The Molecular Mechanisms of Iron Metabolism and Its Role in Cardiac Dysfunction and Cardioprotection

 , , , and
, , , and 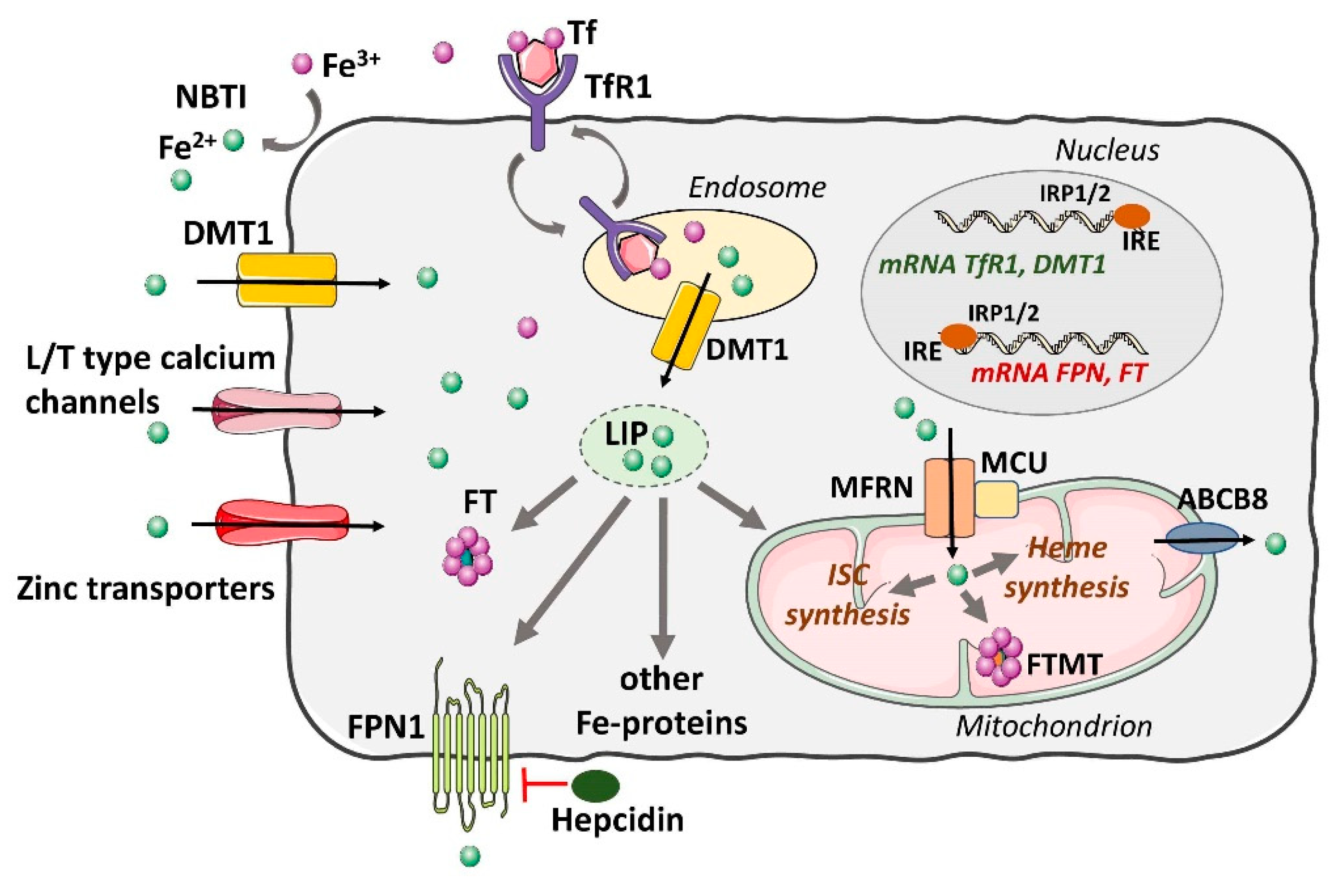{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Systemic Iron Homeostasis
3. Cellular Iron Homeostasis in the Heart
3.1. Uptake and Cellular Regulation of Iron
3.2. The Role of Iron in Heart Mitochondria and Cardiomyocyte Dysfunction
4. Iron and Myocardial I/R Injury
5. Heme Oxygenase System in I/R Injury and Cytoprotection
5.1. Function of Heme Oxygenase
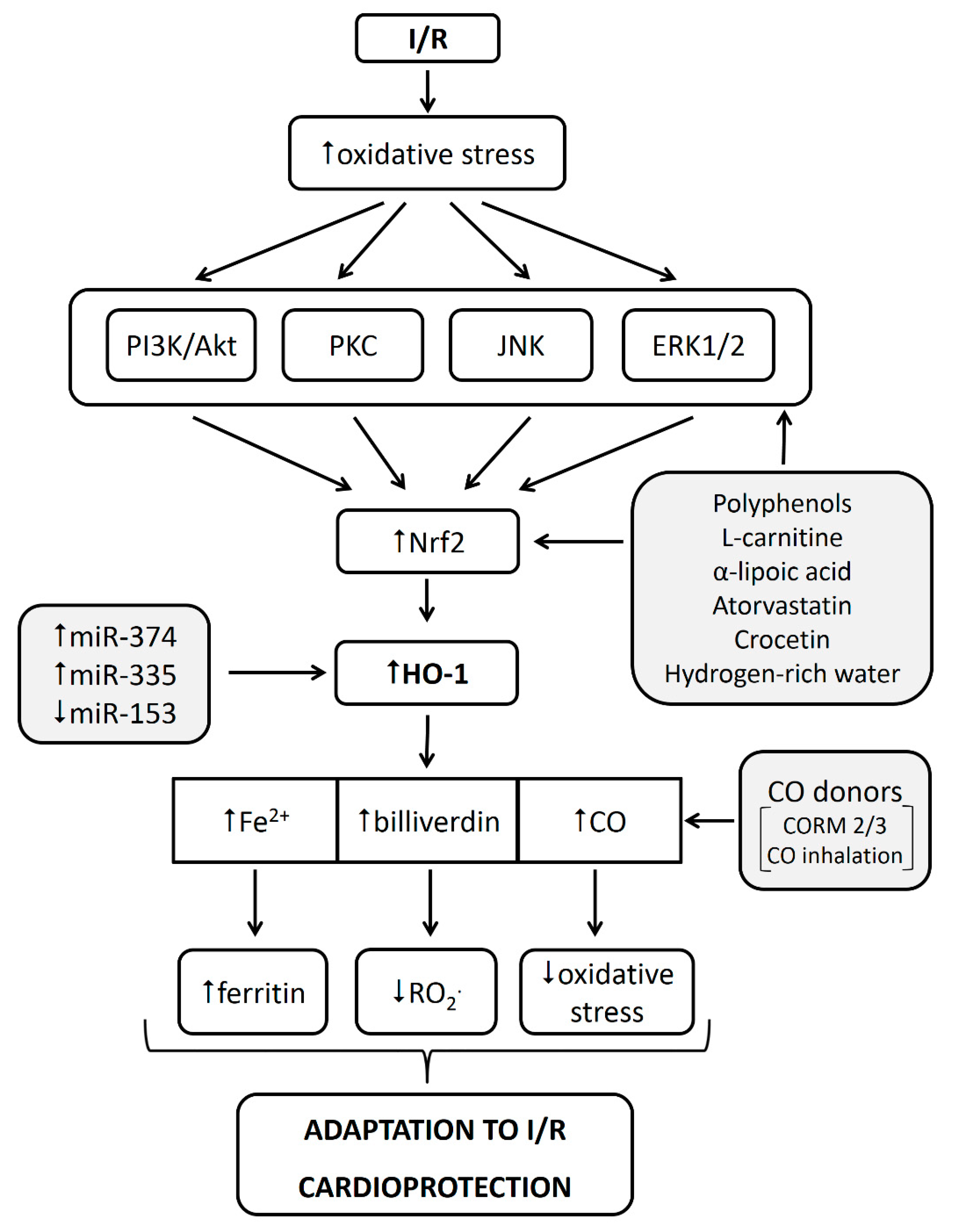
5.2. HO-1 in Cardioprotection against Myocardial I/R Injury
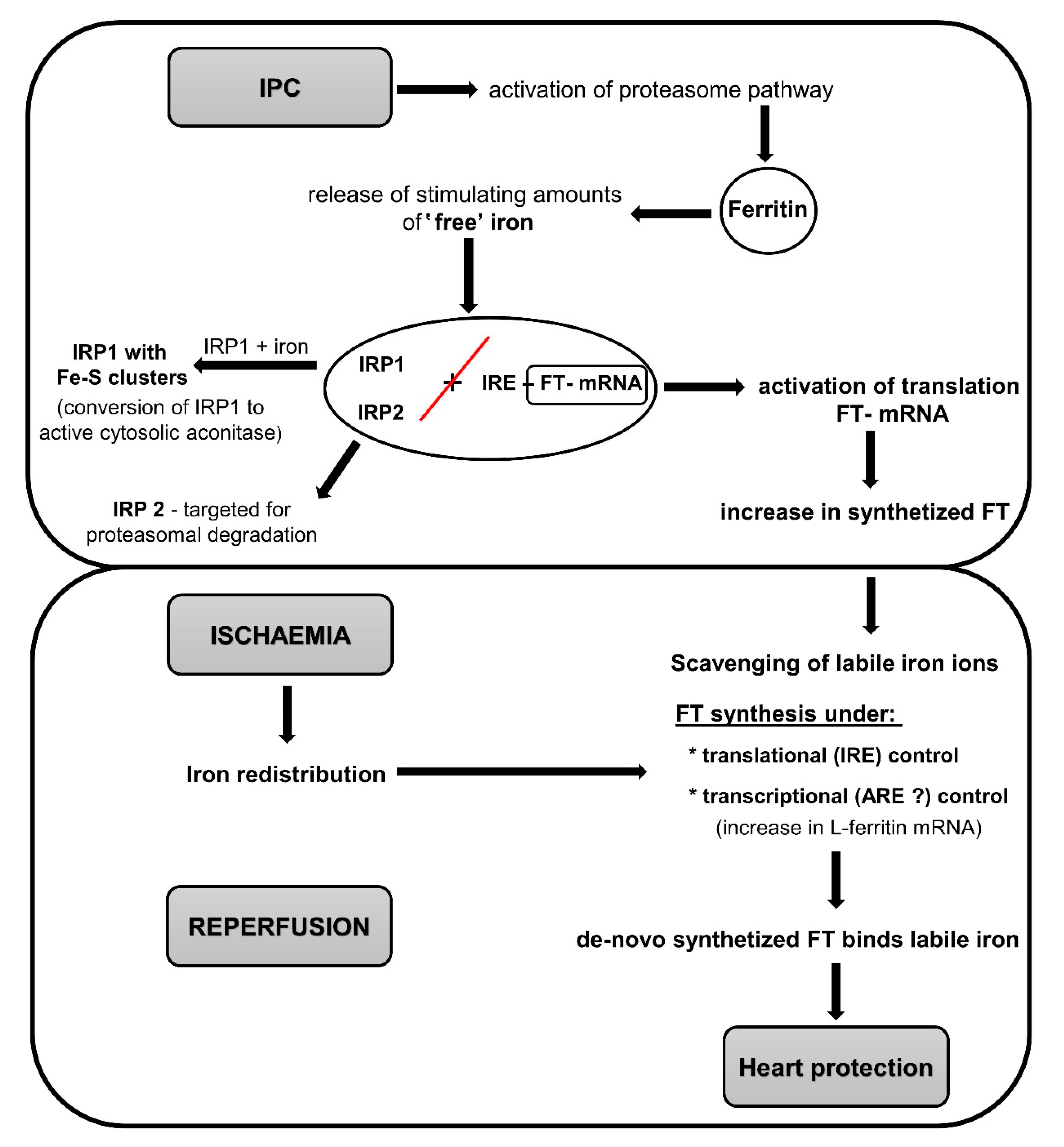
6. Role of Iron and Ferritin in Heart Preconditioning
6.1. Role of Iron and Ferritin in Other Cardioprotective Phenomena
6.2. Ferritin and Protection by NO Donors
7. Iron and Ferroptosis: A Less-Known Form of Cell Death and Its Mechanisms
7.1. Ferroptosis and Post-I/R Myocardial Injury
7.2. Ferroptosis as a Potential Novel Target for Cardioprotection
8. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| ABCB8 | ATP-binding cassette subfamily B member 8 protein |
| ARE | Antioxidant response elements |
| BMP-SMAD | Bone morphogenetic protein/small mothers against decapentaplegic pathway |
| CA | Cytosolic aconitase |
| CO | Carbon monoxide |
| DFO | Deferoxamine |
| DMT-1 | Divalent metal transporter 1 |
| Fe2+, Fe3+ | Ferrous iron, ferric iron |
| FT | Ferritin |
| FTH | Ferritin heavy chain |
| FTL | Ferritin light chain |
| FPN | Ferroportin |
| FTMT | Mitochondria-specific ferritin |
| GPX | Glutathione peroxidase |
| GPX4 | Phospholipid peroxidase glutathione peroxidase 4 |
| GSH | Reduced form of glutathione |
| GSSG | Oxidized form of glutathione |
| HCAECs | Hypothermic preconditioning of human coronary artery endothelial cells |
| HIF | Hypoxia-inducible factor |
| HO. | Hydroxyl radicals |
| HO-1 | Heme oxygenase-1 |
| IPC | Ischemic preconditioning |
| I/R | Ischemia/reperfusion |
| IRE | Iron-Responsive elements |
| IRP1, IRP2 | Iron regulatory proteins 1 and 2 |
| IS | Infarct size |
| ISCs | Iron sulfur clusters |
| LIP | Labile iron pool |
| LOX | Lipoxygenase |
| MCU | Mitochondrial calcium uniporter |
| MFRN | Mitoferrin |
| NO | Nitric oxide |
| Nrf2 | Nuclear factor erythroid 2-related factor 2 |
| NTBI | Nontransferrin-bound iron |
| OH− | Hydroxide ions |
| PBMC | Peripheral blood mononuclear cells |
| RIPC | Remote ischemic preconditioning |
| RO | Alkoxyl radicals |
| RO2 | Peroxyl radicals |
| ROS | Reactive oxygen species |
| Sevo-postC | Postconditioning with sevoflurane |
| SOD | Superoxide dismutase |
| Tf | Transferrin |
| TfR | Tf receptors |
References
- Hirst, J. Mitochondrial complex i. Annu. Rev. Biochem. 2013, 82, 551–575. [Google Scholar] [CrossRef] [PubMed]
- Abbaspour, N.; Hurrell, R.; Kelishadi, R. Review on iron and its importance for human health. J. Res. Med. Sci. 2014, 19, 164–174. [Google Scholar] [PubMed]
- von Haehling, S.; Ebner, N.; Evertz, R.; Ponikowski, P.; Anker, S.D. Iron Deficiency in Heart Failure. JACC Heart Fail. 2019, 7, 36–46. [Google Scholar] [CrossRef] [PubMed]
- Hoes, M.F.; Grote Beverborg, N.; Kijlstra, J.D.; Kuipers, J.; Swinkels, D.W.; Giepmans, B.N.G.; Rodenburg, R.J.; van Veldhuisen, D.J.; de Boer, R.A.; van der Meer, P. Iron deficiency impairs contractility of human cardiomyocytes through decreased mitochondrial function. Eur. J. Heart Fail. 2018, 20, 910–919. [Google Scholar] [CrossRef] [Green Version]
- Das De, S.; Krishna, S.; Jethwa, A. Iron status and its association with coronary heart disease: Systematic review and meta-analysis of prospective studies. Atherosclerosis 2015, 238, 296–303. [Google Scholar] [CrossRef]
- Grammer, T.B.; Scharnagl, H.; Dressel, A.; Kleber, M.E.; Silbernagel, G.; Pilz, S.; Tomaschitz, A.; Koenig, W.; Mueller-Myhsok, B.; März, W.; et al. Iron Metabolism, Hepcidin, and Mortality (the Ludwigshafen Risk and Cardiovascular Health Study). Clin. Chem. 2019, 65, 849–861. [Google Scholar] [CrossRef]
- Pasricha, S.-R.; Drakesmith, H.; Black, J.; Hipgrave, D.; Biggs, B.-A. Control of iron deficiency anemia in low- and middle-income countries. Blood 2013, 121, 2607–2617. [Google Scholar] [CrossRef] [Green Version]
- Pasricha, S.-R.; Drakesmith, H. Iron Deficiency Anemia. Hematol. Oncol. Clin. N. Am. 2016, 30, 309–325. [Google Scholar] [CrossRef]
- Busti, F.; Marchi, G.; Ugolini, S.; Castagna, A.; Girelli, D. Anemia and Iron Deficiency in Cancer Patients: Role of Iron Replacement Therapy. Pharmaceuticals 2018, 11, 94. [Google Scholar] [CrossRef] [Green Version]
- Fraenkel, P.G. Anemia of Inflammation. Med. Clin. N. Am. 2017, 101, 285–296. [Google Scholar] [CrossRef] [Green Version]
- Wen, C.P.; Lee, J.H.; Tai, Y.-P.; Wen, C.; Wu, S.B.; Tsai, M.K.; Hsieh, D.P.H.; Chiang, H.-C.; Hsiung, C.A.; Hsu, C.Y.; et al. High Serum Iron Is Associated with Increased Cancer Risk. Cancer Res. 2014, 74, 6589–6597. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kowdley, K.V.; Brown, K.E.; Ahn, J.; Sundaram, V. ACG Clinical Guideline. Am. J. Gastroenterol. 2019, 114, 1202–1218. [Google Scholar] [CrossRef] [PubMed]
- González-Domínguez, Á.; Visiedo-García, F.M.; Domínguez-Riscart, J.; González-Domínguez, R.; Mateos, R.M.; Lechuga-Sancho, A.M. Iron Metabolism in Obesity and Metabolic Syndrome. Int. J. Mol. Sci. 2020, 21, 5529. [Google Scholar] [CrossRef] [PubMed]
