Diet, Microbiota and Brain Health: Unraveling the Network Intersecting Metabolism and Neurodegeneration

 , ,
, ,
Abstract
:1. Introduction
2. Research Method and Data Collection
3. Nutrients, Microbiota and Brain Health
3.1. Diet-CNS Interactions
3.2. Gut Microbiota-CNS Interactions
3.3. Diet and Microbiota on the Immune System: An Indirect Link to the CNS
4. Roles of Diet and Microbiota in Neurodegenerative Diseases
4.1. Alzheimer’s Disease
4.1.1. AD and Metabolic Disorders
4.1.2. AD and Diet
4.1.3. AD and Microbiota
4.2. Parkinson’s Disease
4.2.1. PD and Metabolic Disorders
4.2.2. PD and Diet
4.2.3. PD and Microbiota
4.3. Amyotrophic Lateral Sclerosis
4.3.1. ALS and Metabolic Disorders
4.3.2. ALS and Diet
4.3.3. ALS and Microbiota
5. Future Perspectives
6. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| AD | Alzheimer’s disease |
| AHR | Aryl hydrocarbon receptor |
| ALS | Amyotrophic lateral sclerosis |
| APOB | Apolipoprotein B |
| APOE | Apolipoprotein E |
| APP | Amyloid precursor protein |
| Aβ | Amyloid-β |
| BBB | Blood–brain barrier |
| BDNF | Brain-derived neurotrophic factor |
| BMI | Body Mass Index |
| C9orf72 | Chromosome 9 open reading frame 72 |
| CIDP | Chronic inflammatory demyelinating polyneuropathy |
| CNS | Central nervous system |
| DHA | Docosahexaenoic acid |
| EAE | Experimental autoimmune encephalomyelitis |
| ENS | Enteric nervous system |
| EPA | Eicosapentaenoic acid |
| FA | Fatty acids |
| FGF21 | fibroblast growth factor 21 |
| FMT | Fecal microbial transplantation |
| FUS | Fused in sarcoma |
| GCS | Glucosylceramide synthase |
| GF | Germ-free |
| GLP-1 | Glucagon-like peptide-1 |
| GM | Gut Microbiota |
| HCAR2 | Hydroxycarboxylic acid receptor 2 |
| IEB | Intestinal epithelial barrier |
| IGF-1 | Insulin-like growth factor-1 |
| IL-1β | Interleukin-1β |
| IL-6 | Interleukin-6 |
| LDL-C | Low density lipoprotein-cholesterol |
| LPS | Lipopolysaccharide |
| LRRK2 | Leucine-rich repeat kinase 2 |
| MAFLD | Metabolic dysfunction-associated fatty liver disease |
| MeD | Mediterranean Diet |
| MS | Multiple sclerosis |
| MTHFD1 | Methylenetetrahydrofolate dehydrogenase-1 |
| NF-kB | Nuclear factor kappa-light-chain-enhancer of activated B cells |
| NLRP3 | Nucleotide-binding oligomerization domain, leucine rich repeat and pyrin domain containing 3 |
| PARK7 | Parkinson disease protein 7 |
| PD | Parkinson’s disease |
| PGC1α | Peroxisome proliferator activated receptor gamma coactivator 1-α |
| RBD | Rapid eye movement sleep behaviour disorder |
| ROS | Reactive oxygen species |
| SCFA | Short-chain fatty acids |
| SLC2A9 | Solute carrier family 2 member 9 |
| SNCA | α-synuclein (gene) |
| SOD1 | Superoxide dismutase 1 |
| T2D | Type 2 diabetes |
| TARDBP | TAR DNA binding protein-43 |
| TDP-43 | Transactive response DNA-binding protein 43 |
| Th | T helper cells |
| TLR | Toll-like receptor |
| TNF-α | Tumor necrosis factor-α |
| Treg | T regulatory cells |
| WT | Wild type |
| α-syn | α-synuclein (protein) |
| ω-3 PUFA | Omega-3 polyunsaturated fatty acids |
References
- Gan, L.; Cookson, M.R.; Petrucelli, L.; La Spada, A.R. Converging pathways in neurodegeneration, from genetics to mechanisms. Nat. Neurosci. 2018, 21, 1300–1309. [Google Scholar] [CrossRef] [PubMed]
- Kivipelto, M.; Mangialasche, F.; Ngandu, T. Lifestyle interventions to prevent cognitive impairment, dementia and Alzheimer disease. Nat. Rev. Neurol. 2018, 14, 653–666. [Google Scholar] [CrossRef] [PubMed]
- Kamel, F. Paths from Pesticides to Parkinson’s. Science 2013, 341, 722–723. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, M.-D.; Little, J.; Gomes, J.; Cashman, N.R.; Krewski, D. Identification of risk factors associated with onset and progression of amyotrophic lateral sclerosis using systematic review and meta-analysis. NeuroToxicology 2017, 61, 101–130. [Google Scholar] [CrossRef] [PubMed]
- Athanasopoulos, D.; Karagiannis, G.; Tsolaki, M. Recent Findings in Alzheimer Disease and Nutrition Focusing on Epigenetics. Adv. Nutr. 2016, 7, 917–927. [Google Scholar] [CrossRef] [PubMed]
- Le Chatelier, E.; Nielsen, T.; Qin, J.; Prifti, E.; Hildebrand, F.; Falony, G.; Almeida, M.; Arumugam, M.; Batto, J.M.; Kennedy, S.; et al. Richness of human gut microbiome correlates with metabolic markers. Nature 2013, 500, 541–546. [Google Scholar] [CrossRef] [PubMed]
- Odamaki, T.; Kato, K.; Sugahara, H.; Hashikura, N.; Takahashi, S.; Xiao, J.Z.; Abe, F.; Osawa, R. Age-related changes in gut microbiota composition from newborn to centenarian: A cross-sectional study. BMC Microbiol. 2016, 16, 90. [Google Scholar] [CrossRef] [Green Version]
- Claesson, M.J.; Jeffery, I.B.; Conde, S.; Power, S.E.; O’Connor, E.M.; Cusack, S.; Harris, H.M.; Coakley, M.; Lakshminarayanan, B.; O’Sullivan, O.; et al. Gut microbiota composition correlates with diet and health in the elderly. Nature 2012, 488, 178–184. [Google Scholar] [CrossRef]
- Kaati, G.; Bygren, L.O.; Edvinsson, S. Cardiovascular and diabetes mortality determined by nutrition during parents’ and grandparents’ slow growth period. Eur. J. Hum. Genet. EJHG 2002, 10, 682–688. [Google Scholar] [CrossRef] [Green Version]
- Kaati, G.; Bygren, L.O.; Pembrey, M.; Sjostrom, M. Transgenerational response to nutrition, early life circumstances and longevity. Eur. J. Hum. Genet. EJHG 2007, 15, 784–790. [Google Scholar] [CrossRef]
- Schonfeld, P.; Reiser, G. Why does brain metabolism not favor burning of fatty acids to provide energy? Reflections on disadvantages of the use of free fatty acids as fuel for brain. J. Cereb. Blood Flow Metab. 2013, 33, 1493–1499. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perera, N.D.; Turner, B.J. AMPK Signalling and Defective Energy Metabolism in Amyotrophic Lateral Sclerosis. Neurochem. Res. 2016, 41, 544–553. [Google Scholar] [CrossRef] [PubMed]
- Loera-Valencia, R.; Goikolea, J.; Parrado-Fernandez, C.; Merino-Serrais, P.; Maioli, S. Alterations in cholesterol metabolism as a risk factor for developing Alzheimer’s disease: Potential novel targets for treatment. J. Steroid Biochem. Mol. Biol. 2019, 190, 104–114. [Google Scholar] [CrossRef] [PubMed]
- Tan, H.-E.; Sisti, A.C.; Jin, H.; Vignovich, M.; Villavicencio, M.; Tsang, K.S.; Goffer, Y.; Zuker, C.S. The gut–brain axis mediates sugar preference. Nature 2020, 580, 511–516. [Google Scholar] [CrossRef] [PubMed]
- Kleinridders, A.; Ferris, H.A.; Cai, W.; Kahn, C.R. Insulin action in brain regulates systemic metabolism and brain function. Diabetes 2014, 63, 2232–2243. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Von Holstein-Rathlou, S.; BonDurant, L.D.; Peltekian, L.; Naber, M.C.; Yin, T.C.; Claflin, K.E.; Urizar, A.I.; Madsen, A.N.; Ratner, C.; Holst, B.; et al. FGF21 Mediates Endocrine Control of Simple Sugar Intake and Sweet Taste Preference by the Liver. Cell Metab. 2016, 23, 335–343. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hill, C.M.; Laeger, T.; Dehner, M.; Albarado, D.C.; Clarke, B.; Wanders, D.; Burke, S.J.; Collier, J.J.; Qualls-Creekmore, E.; Solon-Biet, S.M.; et al. FGF21 Signals Protein Status to the Brain and Adaptively Regulates Food Choice and Metabolism. Cell Rep. 2019, 27, 2934–2947. [Google Scholar] [CrossRef] [Green Version]
- Kullmann, S.; Valenta, V.; Wagner, R.; Tschritter, O.; Machann, J.; Häring, H.-U.; Preissl, H.; Fritsche, A.; Heni, M. Brain insulin sensitivity is linked to adiposity and body fat distribution. Nat. Commun. 2020, 11, 1841. [Google Scholar] [CrossRef] [Green Version]
- Haygood, R.; Fedrigo, O.; Hanson, B.; Yokoyama, K.D.; Wray, G.A. Promoter regions of many neural- and nutrition-related genes have experienced positive selection during human evolution. Nat. Genet. 2007, 39, 1140–1144. [Google Scholar] [CrossRef]
- Isaacs, E.B.; Fischl, B.R.; Quinn, B.T.; Chong, W.K.; Gadian, D.G.; Lucas, A. Impact of Breast Milk on Intelligence Quotient, Brain Size, and White Matter Development. Pediatric Res. 2010, 67, 357–362. [Google Scholar] [CrossRef] [Green Version]
- Victora, C.G.; Horta, B.L.; de Mola, C.L.; Quevedo, L.; Pinheiro, R.T.; Gigante, D.P.; Gonçalves, H.; Barros, F.C. Association between breastfeeding and intelligence, educational attainment, and income at 30 years of age: A prospective birth cohort study from Brazil. Lancet Glob. Health 2015, 3, e199–e205. [Google Scholar] [CrossRef] [Green Version]
- Berger, P.K.; Plows, J.F.; Jones, R.B.; Alderete, T.L.; Yonemitsu, C.; Poulsen, M.; Ryoo, J.H.; Peterson, B.S.; Bode, L.; Goran, M.I. Human milk oligosaccharide 2’-fucosyllactose links feedings at 1 month to cognitive development at 24 months in infants of normal and overweight mothers. PLoS ONE 2020, 15, e0228323. [Google Scholar] [CrossRef] [PubMed]
- Kitajka, K.; Sinclair, A.J.; Weisinger, R.S.; Weisinger, H.S.; Mathai, M.; Jayasooriya, A.P.; Halver, J.E.; Puskas, L.G. Effects of dietary omega-3 polyunsaturated fatty acids on brain gene expression. Proc. Natl. Acad. Sci. USA 2004, 101, 10931–10936. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koletzko, B.; Agostoni, C.; Carlson, S.E.; Clandinin, T.; Hornstra, G.; Neuringer, M.; Uauy, R.; Yamashiro, Y.; Willatts, P. Long chain polyunsaturated fatty acids (LC-PUFA) and perinatal development. Acta Paediatr. 2001, 90, 460–464. [Google Scholar] [CrossRef]
- Williams, C.M.; El Mohsen, M.A.; Vauzour, D.; Rendeiro, C.; Butler, L.T.; Ellis, J.A.; Whiteman, M.; Spencer, J.P. Blueberry-induced changes in spatial working memory correlate with changes in hippocampal CREB phosphorylation and brain-derived neurotrophic factor (BDNF) levels. Free Radic. Biol. Med. 2008, 45, 295–305. [Google Scholar] [CrossRef]
- Lee, S.; Kim, D.H.; Lee, D.H.; Jeon, S.J.; Lee, C.H.; Son, K.H.; Jung, J.W.; Shin, C.Y.; Ryu, J.H. Oroxylin A, a flavonoid, stimulates adult neurogenesis in the hippocampal dentate gyrus region of mice. Neurochem. Res. 2010, 35, 1725–1732. [Google Scholar] [CrossRef]
- Zeisel, S.H. Diet-gene interactions underlie metabolic individuality and influence brain development: Implications for clinical practice derived from studies on choline metabolism. Ann. Nutr. Metab. 2012, 60 S3, 19–25. [Google Scholar] [CrossRef] [Green Version]
- Kohlmeier, M.; da Costa, K.A.; Fischer, L.M.; Zeisel, S.H. Genetic variation of folate-mediated one-carbon transfer pathway predicts susceptibility to choline deficiency in humans. Proc. Natl. Acad. Sci. USA 2005, 102, 16025–16030. [Google Scholar] [CrossRef] [Green Version]
- Scalabrino, G. Vitamin-regulated cytokines and growth factors in the CNS and elsewhere. J. Neurochem. 2009, 111, 1309–1326. [Google Scholar] [CrossRef]
- Psaltopoulou, T.; Sergentanis, T.N.; Panagiotakos, D.B.; Sergentanis, I.N.; Kosti, R.; Scarmeas, N. Mediterranean diet, stroke, cognitive impairment, and depression: A meta-analysis. Ann. Neurol. 2013, 74, 580–591. [Google Scholar] [CrossRef]
- Jacka, F.N.; Cherbuin, N.; Anstey, K.J.; Sachdev, P.; Butterworth, P. Western diet is associated with a smaller hippocampus: A longitudinal investigation. BMC Med. 2015, 13, 215. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Akbaraly, T.; Sexton, C.; Zsoldos, E.; Mahmood, A.; Filippini, N.; Kerleau, C.; Verdier, J.M.; Virtanen, M.; Gabelle, A.; Ebmeier, K.P.; et al. Association of Long-Term Diet Quality with Hippocampal Volume: Longitudinal Cohort Study. Am. J. Med. 2018, 131, 1372–1381. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mattson, M.P. Will caloric restriction and folate protect against AD and PD? Neurology 2003, 60, 690–695. [Google Scholar] [CrossRef] [PubMed]
- Strandwitz, P. Neurotransmitter modulation by the gut microbiota. Brain Res. 2018, 1693, 128–133. [Google Scholar] [CrossRef]
- Goehler, L.E.; Gaykema, R.P.; Opitz, N.; Reddaway, R.; Badr, N.; Lyte, M. Activation in vagal afferents and central autonomic pathways: Early responses to intestinal infection with Campylobacter jejuni. Brain Behav. Immun. 2005, 19, 334–344. [Google Scholar] [CrossRef]
- Han, W.; Tellez, L.A.; Perkins, M.H.; Perez, I.O.; Qu, T.; Ferreira, J.; Ferreira, T.L.; Quinn, D.; Liu, Z.W.; Gao, X.B.; et al. A Neural Circuit for Gut-Induced Reward. Cell 2018, 175, 665–678. [Google Scholar] [CrossRef] [Green Version]
- Teratani, T.; Mikami, Y.; Nakamoto, N.; Suzuki, T.; Harada, Y.; Okabayashi, K.; Hagihara, Y.; Taniki, N.; Kohno, K.; Sibata, S.; et al. The liver-brain-gut neural arc maintains the Treg cell niche in the gut. Nat. Cell. Biol. 2020, 591–596. [Google Scholar] [CrossRef]
- Baganz, N.L.; Blakely, R.D. A dialogue between the immune system and brain, spoken in the language of serotonin. ACS Chem. Neurosci. 2013, 4, 48–63. [Google Scholar] [CrossRef] [Green Version]
