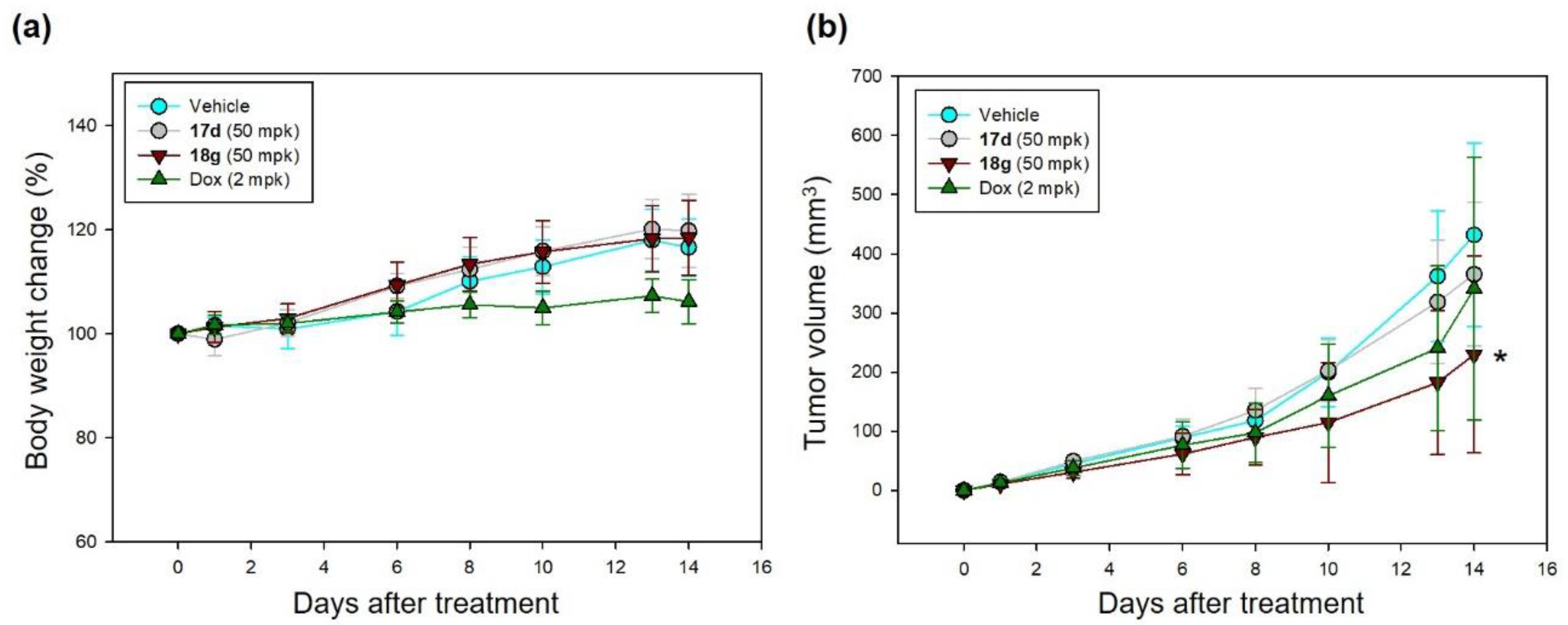
2.5. Effects on Experimental Models of Hematological Malignancy
3. Materials and Methods
3.1. Chemistry
3.1.1. General Procedures for the Synthesis of Compound 6
3.1.2. Acrylic Acid 3-(6-Amino-5-piperazinylpyridin-3-yloxy)phenyl Ester (6a)
3.1.3. Acrylic Acid 3-[6-Amino-5-(4-benzylpiperazinyl)pyridin-3-yloxy]phenyl Ester (6b)
3.1.4. Acrylic Acid 3-{6-Amino-5-[4-(4-fluorobenzyl)piperazinyl]pyridin-3-yloxy}phenyl Ester (6c)
3.1.5. Acrylic Acid 3-{6-Amino-5-[4-(4-chlorobenzyl)piperazinyl]pyridin-3-yloxy}phenyl Ester (6d)
3.1.6. Acrylic Acid 3-[6-Amino-5-(4-phenethylpiperazinyl)pyridin-3-yloxy]phenyl Ester (6e)
3.1.7. Acrylic Acid 3-(6-Amino-5-{4-[2-(4-chlorophenyl)ethyl]piperazinyl}pyridin-3-yloxy)phenyl Ester (6f)
3.1.8. 4-[5-(3-Acryloyloxyphenoxy)-2-aminopyridin-3-yl]piperazine carboxylic Acid Benzyl Ester (6g)
3.1.9. General Procedures for the Synthesis of Compounds 17 and 18
3.1.10. Acrylic Acid 2-[6-Amino-5-(3,4-dihydro-1H-isoquinolin-2-yl)pyridin-3-yloxy]phenyl Ester (17a)
3.1.11. Acrylic Acid 2-[6-Amino-5-(6-benzyloxy-3,4-dihydro-1H-isoquinolin-2-yl)pyridin-3-yloxy]phenyl Ester (17d)
3.1.12. Acrylic Acid 3-[6-Amino-5-(3,4-dihydro-1H-isoquinolin-2-yl)pyridin-3-yloxy]phenyl Ester (18a)
3.1.13. Acrylic Acid 3-[6-Amino-5-(6-propoxy-3,4-dihydro-1H-isoquinolin-2-yl)pyridin-3-yloxy]phenyl Ester (18b)
3.1.14. Acrylic Acid 3-[6-Amino-5-(6-cyclopropylmethoxy-3,4-dihydro-1H-isoquinolin-2-yl)pyridin-3-yloxy]phenyl Ester (18c)
3.1.15. Acrylic Acid 3-[6-Amino-5-(6-benzyloxy-3,4-dihydro-1H-isoquinolin-2-yl)pyridin-3-yloxy]phenyl Ester (18d)
3.1.16. Acrylic Acid 3-{6-Amino-5-[6-(4-fluorobenzyloxy)-3,4-dihydro-1H-isoquinolin-2-yl]pyridin-3-yloxy}phenyl Ester (18e)
3.1.17. Acrylic Acid 3-{6-Amino-5-[6-(4-chlorobenzyloxy)-3,4-dihydro-1H-isoquinolin-2-yl]pyridin-3-yloxy}phenyl Ester (18f)
3.1.18. Acrylic Acid 3-{6-Amino-5-[6-(4-methoxybenzyloxy)-3,4-dihydro-1H-isoquinolin-2-yl]pyridin-3-yloxy}phenyl Ester (18g)
3.2. BTK Kinase Assay
3.2.1. ADP-Glo Assay
3.2.2. HotSpot Kinase Assay
3.3. Molecular Docking Analysis
3.4. In Vitro Growth Inhibition
3.5. In Vivo Xenograft Model