- Vinchi, F.; Porto, G.; Simmelbauer, A.; Altamura, S.; Passos, S.T.; Garbowski, M.; Silva, A.M.N.; Spaich, S.; Seide, S.E.; Sparla, R.; et al. Atherosclerosis is aggravated by iron overload and ameliorated by dietary and pharmacological iron restriction. Eur. Heart J. 2020, 41, 2681–2695. [Google Scholar] [CrossRef]
- Ward, D.M.; Kaplan, J. Ferroportin-mediated iron transport: Expression and regulation. Biochim. Biophys. Acta Mol. Cell Res. 2012, 1823, 1426–1433. [Google Scholar] [CrossRef] [Green Version]
- Drakesmith, H.; Nemeth, E.; Ganz, T. Ironing out Ferroportin. Cell Metab. 2015, 22, 777–787. [Google Scholar] [CrossRef] [Green Version]
- He, H.; Qiao, Y.; Zhou, Q.; Wang, Z.; Chen, X.; Liu, D.; Yin, D.; He, M. Iron Overload Damages the Endothelial Mitochondria via the ROS/ADMA/DDAHII/eNOS/NO Pathway. Oxid. Med. Cell. Longev. 2019, 2019. [Google Scholar] [CrossRef] [Green Version]
- Gammella, E.; Recalcati, S.; Cairo, G. Dual Role of ROS as Signal and Stress Agents: Iron Tips the Balance in favor of Toxic Effects. Oxid. Med. Cell. Longev. 2016, 2016. [Google Scholar] [CrossRef] [Green Version]
- Aisen, P.; Enns, C.; Wessling-Resnick, M. Chemistry and biology of eukaryotic iron metabolism. Int. J. Biochem. Cell Biol. 2001, 33, 940–959. [Google Scholar] [CrossRef]
- Kruszewski, M. Labile iron pool: The main determinant of cellular response to oxidative stress. Mutat. Res. Fundam. Mol. Mech. Mutagen. 2003, 531, 81–92. [Google Scholar] [CrossRef]
- Valko, M.; Jomova, K.; Rhodes, C.J.; Kuča, K.; Musílek, K. Redox- and non-redox-metal-induced formation of free radicals and their role in human disease. Arch. Toxicol. 2016, 90, 1–37. [Google Scholar] [CrossRef]
- Zweier, J.L.; Talukder, M.A.H. The role of oxidants and free radicals in reperfusion injury. Cardiovasc. Res. 2006, 70, 181–190. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chang, H.; Wu, R.; Shang, M.; Sato, T.; Chen, C.; Shapiro, J.S.; Liu, T.; Thakur, A.; Sawicki, K.T.; Prasad, S.V.; et al. Reduction in mitochondrial iron alleviates cardiac damage during injury. EMBO Mol. Med. 2016, 8, 247–267. [Google Scholar] [CrossRef] [PubMed]
- Sullivan, J.L. Iron and the Sex Difference in Heart Disease Risk. Lancet 1981, 317, 1293–1294. [Google Scholar] [CrossRef]
- Shayeghi, M.; Latunde-Dada, G.O.; Oakhill, J.S.; Laftah, A.H.; Takeuchi, K.; Halliday, N.; Khan, Y.; Warley, A.; McCann, F.E.; Hider, R.C.; et al. Identification of an intestinal heme transporter. Cell 2005, 122, 789–801. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lane, D.J.R.; Bae, D.H.; Merlot, A.M.; Sahni, S.; Richardson, D.R. Duodenal cytochrome b (DCYTB) in Iron metabolism: An update on function and regulation. Nutrients 2015, 7, 2274–2296. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Recalcati, S.; Minotti, G.; Cairo, G. Iron regulatory proteins: From molecular mechanisms to drug development. Antioxid. Redox Signal. 2010, 13, 1593–1616. [Google Scholar] [CrossRef] [PubMed]
- Ravasi, G.; Pelucchi, S.; Russo, A.; Mariani, R.; Piperno, A. Ferroportin disease: A novel SLC40A1 mutation. Dig. Liver Dis. 2020, 52, 688–690. [Google Scholar] [CrossRef]
- Brissot, P.; Ropert, M.; Le Lan, C.; Loréal, O. Non-transferrin bound iron: A key role in iron overload and iron toxicity. Biochim. Biophys. Acta Gen. Subj. 2012, 1820, 403–410. [Google Scholar] [CrossRef]
- Gordan, R.; Wongjaikam, S.; Gwathmey, J.K.; Chattipakorn, N.; Chattipakorn, S.C.; Xie, L.H. Involvement of cytosolic and mitochondrial iron in iron overload cardiomyopathy: An update. Heart Fail. Rev. 2018, 23, 801–816. [Google Scholar] [CrossRef]
- Lakhal-Littleton, S.; Wolna, M.; Carr, C.A.; Miller, J.J.J.; Christian, H.C.; Ball, V.; Santos, A.; Diaz, R.; Biggs, D.; Stillion, R.; et al. Cardiac ferroportin regulates cellular iron homeostasis and is important for cardiac function. Proc. Natl. Acad. Sci. USA 2015, 112, 3164–3169. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chang, H.C.; Shapiro, J.S.; Ardehali, H. Getting to the “heart” of Cardiac Disease by Decreasing Mitochondrial Iron. Circ. Res. 2016, 119, 1164–1166. [Google Scholar] [CrossRef] [Green Version]
- Kell, D.B.; Pretorius, E. Serum ferritin is an important inflammatory disease marker, as it is mainly a leakage product from damaged cells. Metallomics 2014, 6, 748–773. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hentze, M.W.; Muckenthaler, M.U.; Galy, B.; Camaschella, C. Two to Tango: Regulation of Mammalian Iron Metabolism. Cell 2010, 142, 24–38. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rishi, G.; Wallace, D.F.; Subramaniam, V.N. Hepcidin: Regulation of the master iron regulator. Biosci. Rep. 2015, 35, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Sangkhae, V.; Nemeth, E. Regulation of the iron homeostatic hormone hepcidin. Adv. Nutr. 2017, 8, 126–136. [Google Scholar] [CrossRef]
- Parrow, N.L.; Fleming, R.E. Liver sinusoidal endothelial cells as iron sensors. Blood 2017, 129, 397–398. [Google Scholar] [CrossRef] [Green Version]
- Yamamoto, K.; Kuragano, T.; Kimura, T.; Nanami, M.; Hasuike, Y.; Nakanishi, T. Interplay of adipocyte and hepatocyte: Leptin upregulates hepcidin. Biochem. Biophys. Res. Commun. 2018, 495, 1548–1554. [Google Scholar] [CrossRef]