- Besser, M.J.; Ganor, Y.; Levite, M. Dopamine by itself activates either D2, D3 or D1/D5 dopaminergic receptors in normal human T-cells and triggers the selective secretion of either IL-10, TNFalpha or both. J. Neuroimmunol. 2005, 169, 161–171. [Google Scholar] [CrossRef]
- Stefano, G.B.; Pilonis, N.; Ptacek, R.; Raboch, J.; Vnukova, M.; Kream, R.M. Gut, Microbiome, and Brain Regulatory Axis: Relevance to Neurodegenerative and Psychiatric Disorders. Cell. Mol. Neurobiol. 2018, 38, 1197–1206. [Google Scholar] [CrossRef] [Green Version]
- Vijay, N.; Morris, M.E. Role of monocarboxylate transporters in drug delivery to the brain. Curr. Pharm. Des. 2014, 20, 1487–1498. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- O’Mahony, S.M.; Felice, V.D.; Nally, K.; Savignac, H.M.; Claesson, M.J.; Scully, P.; Woznicki, J.; Hyland, N.P.; Shanahan, F.; Quigley, E.M.; et al. Disturbance of the gut microbiota in early-life selectively affects visceral pain in adulthood without impacting cognitive or anxiety-related behaviors in male rats. Neuroscience 2014, 277, 885–901. [Google Scholar] [CrossRef] [PubMed]
- Neufeld, K.M.; Kang, N.; Bienenstock, J.; Foster, J.A. Reduced anxiety-like behavior and central neurochemical change in germ-free mice. Neurogastroenterol. Motil. Off. J. Eur. Gastrointest. Motil. Soc. 2011, 23, 255–264. [Google Scholar] [CrossRef] [PubMed]
- Diaz Heijtz, R.; Wang, S.; Anuar, F.; Qian, Y.; Bjorkholm, B.; Samuelsson, A.; Hibberd, M.L.; Forssberg, H.; Pettersson, S. Normal gut microbiota modulates brain development and behavior. Proc. Natl. Acad. Sci. USA 2011, 108, 3047–3052. [Google Scholar] [CrossRef] [Green Version]
- Desbonnet, L.; Clarke, G.; Shanahan, F.; Dinan, T.G.; Cryan, J.F. Microbiota is essential for social development in the mouse. Mol. Psychiatry 2014, 19, 146–148. [Google Scholar] [CrossRef]
- Gareau, M.G.; Wine, E.; Rodrigues, D.M.; Cho, J.H.; Whary, M.T.; Philpott, D.J.; Macqueen, G.; Sherman, P.M. Bacterial infection causes stress-induced memory dysfunction in mice. Gut 2011, 60, 307–317. [Google Scholar] [CrossRef]
- Chu, C.; Murdock, M.H.; Jing, D.; Won, T.H.; Chung, H.; Kressel, A.M.; Tsaava, T.; Addorisio, M.E.; Putzel, G.G.; Zhou, L.; et al. The microbiota regulate neuronal function and fear extinction learning. Nature 2019, 574, 543–548. [Google Scholar] [CrossRef]
- O’Donnell, M.P.; Fox, B.W.; Chao, P.H.; Schroeder, F.C.; Sengupta, P. A neurotransmitter produced by gut bacteria modulates host sensory behaviour. Nature 2020, 583, 415–420. [Google Scholar] [CrossRef]
- Cani, P.D.; Lecourt, E.; Dewulf, E.M.; Sohet, F.M.; Pachikian, B.D.; Naslain, D.; De Backer, F.; Neyrinck, A.M.; Delzenne, N.M. Gut microbiota fermentation of prebiotics increases satietogenic and incretin gut peptide production with consequences for appetite sensation and glucose response after a meal. Am. J. Clin. Nutr. 2009, 90, 1236–1243. [Google Scholar] [CrossRef]
- Aatsinki, A.K.; Lahti, L.; Uusitupa, H.M.; Munukka, E.; Keskitalo, A.; Nolvi, S.; O’Mahony, S.; Pietila, S.; Elo, L.L.; Eerola, E.; et al. Gut microbiota composition is associated with temperament traits in infants. Brain Behav. Immun. 2019, 80, 849–858. [Google Scholar] [CrossRef]
- Johnson, K.V.A. Gut microbiome composition and diversity are related to human personality traits. Hum. Microbiome J. 2020, 15, 100069. [Google Scholar] [CrossRef]
- Luczynski, P.; Whelan, S.O.; O’Sullivan, C.; Clarke, G.; Shanahan, F.; Dinan, T.G.; Cryan, J.F. Adult microbiota-deficient mice have distinct dendritic morphological changes: Differential effects in the amygdala and hippocampus. Eur. J. Neurosci. 2016, 44, 2654–2666. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mohle, L.; Mattei, D.; Heimesaat, M.M.; Bereswill, S.; Fischer, A.; Alutis, M.; French, T.; Hambardzumyan, D.; Matzinger, P.; Dunay, I.R.; et al. Ly6C(hi) Monocytes Provide a Link between Antibiotic-Induced Changes in Gut Microbiota and Adult Hippocampal Neurogenesis. Cell Rep. 2016, 15, 1945–1956. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, H.J.; Leeds, P.; Chuang, D.M. The HDAC inhibitor, sodium butyrate, stimulates neurogenesis in the ischemic brain. J. Neurochem. 2009, 110, 1226–1240. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoban, A.E.; Stilling, R.M.; Ryan, F.J.; Shanahan, F.; Dinan, T.G.; Claesson, M.J.; Clarke, G.; Cryan, J.F. Regulation of prefrontal cortex myelination by the microbiota. Transl. Psychiatry 2016, 6, e774. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matcovitch-Natan, O.; Winter, D.R.; Giladi, A.; Vargas Aguilar, S.; Spinrad, A.; Sarrazin, S.; Ben-Yehuda, H.; David, E.; Zelada Gonzalez, F.; Perrin, P.; et al. Microglia development follows a stepwise program to regulate brain homeostasis. Science 2016, 353, aad8670. [Google Scholar] [CrossRef]
- Erny, D.; Hrabe de Angelis, A.L.; Jaitin, D.; Wieghofer, P.; Staszewski, O.; David, E.; Keren-Shaul, H.; Mahlakoiv, T.; Jakobshagen, K.; Buch, T.; et al. Host microbiota constantly control maturation and function of microglia in the CNS. Nat. Neurosci. 2015, 18, 965–977. [Google Scholar] [CrossRef]
- Rothhammer, V.; Mascanfroni, I.D.; Bunse, L.; Takenaka, M.C.; Kenison, J.E.; Mayo, L.; Chao, C.C.; Patel, B.; Yan, R.; Blain, M.; et al. Type I interferons and microbial metabolites of tryptophan modulate astrocyte activity and central nervous system inflammation via the aryl hydrocarbon receptor. Nat. Med. 2016, 22, 586–597. [Google Scholar] [CrossRef]
- Braniste, V.; Al-Asmakh, M.; Kowal, C.; Anuar, F.; Abbaspour, A.; Tóth, M.; Korecka, A.; Bakocevic, N.; Ng, L.G.; Kundu, P.; et al. The gut microbiota influences blood-brain barrier permeability in mice. Sci. Transl. Med. 2014, 6, 263ra158. [Google Scholar] [CrossRef] [Green Version]
- Hoyles, L.; Snelling, T.; Umlai, U.-K.; Nicholson, J.K.; Carding, S.R.; Glen, R.C.; McArthur, S. Microbiome—Host systems interactions: Protective effects of propionate upon the blood–brain barrier. Microbiome 2018, 6, 55. [Google Scholar] [CrossRef] [Green Version]
- Endo, Y.; Asou, H.K.; Matsugae, N.; Hirahara, K.; Shinoda, K.; Tumes, D.J.; Tokuyama, H.; Yokote, K.; Nakayama, T. Obesity Drives Th17 Cell Differentiation by Inducing the Lipid Metabolic Kinase, ACC1. Cell Rep. 2015, 12, 1042–1055. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haghikia, A.; Jörg, S.; Duscha, A.; Berg, J.; Manzel, A.; Waschbisch, A.; Hammer, A.; Lee, D.H.; May, C.; Wilck, N.; et al. Dietary Fatty Acids Directly Impact Central Nervous System Autoimmunity via the Small Intestine. Immunity 2015, 43, 817–829. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, E.H.; Bantug, G.; Griss, T.; Condotta, S.; Johnson, R.M.; Samborska, B.; Mainolfi, N.; Suri, V.; Guak, H.; Balmer, M.L.; et al. Serine Is an Essential Metabolite for Effector T Cell Expansion. Cell Metab. 2017, 25, 345–357. [Google Scholar] [CrossRef] [PubMed]
- Roy, D.G.; Chen, J.; Mamane, V.; Ma, E.H.; Muhire, B.M.; Sheldon, R.D.; Shorstova, T.; Koning, R.; Johnson, R.M.; Esaulova, E.; et al. Methionine Metabolism Shapes T Helper Cell Responses through Regulation of Epigenetic Reprogramming. Cell Metab. 2020, 31, 250–266. [Google Scholar] [CrossRef]
- Hedstrom, A.K.; Lima Bomfim, I.; Barcellos, L.; Gianfrancesco, M.; Schaefer, C.; Kockum, I.; Olsson, T.; Alfredsson, L. Interaction between adolescent obesity and HLA risk genes in the etiology of multiple sclerosis. Neurology 2014, 82, 865–872. [Google Scholar] [CrossRef] [Green Version]
- Bril, V.; Blanchette, C.M.; Noone, J.M.; Runken, M.C.; Gelinas, D.; Russell, J.W. The dilemma of diabetes in chronic inflammatory demyelinating polyneuropathy. J. Diabetes Its Complicat. 2016, 30, 1401–1407. [Google Scholar] [CrossRef] [Green Version]
- Doneddu, P.E.; Bianchi, E.; Cocito, D.; Manganelli, F.; Fazio, R.; Filosto, M.; Mazzeo, A.; Cosentino, G.; Cortese, A.; Jann, S.; et al. Risk factors for chronic inflammatory demyelinating polyradiculoneuropathy (CIDP): Antecedent events, lifestyle and dietary habits. Data from the Italian CIDP Database. Eur. J. Neurol. 2020, 27, 136–143. [Google Scholar] [CrossRef]
- Li, H.; Limenitakis, J.P.; Greiff, V.; Yilmaz, B.; Scharen, O.; Urbaniak, C.; Zund, M.; Lawson, M.A.E.; Young, I.D.; Rupp, S.; et al. Mucosal or systemic microbiota exposures shape the B cell repertoire. Nature 2020, 1–5. [Google Scholar] [CrossRef]
- Berer, K.; Mues, M.; Koutrolos, M.; Rasbi, Z.A.; Boziki, M.; Johner, C.; Wekerle, H.; Krishnamoorthy, G. Commensal microbiota and myelin autoantigen cooperate to trigger autoimmune demyelination. Nature 2011, 479, 538–541. [Google Scholar] [CrossRef]
- Lee, Y.K.; Menezes, J.S.; Umesaki, Y.; Mazmanian, S.K. Proinflammatory T-cell responses to gut microbiota promote experimental autoimmune encephalomyelitis. Proc. Natl. Acad. Sci. USA 2011, 108 S1, 4615–4622. [Google Scholar] [CrossRef] [Green Version]
- Round, J.L.; Mazmanian, S.K. Inducible Foxp3+ regulatory T-cell development by a commensal bacterium of the intestinal microbiota. Proc. Natl. Acad. Sci. USA 2010, 107, 12204–12209. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, H.; Assmann, J.C.; Krenz, A.; Rahman, M.; Grimm, M.; Karsten, C.M.; Kohl, J.; Offermanns, S.; Wettschureck, N.; Schwaninger, M. Hydroxycarboxylic acid receptor 2 mediates dimethyl fumarate’s protective effect in EAE. J. Clin. Investig. 2014, 124, 2188–2192. [Google Scholar] [CrossRef] [PubMed]
- Parodi, B.; Rossi, S.; Morando, S.; Cordano, C.; Bragoni, A.; Motta, C.; Usai, C.; Wipke, B.T.; Scannevin, R.H.; Mancardi, G.L.; et al. Fumarates modulate microglia activation through a novel HCAR2 signaling pathway and rescue synaptic dysregulation in inflamed CNS. Acta Neuropathol. 2015, 130, 279–295. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qin, J.; Li, Y.; Cai, Z.; Li, S.; Zhu, J.; Zhang, F.; Liang, S.; Zhang, W.; Guan, Y.; Shen, D.; et al. A metagenome-wide association study of gut microbiota in type 2 diabetes. Nature 2012, 490, 55–60. [Google Scholar] [CrossRef]
- Meroni, M.; Longo, M.; Dongiovanni, P. The Role of Probiotics in Nonalcoholic Fatty Liver Disease: A New Insight into Therapeutic Strategies. Nutrients 2019, 11. [Google Scholar] [CrossRef] [Green Version]
- Xu, W.L.; Atti, A.R.; Gatz, M.; Pedersen, N.L.; Johansson, B.; Fratiglioni, L. Midlife overweight and obesity increase late-life dementia risk: A population-based twin study. Neurology 2011, 76, 1568–1574. [Google Scholar] [CrossRef] [Green Version]
- Whitmer, R.A.; Gunderson, E.P.; Barrett-Connor, E.; Quesenberry, C.P., Jr.; Yaffe, K. Obesity in middle age and future risk of dementia: A 27 year longitudinal population based study. BMJ 2005, 330, 1360. [Google Scholar] [CrossRef] [Green Version]
- Kivipelto, M.; Ngandu, T.; Fratiglioni, L.; Viitanen, M.; Kareholt, I.; Winblad, B.; Helkala, E.L.; Tuomilehto, J.; Soininen, H.; Nissinen, A. Obesity and vascular risk factors at midlife and the risk of dementia and Alzheimer disease. Arch. Neurol. 2005, 62, 1556–1560. [Google Scholar] [CrossRef] [Green Version]
- Whitmer, R.A.; Gustafson, D.R.; Barrett-Connor, E.; Haan, M.N.; Gunderson, E.P.; Yaffe, K. Central obesity and increased risk of dementia more than three decades later. Neurology 2008, 71, 1057–1064. [Google Scholar] [CrossRef]
- Levin-Allerhand, J.A.; Lominska, C.E.; Smith, J.D. Increased amyloid-levels in APPSWE transgenic mice treated chronically with a physiological high-fat high-cholesterol diet. J. Nutr. Health Aging 2002, 6, 315–319. [Google Scholar]
- Julien, C.; Tremblay, C.; Phivilay, A.; Berthiaume, L.; Émond, V.; Julien, P.; Calon, F. High-fat diet aggravates amyloid-beta and tau pathologies in the 3xTg-AD mouse model. Neurobiol. Aging 2010, 31, 1516–1531. [Google Scholar] [CrossRef] [PubMed]
- Graham, L.C.; Harder, J.M.; Soto, I.; de Vries, W.N.; John, S.W.; Howell, G.R. Chronic consumption of a western diet induces robust glial activation in aging mice and in a mouse model of Alzheimer’s disease. Sci. Rep. 2016, 6, 21568. [Google Scholar] [CrossRef] [PubMed]
- Mody, N.; Agouni, A.; McIlroy, G.D.; Platt, B.; Delibegovic, M. Susceptibility to diet-induced obesity and glucose intolerance in the APPSWE/PSEN1A246E mouse model of Alzheimer’s disease is associated with increased brain levels of protein tyrosine phosphatase 1B (PTP1B) and retinol-binding protein 4 (RBP4), and basal phosphorylation of S6 ribosomal protein. Diabetologia 2011, 54, 2143–2151. [Google Scholar] [CrossRef] [PubMed]
- Kohjima, M.; Sun, Y.; Chan, L. Increased Food Intake Leads to Obesity and Insulin Resistance in the Tg2576 Alzheimer’s Disease Mouse Model. Endocrinology 2010, 151, 1532–1540. [Google Scholar] [CrossRef]
- Cheng, G.; Huang, C.; Deng, H.; Wang, H. Diabetes as a risk factor for dementia and mild cognitive impairment: A meta-analysis of longitudinal studies. Intern. Med. J. 2012, 42, 484–491. [Google Scholar] [CrossRef]