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| FDA | Food and Drug Administration |
| JAK3 | Janus kinase 3 |
| EGFRK | Epidermal growth factor receptor kinase |
| ATP | Adenosine triphosphate |
| SRC | Proto-oncogene tyrosine-protein kinase Src |
| Boc | tert-Butyloxycarbonyl protecting group |
| TFA | Trifluoroacetic acid |
| DCM | Dichloromethane |
| DMF | Dimethylformamide |
| TEA | Triethylamine |
| PPA | Polyphosphoric acid |
| THF | Tetrahydrofuran |
| DMSO | Dimethyl sulfoxide |
| ADP | Adenosine diphosphate |
| IC50 | The half maximal inhibitory concentration |
| Bn | Benzyl |
| Me | Methyl |
| Et | Ethyl |
| Pr | Propyl |
| Ac | Acetyl |
| GI50 | The half maximal growth inhibitory concentration |
References
- Rawlings, D.J.; Scharenberg, A.M.; Park, H.; Wahl, M.I.; Lin, S.; Kato, R.M.; Fluckiger, A.C.; Witte, O.N.; Kinet, J.P. Activation of BTK by a phosphorylation mechanism initiated by SRC family kinases. Science 1996, 271, 822–825. [Google Scholar] [CrossRef] [PubMed]
- Mohamed, A.J.; Yu, L.; Backesjo, C.M.; Vargas, L.; Faryal, R.; Aints, A.; Christensson, B.; Berglof, A.; Vihinen, M.; Nore, B.F.; et al. Bruton’s tyrosine kinase (Btk): Function, regulation, and transformation with special emphasis on the PH domain. Immunol. Rev. 2009, 228, 58–73. [Google Scholar] [PubMed]
- Satterthwaite, A.B.; Witte, O.N. The role of Bruton’s tyrosine kinase in B-cell development and function: A genetic perspective. Immunol. Rev. 2000, 175, 120–127. [Google Scholar] [CrossRef] [PubMed]
- Buggy, J.J.; Elias, L. Bruton tyrosine kinase (BTK) and its role in B-cell malignancy. Int. Rev. Immunol. 2012, 31, 119–132. [Google Scholar] [CrossRef] [PubMed]
- Roskoski, R., Jr. Properties of FDA-approved small molecule protein kinase inhibitors. Pharmacol. Res. 2019, 144, 19–50. [Google Scholar] [CrossRef] [PubMed]
- Liang, C.; Tian, D.; Ren, X.; Ding, S.; Jia, M.; Xin, M.; Thareja, S. The development of Bruton’s tyrosine kinase (BTK) inhibitors from 2012 to 2017: A mini-review. Eur. J. Med. Chem. 2018, 151, 315–326. [Google Scholar] [CrossRef] [PubMed]
- Kaur, V.; Swami, A. Ibrutinib in CLL: A focus on adverse events, resistance, and novel approaches beyond ibrutinib. Ann. Hematol. 2017, 96, 1175–1184. [Google Scholar] [CrossRef] [PubMed]