- Majzunova, M.; Dovinova, I.; Barancik, M.; Chan, J.Y.H. Redox signaling in pathophysiology of hypertension. J. Biomed. Sci. 2013, 20, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Kerins, M.J.; Ooi, A. The Roles of NRF2 in Modulating Cellular Iron Homeostasis. Antioxid. Redox Signal. 2018, 29, 1756–1773. [Google Scholar] [CrossRef] [Green Version]
- Lim, P.J.; Duarte, T.L.; Arezes, J.; Garcia-Santos, D.; Hamdi, A.; Pasricha, S.R.; Armitage, A.E.; Mehta, H.; Wideman, S.; Santos, A.G.; et al. Nrf2 controls iron homoeostasis in haemochromatosis and thalassaemia via Bmp6 and hepcidin. Nat. Metab. 2019, 1, 519–531. [Google Scholar] [CrossRef] [PubMed]
- Madu, A.J.; Ughasoro, M.D. Anaemia of Chronic Disease: An In-Depth Review. Med. Princ. Pract. 2017, 26, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Oudit, G.Y.; Sun, H.; Trivieri, M.G.; Koch, S.E.; Dawood, F.; Ackerley, C.; Yazdanpanah, M.; Wilson, G.J.; Schwartz, A.; Liu, P.P.; et al. L-type Ca2+ channels provide a major pathway for iron entry into cardiomyocytes in iron-overload cardiomyopathy. Nat. Med. 2003, 9, 1187–1194. [Google Scholar] [CrossRef] [PubMed]
- Liuzzi, J.P.; Aydemir, F.; Nam, H.; Knutson, M.D.; Cousins, R.J. Zip14 (Slc39a14) mediates non-transferrin-bound iron uptake into cells. Proc. Natl. Acad. Sci. USA 2006, 103, 13612–13617. [Google Scholar] [CrossRef] [Green Version]
- Watt, R.K. A Unified Model for Ferritin Iron Loading by the Catalytic Center: Implications for Controlling “Free Iron” during Oxidative Stress. ChemBioChem 2013, 14, 415–419. [Google Scholar] [CrossRef]
- Arosio, P.; Ingrassia, R.; Cavadini, P. Ferritins: A family of molecules for iron storage, antioxidation and more. Biochim. Biophys. Acta Gen. Subj. 2009, 1790, 589–599. [Google Scholar] [CrossRef]
- Kawabata, H. Transferrin and transferrin receptors update. Free Radic. Biol. Med. 2019, 133, 46–54. [Google Scholar] [CrossRef]
- Pan, Y.H.; Sader, K.; Powell, J.J.; Bleloch, A.; Gass, M.; Trinick, J.; Warley, A.; Li, A.; Brydson, R.; Brown, A. 3D morphology of the human hepatic ferritin mineral core: New evidence for a subunit structure revealed by single particle analysis of HAADF-STEM images. J. Struct. Biol. 2009, 166, 22–31. [Google Scholar] [CrossRef] [Green Version]
- Hentze, M.W.; Kühn, L.C. Molecular control of vertebrate iron metabolism: mRNA-based regulatory circuits operated by iron, nitric oxide, and oxidative stress. Proc. Natl. Acad. Sci. USA 1996, 93, 8175–8182. [Google Scholar] [CrossRef] [Green Version]
- Hentze, M.W.; Caughman, S.W.; Rouault, T.A.; Barriocanal, J.G.; Dancis, A.; Harford, J.B.; Klausner, R.D. Identification of the iron-responsive element for the translational regulation of human ferritin mRNA. Science 1987, 238, 1570–1573. [Google Scholar] [CrossRef] [Green Version]
- Haddad, S.; Wang, Y.; Galy, B.; Korf-Klingebiel, M.; Hirsch, V.; Baru, A.M.; Rostami, F.; Reboll, M.R.; Heineke, J.; Flögel, U.; et al. Iron-regulatory proteins secure iron availability in cardiomyocytes to prevent heart failure. Eur. Heart J. 2017, 38, 362–372. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paterek, A.; Mackiewicz, U.; Mączewski, M. Iron and the heart: A paradigm shift from systemic to cardiomyocyte abnormalities. J. Cell. Physiol. 2019, 234, 21613–21629. [Google Scholar] [CrossRef] [PubMed]
- Lakhal-Littleton, S.; Wolna, M.; Chung, Y.J.; Christian, H.C.; Heather, L.C.; Brescia, M.; Ball, V.; Diaz, R.; Santos, A.; Biggs, D.; et al. An essential cell-autonomous role for hepcidin in cardiac iron homeostasis. eLife 2016, 5, 1–25. [Google Scholar] [CrossRef] [PubMed]
- Lakhal-Littleton, S.; Robbins, P.A. The interplay between iron and oxygen homeostasis with a particular focus on the heart. J. Appl. Physiol. 2017, 123, 967–973. [Google Scholar] [CrossRef] [PubMed]
- Horowitz, M.P.; Greenamyre, J.T. Mitochondrial iron metabolism and its role in neurodegeneration. J. Alzheimer’s Dis. 2010, 20, S551–S568. [Google Scholar] [CrossRef] [Green Version]
- Richardson, D.R.; Lane, D.J.R.; Becker, E.M.; Huang, M.L.H.; Whitnall, M.; Rahmanto, Y.S.; Sheftel, A.D.; Ponka, P. Mitochondrial iron trafficking and the integration of iron metabolism between the mitochondrion and cytosol. Proc. Natl. Acad. Sci. USA 2010, 107, 10775–10782. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vela, D. Keeping heart homeostasis in check through the balance of iron metabolism. Acta Physiol. 2020, 228, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Lesnefsky, E.J.; Chen, Q.; Tandler, B.; Hoppel, C.L. Mitochondrial Dysfunction and Myocardial Ischemia-Reperfusion: Implications for Novel Therapies. Annu. Rev. Pharmacol. Toxicol. 2017, 57, 535–565. [Google Scholar] [CrossRef]
- Paul, B.T.; Manz, D.H.; Torti, F.M.; Torti, S.V. Mitochondria and Iron: Current questions. Expert Rev. Hematol. 2017, 10, 65–79. [Google Scholar] [CrossRef] [Green Version]
- Wofford, J.D.; Chakrabarti, M.; Lindahl, P.A. Mössbauer Spectra of Mouse Hearts Reveal Age-dependent Changes in Mitochondrial and Ferritin Iron Levels. J. Biol. Chem. 2017, 292, 5546–5554. [Google Scholar] [CrossRef] [Green Version]
- Chan, S.; Lian, Q.; Chen, M.P.; Jiang, D.; Ho, J.T.K.; Cheung, Y.F.; Chan, G.C.F. Deferiprone inhibits iron overload-induced tissue factor bearing endothelial microparticle generation by inhibition oxidative stress induced mitochondrial injury, and apoptosis. Toxicol. Appl. Pharmacol. 2018, 338, 148–158. [Google Scholar] [CrossRef]