- Cukierman, T.; Gerstein, H.C.; Williamson, J.D. Cognitive decline and dementia in diabetes—Systematic overview of prospective observational studies. Diabetologia 2005, 48, 2460–2469. [Google Scholar] [CrossRef]
- Profenno, L.A.; Porsteinsson, A.P.; Faraone, S.V. Meta-Analysis of Alzheimer’s Disease Risk with Obesity, Diabetes, and Related Disorders. Biol. Psychiatry 2010, 67, 505–512. [Google Scholar] [CrossRef]
- Talbot, K.; Wang, H.Y.; Kazi, H.; Han, L.Y.; Bakshi, K.P.; Stucky, A.; Fuino, R.L.; Kawaguchi, K.R.; Samoyedny, A.J.; Wilson, R.S.; et al. Demonstrated brain insulin resistance in Alzheimer’s disease patients is associated with IGF-1 resistance, IRS-1 dysregulation, and cognitive decline. J. Clin. Investig. 2012, 122, 1316–1338. [Google Scholar] [CrossRef] [Green Version]
- Moloney, A.M.; Griffin, R.J.; Timmons, S.; O’Connor, R.; Ravid, R.; O’Neill, C. Defects in IGF-1 receptor, insulin receptor and IRS-1/2 in Alzheimer’s disease indicate possible resistance to IGF-1 and insulin signalling. Neurobiol. Aging 2010, 31, 224–243. [Google Scholar] [CrossRef]
- Ho, L.; Qin, W.; Pompl, P.N.; Xiang, Z.; Wang, J.; Zhao, Z.; Peng, Y.; Cambareri, G.; Rocher, A.; Mobbs, C.V.; et al. Diet-induced insulin resistance promotes amyloidosis in a transgenic mouse model of Alzheimer’s disease. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2004, 18, 902–904. [Google Scholar] [CrossRef]
- Taylor, M.K.; Sullivan, D.K.; Swerdlow, R.H.; Vidoni, E.D.; Morris, J.K.; Mahnken, J.D.; Burns, J.M. A high-glycemic diet is associated with cerebral amyloid burden in cognitively normal older adults. Am. J. Clin. Nutr. 2017, 106, 1463–1470. [Google Scholar] [CrossRef] [PubMed]
- Kim, B.; Backus, C.; Oh, S.; Hayes, J.M.; Feldman, E.L. Increased tau phosphorylation and cleavage in mouse models of type 1 and type 2 diabetes. Endocrinology 2009, 150, 5294–5301. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bosco, D.; Fava, A.; Plastino, M.; Montalcini, T.; Pujia, A. Possible implications of insulin resistance and glucose metabolism in Alzheimer’s disease pathogenesis. J. Cell Mol. Med. 2011, 15, 1807–1821. [Google Scholar] [CrossRef] [Green Version]
- Orr, M.E.; Salinas, A.; Buffenstein, R.; Oddo, S. Mammalian target of rapamycin hyperactivity mediates the detrimental effects of a high sucrose diet on Alzheimer’s disease pathology. Neurobiol. Aging 2014, 35, 1233–1242. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Son, S.M.; Song, H.; Byun, J.; Park, K.S.; Jang, H.C.; Park, Y.J.; Mook-Jung, I. Accumulation of autophagosomes contributes to enhanced amyloidogenic APP processing under insulin-resistant conditions. Autophagy 2012, 8, 1842–1844. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reitz, C. Dyslipidemia and the risk of Alzheimer’s disease. Curr. Atheroscler. Rep. 2013, 15, 307. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wingo, T.S.; Cutler, D.J.; Wingo, A.P.; Le, N.A.; Rabinovici, G.D.; Miller, B.L.; Lah, J.J.; Levey, A.I. Association of Early-Onset Alzheimer Disease With Elevated Low-Density Lipoprotein Cholesterol Levels and Rare Genetic Coding Variants of APOB. JAMA Neurol. 2019, 76, 809–817. [Google Scholar] [CrossRef] [PubMed]
- Xu, C.; Apostolova, L.G.; Oblak, A.L.; Gao, S. Association of Hypercholesterolemia with Alzheimer’s Disease Pathology and Cerebral Amyloid Angiopathy. J. Alzheimer Dis. JAD 2020, 73, 1305–1311. [Google Scholar] [CrossRef]
- Refolo, L.M.; Malester, B.; LaFrancois, J.; Bryant-Thomas, T.; Wang, R.; Tint, G.S.; Sambamurti, K.; Duff, K.; Pappolla, M.A. Hypercholesterolemia accelerates the Alzheimer’s amyloid pathology in a transgenic mouse model. Neurobiol. Dis. 2000, 7, 321–331. [Google Scholar] [CrossRef]
- Seo, S.W.; Gottesman, R.F.; Clark, J.M.; Hernaez, R.; Chang, Y.; Kim, C.; Ha, K.H.; Guallar, E.; Lazo, M. Nonalcoholic fatty liver disease is associated with cognitive function in adults. Neurology 2016, 86, 1136–1142. [Google Scholar] [CrossRef] [Green Version]
- Weinstein, G.; Zelber-Sagi, S.; Preis, S.R.; Beiser, A.S.; DeCarli, C.; Speliotes, E.K.; Satizabal, C.L.; Vasan, R.S.; Seshadri, S. Association of Nonalcoholic Fatty Liver Disease With Lower Brain Volume in Healthy Middle-aged Adults in the Framingham Study. JAMA Neurol. 2018, 75, 97–104. [Google Scholar] [CrossRef]
- Kim, D.G.; Krenz, A.; Toussaint, L.E.; Maurer, K.J.; Robinson, S.A.; Yan, A.; Torres, L.; Bynoe, M.S. Non-alcoholic fatty liver disease induces signs of Alzheimer’s disease (AD) in wild-type mice and accelerates pathological signs of AD in an AD model. J. Neuroinflamm. 2016, 13, 1. [Google Scholar] [CrossRef] [Green Version]
- Moser, V.A.; Pike, C.J. Obesity Accelerates Alzheimer-Related Pathology in APOE4 but not APOE3 Mice. eNeuro 2017, 4. [Google Scholar] [CrossRef] [Green Version]
- Kypreos, K.E.; Karagiannides, I.; Fotiadou, E.H.; Karavia, E.A.; Brinkmeier, M.S.; Giakoumi, S.M.; Tsompanidi, E.M. Mechanisms of obesity and related pathologies: Role of apolipoprotein E in the development of obesity. FEBS J. 2009, 276, 5720–5728. [Google Scholar] [CrossRef]
- Jones, N.S.; Rebeck, G.W. The Synergistic Effects of APOE Genotype and Obesity on Alzheimer’s Disease Risk. Int. J. Mol. Sci. 2018, 20, 63. [Google Scholar] [CrossRef] [Green Version]
- Morris, M.C.; Evans, D.A.; Bienias, J.L.; Tangney, C.C.; Bennett, D.A.; Aggarwal, N.; Schneider, J.; Wilson, R.S. Dietary fats and the risk of incident Alzheimer disease. Arch. Neurol. 2003, 60, 194–200. [Google Scholar] [CrossRef]
- Kalmijn, S.; Launer, L.J.; Ott, A.; Witteman, J.C.; Hofman, A.; Breteler, M.M. Dietary fat intake and the risk of incident dementia in the Rotterdam Study. Ann. Neurol. 1997, 42, 776–782. [Google Scholar] [CrossRef] [Green Version]
- Luchsinger, J.A.; Tang, M.X.; Shea, S.; Mayeux, R. Caloric intake and the risk of Alzheimer disease. Arch. Neurol. 2002, 59, 1258–1263. [Google Scholar] [CrossRef]
- Laitinen, M.H.; Ngandu, T.; Rovio, S.; Helkala, E.L.; Uusitalo, U.; Viitanen, M.; Nissinen, A.; Tuomilehto, J.; Soininen, H.; Kivipelto, M. Fat intake at midlife and risk of dementia and Alzheimer’s disease: A population-based study. Dement. Geriatr. Cogn. Disord. 2006, 22, 99–107. [Google Scholar] [CrossRef]
- Söderberg, M.; Edlund, C.; Kristensson, K.; Dallner, G. Fatty acid composition of brain phospholipids in aging and in Alzheimer’s disease. Lipids 1991, 26, 421. [Google Scholar] [CrossRef]
- Yassine, H.N.; Feng, Q.; Azizkhanian, I.; Rawat, V.; Castor, K.; Fonteh, A.N.; Harrington, M.G.; Zheng, L.; Reed, B.R.; DeCarli, C.; et al. Association of Serum Docosahexaenoic Acid with Cerebral Amyloidosis. JAMA Neurol. 2016, 73, 1208–1216. [Google Scholar] [CrossRef]
- Barcelo-Coblijn, G.; Hogyes, E.; Kitajka, K.; Puskas, L.G.; Zvara, A.; Hackler, L., Jr.; Nyakas, C.; Penke, Z.; Farkas, T. Modification by docosahexaenoic acid of age-induced alterations in gene expression and molecular composition of rat brain phospholipids. Proc. Natl. Acad. Sci. USA 2003, 100, 11321–11326. [Google Scholar] [CrossRef] [Green Version]
- Puskas, L.G.; Kitajka, K.; Nyakas, C.; Barcelo-Coblijn, G.; Farkas, T. Short-term administration of omega 3 fatty acids from fish oil results in increased transthyretin transcription in old rat hippocampus. Proc. Natl. Acad. Sci. USA 2003, 100, 1580–1585. [Google Scholar] [CrossRef] [Green Version]
- Burdge, G.C.; Lillycrop, K.A. Fatty acids and epigenetics. Curr. Opin. Clin. Nutr. Metab. Care 2014, 17, 156–161. [Google Scholar] [CrossRef]
- Grimm, M.O.; Kuchenbecker, J.; Grosgen, S.; Burg, V.K.; Hundsdorfer, B.; Rothhaar, T.L.; Friess, P.; de Wilde, M.C.; Broersen, L.M.; Penke, B.; et al. Docosahexaenoic acid reduces amyloid beta production via multiple pleiotropic mechanisms. J. Biol. Chem. 2011, 286, 14028–14039. [Google Scholar] [CrossRef] [Green Version]
- Hooijmans, C.R.; Pasker-de Jong, P.C.M.; de Vries, R.B.M.; Ritskes-Hoitinga, M. The Effects of Long-Term Omega-3 Fatty Acid Supplementation on Cognition and Alzheimer’s Pathology in Animal Models of Alzheimer’s Disease: A Systematic Review and Meta-Analysis. J. Alzheimer Dis. 2012, 28, 191–209. [Google Scholar] [CrossRef] [Green Version]
- Hooijmans, C.R.; Rutters, F.; Dederen, P.J.; Gambarota, G.; Veltien, A.; van Groen, T.; Broersen, L.M.; Lütjohann, D.; Heerschap, A.; Tanila, H.; et al. Changes in cerebral blood volume and amyloid pathology in aged Alzheimer APP/PS1 mice on a docosahexaenoic acid (DHA) diet or cholesterol enriched Typical Western Diet (TWD). Neurobiol. Dis. 2007, 28, 16–29. [Google Scholar] [CrossRef]
- Fotuhi, M.; Mohassel, P.; Yaffe, K. Fish consumption, long-chain omega-3 fatty acids and risk of cognitive decline or Alzheimer disease: A complex association. Nat. Clin. Practice. Neurol. 2009, 5, 140–152. [Google Scholar] [CrossRef]
- Kalmijn, S.; van Boxtel, M.P.; Ocke, M.; Verschuren, W.M.; Kromhout, D.; Launer, L.J. Dietary intake of fatty acids and fish in relation to cognitive performance at middle age. Neurology 2004, 62, 275–280. [Google Scholar] [CrossRef]
- Morris, M.C.; Evans, D.A.; Bienias, J.L.; Tangney, C.C.; Bennett, D.A.; Wilson, R.S.; Aggarwal, N.; Schneider, J. Consumption of fish and n-3 fatty acids and risk of incident Alzheimer disease. Arch. Neurol. 2003, 60, 940–946. [Google Scholar] [CrossRef]
- Barberger-Gateau, P.; Raffaitin, C.; Letenneur, L.; Berr, C.; Tzourio, C.; Dartigues, J.F.; Alperovitch, A. Dietary patterns and risk of dementia: The Three-City cohort study. Neurology 2007, 69, 1921–1930. [Google Scholar] [CrossRef]
- Plourde, M.; Vohl, M.-C.; Vandal, M.; Couture, P.; Lemieux, S.; Cunnane, S.C. Plasma n-3 fatty acid response to an n-3 fatty acid supplement is modulated by apoE ε4 but not by the common PPAR-α L162V polymorphism in men. Br. J. Nutr. 2009, 102, 1121–1124. [Google Scholar] [CrossRef] [Green Version]
- Van de Rest, O.; Geleijnse, J.M.; Kok, F.J.; van Staveren, W.A.; Dullemeijer, C.; Olderikkert, M.G.; Beekman, A.T.; de Groot, C.P. Effect of fish oil on cognitive performance in older subjects: A randomized, controlled trial. Neurology 2008, 71, 430–438. [Google Scholar] [CrossRef]
- Quinn, J.F.; Raman, R.; Thomas, R.G.; Yurko-Mauro, K.; Nelson, E.B.; Van Dyck, C.; Galvin, J.E.; Emond, J.; Jack, C.R., Jr.; Weiner, M.; et al. Docosahexaenoic acid supplementation and cognitive decline in Alzheimer disease: A randomized trial. JAMA 2010, 304, 1903–1911. [Google Scholar] [CrossRef]
- Canhada, S.; Castro, K.; Perry, I.S.; Luft, V.C. Omega-3 fatty acids’ supplementation in Alzheimer’s disease: A systematic review. Nutr. Neurosci. 2018, 21, 529–538. [Google Scholar] [CrossRef]
- Arellanes, I.C.; Choe, N.; Solomon, V.; He, X.; Kavin, B.; Martinez, A.E.; Kono, N.; Buennagel, D.P.; Hazra, N.; Kim, G.; et al. Brain delivery of supplemental docosahexaenoic acid (DHA): A randomized placebo-controlled clinical trial. EBioMedicine 2020, 59, 102883. [Google Scholar] [CrossRef]
- Van der Borght, K.; Kohnke, R.; Goransson, N.; Deierborg, T.; Brundin, P.; Erlanson-Albertsson, C.; Lindqvist, A. Reduced neurogenesis in the rat hippocampus following high fructose consumption. Regul. Pept. 2011, 167, 26–30. [Google Scholar] [CrossRef]
- Grimm, M.O.W.; Rothhaar, T.L.; Grösgen, S.; Burg, V.K.; Hundsdörfer, B.; Haupenthal, V.J.; Friess, P.; Kins, S.; Grimm, H.S.; Hartmann, T. Trans fatty acids enhance amyloidogenic processing of the Alzheimer amyloid precursor protein (APP). J. Nutr. Biochem. 2012, 23, 1214–1223. [Google Scholar] [CrossRef]
- Chouliaras, L.; Mastroeni, D.; Delvaux, E.; Grover, A.; Kenis, G.; Hof, P.R.; Steinbusch, H.W.; Coleman, P.D.; Rutten, B.P.; van den Hove, D.L. Consistent decrease in global DNA methylation and hydroxymethylation in the hippocampus of Alzheimer’s disease patients. Neurobiol. Aging 2013, 34, 2091–2099. [Google Scholar] [CrossRef] [Green Version]
- Devore, E.E.; Grodstein, F.; van Rooij, F.J.A.; Hofman, A.; Stampfer, M.J.; Witteman, J.C.M.; Breteler, M.M.B. Dietary Antioxidants and Long-term Risk of Dementia. Arch. Neurol. 2010, 67, 819–825. [Google Scholar] [CrossRef] [Green Version]
- Luchsinger, J.A.; Tang, M.-X.; Shea, S.; Mayeux, R. Antioxidant Vitamin Intake and Risk of Alzheimer Disease. Arch. Neurol. 2003, 60, 203–208. [Google Scholar] [CrossRef] [PubMed]
- Fillenbaum, G.G.; Kuchibhatla, M.N.; Hanlon, J.T.; Artz, M.B.; Pieper, C.F.; Schmader, K.E.; Dysken, M.W.; Gray, S.L. Dementia and Alzheimer’s disease in community-dwelling elders taking vitamin C and/or vitamin E. Ann. Pharmacother. 2005, 39, 2009–2014. [Google Scholar] [CrossRef] [PubMed]
- Petersen, R.C.; Thomas, R.G.; Grundman, M.; Bennett, D.; Doody, R.; Ferris, S.; Galasko, D.; Jin, S.; Kaye, J.; Levey, A.; et al. Vitamin E and donepezil for the treatment of mild cognitive impairment. New Engl. J. Med. 2005, 352, 2379–2388. [Google Scholar] [CrossRef] [Green Version]
