A Novel CCT5 Missense Variant Associated with Early Onset Motor Neuropathy

 , , , , ,
, , , , ,
Abstract
:
1. Introduction
2. Results
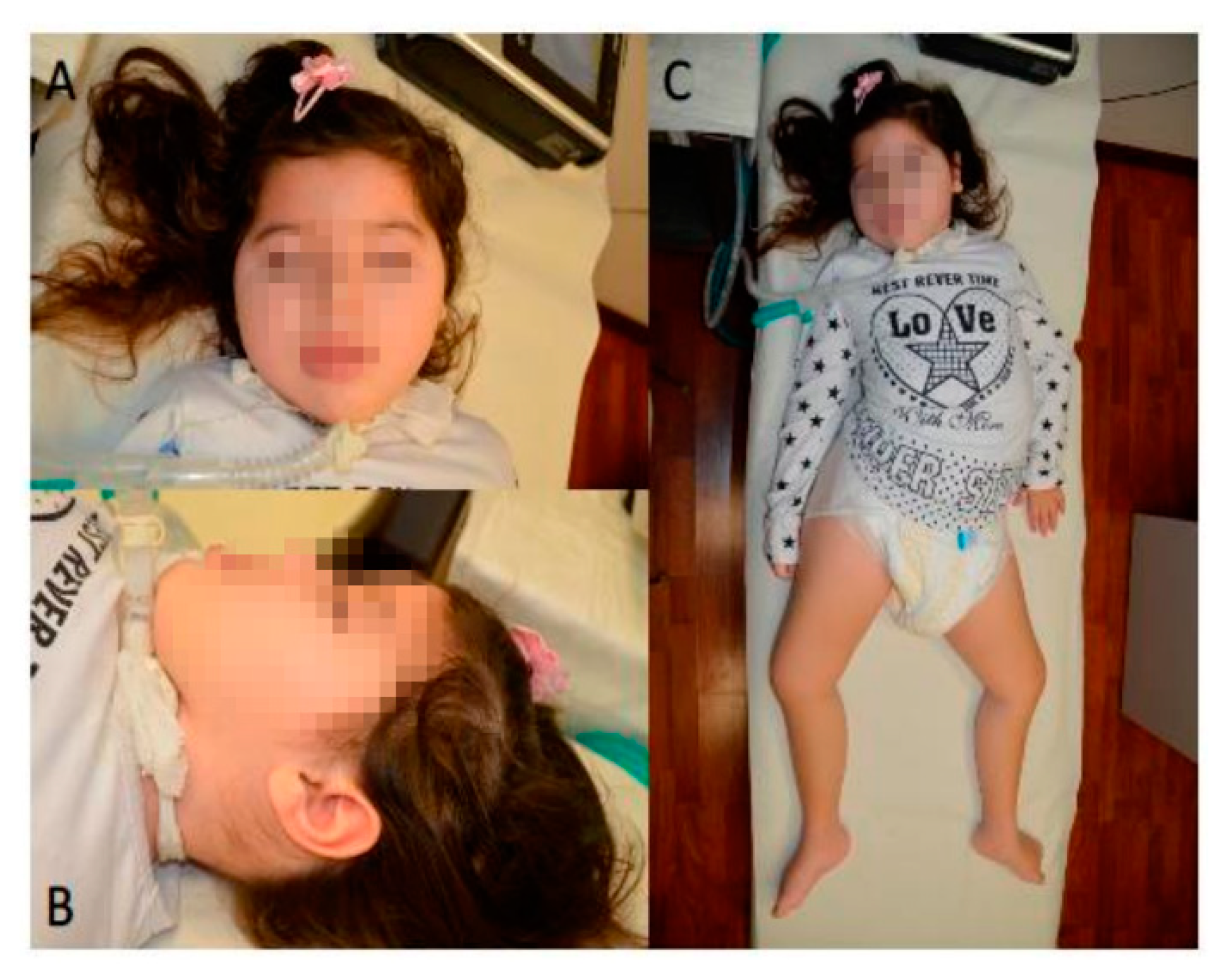
2.1. Clinical History
2.2. Neurological Examination and Electrophysiological Findings
2.3. Motor Evoked Potentials
2.4. Motor Nerve Conduction Study
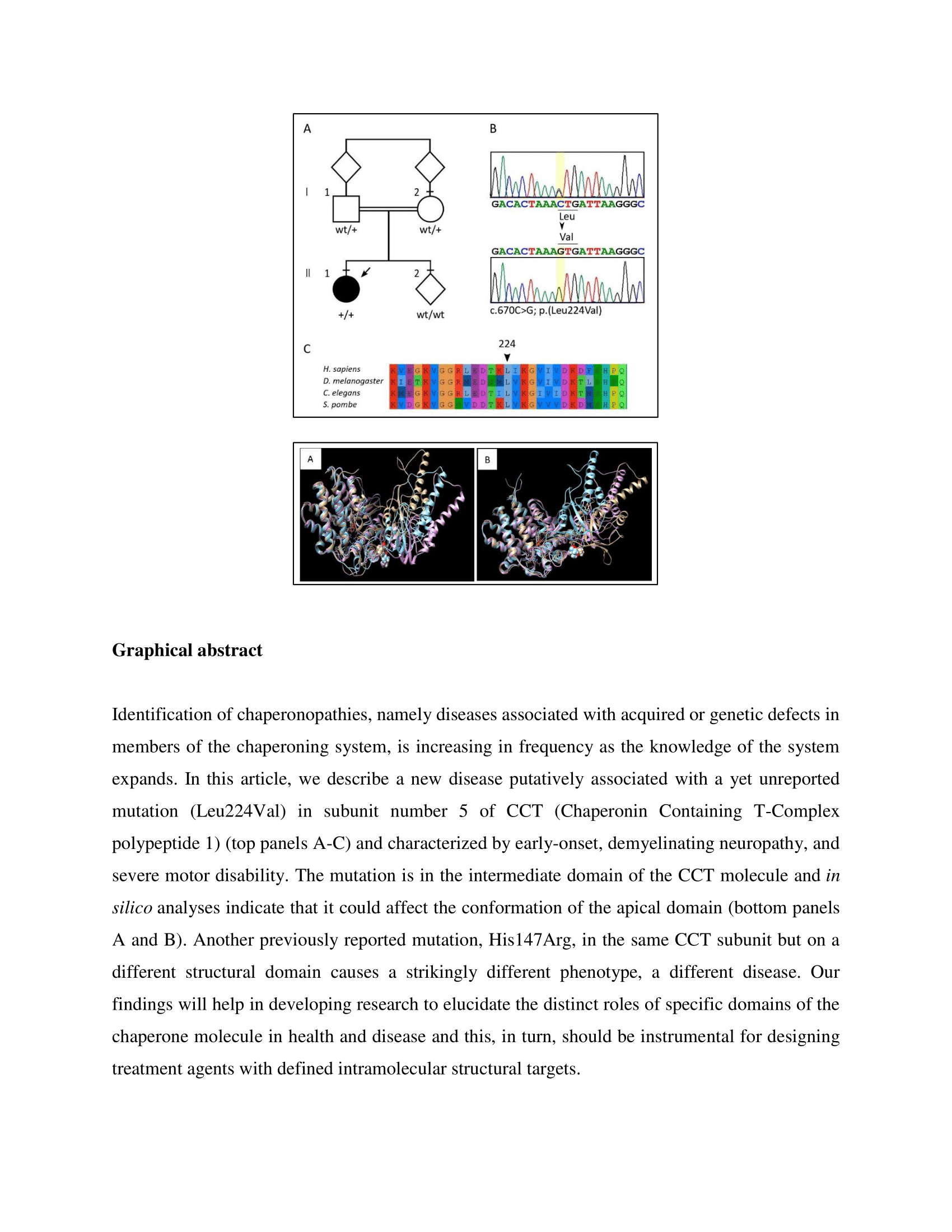
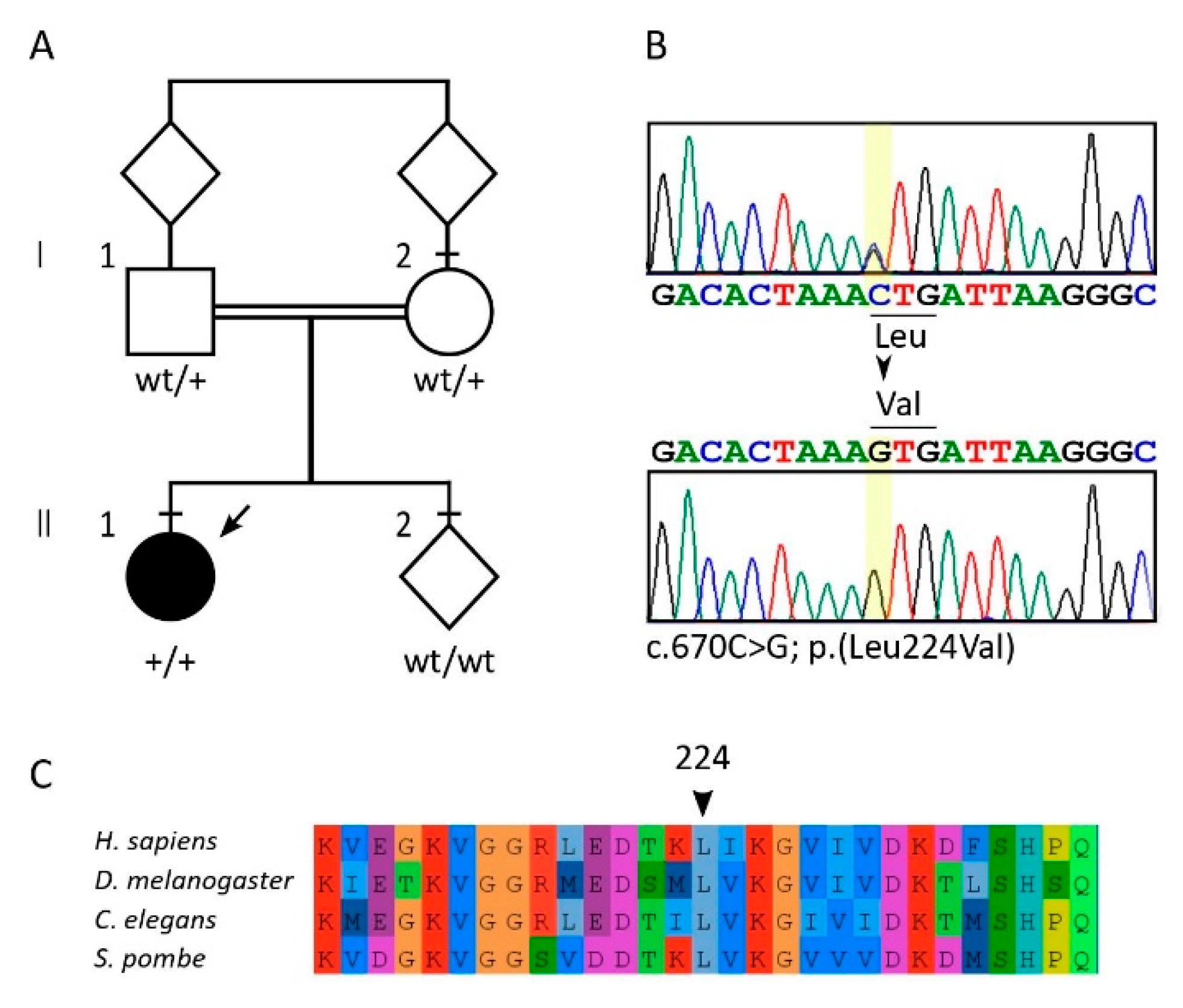
2.5. Laboratory Investigations and Genetic Analyses
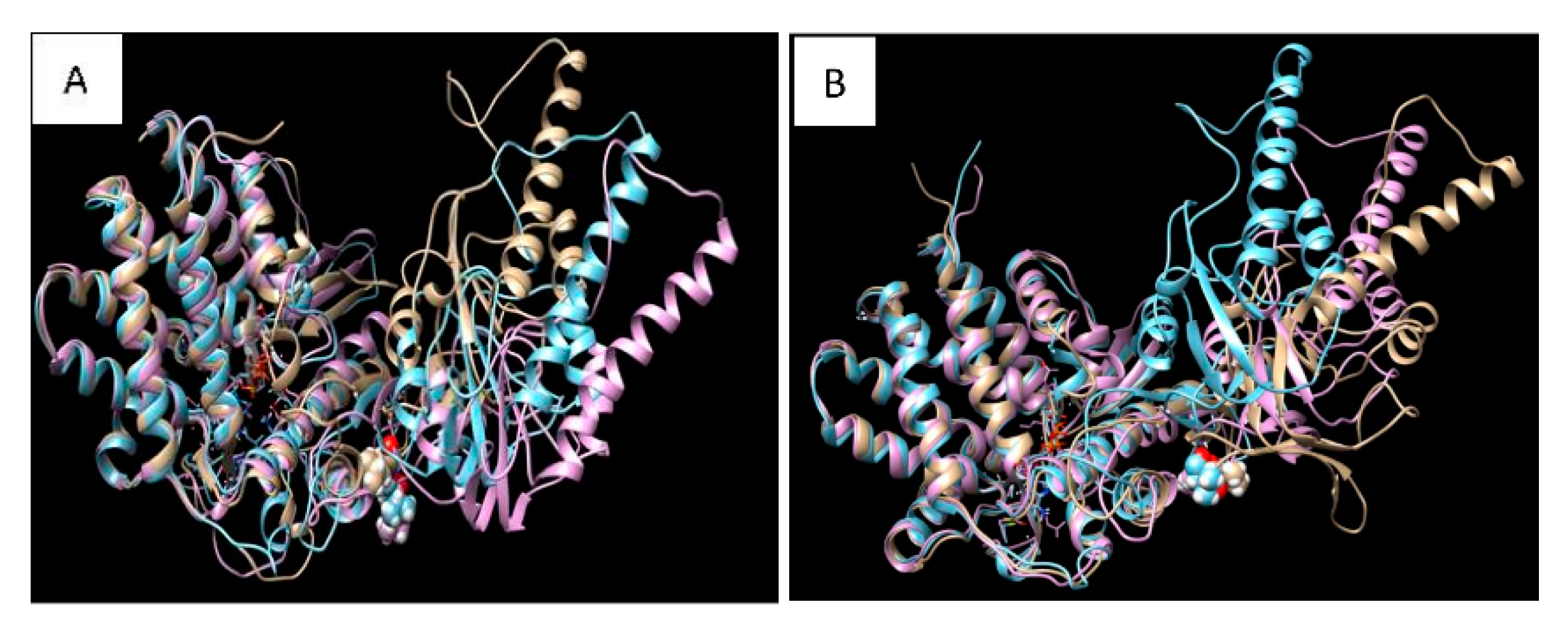
2.6. Molecular Dynamics Simulation and Modelling
3. Discussion
4. Materials and Methods
4.1. Patients
4.2. Motor Evoked Potentials
4.3. Motor Nerve Conduction Study
4.4. Sequencing
4.5. Molecular Dynamics Simulation and Modelling
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Macario, A.J.L.; Conway de Macario, E.; Cappello, F. The Chaperonopathies: Diseases with Defective Molecular Chaperones; Springer: Dordrecht, The Netherlands; Heidelberg, Germany; New York, NY, USA; London, UK, 2013. [Google Scholar]
- Macario, A.J.L.; Conway de Macario, E. Sick Chaperones, Cellular Stress, and Disease. N. Engl. J. Med. 2005, 353, 1489–1501. [Google Scholar] [CrossRef] [PubMed]
- Bouhouche, A.; Benomar, A.; Bouslam, N.; Chkili, T.; Yahyaoui, M. Mutation in the epsilon subunit of the cytosolic chaperonin-containing t-complex peptide-1 (Cct5) gene causes autosomal recessive mutilating sensory neuropathy with spastic paraplegia. J. Med. Genet. 2006, 43, 441–443. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bouhouche, A.; Benomar, A.; Bouslam, N.; Ouazzani, R.; Chkili, T.; Yahyaoui, M. Autosomal recessive mutilating sensory neuropathy with spastic paraplegia maps to chromosome 5p15.31-14.1. Eur. J. Hum. Genet. 2006, 14, 249–252. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morani, F.; Doccini, S.; Sirica, R.; Paterno, M.; Pezzini, F.; Ricca, I.; Simonati, A.; Delledonne, M.; Santorelli, F.M. Functional Transcriptome Analysis in ARSACS KO Cell Model Reveals a Role of Sacsin in Autophagy. Sci. Rep. 2019, 9, 11878. [Google Scholar] [CrossRef] [PubMed]