- Sripetchwandee, J.; Kenknight, S.B.; Sanit, J.; Chattipakorn, S.; Chattipakorn, N. Blockade of mitochondrial calcium uniporter prevents cardiac mitochondrial dysfunction caused by iron overload. Acta Physiol. 2014, 210, 330–341. [Google Scholar] [CrossRef] [PubMed]
- Ichikawa, Y.; Bayeva, M.; Ghanefar, M.; Potini, V.; Sun, L.; Mutharasan, R.K.; Wu, R.; Khechaduri, A.; Naik, T.J.; Ardehali, H. Disruption of ATP-binding cassette B8 in mice leads to cardiomyopathy through a decrease in mitochondrial iron export. Proc. Natl. Acad. Sci. USA 2012, 109, 4152–4157. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, W.; Chang, S.; Wu, Q.; Xu, Z.; Wang, P.; Li, Y.; Yu, P.; Gao, G.; Shi, Z.; Duan, X.; et al. Mitochondrial ferritin protects the murine myocardium from acute exhaustive exercise injury. Cell Death Dis. 2016, 7, e2475. [Google Scholar] [CrossRef] [PubMed]
- Melenovsky, V.; Petrak, J.; Mracek, T.; Benes, J.; Borlaug, B.A.; Nuskova, H.; Pluhacek, T.; Spatenka, J.; Kovalcikova, J.; Drahota, Z.; et al. Myocardial iron content and mitochondrial function in human heart failure: A direct tissue analysis. Eur. J. Heart Fail. 2017, 19, 522–530. [Google Scholar] [CrossRef]
- Kumfu, S.; Chattipakorn, S.; Fucharoen, S.; Chattipakorn, N. Mitochondrial calcium uniporter blocker prevents cardiac mitochondrial dysfunction induced by iron overload in thalassemic mice. BioMetals 2012, 25, 1167–1175. [Google Scholar] [CrossRef]
- Khamseekaew, J.; Kumfu, S.; Wongjaikam, S.; Kerdphoo, S.; Jaiwongkam, T.; Srichairatanakool, S.; Fucharoen, S.; Chattipakorn, S.C.; Chattipakorn, N. Effects of iron overload, an iron chelator and a T-Type calcium channel blocker on cardiac mitochondrial biogenesis and mitochondrial dynamics in thalassemic mice. Eur. J. Pharmacol. 2017, 799, 118–127. [Google Scholar] [CrossRef]
- Berenshtein, E.; Vaisman, B.; Goldberg-Langerman, C.; Kitrossky, N.; Konijn, A.M.; Chevion, M. Roles of ferritin and iron in ischemic preconditioning of the heart. Mol. Cell. Biochem. 2002, 234–235, 283–292. [Google Scholar] [CrossRef]
- Chevion, M.; Jiang, Y.; Har-El, R.; Berenshtein, E.; Uretzky, G.; Kitrossky, N. Copper and iron are mobilized following myocardial ischemia: Possible predictive criteria for tissue injury. Proc. Natl. Acad. Sci. USA 1993, 90, 1102–1106. [Google Scholar] [CrossRef] [Green Version]
- Kramer, J.H.; Lightfoot, F.G.; Weglicki, W.B. Cardiac tissue iron: Effects on post-ischemic function and free radical production, and its possible role during preconditioning. Cell. Mol. Biol. (Noisy-le-Grand) 2000, 46, 1313–1327. [Google Scholar]
- Williams, R.E.; Zweier, J.L.; Flaherty, J.T. Treatment with deferoxamine during ischemia improves functional and metabolic recovery and reduces reperfusion-induced oxygen radica lgeneration in rabbit hearts. Circulation 1991, 83, 1006–1014. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Drossos, G.; Lazou, A.; Panagopoulos, P.; Westaby, S. Deferoxamine cardioplegia reduces superoxide radical production in human myocardium. Ann. Thorac. Surg. 1995, 59, 169–172. [Google Scholar] [CrossRef]
- Tang, W.H.; Wu, S.; Wong, T.M.; Chung, S.K.; Chung, S.S.M. Polyol pathway mediates iron-induced oxidative injury in ischemic-reperfused rat heart. Free Radic. Biol. Med. 2008, 45, 602–610. [Google Scholar] [CrossRef] [PubMed]
- Omiya, S.; Hikoso, S.; Imanishi, Y.; Saito, A.; Yamaguchi, O.; Takeda, T.; Mizote, I.; Oka, T.; Taneike, M.; Nakano, Y.; et al. Downregulation of ferritin heavy chain increases labile iron pool, oxidative stress and cell death in cardiomyocytes. J. Mol. Cell. Cardiol. 2009, 46, 59–66. [Google Scholar] [CrossRef] [PubMed]
- Ramesh Reddy, B.; Kloner, R.A.; Przyklenk, K. Early treatment with deferoxamine limits myocardial ischemic/reperfusion injury. Free Radic. Biol. Med. 1989, 7, 45–52. [Google Scholar] [CrossRef]
- Deboer, D.A.; Clark, R.E. Iron chelation in myocardial preservation after ischemia-reperfusion injury: The importance of pretreatment and toxicity. Ann. Thorac. Surg. 1992, 53, 412–418. [Google Scholar] [CrossRef]
- Fang, X.; Wang, H.; Han, D.; Xie, E.; Yang, X.; Wei, J.; Gu, S.; Gao, F.; Zhu, N.; Yin, X.; et al. Ferroptosis as a target for protection against cardiomyopathy. Proc. Natl. Acad. Sci. USA 2019, 116, 2672–2680. [Google Scholar] [CrossRef] [Green Version]
- Watanabe, B.I.; Limm, W.; Suehiro, A.; Suehiro, G.; Premaratne, S.; McNamara, J.J. Failure of deferoxamine to reduce myocardial infarct size in a primate model of ischemia-reperfusion injury. J. Surg. Res. 1993, 55, 537–542. [Google Scholar] [CrossRef]
- Lesnefsky, E.J.; Hedlund, B.E.; Hallaway, P.E.; Horwitz, L.D. High-Dose Iron-Chelator Therapy During Reperfusion with Deferoxamine-Hydroxyethyl Starch Conjugate Fails to Reduce Canine Infarct Size. J. Cardiovasc. Pharmacol. 1990, 16, 523–528. [Google Scholar] [CrossRef]
- Chatziathanasiou, G.N.; Nikas, D.N.; Katsouras, C.S.; Kazakos, N.D.; Bouba, V.; Vougiouklakis, T.; Naka, K.K.; Michalis, L.K. Combined intravenous treatment with ascorbic acid and desferrioxamine to reduce myocardial reperfusion injury in an experimental model resembling the clinical setting of primary PCI. Hellenic J. Cardiol. 2012, 53, 195–204. [Google Scholar]
- Doulias, P.-T.; Christoforidis, S.; Brunk, U.T.; Galaris, D. Endosomal and lysosomal effects of desferrioxamine: Protection of HeLa cells from hydrogen peroxide-induced DNA damage and induction of cell-cycle arrest. Free Radic. Biol. Med. 2003, 35, 719–728. [Google Scholar] [CrossRef]