- Farina, N.; Llewellyn, D.; Isaac, M.; Tabet, N. Vitamin E for Alzheimer’s dementia and mild cognitive impairment. Cochrane Database Syst. Rev. 2017, 4, CD002854. [Google Scholar] [CrossRef] [PubMed]
- Holland, T.M.; Agarwal, P.; Wang, Y.; Leurgans, S.E.; Bennett, D.A.; Booth, S.L.; Morris, M.C. Dietary flavonols and risk of Alzheimer dementia. Neurology 2020, 94. [Google Scholar] [CrossRef]
- Carman, A.J.; Dacks, P.A.; Lane, R.F.; Shineman, D.W.; Fillit, H.M. Current evidence for the use of coffee and caffeine to prevent age-related cognitive decline and Alzheimer’s disease. J. Nutr. Health Aging 2014, 18, 383–392. [Google Scholar] [CrossRef] [PubMed]
- Larsson, S.C.; Orsini, N. Coffee Consumption and Risk of Dementia and Alzheimer’s Disease: A Dose-Response Meta-Analysis of Prospective Studies. Nutrients 2018, 10. [Google Scholar] [CrossRef] [Green Version]
- Gardener, S.; Gu, Y.; Rainey-Smith, S.R.; Keogh, J.B.; Clifton, P.M.; Mathieson, S.L.; Taddei, K.; Mondal, A.; Ward, V.K.; Scarmeas, N.; et al. Adherence to a Mediterranean diet and Alzheimer’s disease risk in an Australian population. Transl. Psychiatry 2012, 2, e164. [Google Scholar] [CrossRef] [Green Version]
- Scarmeas, N.; Stern, Y.; Tang, M.X.; Mayeux, R.; Luchsinger, J.A. Mediterranean diet and risk for Alzheimer’s disease. Ann. Neurol. 2006, 59, 912–921. [Google Scholar] [CrossRef] [Green Version]
- Karstens, A.J.; Tussing-Humphreys, L.; Zhan, L.; Rajendran, N.; Cohen, J.; Dion, C.; Zhou, X.J.; Lamar, M. Associations of the Mediterranean diet with cognitive and neuroimaging phenotypes of dementia in healthy older adults. Am. J. Clin. Nutr. 2019, 109, 361–368. [Google Scholar] [CrossRef]
- Scarmeas, N.; Luchsinger, J.A.; Mayeux, R.; Stern, Y. Mediterranean diet and Alzheimer disease mortality. Neurology 2007, 69, 1084–1093. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chung, K.W.; Kim, D.H.; Park, M.H.; Choi, Y.J.; Kim, N.D.; Lee, J.; Yu, B.P.; Chung, H.Y. Recent advances in calorie restriction research on aging. Exp. Gerontol. 2013, 48, 1049–1053. [Google Scholar] [CrossRef] [PubMed]
- Vogt, N.M.; Kerby, R.L.; Dill-McFarland, K.A.; Harding, S.J.; Merluzzi, A.P.; Johnson, S.C.; Carlsson, C.M.; Asthana, S.; Zetterberg, H.; Blennow, K.; et al. Gut microbiome alterations in Alzheimer’s disease. Sci. Rep. 2017, 7, 13537. [Google Scholar] [CrossRef] [PubMed]
- Haran, J.P.; Bhattarai, S.K.; Foley, S.E.; Dutta, P.; Ward, D.V.; Bucci, V.; McCormick, B.A. Alzheimer’s Disease Microbiome Is Associated with Dysregulation of the Anti-Inflammatory P-Glycoprotein Pathway. mBio 2019, 10. [Google Scholar] [CrossRef] [Green Version]
- Cattaneo, A.; Cattane, N.; Galluzzi, S.; Provasi, S.; Lopizzo, N.; Festari, C.; Ferrari, C.; Guerra, U.P.; Paghera, B.; Muscio, C.; et al. Association of brain amyloidosis with pro-inflammatory gut bacterial taxa and peripheral inflammation markers in cognitively impaired elderly. Neurobiol. Aging 2017, 49, 60–68. [Google Scholar] [CrossRef] [Green Version]
- Sun, J.; Xu, J.; Ling, Y.; Wang, F.; Gong, T.; Yang, C.; Ye, S.; Ye, K.; Wei, D.; Song, Z.; et al. Fecal microbiota transplantation alleviated Alzheimer’s disease-like pathogenesis in APP/PS1 transgenic mice. Transl. Psychiatry 2019, 9, 189. [Google Scholar] [CrossRef] [Green Version]
- Brandscheid, C.; Schuck, F.; Reinhardt, S.; Schafer, K.H.; Pietrzik, C.U.; Grimm, M.; Hartmann, T.; Schwiertz, A.; Endres, K. Altered Gut Microbiome Composition and Tryptic Activity of the 5xFAD Alzheimer’s Mouse Model. J. Alzheimer Dis. JAD 2017, 56, 775–788. [Google Scholar] [CrossRef]
- Shen, L.; Liu, L.; Ji, H.F. Alzheimer’s Disease Histological and Behavioral Manifestations in Transgenic Mice Correlate with Specific Gut Microbiome State. J. Alzheimer Dis. JAD 2017, 56, 385–390. [Google Scholar] [CrossRef]
- Li, Z.; Zhu, H.; Guo, Y.; Du, X.; Qin, C. Gut microbiota regulate cognitive deficits and amyloid deposition in a model of Alzheimer’s disease. J. Neurochem. 2020. [Google Scholar] [CrossRef]
- Honarpisheh, P.; Reynolds, C.R.; Blasco Conesa, M.P.; Moruno Manchon, J.F.; Putluri, N.; Bhattacharjee, M.B.; Urayama, A.; McCullough, L.D.; Ganesh, B.P. Dysregulated Gut Homeostasis Observed Prior to the Accumulation of the Brain Amyloid-beta in Tg2576 Mice. Int. J. Mol. Sci. 2020, 21. [Google Scholar] [CrossRef] [Green Version]
- Harach, T.; Marungruang, N.; Duthilleul, N.; Cheatham, V.; Mc Coy, K.D.; Frisoni, G.; Neher, J.J.; Fak, F.; Jucker, M.; Lasser, T.; et al. Reduction of Abeta amyloid pathology in APPPS1 transgenic mice in the absence of gut microbiota. Sci. Rep. 2017, 7, 41802. [Google Scholar] [CrossRef] [PubMed]
- Minter, M.R.; Zhang, C.; Leone, V.; Ringus, D.L.; Zhang, X.; Oyler-Castrillo, P.; Musch, M.W.; Liao, F.; Ward, J.F.; Holtzman, D.M.; et al. Antibiotic-induced perturbations in gut microbial diversity influences neuro-inflammation and amyloidosis in a murine model of Alzheimer’s disease. Sci. Rep. 2016, 6, 30028. [Google Scholar] [CrossRef] [PubMed]
- Shen, H.; Guan, Q.; Zhang, X.; Yuan, C.; Tan, Z.; Zhai, L.; Hao, Y.; Gu, Y.; Han, C. New mechanism of neuroinflammation in Alzheimer’s disease: The activation of NLRP3 inflammasome mediated by gut microbiota. Prog. Neuropsychopharmacol. Biol. Psychiatry 2020, 100, 109884. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.S.; Kim, Y.; Choi, H.; Kim, W.; Park, S.; Lee, D.; Kim, D.K.; Kim, H.J.; Choi, H.; Hyun, D.W.; et al. Transfer of a healthy microbiota reduces amyloid and tau pathology in an Alzheimer’s disease animal model. Gut 2020, 69, 283–294. [Google Scholar] [CrossRef]
- Parikh, I.J.; Estus, J.L.; Zajac, D.J.; Malik, M.; Maldonado Weng, J.; Tai, L.M.; Chlipala, G.E.; LaDu, M.J.; Green, S.J.; Estus, S. Murine Gut Microbiome Association with APOE Alleles. Front. Immunol. 2020, 11, 200. [Google Scholar] [CrossRef]
- Zhan, X.; Stamova, B.; Jin, L.W.; DeCarli, C.; Phinney, B.; Sharp, F.R. Gram-negative bacterial molecules associate with Alzheimer disease pathology. Neurology 2016, 87, 2324–2332. [Google Scholar] [CrossRef] [Green Version]
- Asti, A.; Gioglio, L. Can a bacterial endotoxin be a key factor in the kinetics of amyloid fibril formation? J. Alzheimer Dis. JAD 2014, 39, 169–179. [Google Scholar] [CrossRef]
- Lukiw, W.J. Bacteroides fragilis Lipopolysaccharide and Inflammatory Signaling in Alzheimer’s Disease. Front. Microbiol 2016, 7, 1544. [Google Scholar] [CrossRef] [Green Version]
- Jaeger, L.B.; Dohgu, S.; Sultana, R.; Lynch, J.L.; Owen, J.B.; Erickson, M.A.; Shah, G.N.; Price, T.O.; Fleegal-Demotta, M.A.; Butterfield, D.A.; et al. Lipopolysaccharide alters the blood-brain barrier transport of amyloid beta protein: A mechanism for inflammation in the progression of Alzheimer’s disease. Brain Behav. Immun. 2009, 23, 507–517. [Google Scholar] [CrossRef] [Green Version]
- Zhu, B.; Wang, Z.G.; Ding, J.; Liu, N.; Wang, D.M.; Ding, L.C.; Yang, C. Chronic lipopolysaccharide exposure induces cognitive dysfunction without affecting BDNF expression in the rat hippocampus. Exp. Ther. Med. 2014, 7, 750–754. [Google Scholar] [CrossRef] [Green Version]
- Kahn, M.S.; Kranjac, D.; Alonzo, C.A.; Haase, J.H.; Cedillos, R.O.; McLinden, K.A.; Boehm, G.W.; Chumley, M.J. Prolonged elevation in hippocampal Abeta and cognitive deficits following repeated endotoxin exposure in the mouse. Behav. Brain Res. 2012, 229, 176–184. [Google Scholar] [CrossRef] [PubMed]
- Hauss-Wegrzyniak, B.; Vraniak, P.D.; Wenk, G.L. LPS-induced neuroinflammatory effects do not recover with time. Neuroreport 2000, 11, 1759–1763. [Google Scholar] [CrossRef] [PubMed]
- Pistollato, F.; Sumalla Cano, S.; Elio, I.; Masias Vergara, M.; Giampieri, F.; Battino, M. Role of gut microbiota and nutrients in amyloid formation and pathogenesis of Alzheimer disease. Nutr. Rev. 2016, 74, 624–634. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, S.G.; Stribinskis, V.; Rane, M.J.; Demuth, D.R.; Gozal, E.; Roberts, A.M.; Jagadapillai, R.; Liu, R.; Choe, K.; Shivakumar, B.; et al. Exposure to the Functional Bacterial Amyloid Protein Curli Enhances Alpha-Synuclein Aggregation in Aged Fischer 344 Rats and Caenorhabditis elegans. Sci. Rep. 2016, 6, 34477. [Google Scholar] [CrossRef] [PubMed]
- Lundmark, K.; Westermark, G.T.; Olsen, A.; Westermark, P. Protein fibrils in nature can enhance amyloid protein A amyloidosis in mice: Cross-seeding as a disease mechanism. Proc. Natl. Acad. Sci. USA 2005, 102, 6098–6102. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Galloway, S.; Jian, L.; Johnsen, R.; Chew, S.; Mamo, J.C. beta-amyloid or its precursor protein is found in epithelial cells of the small intestine and is stimulated by high-fat feeding. J. Nutr. Biochem. 2007, 18, 279–284. [Google Scholar] [CrossRef] [PubMed]
- Puig, K.L.; Brose, S.A.; Zhou, X.; Sens, M.A.; Combs, G.F.; Jensen, M.D.; Golovko, M.Y.; Combs, C.K. Amyloid precursor protein modulates macrophage phenotype and diet-dependent weight gain. Sci. Rep. 2017, 7, 43725. [Google Scholar] [CrossRef]
- Eisele, Y.S.; Bolmont, T.; Heikenwalder, M.; Langer, F.; Jacobson, L.H.; Yan, Z.X.; Roth, K.; Aguzzi, A.; Staufenbiel, M.; Walker, L.C.; et al. Induction of cerebral beta-amyloidosis: Intracerebral versus systemic Abeta inoculation. Proc. Natl. Acad. Sci. USA 2009, 106, 12926–12931. [Google Scholar] [CrossRef] [Green Version]
- Eisele, Y.S.; Obermuller, U.; Heilbronner, G.; Baumann, F.; Kaeser, S.A.; Wolburg, H.; Walker, L.C.; Staufenbiel, M.; Heikenwalder, M.; Jucker, M. Peripherally applied Abeta-containing inoculates induce cerebral beta-amyloidosis. Science 2010, 330, 980–982. [Google Scholar] [CrossRef] [Green Version]
- Cintron, A.F.; Dalal, N.V.; Dooyema, J.; Betarbet, R.; Walker, L.C. Transport of cargo from periphery to brain by circulating monocytes. Brain Res. 2015, 1622, 328–338. [Google Scholar] [CrossRef] [Green Version]
- Ikeda, S.-I.; Yanagisawa, N.; Hongo, M.; Ito, N. Vagus nerve and celiac ganglion lesions in generalized amyloidosis: A correlative study of familial amyloid polyneuropathy and AL-amyloidosis. J. Neurol. Sci. 1987, 79, 129–139. [Google Scholar] [CrossRef]
- Sun, Y.; Sommerville, N.R.; Liu, J.Y.H.; Ngan, M.P.; Poon, D.; Ponomarev, E.D.; Lu, Z.; Kung, J.S.C.; Rudd, J.A. Intra-gastrointestinal amyloid-beta1-42 oligomers perturb enteric function and induce Alzheimer’s disease pathology. J. Physiol. 2020. [Google Scholar] [CrossRef] [PubMed]
- Xu, M.; Mo, X.; Huang, H.; Chen, X.; Liu, H.; Peng, Z.; Chen, L.; Rong, S.; Yang, W.; Xu, S.; et al. Yeast beta-glucan alleviates cognitive deficit by regulating gut microbiota and metabolites in Abeta1-42-induced AD-like mice. Int. J. Biol. Macromol. 2020, 161, 258–270. [Google Scholar] [CrossRef] [PubMed]
- Ho, L.; Ono, K.; Tsuji, M.; Mazzola, P.; Singh, R.; Pasinetti, G.M. Protective roles of intestinal microbiota derived short chain fatty acids in Alzheimer’s disease-type beta-amyloid neuropathological mechanisms. Expert Rev. Neurother. 2018, 18, 83–90. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, S.Y.; Chae, C.W.; Lee, H.J.; Jung, Y.H.; Choi, G.E.; Kim, J.S.; Lim, J.R.; Lee, J.E.; Cho, J.H.; Park, H.; et al. Sodium butyrate inhibits high cholesterol-induced neuronal amyloidogenesis by modulating NRF2 stabilization-mediated ROS levels: Involvement of NOX2 and SOD1. Cell Death Dis. 2020, 11, 469. [Google Scholar] [CrossRef]
- Wenzel, T.J.; Gates, E.J.; Ranger, A.L.; Klegeris, A. Short-chain fatty acids (SCFAs) alone or in combination regulate select immune functions of microglia-like cells. Mol. Cell. Neurosci. 2020, 105, 103493. [Google Scholar] [CrossRef]
- Sun, J.; Xu, J.; Yang, B.; Chen, K.; Kong, Y.; Fang, N.; Gong, T.; Wang, F.; Ling, Z.; Liu, J. Effect of Clostridium butyricum against Microglia-Mediated Neuroinflammation in Alzheimer’s Disease via Regulating Gut Microbiota and Metabolites Butyrate. Mol. Nutr. Food Res. 2020, 64, e1900636. [Google Scholar] [CrossRef]
- Wang, F.; Xu, T.; Zhang, Y.; Zheng, T.; He, Y.; He, F.; Jiang, Y. Long-term combined administration of Bifidobacterium bifidum TMC3115 and Lactobacillus plantarum 45 alleviates spatial memory impairment and gut dysbiosis in APP/PS1 mice. FEMS Microbiol. Lett. 2020, 367. [Google Scholar] [CrossRef]
- Wang, Q.J.; Shen, Y.E.; Wang, X.; Fu, S.; Zhang, X.; Zhang, Y.N.; Wang, R.T. Concomitant memantine and Lactobacillus plantarum treatment attenuates cognitive impairments in APP/PS1 mice. Aging 2020, 12, 628–649. [Google Scholar] [CrossRef]
- Bonfili, L.; Cecarini, V.; Berardi, S.; Scarpona, S.; Suchodolski, J.S.; Nasuti, C.; Fiorini, D.; Boarelli, M.C.; Rossi, G.; Eleuteri, A.M. Microbiota modulation counteracts Alzheimer’s disease progression influencing neuronal proteolysis and gut hormones plasma levels. Sci. Rep. 2017, 7, 2426. [Google Scholar] [CrossRef]