- Flex, E.; Niceta, M.; Cecchetti, S.; Thiffault, I.; Au, M.G.; Capuano, A.; Piermarini, E.; Ivanova, A.A.; Francis, J.W.; Chillemi, G.; et al. Biallelic Mutations in TBCD, Encoding the Tubulin Folding Cofactor D, Perturb Microtubule Dynamics and Cause Early-Onset Encephalopathy. Am. J. Hum. Genet. 2016, 99, 962–973. [Google Scholar] [CrossRef] [Green Version]
- Macario, A.J.L.; Conway de Macario, E. Molecular mechanisms in chaperonopathies: Clues to understanding the histopathological abnormalities and developing novel therapies. J. Pathol. 2020, 250, 9–18. [Google Scholar] [CrossRef] [Green Version]
- Kalisman, N.; Schröder, G.F.; Levitt, M. The crystal structures of the eukaryotic chaperonin CCT reveal its functional partitioning. Structure 2013, 21, 540–549. [Google Scholar] [CrossRef] [Green Version]
- Willison, K.R. The structure and evolution of eukaryotic chaperonin-containing TCP-1 and its mechanism that folds actin into a protein spring. Biochem. J. 2018, 475, 3009–3034. [Google Scholar] [CrossRef]
- Kubota, H.; Hynes, G.; Willison, K. The chaperonin containing t-complex polypeptide 1 (TCP-1). Multisubunit machinery assisting in protein folding and assembly in the eukaryotic cytosol. Eur. J. Biochem. 1995, 230, 3–16. [Google Scholar] [CrossRef]
- Lopez, T.; Dalton, K.; Frydman, J. The Mechanism and Function of Group II Chaperonins. J. Mol. Biol. 2015, 427, 2919–2930. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Balchin, D.; Miličić, G.; Strauss, M.; Hayer-Hartl, M.; Hartl, F.U. Pathway of Actin Folding Directed by the Eukaryotic Chaperonin TRiC. Cell 2018, 174, 1507–1521. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Niceta, M.; Stellacci, E.; Gripp, K.W.; Zampino, G.; Kousi, M.; Anselmi, M.; Traversa, A.; Ciolfi, A.; Stabley, D.; Bruselles, A.; et al. Mutations Impairing GSK3-Mediated MAF Phosphorylation Cause Cataract, Deafness, Intellectual Disability, Seizures, and a Down Syndrome-like Facies. Am. J. Hum. Genet. 2015, 96, 816–825. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kircher, M.; Witten, D.M.; Jain, P.; O’Roak, B.J.; Cooper, G.M.; Shendure, J. A general framework for estimating the relative pathogenicity of human genetic variants. Nat. Genet. 2014, 46, 310–315. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jagadeesh, K.A.; Wenger, A.M.; Berger, M.J.; Guturu, H.; Stenson, P.D.; Cooper, D.N.; Bernstein, J.A.; Bejerano, G. M-CAP eliminates a majority of variants of uncertain significance in clinical exomes at high sensitivity. Nat. Genet. 2016, 48, 1581–1586. [Google Scholar] [CrossRef]
- Li, Q.; Wang, K. InterVar: Clinical interpretation of genetic variants by the 2015 ACMG-AMP guidelines. Am. J. Hum. Genet. 2017, 100, 267–280. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seelow, D.; Schuelke, M. HomozygosityMapper2012—Bridging the gap between homozygosity mapping and deep sequencing. Nucleic Acids Res. 2012, 40, W516–W520. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Jian, X.; Boerwinkle, E. dbNSFP v2.0: A database of human non-synonymous SNVs and their functional predictions and annotations. Hum. Mutat. 2013, 34, E2393–E2402. [Google Scholar] [CrossRef] [Green Version]
- Pereira, J.H.; McAndrew, R.P.; Sergeeva, O.A.; Ralston, C.Y.; King, J.A.; Adams, P.D. Structure of the human TRiC/CCT Subunit 5 associated with hereditary sensory neuropathy. Sci. Rep. 2017, 7, 3673. [Google Scholar] [CrossRef] [Green Version]
- Van Der Spoel, D.; Lindahl, E.; Hess, B.; Groenhof, G.; Mark, A.E.; Berendsen, H.J.V. GROMACS: Fast, flexible, and free. J. Comp. Chem. 2005, 26, 1701–1718. [Google Scholar] [CrossRef]