- Chan, W.; Taylor, A.J.; Ellims, A.H.; Lefkovits, L.; Wong, C.; Kingwell, B.A.; Natoli, A.; Croft, K.D.; Mori, T.; Kaye, D.M.; et al. Effect of Iron Chelation on Myocardial Infarct Size and Oxidative Stress in ST-Elevation–Myocardial Infarction. Circ. Cardiovasc. Interv. 2012, 5, 270–278. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paraskevaidis, I.A.; Iliodromitis, E.K.; Vlahakos, D.; Tsiapras, D.P.; Nikolaidis, A.; Marathias, A.; Michalis, A.; Kremastinos, D.T. Deferoxamine infusion during coronary artery bypass grafting ameliorates lipid peroxidation and protects the myocardium against reperfusion injury: Immediate and long-term significance. Eur. Heart J. 2005, 26, 263–270. [Google Scholar] [CrossRef] [PubMed]
- Duffy, S.J.; Biegelsen, E.S.; Holbrook, M.; Russell, J.D.; Gokce, N.; Keaney, J.F.; Vita, J.A. Iron Chelation Improves Endothelial Function in Patients With Coronary Artery Disease. Circulation 2001, 103, 2799–2804. [Google Scholar] [CrossRef] [Green Version]
- Lamas, G.A.; Goertz, C.; Boineau, R.; Mark, D.B.; Rozema, T.; Nahin, R.L.; Lindblad, L.; Lewis, E.F.; Drisko, J.; Lee, K.L.; et al. Effect of Disodium EDTA Chelation Regimen on Cardiovascular Events in Patients With Previous Myocardial Infarction. JAMA 2013, 309, 1241. [Google Scholar] [CrossRef] [Green Version]
- Muñoz-Sánchez, J.; Chánez-Cárdenas, M.E. A review on hemeoxygenase-2: Focus on cellular protection and oxygen response. Oxid. Med. Cell. Longev. 2014, 2014, 25–28. [Google Scholar] [CrossRef] [Green Version]
- Tsuchihashi, S.; Fondevila, C.; Kupiec-Weglinski, J.W. Heme oxygenase system in ischemia and reperfusion injury. Ann. Transplant. 2004, 9, 84–87. [Google Scholar]
- Otterbein, L.E.; Foresti, R.; Motterlini, R. Heme Oxygenase-1 and Carbon Monoxide in the Heart: The Balancing Act between Danger Signaling and Pro-Survival. Circ. Res. 2016, 118, 1940–1959. [Google Scholar] [CrossRef] [Green Version]
- Cheng, Y.; Rong, J. Therapeutic Potential of Heme Oxygenase-1/carbon Monoxide System Against Ischemia-Reperfusion Injury. Curr. Pharm. Des. 2017, 23, 3884–3898. [Google Scholar] [CrossRef]
- Alam, J.; Stewart, D.; Touchard, C.; Boinapally, S.; Choi, A.M.K.; Cook, J.L. Nrf2, a Cap‘n’Collar Transcription Factor, Regulates Induction of the Heme Oxygenase-1 Gene. J. Biol. Chem. 1999, 274, 26071–26078. [Google Scholar] [CrossRef] [Green Version]
- Kumada, Y.; Takahashi, T.; Shimizu, H.; Nakamura, R.; Omori, E.; Inoue, K.; Morimatsu, H. Therapeutic effect of carbon monoxide-releasing molecule-3 on acute lung injury after hemorrhagic shock and resuscitation. Exp. Ther. Med. 2019, 17, 3429–3440. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berberat, P.O.; Katori, M.; Kaczmarek, E.; Anselmo, D.; Lassman, C.; Ke, B.; Shen, X.; Busuttil, R.W.; Yamashita, K.; Csizmadia, E.; et al. Heavy chain ferritin acts as an antiapoptotic gene that protects livers from ischemia reperfusion injury. FASEB J. 2003, 17, 1724–1726. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaga, S.; Zhan, L.; Matsumoto, M.; Maulik, N. Resveratrol enhances neovascularization in the infarcted rat myocardium through the induction of thioredoxin-1, heme oxygenase-1 and vascular endothelial growth factor. J. Mol. Cell. Cardiol. 2005, 39, 813–822. [Google Scholar] [CrossRef] [PubMed]
- Thirunavukkarasu, M.; Penumathsa, S.V.; Koneru, S.; Juhasz, B.; Zhan, L.; Otani, H.; Bagchi, D.; Das, D.K.; Maulik, N. Resveratrol alleviates cardiac dysfunction in streptozotocin-induced diabetes: Role of nitric oxide, thioredoxin, and heme oxygenase. Free Radic. Biol. Med. 2007, 43, 720–729. [Google Scholar] [CrossRef] [Green Version]
- Penumathsa, S.V.; Koneru, S.; Samuel, S.M.; Maulik, G.; Bagchi, D.; Yet, S.-F.; Menon, V.P.; Maulik, N. Strategic targets to induce neovascularization by resveratrol in hypercholesterolemic rat myocardium: Role of caveolin-1, endothelial nitric oxide synthase, hemeoxygenase-1, and vascular endothelial growth factor. Free Radic. Biol. Med. 2008, 45, 1027–1034. [Google Scholar] [CrossRef] [Green Version]
- Cheng, L.; Jin, Z.; Zhao, R.; Ren, K.; Deng, C.; Yu, S. Resveratrol attenuates inflammation and oxidative stress induced by myocardial ischemia-reperfusion injury: Role of Nrf2/ARE pathway. Int. J. Clin. Exp. Med. 2015, 8, 10420–10428. [Google Scholar]
- Xu, D.; Hu, M.-J.; Wang, Y.-Q.; Cui, Y.-L. Antioxidant Activities of Quercetin and Its Complexes for Medicinal Application. Molecules 2019, 24, 1123. [Google Scholar] [CrossRef] [Green Version]
- Sun, Z.-M.; Guan, P.; Luo, L.-F.; Qin, L.-Y.; Wang, N.; Zhao, Y.-S.; Ji, E.-S. Resveratrol protects against CIH-induced myocardial injury by targeting Nrf2 and blocking NLRP3 inflammasome activation. Life Sci. 2020, 245, 117362. [Google Scholar] [CrossRef]
- Zhao, T.; Chen, S.; Wang, B.; Cai, D. L-carnitine reduces myocardial oxidative stress and alleviates myocardial ischemia-reperfusion injury by activating nuclear transcription-Related Factor 2 (Nrf2)/Heme oxygenase-1 (HO-1) signaling pathway. Med. Sci. Monit. 2020, 26, e923251-1. [Google Scholar] [CrossRef]
- Hou, X.; Fu, M.; Cheng, B.; Kang, Y.; Xie, D. Galanthamine improves myocardial ischemia-reperfusion-induced cardiac dysfunction, endoplasmic reticulum stress-related apoptosis, and myocardial fibrosis by suppressing AMPK/Nrf2 pathway in rats. Ann. Transl. Med. 2019, 7, 634. [Google Scholar] [CrossRef]