- Kaur, H.; Nagamoto-Combs, K.; Golovko, S.; Golovko, M.Y.; Klug, M.G.; Combs, C.K. Probiotics ameliorate intestinal pathophysiology in a mouse model of Alzheimer’s disease. Neurobiol. Aging 2020, 92, 114–134. [Google Scholar] [CrossRef] [PubMed]
- Bonfili, L.; Cecarini, V.; Gogoi, O.; Berardi, S.; Scarpona, S.; Angeletti, M.; Rossi, G.; Eleuteri, A.M. Gut microbiota manipulation through probiotics oral administration restores glucose homeostasis in a mouse model of Alzheimer’s disease. Neurobiol. Aging 2020, 87, 35–43. [Google Scholar] [CrossRef] [PubMed]
- Kruger, J.F.; Hillesheim, E.; Pereira, A.; Camargo, C.Q.; Rabito, E.I. Probiotics for dementia: A systematic review and meta-analysis of randomized controlled trials. Nutr. Rev. 2020. [Google Scholar] [CrossRef] [PubMed]
- Abbott, R.D.; Ross, G.W.; White, L.R.; Nelson, J.S.; Masaki, K.H.; Tanner, C.M.; Curb, J.D.; Blanchette, P.L.; Popper, J.S.; Petrovitch, H. Midlife adiposity and the future risk of Parkinson’s disease. Neurology 2002, 59, 1051–1057. [Google Scholar] [CrossRef]
- Chen, H.; Zhang, S.M.; Schwarzschild, M.A.; Hernán, M.A.; Willett, W.C.; Ascherio, A. Obesity and the Risk of Parkinson’s Disease. Am. J. Epidemiol. 2004, 159, 547–555. [Google Scholar] [CrossRef]
- Palacios, N.; Gao, X.; McCullough, M.L.; Jacobs, E.J.; Patel, A.V.; Mayo, T.; Schwarzschild, M.A.; Ascherio, A. Obesity, diabetes, and risk of Parkinson’s disease. Mov. Disord. Off. J. Mov. Disord. Soc. 2011, 26, 2253–2259. [Google Scholar] [CrossRef] [Green Version]
- Kyrozis, A.; Ghika, A.; Stathopoulos, P.; Vassilopoulos, D.; Trichopoulos, D.; Trichopoulou, A. Dietary and lifestyle variables in relation to incidence of Parkinson’s disease in Greece. Eur. J. Epidemiol. 2013, 28, 67–77. [Google Scholar] [CrossRef]
- Hu, G.; Jousilahti, P.; Nissinen, A.; Antikainen, R.; Kivipelto, M.; Tuomilehto, J. Body mass index and the risk of Parkinson disease. Neurology 2006, 67, 1955–1959. [Google Scholar] [CrossRef]
- Abbott, R.D.; Ross, G.W.; White, L.R.; Sanderson, W.T.; Burchfiel, C.M.; Kashon, M.; Sharp, D.S.; Masaki, K.H.; Curb, J.D.; Petrovitch, H. Environmental, life-style, and physical precursors of clinical Parkinson’s disease: Recent findings from the Honolulu-Asia Aging Study. J. Neurol. 2003, 250 S3, III30–39. [Google Scholar] [CrossRef]
- Xu, Q.; Park, Y.; Huang, X.; Hollenbeck, A.; Blair, A.; Schatzkin, A.; Chen, H. Diabetes and Risk of Parkinson’s Disease. Diabetes Care 2011, 34, 910–915. [Google Scholar] [CrossRef] [Green Version]
- Cereda, E.; Barichella, M.; Pedrolli, C.; Klersy, C.; Cassani, E.; Caccialanza, R.; Pezzoli, G. Diabetes and risk of Parkinson’s disease: A systematic review and meta-analysis. Diabetes Care 2011, 34, 2614–2623. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Simon, K.C.; Chen, H.; Schwarzschild, M.; Ascherio, A. Hypertension, hypercholesterolemia, diabetes, and risk of Parkinson disease. Neurology 2007, 69, 1688–1695. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, C.; Hsieh, M.-C.; Chang, S.-J. Metabolic syndrome, diabetes, and hyperuricemia. Curr. Opin. Rheumatol. 2013, 25, 210–216. [Google Scholar] [CrossRef] [PubMed]
- Kotagal, V.; Albin, R.L.; Müller, M.L.T.M.; Koeppe, R.A.; Frey, K.A.; Bohnen, N.I. Diabetes is associated with postural instability and gait difficulty in Parkinson disease. Parkinsonism Relat. Disord. 2013, 19, 522–526. [Google Scholar] [CrossRef] [Green Version]
- Santiago, J.A.; Potashkin, J.A. Shared dysregulated pathways lead to Parkinson’s disease and diabetes. Trends Mol. Med. 2013, 19, 176–186. [Google Scholar] [CrossRef]
- Aviles-Olmos, I.; Limousin, P.; Lees, A.; Foltynie, T. Parkinson’s disease, insulin resistance and novel agents of neuroprotection. Brain J. Neurol. 2012, 136, 374–384. [Google Scholar] [CrossRef] [Green Version]
- Svenningsson, P.; Wirdefeldt, K.; Yin, L.; Fang, F.; Markaki, I.; Efendic, S.; Ludvigsson, J.F. Reduced incidence of Parkinson’s disease after dipeptidyl peptidase-4 inhibitors—A nation wide case-control study. Mov. Disord. 2016, 31, 1422–1423. [Google Scholar] [CrossRef]
- Wahlqvist, M.L.; Lee, M.-S.; Hsu, C.-C.; Chuang, S.-Y.; Lee, J.-T.; Tsai, H.-N. Metformin-inclusive sulfonylurea therapy reduces the risk of Parkinson’s disease occurring with Type 2 diabetes in a Taiwanese population cohort. Parkinsonism Relat. Disord. 2012, 18, 753–758. [Google Scholar] [CrossRef]
- Ascherio, A.; Schwarzschild, M.A. The epidemiology of Parkinson’s disease: Risk factors and prevention. Lancet Neurol. 2016, 15, 1257–1272. [Google Scholar] [CrossRef]
- Fu, X.; Wang, Y.; He, X.; Li, H.; Liu, H.; Zhang, X. A systematic review and meta-analysis of serum cholesterol and triglyceride levels in patients with Parkinson’s disease. Lipids Health Dis. 2020, 19, 97. [Google Scholar] [CrossRef]
- Huang, X.; Auinger, P.; Eberly, S.; Oakes, D.; Schwarzschild, M.; Ascherio, A.; Mailman, R.; Chen, H. Parkinson Study Group, DATATOP Investigators. Serum cholesterol and the progression of Parkinson’s disease: Results from DATATOP. PLoS ONE 2011, 6. [Google Scholar] [CrossRef] [Green Version]
- Du, G.; Lewis, M.M.; Shaffer, M.L.; Chen, H.; Yang, Q.X.; Mailman, R.B.; Huang, X. Serum cholesterol and nigrostriatal R2* values in Parkinson’s disease. PLoS ONE 2012, 7, e35397. [Google Scholar] [CrossRef] [PubMed]
- Paul, R.; Choudhury, A.; Kumar, S.; Giri, A.; Sandhir, R.; Borah, A. Cholesterol contributes to dopamine-neuronal loss in MPTP mouse model of Parkinson’s disease: Involvement of mitochondrial dysfunctions and oxidative stress. PLoS ONE 2017, 12, e0171285. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paul, R.; Choudhury, A.; Chandra Boruah, D.; Devi, R.; Bhattacharya, P.; Choudhury, M.D.; Borah, A. Hypercholesterolemia causes psychomotor abnormalities in mice and alterations in cortico-striatal biogenic amine neurotransmitters: Relevance to Parkinson’s disease. Neurochem. Int. 2017, 108, 15–26. [Google Scholar] [CrossRef] [PubMed]
- Talim, M. Prevalence of nonalcoholic fatty liver disease and its association with cardiovascular disease among type 2 diabetic patients: Response to Targher et al. and Hu et al. Diabetes Care 2007, 30, e55–e56. [Google Scholar] [CrossRef] [Green Version]
- Vural, S. Prevalence of Non Alcoholic Fatty Liver Disease in patients with Parkinson Disease. Anatol. J. Fam. Med. 2019, 38–40. [Google Scholar] [CrossRef]
- Repici, M.; Giorgini, F. DJ-1 in Parkinson’s Disease: Clinical Insights and Therapeutic Perspectives. J. Clin. Med. 2019, 8. [Google Scholar] [CrossRef] [Green Version]
- Xu, M.; Chen, L.; Li, J.; Wu, H.; Xia, Q.; Kong, X. Emerging roles of DJ-1 in liver diseases through regulation of oxidative stress and immune response. Liver Res. 2018, 2, 87–91. [Google Scholar] [CrossRef]
- Burkhard, P.R.; Delavelle, J.; Du Pasquier, R.; Spahr, L. Chronic parkinsonism associated with cirrhosis: A distinct subset of acquired hepatocerebral degeneration. Arch. Neurol. 2003, 60, 521–528. [Google Scholar] [CrossRef] [Green Version]
- Chen, H.; O’Reilly, E.; McCullough, M.L.; Rodriguez, C.; Schwarzschild, M.A.; Calle, E.E.; Thun, M.J.; Ascherio, A. Consumption of dairy products and risk of Parkinson’s disease. Am. J. Epidemiol. 2007, 165, 998–1006. [Google Scholar] [CrossRef]
- Park, M.; Ross, G.W.; Petrovitch, H.; White, L.R.; Masaki, K.H.; Nelson, J.S.; Tanner, C.M.; Curb, J.D.; Blanchette, P.L.; Abbott, R.D. Consumption of milk and calcium in midlife and the future risk of Parkinson disease. Neurology 2005, 64, 1047–1051. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Zhang, S.M.; Hernan, M.A.; Willett, W.C.; Ascherio, A. Diet and Parkinson’s disease: A potential role of dairy products in men. Ann. Neurol. 2002, 52, 793–801. [Google Scholar] [CrossRef] [PubMed]
- Liu, R.; Guo, X.; Park, Y.; Huang, X.; Sinha, R.; Freedman, N.D.; Hollenbeck, A.R.; Blair, A.; Chen, H. Caffeine intake, smoking, and risk of Parkinson disease in men and women. Am. J. Epidemiol. 2012, 175, 1200–1207. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ross, G.W.; Abbott, R.D.; Petrovitch, H.; Morens, D.M.; Grandinetti, A.; Tung, K.-H.; Tanner, C.M.; Masaki, K.H.; Blanchette, P.L.; Curb, J.D.; et al. Association of Coffee and Caffeine Intake With the Risk of Parkinson Disease. JAMA 2000, 283, 2674–2679. [Google Scholar] [CrossRef] [PubMed]
- Ascherio, A.; Zhang, S.M.; Hernán, M.A.; Kawachi, I.; Colditz, G.A.; Speizer, F.E.; Willett, W.C. Prospective study of caffeine consumption and risk of Parkinson’s disease in men and women. Ann. Neurol. 2001, 50, 56–63. [Google Scholar] [CrossRef]
- Kachroo, A.; Irizarry, M.C.; Schwarzschild, M.A. Caffeine protects against combined paraquat and maneb-induced dopaminergic neuron degeneration. Exp. Neurol. 2010, 223, 657–661. [Google Scholar] [CrossRef] [Green Version]
- Seidl, S.E.; Santiago, J.A.; Bilyk, H.; Potashkin, J.A. The emerging role of nutrition in Parkinson’s disease. Front. Aging Neurosci. 2014, 6, 36. [Google Scholar] [CrossRef] [Green Version]
- Altman, R.D.; Lang, A.E.; Postuma, R.B. Caffeine in Parkinson’s disease: A pilot open-label, dose-escalation study. Mov. Disord. Off. J. Mov. Disord. Soc. 2011, 26, 2427–2431. [Google Scholar] [CrossRef]
- Postuma, R.B.; Lang, A.E.; Munhoz, R.P.; Charland, K.; Pelletier, A.; Moscovich, M.; Filla, L.; Zanatta, D.; Rios Romenets, S.; Altman, R.; et al. Caffeine for treatment of Parkinson disease: A randomized controlled trial. Neurology 2012, 79, 651–658. [Google Scholar] [CrossRef]
- LeWitt, P.A.; Guttman, M.; Tetrud, J.W.; Tuite, P.J.; Mori, A.; Chaikin, P.; Sussman, N.M.; 6002-US-005 Study Group. Adenosine A2A receptor antagonist istradefylline (KW-6002) reduces “off” time in Parkinson’s disease: A double-blind, randomized, multicenter clinical trial (6002-US-005). Ann. Neurol. 2008, 63, 295–302. [Google Scholar] [CrossRef]
- Mizuno, Y.; Hasegawa, K.; Kondo, T.; Kuno, S.; Yamamoto, M. Japanese Istradefylline Study Group. Clinical efficacy of istradefylline (KW-6002) in Parkinson’s disease: A randomized, controlled study. Mov. Disord. Off. J. Mov. Disord. Soc. 2010, 25, 1437–1443. [Google Scholar] [CrossRef] [PubMed]
- Vorovenci, R.J.; Antonini, A. The efficacy of oral adenosine A(2A) antagonist istradefylline for the treatment of moderate to severe Parkinson’s disease. Expert Rev. Neurother. 2015, 15, 1383–1390. [Google Scholar] [CrossRef] [PubMed]
- Weisskopf, M.G.; O’Reilly, E.; Chen, H.; Schwarzschild, M.A.; Ascherio, A. Plasma urate and risk of Parkinson’s disease. Am. J. Epidemiol. 2007, 166, 561–567. [Google Scholar] [CrossRef] [PubMed]
- Facheris, M.F.; Hicks, A.A.; Minelli, C.; Hagenah, J.M.; Kostic, V.; Campbell, S.; Hayward, C.; Volpato, C.B.; Pattaro, C.; Vitart, V.; et al. Variation in the Uric Acid Transporter Gene SLC2A9 and Its Association with AAO of Parkinson’s Disease. J. Mol. Neurosci. 2011, 43, 246–250. [Google Scholar] [CrossRef]
- Ascherio, A.; LeWitt, P.A.; Xu, K.; Eberly, S.; Watts, A.; Matson, W.R.; Marras, C.; Kieburtz, K.; Rudolph, A.; Bogdanov, M.B.; et al. Urate as a predictor of the rate of clinical decline in Parkinson disease. Arch. Neurol. 2009, 66, 1460–1468. [Google Scholar] [CrossRef] [Green Version]
- Bakshi, R.; Zhang, H.; Logan, R.; Joshi, I.; Xu, Y.; Chen, X.; Schwarzschild, M.A. Neuroprotective effects of urate are mediated by augmenting astrocytic glutathione synthesis and release. Neurobiol. Dis. 2015, 82, 574–579. [Google Scholar] [CrossRef] [Green Version]
- Chen, H.; Zhang, S.M.; Hernan, M.A.; Willett, W.C.; Ascherio, A. Dietary intakes of fat and risk of Parkinson’s disease. Am. J. Epidemiol. 2003, 157, 1007–1014. [Google Scholar] [CrossRef] [Green Version]
- Tan, L.C.; Koh, W.P.; Yuan, J.M.; Wang, R.; Au, W.L.; Tan, J.H.; Tan, E.K.; Yu, M.C. Differential effects of black versus green tea on risk of Parkinson’s disease in the Singapore Chinese Health Study. Am. J. Epidemiol. 2008, 167, 553–560. [Google Scholar] [CrossRef] [Green Version]
- De Lau, L.M.; Bornebroek, M.; Witteman, J.C.; Hofman, A.; Koudstaal, P.J.; Breteler, M.M. Dietary fatty acids and the risk of Parkinson disease: The Rotterdam study. Neurology 2005, 64, 2040–2045. [Google Scholar] [CrossRef]
- Samadi, P.; Grégoire, L.; Rouillard, C.; Bédard, P.J.; Di Paolo, T.; Lévesque, D. Docosahexaenoic acid reduces levodopa-induced dyskinesias in 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine monkeys. Ann. Neurol. 2006, 59, 282–288. [Google Scholar] [CrossRef]
- Erro, R.; Brigo, F.; Tamburin, S.; Zamboni, M.; Antonini, A.; Tinazzi, M. Nutritional habits, risk, and progression of Parkinson disease. J. Neurol. 2018, 265, 12–23. [Google Scholar] [CrossRef] [PubMed]
- Crevoisier, C.; Zerr, P.; Calvi-Gries, F.; Nilsen, T. Effects of food on the pharmacokinetics of levodopa in a dual-release formulation. Eur. J. Pharm. Biopharm. 2003, 55, 71–76. [Google Scholar] [CrossRef]