- Hess, B.; Kutzner, C.; Van Der Spoel, D.; Lindahl, E. GRGMACS 4: Algorithms for highly efficient, load-balanced, and scalable molecular simulation. J. Chem. Theory Comput. 2008, 4, 435–447. [Google Scholar] [CrossRef] [Green Version]
- Foloppe, N.; MacKerell, A.D., Jr. All-atom empirical force field for nucleic acids: I. Parameter optimization based on small molecule and condensed phase macromolecular target data. J. Comput. Chem. 2000, 21, 86–104. [Google Scholar] [CrossRef]
- MacKerell, A.D.; Banavali, N.K. All-atom empirical force field for nucleic acids: II. Application to molecular dynamics simulations of DNA and RNA in solution. J. Comput. Chem. 2000, 21, 105–120. [Google Scholar] [CrossRef]
- Bussi, G.; Donadio, D.; Parrinello, M. Canonical sampling through velocity rescaling. J. Chem. Phys. 2007, 126, 014101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parrinello, M.; Rahman, A. Polymorphic transitions in single crystals: A new molecular dynamics method. J. Appl. Phys. 1981, 52, 7182–7190. [Google Scholar] [CrossRef]
- Darden, T.; York, D.; Pedersen, L. Particle mesh Ewald: An N·log(N) method for Ewald sums in large systems. J. Chem. Phys. 1993, 98, 10089–10092. [Google Scholar] [CrossRef] [Green Version]
- Essmann, U.; Perera, L.; Berkowitz, M.L. A smooth particle mesh Ewald method. J. Chem. Phys. 1995, 103, 8577–8593. [Google Scholar] [CrossRef] [Green Version]
- Giancarlo, R.; Lo Bosco, G.; Pinello, L.; Utro, F. The Three Steps of Clustering in the Post-Genomic Era. In Biological Knowledge Discovery Handbook; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2013; pp. 519–556. [Google Scholar] [CrossRef]
- Giancarlo, R.; Lo Bosco, G.; Utro, F. Bayesian versus data driven model selection for microarray data. Nat. Comput. 2015, 14, 393–402. [Google Scholar] [CrossRef]