- Sun, G.; Li, Y.; Ji, Z. Atorvastatin attenuates inflammation and oxidative stress induced by ischemia/reperfusion in rat heart via the Nrf2 transcription factor. Int. J. Clin. Exp. Med. 2015, 8, 14837–14845. [Google Scholar] [PubMed]
- Yin, W.; Wang, C.; Peng, Y.; Yuan, W.; Zhang, Z.; Liu, H.; Xia, Z.; Ren, C.; Qian, J. Dexmedetomidine alleviates H2O2-induced oxidative stress and cell necroptosis through activating of α2-adrenoceptor in H9C2 cells. Mol. Biol. Rep. 2020, 47, 3629–3639. [Google Scholar] [CrossRef] [PubMed]
- Yang, M.; Mao, G.; Ouyang, L.; Shi, C.; Hu, P.; Huang, S. Crocetin alleviates myocardial ischemia/reperfusion injury by regulating inflammation and the unfolded protein response. Mol. Med. Rep. 2020, 21, 641–648. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Liu, T.; Liu, L.; Li, S.; Zhang, Z.; Zhang, R.; Zhou, Y.; Liu, F. Effect of hydrogen-rich water on the Nrf2/ARE signaling pathway in rats with myocardial ischemia-reperfusion injury. J. Bioenerg. Biomembr. 2019, 51, 393–402. [Google Scholar] [CrossRef] [PubMed]
- Juhasz, B.; Varga, B.; Czompa, A.; Bak, I.; Lekli, I.; Gesztelyi, R.; Zsuga, J.; Kemeny-Beke, A.; Antal, M.; Szendrei, L.; et al. Postischemic cardiac recovery in heme oxygenase-1 transgenic ischemic/reperfused mouse myocardium. J. Cell. Mol. Med. 2011, 15, 1973–1982. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fujisaki, N.; Kohama, K.; Nishimura, T.; Yamashita, H.; Ishikawa, M.; Kanematsu, A.; Yamada, T.; Lee, S.; Yumoto, T.; Tsukahara, K.; et al. Donor pretreatment with carbon monoxide prevents ischemia/reperfusion injury following heart transplantation in rats. Med. Gas Res. 2016, 6, 122. [Google Scholar] [PubMed] [Green Version]
- Meng, C.; Ma, L.; Liu, J.; Cui, X.; Liu, R.; Xing, J.; Zhou, H. Inflation with carbon monoxide in rat donor lung during cold ischemia phase ameliorates graft injury. Exp. Biol. Med. 2016, 241, 246–254. [Google Scholar] [CrossRef] [Green Version]
- Zhao, S.; Lin, Q.; Li, H.; He, Y.; Fang, X.; Chen, F.; Chen, C.; Huang, Z. Carbon monoxide releasing molecule-2 attenuated ischemia/reperfusion-induced apoptosis in cardiomyocytes via a mitochondrial pathway. Mol. Med. Rep. 2014, 9, 754–762. [Google Scholar] [CrossRef]
- Stein, A.B.; Bolli, R.; Dawn, B.; Sanganalmath, S.K.; Zhu, Y.; Wang, O.-L.; Guo, Y.; Motterlini, R.; Xuan, Y.-T. Carbon monoxide induces a late preconditioning-mimetic cardioprotective and antiapoptotic milieu in the myocardium. J. Mol. Cell. Cardiol. 2012, 52, 228–236. [Google Scholar] [CrossRef] [Green Version]
- Yellon, D.M.; Downey, J.M. Preconditioning the myocardium: From cellular physiology to clinical cardiology. Physiol. Rev. 2003, 83, 1113–1151. [Google Scholar] [CrossRef]
- Murry, C.E.; Jennings, R.B.; Reimer, K.A. Preconditioning with ischemia: A delay of lethal cell injury in ischemic myocardium. Circulation 1986, 74, 1124–1136. [Google Scholar]
- Ponka, P. Cellular iron metabolism. Kidney Int. Suppl. 1999, 55, 2–11. [Google Scholar]
- Galleano, M.; Tapia, G.; Puntarulo, S.; Varela, P.; Videla, L.A.; Fernández, V. Liver preconditioning induced by iron in a rat model of ischemia/reperfusion. Life Sci. 2011, 89, 221–228. [Google Scholar]
- Chevion, M.; Leibowitz, S.; Aye, N.N.; Novogrodsky, O.; Singer, A.; Avizemer, O.; Bulvik, B.; Konijn, A.M.; Berenshtein, E. Heart protection by ischemic preconditioning: A novel pathway initiated by iron and mediated by ferritin. J. Mol. Cell. Cardiol. 2008, 45, 839–845. [Google Scholar] [PubMed]
- Bulvik, B.E.; Berenshtein, E.; Meyron-Holtz, E.G.; Konijn, A.M.; Chevion, M. Cardiac Protection by Preconditioning Is Generated via an Iron-Signal Created by Proteasomal Degradation of Iron Proteins. PLoS ONE 2012, 7, e48947. [Google Scholar]
- Zieger, M.A.J.; Gupta, M.P. Hypothermic preconditioning of endothelial cells attenuates cold-induced injury by a ferritin-dependent process. Free Radic. Biol. Med. 2009, 46, 680–691. [Google Scholar] [PubMed]
- Li, Y.; Zhou, Y.; Zhang, D.; Wu, W.Y.; Kang, X.; Wu, Q.; Wang, P.; Liu, X.; Gao, G.; Zhou, Y.; et al. Hypobaric hypoxia regulates iron metabolism in rats. J. Cell. Biochem. 2019, 120, 14076–14087. [Google Scholar]
- Ravingerová, T.; Neckář, J.; Kolář, F. Ischemic tolerance of rat hearts in acute and chronic phases of experimental diabetes. Mol. Cell. Biochem. 2003, 249, 167–174. [Google Scholar]
- Chen, H.; Shen, W.L.; Wang, X.H.; Chen, H.Z.; Gu, J.Z.; Fu, J.; Ni, Y.F.; Gao, P.J.; Zhu, D.L.; Higashino, H. Paradoxically enhanced heart tolerance to ischaemia in type 1 diabetes and role of increased osmolarity. Clin. Exp. Pharmacol. Physiol. 2006, 33, 910–916. [Google Scholar] [PubMed]
- Ravingerová, T.; Adameová, A.; Matejíková, J.; Kelly, T.; Nemčeková, M.; Kucharská, J.; Pecháňová, O.; Lazou, A. Subcellular mechanisms of adaptation in the diabetic myocardium: Relevance to ischemic preconditioning in the nondiseased heart. Exp. Clin. Cardiol. 2010, 15, 68–76. [Google Scholar]
- Muráriková, M.; Ferko, M.; Waczulíková, I.; Jašová, M.; Kancirová, I.; Murínová, J.; Ravingerová, T. Changes in mitochondrial properties may contribute to enhanced resistance to ischemia-reperfusion injury in the diabetic rat heart. Can. J. Physiol. Pharmacol. 2017, 95, 969–976. [Google Scholar] [CrossRef] [PubMed]
- Andelova, N.; Waczulikova, I.; Talian, I.; Sykora, M.; Ferko, M. mPTP proteins regulated by streptozotocin-induced diabetes mellitus are effectively involved in the processes of maintaining myocardial metabolic adaptation. Int. J. Mol. Sci. 2020, 21, 2622. [Google Scholar]