- Pincus, J.H.; Barry, K. Influence of dietary protein on motor fluctuations in Parkinson’s disease. Arch. Neurol. 1987, 44, 270–272. [Google Scholar] [CrossRef] [PubMed]
- Wang, A.; Lin, Y.; Wu, Y.; Zhang, D. Macronutrients intake and risk of Parkinson’s disease: A meta-analysis. Geriatr. Gerontol. Int. 2015, 15, 606–616. [Google Scholar] [CrossRef]
- Gao, X.; Chen, H.; Fung, T.T.; Logroscino, G.; Schwarzschild, M.A.; Hu, F.B.; Ascherio, A. Prospective study of dietary pattern and risk of Parkinson disease. Am. J. Clin. Nutr. 2007, 86, 1486–1494. [Google Scholar] [CrossRef] [Green Version]
- Sofi, F.; Cesari, F.; Abbate, R.; Gensini, G.F.; Casini, A. Adherence to Mediterranean diet and health status: Meta-analysis. BMJ 2008, 337, a1344. [Google Scholar] [CrossRef] [Green Version]
- Nishiwaki, H.; Ito, M.; Ishida, T.; Hamaguchi, T.; Maeda, T.; Kashihara, K.; Tsuboi, Y.; Ueyama, J.; Shimamura, T.; Mori, H.; et al. Meta-Analysis of Gut Dysbiosis in Parkinson’s Disease. Mov. Disord. Off. J. Mov. Disord. Soc. 2020. [Google Scholar] [CrossRef]
- Nuzum, N.D.; Loughman, A.; Szymlek-Gay, E.A.; Hendy, A.; Teo, W.P.; Macpherson, H. Gut microbiota differences between healthy older adults and individuals with Parkinson’s disease: A systematic review. Neurosci. Biobehav. Rev. 2020, 112, 227–241. [Google Scholar] [CrossRef]
- Wallen, Z.D.; Appah, M.; Dean, M.N.; Sesler, C.L.; Factor, S.A.; Molho, E.; Zabetian, C.P.; Standaert, D.G.; Payami, H. Characterizing dysbiosis of gut microbiome in PD: Evidence for overabundance of opportunistic pathogens. NPJ Parkinson Dis. 2020, 6, 11. [Google Scholar] [CrossRef]
- Heintz-Buschart, A.; Pandey, U.; Wicke, T.; Sixel-Doring, F.; Janzen, A.; Sittig-Wiegand, E.; Trenkwalder, C.; Oertel, W.H.; Mollenhauer, B.; Wilmes, P. The nasal and gut microbiome in Parkinson’s disease and idiopathic rapid eye movement sleep behavior disorder. Mov. Disord. Off. J. Mov. Disord. Soc. 2018, 33, 88–98. [Google Scholar] [CrossRef] [Green Version]
- Aho, V.T.E.; Pereira, P.A.B.; Voutilainen, S.; Paulin, L.; Pekkonen, E.; Auvinen, P.; Scheperjans, F. Gut microbiota in Parkinson’s disease: Temporal stability and relations to disease progression. EBioMedicine 2019, 44, 691–707. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weis, S.; Schwiertz, A.; Unger, M.M.; Becker, A.; Fassbender, K.; Ratering, S.; Kohl, M.; Schnell, S.; Schafer, K.H.; Egert, M. Effect of Parkinson’s disease and related medications on the composition of the fecal bacterial microbiota. NPJ Parkinson Dis. 2019, 5, 28. [Google Scholar] [CrossRef] [PubMed]
- Maini Rekdal, V.; Bess, E.N.; Bisanz, J.E.; Turnbaugh, P.J.; Balskus, E.P. Discovery and inhibition of an interspecies gut bacterial pathway for Levodopa metabolism. Science 2019, 364, eaau6323. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lai, F.; Jiang, R.; Xie, W.; Liu, X.; Tang, Y.; Xiao, H.; Gao, J.; Jia, Y.; Bai, Q. Intestinal Pathology and Gut Microbiota Alterations in a Methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) Mouse Model of Parkinson’s Disease. Neurochem. Res. 2018, 43, 1986–1999. [Google Scholar] [CrossRef] [PubMed]
- Gorecki, A.M.; Preskey, L.; Bakeberg, M.C.; Kenna, J.E.; Gildenhuys, C.; MacDougall, G.; Dunlop, S.A.; Mastaglia, F.L.; Akkari, P.A.; Koengten, F.; et al. Altered Gut Microbiome in Parkinson’s Disease and the Influence of Lipopolysaccharide in a Human alpha-Synuclein Over-Expressing Mouse Model. Front. Neurosci. 2019, 13, 839. [Google Scholar] [CrossRef] [Green Version]
- Koutzoumis, D.N.; Vergara, M.; Pino, J.; Buddendorff, J.; Khoshbouei, H.; Mandel, R.J.; Torres, G.E. Alterations of the gut microbiota with antibiotics protects dopamine neuron loss and improve motor deficits in a pharmacological rodent model of Parkinson’s disease. Exp. Neurol. 2020, 325, 113159. [Google Scholar] [CrossRef]
- Sampson, T.R.; Debelius, J.W.; Thron, T.; Janssen, S.; Shastri, G.G.; Ilhan, Z.E.; Challis, C.; Schretter, C.E.; Rocha, S.; Gradinaru, V.; et al. Gut Microbiota Regulate Motor Deficits and Neuroinflammation in a Model of Parkinson’s Disease. Cell 2016, 167, 1469–1480. [Google Scholar] [CrossRef] [Green Version]
- Sun, M.F.; Zhu, Y.L.; Zhou, Z.L.; Jia, X.B.; Xu, Y.D.; Yang, Q.; Cui, C.; Shen, Y.Q. Neuroprotective effects of fecal microbiota transplantation on MPTP-induced Parkinson’s disease mice: Gut microbiota, glial reaction and TLR4/TNF-alpha signaling pathway. Brain Behav. Immun. 2018, 70, 48–60. [Google Scholar] [CrossRef]
- Bedarf, J.R.; Hildebrand, F.; Coelho, L.P.; Sunagawa, S.; Bahram, M.; Goeser, F.; Bork, P.; Wullner, U. Functional implications of microbial and viral gut metagenome changes in early stage L-DOPA-naive Parkinson’s disease patients. Genome Med. 2017, 9, 39. [Google Scholar] [CrossRef]
- Cirstea, M.S.; Yu, A.C.; Golz, E.; Sundvick, K.; Kliger, D.; Radisavljevic, N.; Foulger, L.H.; Mackenzie, M.; Huan, T.; Finlay, B.B.; et al. Microbiota Composition and Metabolism Are Associated With Gut Function in Parkinson’s Disease. Mov. Disord. Off. J. Mov. Disord. Soc. 2020, 35, 1208–1217. [Google Scholar] [CrossRef]
- Hertel, J.; Harms, A.C.; Heinken, A.; Baldini, F.; Thinnes, C.C.; Glaab, E.; Vasco, D.A.; Pietzner, M.; Stewart, I.D.; Wareham, N.J.; et al. Integrated Analyses of Microbiome and Longitudinal Metabolome Data Reveal Microbial-Host Interactions on Sulfur Metabolism in Parkinson’s Disease. Cell Rep. 2019, 291, 1767–1777. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Keshavarzian, A.; Green, S.J.; Engen, P.A.; Voigt, R.M.; Naqib, A.; Forsyth, C.B.; Mutlu, E.; Shannon, K.M. Colonic bacterial composition in Parkinson’s disease. Mov. Disord. Off. J. Mov. Disord. Soc. 2015, 30, 1351–1360. [Google Scholar] [CrossRef] [PubMed]
- Forsyth, C.B.; Shannon, K.M.; Kordower, J.H.; Voigt, R.M.; Shaikh, M.; Jaglin, J.A.; Estes, J.D.; Dodiya, H.B.; Keshavarzian, A. Increased intestinal permeability correlates with sigmoid mucosa alpha-synuclein staining and endotoxin exposure markers in early Parkinson’s disease. PLoS ONE 2011, 6, e28032. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Devos, D.; Lebouvier, T.; Lardeux, B.; Biraud, M.; Rouaud, T.; Pouclet, H.; Coron, E.; Bruley des Varannes, S.; Naveilhan, P.; Nguyen, J.M.; et al. Colonic inflammation in Parkinson’s disease. Neurobiol. Dis. 2013, 50, 42–48. [Google Scholar] [CrossRef]
- Villumsen, M.; Aznar, S.; Pakkenberg, B.; Jess, T.; Brudek, T. Inflammatory bowel disease increases the risk of Parkinson’s disease: A Danish nationwide cohort study 1977-2014. Gut 2019, 68, 18–24. [Google Scholar] [CrossRef]
- Hui, K.Y.; Fernandez-Hernandez, H.; Hu, J.; Schaffner, A.; Pankratz, N.; Hsu, N.Y.; Chuang, L.S.; Carmi, S.; Villaverde, N.; Li, X.; et al. Functional variants in the LRRK2 gene confer shared effects on risk for Crohn’s disease and Parkinson’s disease. Sci. Transl. Med. 2018, 10. [Google Scholar] [CrossRef] [Green Version]
- Kishimoto, Y.; Zhu, W.; Hosoda, W.; Sen, J.M.; Mattson, M.P. Chronic Mild Gut Inflammation Accelerates Brain Neuropathology and Motor Dysfunction in Alpha-Synuclein Mutant Mice. Neuromolecular Med. 2019, 21, 239–249. [Google Scholar] [CrossRef]
- Dutta, G.; Zhang, P.; Liu, B. The lipopolysaccharide Parkinson’s disease animal model: Mechanistic studies and drug discovery. Fundam. Clin. Pharmacol. 2008, 22, 453–464. [Google Scholar] [CrossRef]
- Liu, Y.; Qin, L.; Wilson, B.; Wu, X.; Qian, L.; Granholm, A.C.; Crews, F.T.; Hong, J.S. Endotoxin induces a delayed loss of TH-IR neurons in substantia nigra and motor behavioral deficits. Neurotoxicology 2008, 29, 864–870. [Google Scholar] [CrossRef] [Green Version]
- Qin, L.; Wu, X.; Block, M.L.; Liu, Y.; Breese, G.R.; Hong, J.S.; Knapp, D.J.; Crews, F.T. Systemic LPS causes chronic neuroinflammation and progressive neurodegeneration. Glia 2007, 55, 453–462. [Google Scholar] [CrossRef] [Green Version]
- Perez-Pardo, P.; Dodiya, H.B.; Engen, P.A.; Forsyth, C.B.; Huschens, A.M.; Shaikh, M.; Voigt, R.M.; Naqib, A.; Green, S.J.; Kordower, J.H.; et al. Role of TLR4 in the gut-brain axis in Parkinson’s disease: A translational study from men to mice. Gut 2019, 68, 829–843. [Google Scholar] [CrossRef] [PubMed]
- Kim, W.G.; Mohney, R.P.; Wilson, B.; Jeohn, G.H.; Liu, B.; Hong, J.S. Regional difference in susceptibility to lipopolysaccharide-induced neurotoxicity in the rat brain: Role of microglia. J. Neurosci. Off. J. Soc. Neurosci. 2000, 20, 6309–6316. [Google Scholar] [CrossRef]
- Banks, W.A.; Gray, A.M.; Erickson, M.A.; Salameh, T.S.; Damodarasamy, M.; Sheibani, N.; Meabon, J.S.; Wing, E.E.; Morofuji, Y.; Cook, D.G.; et al. Lipopolysaccharide-induced blood-brain barrier disruption: Roles of cyclooxygenase, oxidative stress, neuroinflammation, and elements of the neurovascular unit. J. Neuroinflammation 2015, 12, 223. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wakabayashi, K.; Takahashi, H.; Takeda, S.; Ohama, E.; Ikuta, F. Parkinson’s disease: The presence of Lewy bodies in Auerbach’s and Meissner’s plexuses. Acta Neuropathol. 1988, 76, 217–221. [Google Scholar] [CrossRef]
- Beach, T.G.; Adler, C.H.; Sue, L.I.; Vedders, L.; Lue, L.; White Iii, C.L.; Akiyama, H.; Caviness, J.N.; Shill, H.A.; Sabbagh, M.N.; et al. Multi-organ distribution of phosphorylated alpha-synuclein histopathology in subjects with Lewy body disorders. Acta Neuropathol. 2010, 119, 689–702. [Google Scholar] [CrossRef] [Green Version]
- Gelpi, E.; Navarro-Otano, J.; Tolosa, E.; Gaig, C.; Compta, Y.; Rey, M.J.; Marti, M.J.; Hernandez, I.; Valldeoriola, F.; Rene, R.; et al. Multiple organ involvement by alpha-synuclein pathology in Lewy body disorders. Mov. Disord. Off. J. Mov. Disord. Soc. 2014, 29, 1010–1018. [Google Scholar] [CrossRef]
- Hilton, D.; Stephens, M.; Kirk, L.; Edwards, P.; Potter, R.; Zajicek, J.; Broughton, E.; Hagan, H.; Carroll, C. Accumulation of alpha-synuclein in the bowel of patients in the pre-clinical phase of Parkinson’s disease. Acta Neuropathol. 2014, 127, 235–241. [Google Scholar] [CrossRef]
- Stokholm, M.G.; Danielsen, E.H.; Hamilton-Dutoit, S.J.; Borghammer, P. Pathological alpha-synuclein in gastrointestinal tissues from prodromal Parkinson disease patients. Ann. Neurol. 2016, 79, 940–949. [Google Scholar] [CrossRef]
- Lebouvier, T.; Neunlist, M.; Bruley des Varannes, S.; Coron, E.; Drouard, A.; N’Guyen, J.M.; Chaumette, T.; Tasselli, M.; Paillusson, S.; Flamand, M.; et al. Colonic biopsies to assess the neuropathology of Parkinson’s disease and its relationship with symptoms. PLoS ONE 2010, 5, e12728. [Google Scholar] [CrossRef] [Green Version]
- Stolzenberg, E.; Berry, D.; Yang; Lee, E.Y.; Kroemer, A.; Kaufman, S.; Wong, G.C.L.; Oppenheim, J.J.; Sen, S.; Fishbein, T.; et al. A Role for Neuronal Alpha-Synuclein in Gastrointestinal Immunity. J. Innate Immun. 2017, 9, 456–463. [Google Scholar] [CrossRef]
- Fellner, L.; Irschick, R.; Schanda, K.; Reindl, M.; Klimaschewski, L.; Poewe, W.; Wenning, G.K.; Stefanova, N. Toll-like receptor 4 is required for alpha-synuclein dependent activation of microglia and astroglia. Glia 2013, 61, 349–360. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gordon, R.; Albornoz, E.A.; Christie, D.C.; Langley, M.R.; Kumar, V.; Mantovani, S.; Robertson, A.A.B.; Butler, M.S.; Rowe, D.B.; O’Neill, L.A.; et al. Inflammasome inhibition prevents alpha-synuclein pathology and dopaminergic neurodegeneration in mice. Sci. Transl. Med. 2018, 10. [Google Scholar] [CrossRef] [Green Version]
- Sampson, T.R.; Challis, C.; Jain, N.; Moiseyenko, A.; Ladinsky, M.S.; Shastri, G.G.; Thron, T.; Needham, B.D.; Horvath, I.; Debelius, J.W.; et al. A gut bacterial amyloid promotes alpha-synuclein aggregation and motor impairment in mice. eLife 2020, 9. [Google Scholar] [CrossRef]
- Chandra, R.; Hiniker, A.; Kuo, Y.M.; Nussbaum, R.L.; Liddle, R.A. alpha-Synuclein in gut endocrine cells and its implications for Parkinson’s disease. JCI Insight 2017, 2. [Google Scholar] [CrossRef] [Green Version]
- Braak, H.; Rub, U.; Gai, W.P.; Del Tredici, K. Idiopathic Parkinson’s disease: Possible routes by which vulnerable neuronal types may be subject to neuroinvasion by an unknown pathogen. J. Neural. Transm. 2003, 110, 517–536. [Google Scholar] [CrossRef] [PubMed]
- Challis, C.; Hori, A.; Sampson, T.R.; Yoo, B.B.; Challis, R.C.; Hamilton, A.M.; Mazmanian, S.K.; Volpicelli-Daley, L.A.; Gradinaru, V. Gut-seeded alpha-synuclein fibrils promote gut dysfunction and brain pathology specifically in aged mice. Nat. Neurosci. 2020, 23, 327–336. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.; Kwon, S.H.; Kam, T.I.; Panicker, N.; Karuppagounder, S.S.; Lee, S.; Lee, J.H.; Kim, W.R.; Kook, M.; Foss, C.A.; et al. Transneuronal Propagation of Pathologic alpha-Synuclein from the Gut to the Brain Models Parkinson’s Disease. Neuron 2019, 103, 627–641. [Google Scholar] [CrossRef] [PubMed]