- Camastra, F.; Di Taranto, M.D.; Staiano, A. Statistical and Computational Methods for Genetic Diseases: An Overview. Comput. Math. Method. Med. 2015. [Google Scholar] [CrossRef] [PubMed]
- Söderhjelm, P.; Tribello, G.A.; Parrinello, M. Locating binding poses in protein-ligand systems using reconnaissance metadynamics. Proc. Natl. Acad. Sci. USA 2012, 109, 5170–5175. [Google Scholar] [CrossRef] [Green Version]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera—A visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef] [Green Version]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Leu224Val | His147Arg 1 | |
|---|---|---|
| Gender; Age | Female; 8 years. | Males; 34, 22, 19, and 7 years. |
| Age at onset | Eight months of age. | Early infancy, range from 1 to 5 years. |
| Major clinical signs and symptoms | Progressive hypotonia and hyposthenia. Absent osteotendinous reflexes. Bilateral clonus of the foot. Dystonic crisis with extrapyramidal hypertonia, dystonic posturing of the extremities, fine tremors, and up-rolling of the eyes suggestive of seizures. Absent eye contact, horizontal nystagmus, tetra paresis, marked axial and segmental hypotonia, incomplete head control. | Spasticity in walking with legs in form of X. Increased tendon reflexes with ankle clonus, Babinsky sign, and discreet distal amyotrophy. Normal motor function. Sensory loss for all modalities in both upper and lower limbs, particularly in foot. Normal cranial nerves, coordination, and cognitive functions. |
| General disease progression | Progressive worsening of dystonic crisis with neuronal impairment. Severe hypotonia and feeding problems (poor suck, swallowing difficulties, gastroesophageal reflux). Difficulties getting an upright position and inability to walk. Dyspnea, fever, and acute respiratory failure. Absence of sphincter function control. Expressive language absent. Jaw dystonia. Severe impairment of intelligence quotient. | Oldest patient: scars of healed ulcers in hands and osteomyelitis of the feet leading to amputation of his two legs up to the thighs. |