- Barf, T.; Kaptein, A. Irreversible protein kinase inhibitors: Balancing the benefits and risks. J. Med. Chem. 2012, 55, 6243–6262. [Google Scholar] [CrossRef] [PubMed]
- Wu, P.; Nielsen, T.E.; Clausen, M.H. FDA-approved small-molecule kinase inhibitors. Trends Pharmacol. Sci. 2015, 36, 422–439. [Google Scholar] [PubMed] [Green Version]
- Lou, Y.; Han, X.; Kuglstatter, A.; Kondru, R.K.; Sweeney, Z.K.; Soth, M.; McIntosh, J.; Litman, R.; Suh, J.; Kocer, B.; et al. Structure-based drug design of RN486, a potent and selective Bruton’s tyrosine kinase (BTK) inhibitor, for the treatment of rheumatoid arthritis. J. Med. Chem. 2015, 58, 512–516. [Google Scholar] [CrossRef] [PubMed]
- Liang, Q.; Chen, Y.; Yu, K.; Chen, C.; Zhang, S.; Wang, A.; Wang, W.; Wu, H.; Liu, X.; Wang, B.; et al. Discovery of N-(3-(5-((3-acrylamido-4-(morpholine-4-carbonyl)phenyl)amino)-1-methyl-6-oxo-1,6- dihydropyridin-3-yl)-2-methylphenyl)-4-(tert-butyl)benzamide (CHMFL-BTK-01) as a highly selective irreversible Bruton’s tyrosine kinase (BTK) inhibitor. Eur. J. Med. Chem. 2017, 131, 107–125. [Google Scholar] [CrossRef] [PubMed]
- Marcotte, D.J.; Liu, Y.T.; Arduini, R.M.; Hession, C.A.; Miatkowski, K.; Wildes, C.P.; Cullen, P.F.; Hong, V.; Hopkins, B.T.; Mertsching, E.; et al. Structures of human Bruton’s tyrosine kinase in active and inactive conformations suggest a mechanism of activation for TEC family kinases. Protein Sci. A Publ. Protein Soc. 2010, 19, 429–439. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barf, T.; Covey, T.; Izumi, R.; van de Kar, B.; Gulrajani, M.; van Lith, B.; van Hoek, M.; de Zwart, E.; Mittag, D.; Demont, D.; et al. Acalabrutinib (ACP-196): A Covalent Bruton Tyrosine Kinase Inhibitor with a Differentiated Selectivity and In Vivo Potency Profile. J. Pharmacol. Exp. Ther. 2017, 363, 240–252. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Gray, N.S. Rational design of inhibitors that bind to inactive kinase conformations. Nat. Chem. Biol. 2006, 2, 358–364. [Google Scholar] [CrossRef] [PubMed]
- RCSB Protein Data Bank. Available online: https://www.rcsb.org/ (accessed on 10 August 2020).