- Vinokur, V.; Berenshtein, E.; Bulvik, B.; Grinberg, L.; Eliashar, R.; Chevion, M. The Bitter Fate of the Sweet Heart: Impairment of Iron Homeostasis in Diabetic Heart Leads to Failure in Myocardial Protection by Preconditioning. PLoS ONE 2013, 8, e62948. [Google Scholar] [CrossRef] [PubMed]
- Grievink, H.; Kuzmina, N.; Chevion, M.; Drenger, B. Sevoflurane postconditioning is not mediated by ferritin accumulation and cannot be rescued by simvastatin in isolated streptozotocin-induced diabetic rat hearts. PLoS ONE 2019, 14, e0211238. [Google Scholar] [CrossRef]
- Mieszkowski, J.; Kochanowicz, M.; Zychowska, M.; Kochanowicz, A.; Grzybkowska, A.; Anczykowska, K.; Sawicki, P.; Borkowska, A.; Niespodzinski, B.; Antosiewicz, J. Ferritin genes overexpression in pbmc and a rise in exercise performance as an adaptive response to ischaemic preconditioning in young men. BioMed Res. Int. 2019, 2019. [Google Scholar] [CrossRef] [Green Version]
- Grievink, H.; Zeltcer, G.; Drenger, B.; Berenshtein, E.; Chevion, M. Protection by nitric oxide donors of isolated rat hearts is associated with activation of redox metabolism and ferritin accumulation. PLoS ONE 2016, 11, e0159951. [Google Scholar] [CrossRef] [Green Version]
- Bertrand, R.L. Iron accumulation, glutathione depletion, and lipid peroxidation must occur simultaneously during ferroptosis and are mutually amplifying events. Med. Hypotheses 2017, 101, 69–74. [Google Scholar] [CrossRef]
- Dixon, S.J.; Lemberg, K.M.; Lamprecht, M.R.; Skouta, R.; Zaitsev, E.M.; Gleason, C.E.; Patel, D.N.; Bauer, A.J.; Cantley, A.M.; Yang, W.S.; et al. Ferroptosis: An iron-dependent form of nonapoptotic cell death. Cell 2012, 149, 1060–1072. [Google Scholar] [CrossRef] [Green Version]
- Feng, Y.; Madungwe, N.B.; Imam Aliagan, A.D.; Tombo, N.; Bopassa, J.C. Liproxstatin-1 protects the mouse myocardium against ischemia/reperfusion injury by decreasing VDAC1 levels and restoring GPX4 levels. Biochem. Biophys. Res. Commun. 2019, 520, 606–611. [Google Scholar] [CrossRef]
- Wang, C.; Zhu, L.; Yuan, W.; Sun, L.; Xia, Z.; Zhang, Z.; Yao, W. Diabetes aggravates myocardial ischaemia reperfusion injury via activating Nox2-related programmed cell death in an AMPK-dependent manner. J. Cell. Mol. Med. 2020, 24, 6670–6679. [Google Scholar] [CrossRef]
- Li, J. Ferroptosis: Past, present and future. Cell Death Dis. 2020, 11, 1–13. [Google Scholar] [CrossRef]
- Park, T.J.; Park, J.H.; Lee, G.S.; Lee, J.Y.; Shin, J.H.; Kim, M.W.; Kim, Y.S.; Kim, J.Y.; Oh, K.J.; Han, B.S.; et al. Quantitative proteomic analyses reveal that GPX4 downregulation during myocardial infarction contributes to ferroptosis in cardiomyocytes. Cell Death Dis. 2019, 10, 1–15. [Google Scholar] [CrossRef] [Green Version]
- Wang, C.; Yuan, W.; Hu, A.; Lin, J.; Xia, Z.; Yang, C.; Li, Y.; Zhang, Z. Dexmedetomidine alleviated sepsis-induced myocardial ferroptosis and septic heart injury. Mol. Med. Rep. 2020, 22, 175–184. [Google Scholar] [CrossRef] [PubMed]
- Xie, Y.; Hou, W.; Song, X.; Yu, Y.; Huang, J.; Sun, X.; Kang, R.; Tang, D. Ferroptosis: Process and function. Cell Death Differ. 2016, 23, 369–379. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stockwell, B.R.; Friedmann Angeli, J.P.; Bayir, H.; Bush, A.I.; Conrad, M.; Dixon, S.J.; Fulda, S.; Gascón, S.; Hatzios, S.K.; Kagan, V.E.; et al. Ferroptosis: A Regulated Cell Death Nexus Linking Metabolism, Redox Biology, and Disease. Cell 2017, 171, 273–285. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, W.S.; Sriramaratnam, R.; Welsch, M.E.; Shimada, K.; Skouta, R.; Viswanathan, V.S.; Cheah, J.H.; Clemons, P.A.; Shamji, A.F.; Clish, C.B.; et al. Regulation of ferroptotic cancer cell death by GPX4. Cell 2014, 156, 317–331. [Google Scholar] [CrossRef] [Green Version]
- Forcina, G.C.; Dixon, S.J. GPX4 at the Crossroads of Lipid Homeostasis and Ferroptosis. Proteomics 2019, 19, 1800311. [Google Scholar] [CrossRef]
- Haeggström, J.Z.; Funk, C.D. Lipoxygenase and leukotriene pathways: Biochemistry, biology, and roles in disease. Chem. Rev. 2011, 111, 5866–5896. [Google Scholar] [CrossRef]
- Kagan, V.E.; Mao, G.; Qu, F.; Angeli, J.P.F.; Doll, S.; Croix, C.S.; Dar, H.H.; Liu, B.; Tyurin, V.A.; Ritov, V.B.; et al. Oxidized arachidonic and adrenic PEs navigate cells to ferroptosis. Nat. Chem. Biol. 2017, 13, 81–90. [Google Scholar] [CrossRef]
- Gao, M.; Yi, J.; Zhu, J.; Minikes, A.M.; Monian, P.; Thompson, C.B.; Jiang, X. Role of Mitochondria in Ferroptosis. Mol. Cell 2019, 73, 354–363.e3. [Google Scholar] [CrossRef] [Green Version]
- Torii, S.; Shintoku, R.; Kubota, C.; Yaegashi, M.; Torii, R.; Sasaki, M.; Suzuki, T.; Mori, M.; Yoshimoto, Y.; Takeuchi, T.; et al. An essential role for functional lysosomes in ferroptosis of cancer cells. Biochem. J. 2016, 473, 769–777. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Magtanong, L.; Ko, P.J.; To, M.; Cao, J.Y.; Forcina, G.C.; Tarangelo, A.; Ward, C.C.; Cho, K.; Patti, G.J.; Nomura, D.K.; et al. Exogenous Monounsaturated Fatty Acids Promote a Ferroptosis-Resistant Cell State. Cell Chem. Biol. 2019, 26, 420–432.e9. [Google Scholar] [CrossRef] [PubMed]
- Friedmann Angeli, J.P.; Conrad, M. Selenium and GPX4, a vital symbiosis. Free Radic. Biol. Med. 2018, 127, 153–159. [Google Scholar] [CrossRef] [Green Version]