| Data Source | Leu224Val | His147Arg 1 |
|---|---|---|
| MRI 2 | Mild delay of white matter myelination of the posterior regions. Thin corpus callosum, signs of ventricular enlargement and a pattern of diffuse hyperintensity of cerebral white matter, consistent with diffuse hypo/demyelination, enlargement of sulci and subarachnoid spaces mainly in the frontal and parietal areas. | Severe atrophy of the spinal cord predominantly in the posterior tract. |
| EEG | Disorganized brain electrical activity, with theta and delta activity on left temporo-occipital derivations. Asymmetric pattern (generalized seizures). Irregular background rhythm characterized by slow diffuse waves; high voltage delta waves prevalent in posterior head regions. Absence of physiological figures of sleep. Presence of rapid pharmacological activity. | Not reported |
| Motor nerve conduction studies (see also Table 3) | Signs of both axonal and demyelinating motor neuropathy upon stimulation of the median and peroneal nerve. Presence of reduced motor amplitude and delayed motor latency responses after stimulation of distal sites of the median and peroneal nerves. No compound motor evoked potentials were elicited by stimulation of proximal nerve sites. | Normal or slightly reduced motor conduction velocity of the median and the peroneal nerves. |
| Motor evoked potentials (see also Table 3) | Absent motor evoked potentials following cortical stimulation in either the APB or TA muscles. | Not reported |
| Ultrasound scanning | Normal cardiac, renal, hepatic, and urogenital ultrasound results. | Not reported. |
| X-rays | Chest X rays, lumbar spine scoliosis. | Not reported |
| Karyotype | Normal | Not reported |
| Array CGH | Normal | Not reported |