- Kuglstatter, A.; Wong, A.; Tsing, S.; Lee, S.W.; Lou, Y.; Villasenor, A.G.; Bradshaw, J.M.; Shaw, D.; Barnett, J.W.; Browner, M.F. Insights into the conformational flexibility of Bruton’s tyrosine kinase from multiple ligand complex structures. Protein Sci. A Publ. Protein Soc. 2011, 20, 428–436. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Young, W.B.; Barbosa, J.; Blomgren, P.; Bremer, M.C.; Crawford, J.J.; Dambach, D.; Gallion, S.; Hymowitz, S.G.; Kropf, J.E.; Lee, S.H.; et al. Potent and selective Bruton’s tyrosine kinase inhibitors: Discovery of GDC-0834. Bioorganic Med. Chem. Lett. 2015, 25, 1333–1337. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anastassiadis, T.; Deacon, S.W.; Devarajan, K.; Ma, H.; Peterson, J.R. Comprehensive assay of kinase catalytic activity reveals features of kinase inhibitor selectivity. Nat. Biotechnol. 2011, 29, 1039–1045. [Google Scholar] [PubMed] [Green Version]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | R1 | % Inhibition 1 | IC50 (μM) 2 |
|---|---|---|---|
| 6a | H | 14 | ND 3 |
| 6b | Bn | 85 | 3.17 |
| 6c | p-FBn | 83 | ND |
| 6d | p-ClBn | 92 | 1.54 |
| 6e | PhEt | 89 | 5.01 |
| 6f | p-ClPhEt | 52 | ND |
| 6g | benzyloxycarbonyl | 77 | ND |
| Ibrutinib | 0.0002 |
| Compound | Position | R3 | % Inhibition 1 | IC50 (μM) 2 |
|---|---|---|---|---|
| 17a | 2 | H | 98 | 0.63 |
| 17d | 2 | OBn | 98 | 0.46 |
| 18a | 3 | H | 98 | 2.20 |
| 18b | 3 | OnPr | 99 | 0.97 |
| 18c | 3 | OCH2cPr | 96 | 1.20 |
| 18d | 3 | OBn | 97 | 0.10 |
| 18e | 3 | Op-FBn | 53 * | 8.53 |
| 18f | 3 | Op-ClBn | 98 | 1.00 |
| 18g | 3 | Op-OMeBn | 92 * | 0.19 |
| Kinase | % Inhibition 1 | ||
|---|---|---|---|
| 17d | 18d | 18g | |
| BMX/ETK | 98.7 ± 0.6 | 95.5 ± 0.1 | 96.0 ± 0.1 |
| EGFRK | 87.0 ± 0.3 | 60.5 ± 4.4 | 33.5 ± 0.5 |
| ERBB2 | 43.4 ± 1.0 | 82.3 ± 1.6 | 78.8 ± 0.7 |
| ERBB4 | 95.0 ± 0.1 | 96.2 ± 0.7 | 95.0 ± 0.1 |
| ITK | 75.1 ± 0.0 | 52.8 ± 1.8 | 46.0 ± 2.3 |
| JAK3 | 64.8 ± 0.3 | 46.2 ± 4.2 | 77.1 ± 3.0 |
| TEC | 84.1 ± 0.2 | 71.3 ± 1.0 | 66.4 ± 0.9 |
| TXK | 88.8 ± 0.1 | 37.5 ± 3.3 | 49.7 ± 2.5 |
| Compound | GI50 (μM) 1 | |||
|---|---|---|---|---|
| Raji 2 | Ramos 2 | MRC-5 3 | MCF10A 3 | |
| 17d | 16.1 ± 0.5 | 12.5 ± 0.1 | 28.4 ± 4.0 | 33.4 ± 2.2 |
| 18g | 25.4 ± 1.4 | 16.2 ± 1.2 | >100 | >100 |
| Ibrutinib | 9.5 ± 0.2 | 6.3 ± 4.8 | 40.4 ± 0.9 | 4.6 ± 1.1 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lee, E.; Cho, H.; Lee, D.K.; Ha, J.; Choi, B.J.; Jeong, J.H.; Ryu, J.-H.; Kang, J.S.; Jeon, R. Discovery of 5-Phenoxy-2-aminopyridine Derivatives as Potent and Selective Irreversible Inhibitors of Bruton’s Tyrosine Kinase. Int. J. Mol. Sci. 2020, 21, 8006. https://doi.org/10.3390/ijms21218006
Lee E, Cho H, Lee DK, Ha J, Choi BJ, Jeong JH, Ryu J-H, Kang JS, Jeon R. Discovery of 5-Phenoxy-2-aminopyridine Derivatives as Potent and Selective Irreversible Inhibitors of Bruton’s Tyrosine Kinase. International Journal of Molecular Sciences. 2020; 21(21):8006. https://doi.org/10.3390/ijms21218006
Chicago/Turabian StyleLee, Eun, Hyewon Cho, Da Kyung Lee, JuHyun Ha, Byeong Jo Choi, Ji Hye Jeong, Jae-Ha Ryu, Jong Soon Kang, and Raok Jeon. 2020. "Discovery of 5-Phenoxy-2-aminopyridine Derivatives as Potent and Selective Irreversible Inhibitors of Bruton’s Tyrosine Kinase" International Journal of Molecular Sciences 21, no. 21: 8006. https://doi.org/10.3390/ijms21218006