- Shimada, K.; Hayano, M.; Pagano, N.C.; Stockwell, B.R. Cell-Line Selectivity Improves the Predictive Power of Pharmacogenomic Analyses and Helps Identify NADPH as Biomarker for Ferroptosis Sensitivity. Cell Chem. Biol. 2016, 23, 225–235. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shimada, K.; Skouta, R.; Kaplan, A.; Yang, W.S.; Hayano, M.; Dixon, S.J.; Brown, L.M.; Valenzuela, C.A.; Wolpaw, A.J.; Stockwell, B.R. Global survey of cell death mechanisms reveals metabolic regulation of ferroptosis. Nat. Chem. Biol. 2016, 12, 497–503. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ganame, J.; Messalli, G.; Dymarkowski, S.; Rademakers, F.E.; Desmet, W.; Van de Werf, F.; Bogaert, J. Impact of myocardial haemorrhage on left ventricular function and remodelling in patients with reperfused acute myocardial infarction. Eur. Heart J. 2009, 30, 1440–1449. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carrick, D.; Haig, C.; Ahmed, N.; McEntegart, M.; Petrie, M.C.; Eteiba, H.; Hood, S.; Watkins, S.; Lindsay, M.M.; Davie, A.; et al. Myocardial Hemorrhage After Acute Reperfused ST-Segment–Elevation Myocardial Infarction. Circ. Cardiovasc. Imaging 2016, 9, e004148. [Google Scholar] [CrossRef] [Green Version]
- Bulluck, H.; Rosmini, S.; Abdel-Gadir, A.; White, S.K.; Bhuva, A.N.; Treibel, T.A.; Fontana, M.; Ramlall, M.; Hamarneh, A.; Sirker, A.; et al. Residual Myocardial Iron Following Intramyocardial Hemorrhage During the Convalescent Phase of Reperfused ST-Segment–Elevation Myocardial Infarction and Adverse Left Ventricular Remodeling. Circ. Cardiovasc. Imaging 2016, 9, e004940. [Google Scholar] [CrossRef] [Green Version]
- Baba, Y.; Higa, J.K.; Shimada, B.K.; Horiuchi, K.M.; Suhara, T.; Kobayashi, M.; Woo, J.D.; Aoyagi, H.; Marh, K.S.; Kitaoka, H.; et al. Protective effects of the mechanistic target of rapamycin against excess iron and ferroptosis in cardiomyocytes. Am. J. Physiol. Circ. Physiol. 2018, 314, H659–H668. [Google Scholar] [CrossRef] [Green Version]
- Robbers, L.F.H.J.; Eerenberg, E.S.; Teunissen, P.F.A.; Jansen, M.F.; Hollander, M.R.; Horrevoets, A.J.G.; Knaapen, P.; Nijveldt, R.; Heymans, M.W.; Levi, M.M.; et al. Magnetic resonance imaging-defined areas of microvascular obstruction after acute myocardial infarction represent microvascular destruction and haemorrhage. Eur. Heart J. 2013, 34, 2346–2353. [Google Scholar] [CrossRef] [Green Version]
- Fang, X.; Cai, Z.; Wang, H.; Han, D.; Cheng, Q.; Zhang, P.; Gao, F.; Yu, Y.; Song, Z.; Wu, Q.; et al. Loss of Cardiac Ferritin H Facilitates Cardiomyopathy via Slc7a11-Mediated Ferroptosis. Circ. Res. 2020, 127, 486–501. [Google Scholar] [CrossRef] [PubMed]
- Ramu, E.; Korach, A.; Houminer, E.; Schneider, A.; Elami, A.; Schwalb, H. Dexrazoxane Prevents Myocardial Ischemia/Reperfusion-Induced Oxidative Stress in the Rat Heart. Cardiovasc. Drugs Ther. 2006, 20, 343–348. [Google Scholar] [CrossRef] [PubMed]
- Philipp, S.; Cui, L.; Ludolph, B.; Kelm, M.; Schulz, R.; Cohen, M.V.; Downey, J.M. Desferoxamine and ethyl-3,4-dihydroxybenzoate protect myocardium by activating NOS and generating mitochondrial ROS. Am. J. Physiol. Circ. Physiol. 2006, 290, H450–H457. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miotto, G.; Rossetto, M.; Di Paolo, M.L.; Orian, L.; Venerando, R.; Roveri, A.; Vučković, A.M.; Bosello Travain, V.; Zaccarin, M.; Zennaro, L.; et al. Insight into the mechanism of ferroptosis inhibition by ferrostatin-1. Redox Biol. 2020, 28, 101328. [Google Scholar] [CrossRef] [PubMed]
- Adameova, A.; Goncalvesova, E.; Szobi, A.; Dhalla, N.S. Necroptotic cell death in failing heart: Relevance and proposed mechanisms. Heart Fail. Rev. 2016, 21, 213–221. [Google Scholar] [CrossRef] [PubMed]
- Adameova, A.; Hrdlicka, J.; Szobi, A.; Farkasova, V.; Kopaskova, K.; Murarikova, M.; Neckar, J.; Kolar, F.; Ravingerova, T.; Dhalla, N.S. Evidence of necroptosis in hearts subjected to various forms of ischemic insults. Can. J. Physiol. Pharmacol. 2017, 95, 1163–1169. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lichý, M.; Szobi, A.; Hrdlička, J.; Horváth, C.; Kormanová, V.; Rajtík, T.; Neckář, J.; Kolář, F.; Adameová, A. Different signalling in infarcted and non-infarcted areas of rat failing hearts: A role of necroptosis and inflammation. J. Cell. Mol. Med. 2019, 23, 6429–6441. [Google Scholar] [CrossRef] [Green Version]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ravingerová, T.; Kindernay, L.; Barteková, M.; Ferko, M.; Adameová, A.; Zohdi, V.; Bernátová, I.; Ferenczyová, K.; Lazou, A. The Molecular Mechanisms of Iron Metabolism and Its Role in Cardiac Dysfunction and Cardioprotection. Int. J. Mol. Sci. 2020, 21, 7889. https://doi.org/10.3390/ijms21217889
Ravingerová T, Kindernay L, Barteková M, Ferko M, Adameová A, Zohdi V, Bernátová I, Ferenczyová K, Lazou A. The Molecular Mechanisms of Iron Metabolism and Its Role in Cardiac Dysfunction and Cardioprotection. International Journal of Molecular Sciences. 2020; 21(21):7889. https://doi.org/10.3390/ijms21217889
Chicago/Turabian StyleRavingerová, Tanya, Lucia Kindernay, Monika Barteková, Miroslav Ferko, Adriana Adameová, Vladislava Zohdi, Iveta Bernátová, Kristina Ferenczyová, and Antigone Lazou. 2020. "The Molecular Mechanisms of Iron Metabolism and Its Role in Cardiac Dysfunction and Cardioprotection" International Journal of Molecular Sciences 21, no. 21: 7889. https://doi.org/10.3390/ijms21217889