- Svensson, E.; Horvath-Puho, E.; Thomsen, R.W.; Djurhuus, J.C.; Pedersen, L.; Borghammer, P.; Sorensen, H.T. Vagotomy and subsequent risk of Parkinson’s disease. Ann. Neurol. 2015, 78, 522–529. [Google Scholar] [CrossRef]
- Sumikura, H.; Takao, M.; Hatsuta, H.; Ito, S.; Nakano, Y.; Uchino, A.; Nogami, A.; Saito, Y.; Mochizuki, H.; Murayama, S. Distribution of alpha-synuclein in the spinal cord and dorsal root ganglia in an autopsy cohort of elderly persons. Acta Neuropathol. Commun. 2015, 3, 57. [Google Scholar] [CrossRef] [Green Version]
- Peelaerts, W.; Bousset, L.; Van der Perren, A.; Moskalyuk, A.; Pulizzi, R.; Giugliano, M.; Van den Haute, C.; Melki, R.; Baekelandt, V. alpha-Synuclein strains cause distinct synucleinopathies after local and systemic administration. Nature 2015, 522, 340–344. [Google Scholar] [CrossRef]
- Visanji, N.P.; Marras, C.; Kern, D.S.; Al Dakheel, A.; Gao, A.; Liu, L.W.; Lang, A.E.; Hazrati, L.N. Colonic mucosal a-synuclein lacks specificity as a biomarker for Parkinson disease. Neurology 2015, 84, 609–616. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Phillips, R.J.; Walter, G.C.; Ringer, B.E.; Higgs, K.M.; Powley, T.L. Alpha-synuclein immunopositive aggregates in the myenteric plexus of the aging Fischer 344 rat. Exp. Neurol. 2009, 220, 109–119. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Unger, M.M.; Spiegel, J.; Dillmann, K.U.; Grundmann, D.; Philippeit, H.; Burmann, J.; Fassbender, K.; Schwiertz, A.; Schafer, K.H. Short chain fatty acids and gut microbiota differ between patients with Parkinson’s disease and age-matched controls. Parkinsonism Relat. Disord. 2016, 32, 66–72. [Google Scholar] [CrossRef] [PubMed]
- Qiao, C.M.; Sun, M.F.; Jia, X.B.; Shi, Y.; Zhang, B.P.; Zhou, Z.L.; Zhao, L.P.; Cui, C.; Shen, Y.Q. Sodium butyrate causes alpha-synuclein degradation by an Atg5-dependent and PI3K/Akt/mTOR-related autophagy pathway. Exp. Cell Res. 2020, 387, 111772. [Google Scholar] [CrossRef]
- Cantu-Jungles, T.M.; Rasmussen, H.E.; Hamaker, B.R. Potential of Prebiotic Butyrogenic Fibers in Parkinson’s Disease. Front. Neurol. 2019, 10, 663. [Google Scholar] [CrossRef] [Green Version]
- Gazerani, P. Probiotics for Parkinson’s Disease. Int J. Mol. Sci 2019, 20. [Google Scholar] [CrossRef] [Green Version]
- Barichella, M.; Pacchetti, C.; Bolliri, C.; Cassani, E.; Iorio, L.; Pusani, C.; Pinelli, G.; Privitera, G.; Cesari, I.; Faierman, S.A.; et al. Probiotics and prebiotic fiber for constipation associated with Parkinson disease: An RCT. Neurology 2016, 87, 1274–1280. [Google Scholar] [CrossRef]
- Tamtaji, O.R.; Taghizadeh, M.; Daneshvar Kakhaki, R.; Kouchaki, E.; Bahmani, F.; Borzabadi, S.; Oryan, S.; Mafi, A.; Asemi, Z. Clinical and metabolic response to probiotic administration in people with Parkinson’s disease: A randomized, double-blind, placebo-controlled trial. Clin. Nutr. 2019, 38, 1031–1035. [Google Scholar] [CrossRef]
- Huang, R.; Wang, K.; Hu, J. Effect of Probiotics on Depression: A Systematic Review and Meta-Analysis of Randomized Controlled Trials. Nutrients 2016, 8. [Google Scholar] [CrossRef]
- Goya, M.E.; Xue, F.; Sampedro-Torres-Quevedo, C.; Arnaouteli, S.; Riquelme-Dominguez, L.; Romanowski, A.; Brydon, J.; Ball, K.L.; Stanley-Wall, N.R.; Doitsidou, M. Probiotic Bacillus subtilis Protects against alpha-Synuclein Aggregation in C. elegans. Cell Rep. 2020, 30, 367–380. [Google Scholar] [CrossRef]
- Riva, N.; Agosta, F.; Lunetta, C.; Filippi, M.; Quattrini, A. Recent advances in amyotrophic lateral sclerosis. J. Neurol. 2016, 263, 1241–1254. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hardiman, O.; Al-Chalabi, A.; Chio, A.; Corr, E.M.; Logroscino, G.; Robberecht, W.; Shaw, P.J.; Simmons, Z.; van den Berg, L.H. Amyotrophic lateral sclerosis. Nat. Reviews. Dis. Primers 2017, 3, 17071. [Google Scholar] [CrossRef] [PubMed]
- Pupillo, E.; Poloni, M.; Bianchi, E.; Giussani, G.; Logroscino, G.; Zoccolella, S.; Chio, A.; Calvo, A.; Corbo, M.; Lunetta, C.; et al. Trauma and amyotrophic lateral sclerosis: A European population-based case-control study from the EURALS consortium. Amyotroph. Lateral Scler. Front. Degener. 2018, 19, 118–125. [Google Scholar] [CrossRef] [PubMed]
- Peters, S.; Visser, A.E.; D’Ovidio, F.; Vlaanderen, J.; Portengen, L.; Beghi, E.; Chio, A.; Logroscino, G.; Hardiman, O.; Pupillo, E.; et al. Effect modification of the association between total cigarette smoking and ALS risk by intensity, duration and time-since-quitting: Euro-MOTOR. J. Neurol. Neurosurg. Psychiatry 2020, 91, 33–39. [Google Scholar] [CrossRef]
- Visser, A.E.; Rooney, J.P.K.; D’Ovidio, F.; Westeneng, H.J.; Vermeulen, R.C.H.; Beghi, E.; Chio, A.; Logroscino, G.; Hardiman, O.; Veldink, J.H.; et al. Multicentre, cross-cultural, population-based, case-control study of physical activity as risk factor for amyotrophic lateral sclerosis. J. Neurol. Neurosurg. Psychiatry 2018, 89, 797–803. [Google Scholar] [CrossRef]
- O’Reilly, E.J.; Wang, H.; Weisskopf, M.G.; Fitzgerald, K.C.; Falcone, G.; McCullough, M.L.; Thun, M.; Park, Y.; Kolonel, L.N.; Ascherio, A. Premorbid body mass index and risk of amyotrophic lateral sclerosis. Amyotroph. Lateral Scler. Front. Degener. 2013, 14, 205–211. [Google Scholar] [CrossRef] [Green Version]
- Gallo, V.; Wark, P.A.; Jenab, M.; Pearce, N.; Brayne, C.; Vermeulen, R.; Andersen, P.M.; Hallmans, G.; Kyrozis, A.; Vanacore, N.; et al. Prediagnostic body fat and risk of death from amyotrophic lateral sclerosis: The EPIC cohort. Neurology 2013, 80, 829–838. [Google Scholar] [CrossRef] [Green Version]
- Paganoni, S.; Deng, J.; Jaffa, M.; Cudkowicz, M.E.; Wills, A.-M. Body mass index, not dyslipidemia, is an independent predictor of survival in amyotrophic lateral sclerosis. Muscle Nerve 2011, 44, 20–24. [Google Scholar] [CrossRef] [Green Version]
- Calvo, A.; Moglia, C.; Lunetta, C.; Marinou, K.; Ticozzi, N.; Ferrante, G.D.; Scialo, C.; Soraru, G.; Trojsi, F.; Conte, A.; et al. Factors predicting survival in ALS: A multicenter Italian study. J. Neurol. 2017, 264, 54–63. [Google Scholar] [CrossRef]
- Dupuis, L.; Pradat, P.F.; Ludolph, A.C.; Loeffler, J.P. Energy metabolism in amyotrophic lateral sclerosis. Lancet. Neurol. 2011, 10, 75–82. [Google Scholar] [CrossRef]
- Desport, J.C.; Torny, F.; Lacoste, M.; Preux, P.M.; Couratier, P. Hypermetabolism in ALS: Correlations with clinical and paraclinical parameters. Neurodegener. Dis. 2005, 2, 202–207. [Google Scholar] [CrossRef] [PubMed]
- Funalot, B.; Desport, J.C.; Sturtz, F.; Camu, W.; Couratier, P. High metabolic level in patients with familial amyotrophic lateral sclerosis. Amyotroph. Lateral Scler. 2009, 10, 113–117. [Google Scholar] [CrossRef] [PubMed]
- Dupuis, L.; Oudart, H.; Rene, F.; Gonzalez de Aguilar, J.L.; Loeffler, J.P. Evidence for defective energy homeostasis in amyotrophic lateral sclerosis: Benefit of a high-energy diet in a transgenic mouse model. Proc. Natl. Acad. Sci. USA 2004, 101, 11159–11164. [Google Scholar] [CrossRef] [Green Version]
- Henriques, A.; Croixmarie, V.; Priestman, D.A.; Rosenbohm, A.; Dirrig-Grosch, S.; D’Ambra, E.; Huebecker, M.; Hussain, G.; Boursier-Neyret, C.; Echaniz-Laguna, A.; et al. Amyotrophic lateral sclerosis and denervation alter sphingolipids and up-regulate glucosylceramide synthase. Hum. Mol. Genet. 2015, 24, 7390–7405. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karpati, G.; Klassen, G.; Tanser, P. The effects of partial chronic denervation on forearm metabolism. Can. J. Neurol. Sciences 1979, 6, 105–112. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lunetta, C.; Lizio, A.; Tremolizzo, L.; Ruscica, M.; Macchi, C.; Riva, N.; Weydt, P.; Corradi, E.; Magni, P.; Sansone, V. Serum irisin is upregulated in patients affected by amyotrophic lateral sclerosis and correlates with functional and metabolic status. J. Neurol. 2018, 265, 3001–3008. [Google Scholar] [CrossRef]
- D’Ovidio, F.; d’Errico, A.; Carna, P.; Calvo, A.; Costa, G.; Chio, A. The role of pre-morbid diabetes on developing amyotrophic lateral sclerosis. Eur. J. Neurol. 2018, 25, 164–170. [Google Scholar] [CrossRef]
- Kioumourtzoglou, M.A.; Rotem, R.S.; Seals, R.M.; Gredal, O.; Hansen, J.; Weisskopf, M.G. Diabetes Mellitus, Obesity, and Diagnosis of Amyotrophic Lateral Sclerosis: A Population-Based Study. JAMA Neurol 2015, 72, 905–911. [Google Scholar] [CrossRef] [Green Version]
- Sun, Y.; Lu, C.J.; Chen, R.C.; Hou, W.H.; Li, C.Y. Risk of Amyotrophic Lateral Sclerosis in Patients with Diabetes: A Nationwide Population-Based Cohort Study. J. Epidemiol. 2015, 25, 445–451. [Google Scholar] [CrossRef] [Green Version]
- Wei, Q.Q.; Chen, Y.; Cao, B.; Ou, R.W.; Zhang, L.; Hou, Y.; Gao, X.; Shang, H. Blood hemoglobin A1c levels and amyotrophic lateral sclerosis survival. Mol. Neurodegener. 2017, 12, 69. [Google Scholar] [CrossRef] [Green Version]
- Paganoni, S.; Hyman, T.; Shui, A.; Allred, P.; Harms, M.; Liu, J.; Maragakis, N.; Schoenfeld, D.; Yu, H.; Atassi, N.; et al. Pre-morbid type 2 diabetes mellitus is not a prognostic factor in amyotrophic lateral sclerosis. Muscle Nerve 2015, 52, 339–343. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mariosa, D.; Hammar, N.; Malmstrom, H.; Ingre, C.; Jungner, I.; Ye, W.; Fang, F.; Walldius, G. Blood biomarkers of carbohydrate, lipid, and apolipoprotein metabolisms and risk of amyotrophic lateral sclerosis: A more than 20-year follow-up of the Swedish AMORIS cohort. Ann. Neurol. 2017, 81, 718–728. [Google Scholar] [CrossRef] [PubMed]
- Rosenbohm, A.; Nagel, G.; Peter, R.S.; Brehme, T.; Koenig, W.; Dupuis, L.; Rothenbacher, D.; Ludolph, A.C. ALS Registry Study Group. Association of Serum Retinol-Binding Protein 4 Concentration with Risk for and Prognosis of Amyotrophic Lateral Sclerosis. JAMA Neurol. 2018, 75, 600–607. [Google Scholar] [CrossRef] [PubMed]
- Pradat, P.F.; Bruneteau, G.; Gordon, P.H.; Dupuis, L.; Bonnefont-Rousselot, D.; Simon, D.; Salachas, F.; Corcia, P.; Frochot, V.; Lacorte, J.M.; et al. Impaired glucose tolerance in patients with amyotrophic lateral sclerosis. Amyotroph. Lateral Scler. 2010, 11, 166–171. [Google Scholar] [CrossRef] [PubMed]
- Doré, S.; Krieger, C.; Kar, S.; Quirion, R. Distribution and levels of insulin-like growth factor (IGF-I and IGF-II) and insulin receptor binding sites in the spinal cords of amyotrophic lateral sclerosis (ALS) patients. Mol. Brain Res. 1996, 41, 128–133. [Google Scholar] [CrossRef]
- Lunetta, C.; Serafini, M.; Prelle, A.; Magni, P.; Dozio, E.; Ruscica, M.; Sassone, J.; Colciago, C.; Moggio, M.; Corbo, M.; et al. Impaired expression of insulin-like growth factor-1 system in skeletal muscle of amyotrophic lateral sclerosis patients. Muscle Nerve 2012, 45, 200–208. [Google Scholar] [CrossRef] [Green Version]
- Ngo, S.T.; Steyn, F.J. The interplay between metabolic homeostasis and neurodegeneration: Insights into the neurometabolic nature of amyotrophic lateral sclerosis. Cell Regen. 2015, 4, 5. [Google Scholar] [CrossRef] [Green Version]
- Pagani, M.; Chio, A.; Valentini, M.C.; Oberg, J.; Nobili, F.; Calvo, A.; Moglia, C.; Bertuzzo, D.; Morbelli, S.; De Carli, F.; et al. Functional pattern of brain FDG-PET in amyotrophic lateral sclerosis. Neurology 2014, 83, 1067–1074. [Google Scholar] [CrossRef] [Green Version]
- Stallings, N.R.; Puttaparthi, K.; Dowling, K.J.; Luther, C.M.; Burns, D.K.; Davis, K.; Elliott, J.L. TDP-43, an ALS linked protein, regulates fat deposition and glucose homeostasis. PLoS ONE 2013, 8, e71793. [Google Scholar] [CrossRef] [Green Version]
- Torres-Aleman, I.; Barrios, V.; Berciano, J. The peripheral insulin-like growth factor system in amyotrophic lateral sclerosis and in multiple sclerosis. Neurology 1998, 50, 772–776. [Google Scholar] [CrossRef]
- Araki, K.; Araki, A.; Honda, D.; Izumoto, T.; Hashizume, A.; Hijikata, Y.; Yamada, S.; Iguchi, Y.; Hara, A.; Ikumi, K.; et al. TDP-43 regulates early-phase insulin secretion via CaV1.2-mediated exocytosis in islets. J. Clin. Investig. 2019, 129, 3578–3593. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bjornevik, K.; Zhang, Z.; O’Reilly, E.J.; Berry, J.D.; Clish, C.B.; Deik, A.; Jeanfavre, S.; Kato, I.; Kelly, R.S.; Kolonel, L.N.; et al. Prediagnostic plasma metabolomics and the risk of amyotrophic lateral sclerosis. Neurology 2019, 92, e2089–e2100. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bandres-Ciga, S.; Noyce, A.J.; Hemani, G.; Nicolas, A.; Calvo, A.; Mora, G.; Consortium, I.; International, A.L.S.G.C.; Tienari, P.J.; Stone, D.J.; et al. Shared polygenic risk and causal inferences in amyotrophic lateral sclerosis. Ann. Neurol. 2019, 85, 470–481. [Google Scholar] [CrossRef] [PubMed]