| Biochemical measurements (see also Table S1). | Normal levels of Apo B lipoprotein, total cholesterol, and triglycerides. Other: normal values of organic acids, carnitine, congenital glycosylation disorders, VLCFA, full plasma amino acid profile, purine and pyrimidine, serum lactate, ammonia, glycemia, beta galactosidase and transaminases. | Decreased levels of Apo B lipoprotein, total cholesterol, and triglycerides. Other: not reported. |
| Transcranial magnetic stimulation | Target Muscle | Latency | Amplitude | ||||
| APB 1 | Absent Response | Absent Response | |||||
| TA | Absent Response | Absent Response | |||||
| Nerve conduction study | Nerve | Distal Latency (ms) | Proximal Latency (ms) | Distal Amplitude (mV) | Proximal Amplitude (mV) | ||
| median | 5.1 | absent response | 0.9 | 0 | |||
| peroneal | 7.2 | absent response | 1.2 | 0 | |||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Antona, V.; Scalia, F.; Giorgio, E.; Radio, F.C.; Brusco, A.; Oliveri, M.; Corsello, G.; Lo Celso, F.; Vadalà, M.; Conway de Macario, E.; et al. A Novel CCT5 Missense Variant Associated with Early Onset Motor Neuropathy. Int. J. Mol. Sci. 2020, 21, 7631. https://doi.org/10.3390/ijms21207631
Antona V, Scalia F, Giorgio E, Radio FC, Brusco A, Oliveri M, Corsello G, Lo Celso F, Vadalà M, Conway de Macario E, et al. A Novel CCT5 Missense Variant Associated with Early Onset Motor Neuropathy. International Journal of Molecular Sciences. 2020; 21(20):7631. https://doi.org/10.3390/ijms21207631
Chicago/Turabian StyleAntona, Vincenzo, Federica Scalia, Elisa Giorgio, Francesca C. Radio, Alfredo Brusco, Massimiliano Oliveri, Giovanni Corsello, Fabrizio Lo Celso, Maria Vadalà, Everly Conway de Macario, and et al. 2020. "A Novel CCT5 Missense Variant Associated with Early Onset Motor Neuropathy" International Journal of Molecular Sciences 21, no. 20: 7631. https://doi.org/10.3390/ijms21207631