- Zeng, P.; Zhou, X. Causal effects of blood lipids on amyotrophic lateral sclerosis: A Mendelian randomization study. Hum. Mol. Genet. 2019, 28, 688–697. [Google Scholar] [CrossRef] [Green Version]
- Dupuis, L.; Corcia, P.; Fergani, A.; Gonzalez De Aguilar, J.L.; Bonnefont-Rousselot, D.; Bittar, R.; Seilhean, D.; Hauw, J.J.; Lacomblez, L.; Loeffler, J.P.; et al. Dyslipidemia is a protective factor in amyotrophic lateral sclerosis. Neurology 2008, 70, 1004–1009. [Google Scholar] [CrossRef]
- Dorst, J.; Kuhnlein, P.; Hendrich, C.; Kassubek, J.; Sperfeld, A.D.; Ludolph, A.C. Patients with elevated triglyceride and cholesterol serum levels have a prolonged survival in amyotrophic lateral sclerosis. J. Neurol. 2011, 258, 613–617. [Google Scholar] [CrossRef]
- Ingre, C.; Chen, L.; Zhan, Y.; Termorshuizen, J.; Yin, L.; Fang, F. Lipids, apolipoproteins, and prognosis of amyotrophic lateral sclerosis. Neurology 2020, 94, e1835–e1844. [Google Scholar] [CrossRef] [Green Version]
- Szelechowski, M.; Amoedo, N.; Obre, E.; Leger, C.; Allard, L.; Bonneu, M.; Claverol, S.; Lacombe, D.; Oliet, S.; Chevallier, S.; et al. Metabolic Reprogramming in Amyotrophic Lateral Sclerosis. Sci. Rep. 2018, 8, 3953. [Google Scholar] [CrossRef] [Green Version]
- Palamiuc, L.; Schlagowski, A.; Ngo, S.T.; Vernay, A.; Dirrig-Grosch, S.; Henriques, A.; Boutillier, A.L.; Zoll, J.; Echaniz-Laguna, A.; Loeffler, J.P.; et al. A metabolic switch toward lipid use in glycolytic muscle is an early pathologic event in a mouse model of amyotrophic lateral sclerosis. EMBO Mol. Med. 2015, 7, 526–546. [Google Scholar] [CrossRef]
- Chaves-Filho, A.B.; Pinto, I.F.D.; Dantas, L.S.; Xavier, A.M.; Inague, A.; Faria, R.L.; Medeiros, M.H.G.; Glezer, I.; Yoshinaga, M.Y.; Miyamoto, S. Alterations in lipid metabolism of spinal cord linked to amyotrophic lateral sclerosis. Sci. Rep. 2019, 9, 11642. [Google Scholar] [CrossRef] [Green Version]
- Lindauer, E.; Dupuis, L.; Muller, H.P.; Neumann, H.; Ludolph, A.C.; Kassubek, J. Adipose Tissue Distribution Predicts Survival in Amyotrophic Lateral Sclerosis. PLoS ONE 2013, 8, e67783. [Google Scholar] [CrossRef]
- Pennetta, G.; Welte, M.A. Emerging Links between Lipid Droplets and Motor Neuron Diseases. Dev. Cell 2018, 45, 427–432. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chiang, P.M.; Ling, J.; Jeong, Y.H.; Price, D.L.; Aja, S.M.; Wong, P.C. Deletion of TDP-43 down-regulates Tbc1d1, a gene linked to obesity, and alters body fat metabolism. Proc. Natl. Acad. Sci. USA 2010, 107, 16320–16324. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nodera, H.; Takamatsu, N.; Muguruma, N.; Ukimoto, K.; Nishio, S.; Oda, M.; Izumi, Y.; Kaji, R. Frequent hepatic steatosis in amyotrophic lateral sclerosis: Implication for systemic involvement. Neurol. Clin. Neurosci. 2015, 3, 58–62. [Google Scholar] [CrossRef]
- Morozova, N.; Weisskopf, M.G.; McCullough, M.L.; Munger, K.L.; Calle, E.E.; Thun, M.J.; Ascherio, A. Diet and amyotrophic lateral sclerosis. Epidemiology 2008, 19, 324–337. [Google Scholar] [CrossRef]
- Fondell, E.; O’Reilly, E.I.; Fitzgerald, K.C.; Falcone, G.J.; Kolonel, L.N.; Park, Y.; Gapstur, S.M.; Ascherio, A. Intakes of caffeine, coffee and tea and risk of amyotrophic lateral sclerosis: Results from five cohort studies. Amyotroph. Lateral Scler. Front. Degener. 2015, 16, 366–371. [Google Scholar] [CrossRef] [Green Version]
- Nelson, L.M.; Matkin, C.; Longstreth, W.T., Jr.; McGuire, V. Population-based case-control study of amyotrophic lateral sclerosis in western Washington State. II. Diet. Am. J. Epidemiol. 2000, 151, 164–173. [Google Scholar] [CrossRef]
- Kim, B.; Jin, Y.; Kim, S.H.; Park, Y. Association between macronutrient intake and amyotrophic lateral sclerosis prognosis. Nutr. Neurosci. 2020, 23, 8–15. [Google Scholar] [CrossRef]
- Wills, A.-M.; Hubbard, J.; Macklin, E.A.; Glass, J.; Tandan, R.; Simpson, E.P.; Brooks, B.; Gelinas, D.; Mitsumoto, H.; Mozaffar, T.; et al. Hypercaloric enteral nutrition in patients with amyotrophic lateral sclerosis: A randomised, double-blind, placebo-controlled phase 2 trial. Lancet 2014, 383, 2065–2072. [Google Scholar] [CrossRef] [Green Version]
- Dorst, J.; Cypionka, J.; Ludolph, A.C. High-caloric food supplements in the treatment of amyotrophic lateral sclerosis: A prospective interventional study. Amyotroph. Lateral Scler. Front. Degener. 2013, 14, 533–536. [Google Scholar] [CrossRef]
- Ludolph, A.C.; Dorst, J.; Dreyhaupt, J.; Weishaupt, J.H.; Kassubek, J.; Weiland, U.; Meyer, T.; Petri, S.; Hermann, A.; Emmer, A.; et al. Effect of High-Caloric Nutrition on Survival in Amyotrophic Lateral Sclerosis. Ann. Neurol. 2020, 87, 206–216. [Google Scholar] [CrossRef] [PubMed]
- Fitzgerald, K.C.; O’Reilly, E.J.; Falcone, G.J.; McCullough, M.L.; Park, Y.; Kolonel, L.N.; Ascherio, A. Dietary omega-3 polyunsaturated fatty acid intake and risk for amyotrophic lateral sclerosis. JAMA Neurol 2014, 71, 1102–1110. [Google Scholar] [CrossRef] [PubMed]
- O’Reilly, É.J.; Bjornevik, K.; Furtado, J.D.; Kolonel, L.N.; Le Marchand, L.; McCullough, M.L.; Stevens, V.L.; Shadyab, A.H.; Snetselaar, L.; Manson, J.E.; et al. Prediagnostic plasma polyunsaturated fatty acids and the risk of amyotrophic lateral sclerosis. Neurology 2020, 94, e811–e819. [Google Scholar] [CrossRef] [PubMed]
- Ilieva, E.V.; Ayala, V.; Jove, M.; Dalfo, E.; Cacabelos, D.; Povedano, M.; Bellmunt, M.J.; Ferrer, I.; Pamplona, R.; Portero-Otin, M. Oxidative and endoplasmic reticulum stress interplay in sporadic amyotrophic lateral sclerosis. Brain J. Neurol. 2007, 130, 3111–3123. [Google Scholar] [CrossRef] [PubMed]
- Torres, P.; Cacabelos, D.; Pairada, J.; Bauer, K.C.; Boada, J.; Fontdevila, L.; Rossi, C.; Povedano, M.; Ferrer, I.; Pamplona, R.; et al. Gender-Specific Beneficial Effects of Docosahexaenoic Acid Dietary Supplementation in G93A-SOD1 Amyotrophic Lateral Sclerosis Mice. Neurother. J. Am. Soc. Exp. Neurother. 2020, 17, 269–281. [Google Scholar] [CrossRef]
- Cacabelos, D.; Ayala, V.; Granado-Serrano, A.B.; Jove, M.; Torres, P.; Boada, J.; Cabre, R.; Ramirez-Nunez, O.; Gonzalo, H.; Soler-Cantero, A.; et al. Interplay between TDP-43 and docosahexaenoic acid-related processes in amyotrophic lateral sclerosis. Neurobiol. Dis. 2016, 88, 148–160. [Google Scholar] [CrossRef]
- Yip, P.K.; Pizzasegola, C.; Gladman, S.; Biggio, M.L.; Marino, M.; Jayasinghe, M.; Ullah, F.; Dyall, S.C.; Malaspina, A.; Bendotti, C.; et al. The omega-3 fatty acid eicosapentaenoic acid accelerates disease progression in a model of amyotrophic lateral sclerosis. PLoS ONE 2013, 8, e61626. [Google Scholar] [CrossRef] [Green Version]
- Kim, Y.J.; Nakatomi, R.; Akagi, T.; Hashikawa, T.; Takahashi, R. Unsaturated fatty acids induce cytotoxic aggregate formation of amyotrophic lateral sclerosis-linked superoxide dismutase 1 mutants. J. Biol. Chem. 2005, 280, 21515–21521. [Google Scholar] [CrossRef] [Green Version]
- Veldink, J.H.; Kalmijn, S.; Groeneveld, G.-J.; Wunderink, W.; Koster, A.; de Vries, J.H.M.; Van der Luyt, J.; Wokke, J.H.J.; Van den Berg, L.H. Intake of polyunsaturated fatty acids and vitamin E reduces the risk of developing amyotrophic lateral sclerosis. J. Neurol. Neurosurg. Psychiatry 2007, 78, 367–371. [Google Scholar] [CrossRef] [Green Version]
- Pupillo, E.; Bianchi, E.; Chio, A.; Casale, F.; Zecca, C.; Tortelli, R.; Beghi, E.; Group, S.; Group, P.; Group, S. Amyotrophic lateral sclerosis and food intake. Amyotroph. Lateral Scler. Front. Degener. 2018, 19, 267–274. [Google Scholar] [CrossRef]
- Gentile, F.; Scarlino, S.; Falzone, Y.M.; Lunetta, C.; Tremolizzo, L.; Quattrini, A.; Riva, N. The Peripheral Nervous System in Amyotrophic Lateral Sclerosis: Opportunities for Translational Research. Front. Neurosci. 2019, 13, 601. [Google Scholar] [CrossRef] [PubMed]
- Fitzgerald, K.C.; O’Reilly, E.J.; Fondell, E.; Falcone, G.J.; McCullough, M.L.; Park, Y.; Kolonel, L.N.; Ascherio, A. Intakes of vitamin C and carotenoids and risk of amyotrophic lateral sclerosis: Pooled results from 5 cohort studies. Ann. Neurol. 2013, 73, 236–245. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ascherio, A.; Weisskopf, M.G.; O’Reilly, E., J.; Jacobs, E.J.; McCullough, M.L.; Calle, E.E.; Cudkowicz, M.; Thun, M.J. Vitamin E intake and risk of amyotrophic lateral sclerosis. Ann. Neurol. 2005, 57, 104–110. [Google Scholar] [CrossRef] [PubMed]
- Seelen, M.; Huisman, M.; de Jong, S.; van Doormaal, P.; de Vries, J.; Van Der Kooi, A.; De Visser, M.; Schelhaas, H.; Van den Berg, L.; Veldink, J. Mediterranean Diet Modifies Risk of Amyotrophic Lateral Sclerosis (P05.075). Neurology 2013, 80. [Google Scholar]
- Nieves, J.W.; Gennings, C.; Factor-Litvak, P.; Hupf, J.; Singleton, J.; Sharf, V.; Oskarsson, B.; Fernandes Filho, J.A.; Sorenson, E.J.; D’Amico, E.; et al. Association Between Dietary Intake and Function in Amyotrophic Lateral Sclerosis. JAMA Neurol 2016, 73, 1425–1432. [Google Scholar] [CrossRef] [Green Version]
- Sala, G.; Arosio, A.; Conti, E.; Beretta, S.; Lunetta, C.; Riva, N.; Ferrarese, C.; Tremolizzo, L. Riluzole Selective Antioxidant Effects in Cell Models Expressing Amyotrophic Lateral Sclerosis Endophenotypes. Clin. Psychopharmacol. Neurosci. 2019, 17, 438–442. [Google Scholar] [CrossRef] [Green Version]
- Beghi, E.; Pupillo, E.; Messina, P.; Giussani, G.; Chio, A.; Zoccolella, S.; Moglia, C.; Corbo, M.; Logroscino, G. Coffee and amyotrophic lateral sclerosis: A possible preventive role. Am. J. Epidemiol. 2011, 174, 1002–1008. [Google Scholar] [CrossRef] [Green Version]
- Pedersen, W.A.; Mattson, M.P. No benefit of dietary restriction on disease onset or progression in amyotrophic lateral sclerosis Cu/Zn-superoxide dismutase mutant mice. Brain Res. 1999, 833, 117–120. [Google Scholar] [CrossRef]
- Erber, A.C.; Cetin, H.; Berry, D.; Schernhammer, E.S. The role of gut microbiota, butyrate and proton pump inhibitors in amyotrophic lateral sclerosis: A systematic review. Int. J. Neurosci. 2019, 1–9. [Google Scholar] [CrossRef]
- Di Gioia, D.; Bozzi Cionci, N.; Baffoni, L.; Amoruso, A.; Pane, M.; Mogna, L.; Gaggia, F.; Lucenti, M.A.; Bersano, E.; Cantello, R.; et al. A prospective longitudinal study on the microbiota composition in amyotrophic lateral sclerosis. BMC Med. 2020, 18, 153. [Google Scholar] [CrossRef]
- Zeng, Q.; Shen, J.; Chen, K.; Zhou, J.; Liao, Q.; Lu, K.; Yuan, J.; Bi, F. The alteration of gut microbiome and metabolism in amyotrophic lateral sclerosis patients. Sci. Rep. 2020, 10, 12998. [Google Scholar] [CrossRef]
- Blacher, E.; Bashiardes, S.; Shapiro, H.; Rothschild, D.; Mor, U.; Dori-Bachash, M.; Kleimeyer, C.; Moresi, C.; Harnik, Y.; Zur, M.; et al. Potential roles of gut microbiome and metabolites in modulating ALS in mice. Nature 2019, 572, 474–480. [Google Scholar] [CrossRef] [PubMed]
- Figueroa-Romero, C.; Guo, K.; Murdock, B.J.; Paez-Colasante, X.; Bassis, C.M.; Mikhail, K.A.; Raue, K.D.; Evans, M.C.; Taubman, G.F.; McDermott, A.J.; et al. Temporal evolution of the microbiome, immune system and epigenome with disease progression in ALS mice. Dis. Models Mech. 2019, 13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, S.; Yi, J.; Zhang, Y.G.; Zhou, J.; Sun, J. Leaky intestine and impaired microbiome in an amyotrophic lateral sclerosis mouse model. Physiol. Rep. 2015, 3, e12356. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, J.; Zhan, Y.; Mariosa, D.; Larsson, H.; Almqvist, C.; Ingre, C.; Zagai, U.; Pawitan, Y.; Fang, F. Antibiotics use and risk of amyotrophic lateral sclerosis in Sweden. Eur. J. Neurol. 2019, 26, 1355–1361. [Google Scholar] [CrossRef] [PubMed]
- Rowin, J.; Xia, Y.; Jung, B.; Sun, J. Gut inflammation and dysbiosis in human motor neuron disease. Physiol. Rep. 2017, 5. [Google Scholar] [CrossRef] [PubMed]
- Zhang, R.; Miller, R.G.; Gascon, R.; Champion, S.; Katz, J.; Lancero, M.; Narvaez, A.; Honrada, R.; Ruvalcaba, D.; McGrath, M.S. Circulating endotoxin and systemic immune activation in sporadic amyotrophic lateral sclerosis (sALS). J. Neuroimmunol. 2009, 206, 121–124. [Google Scholar] [CrossRef] [Green Version]
- Burberry, A.; Wells, M.F.; Limone, F.; Couto, A.; Smith, K.S.; Keaney, J.; Gillet, G.; van Gastel, N.; Wang, J.Y.; Pietilainen, O.; et al. C9orf72 suppresses systemic and neural inflammation induced by gut bacteria. Nature 2020, 582, 89–94. [Google Scholar] [CrossRef]
- Piccione, E.A.; Sletten, D.M.; Staff, N.P.; Low, P.A. Autonomic system and amyotrophic lateral sclerosis. Muscle Nerve 2015, 51, 676–679. [Google Scholar] [CrossRef] [Green Version]
- Esmaeili, M.A.; Panahi, M.; Yadav, S.; Hennings, L.; Kiaei, M. Premature death of TDP-43 (A315T) transgenic mice due to gastrointestinal complications prior to development of full neurological symptoms of amyotrophic lateral sclerosis. Int. J. Exp. Pathol. 2013, 94, 56–64. [Google Scholar] [CrossRef]
- Guo, Y.; Wang, Q.; Zhang, K.; An, T.; Shi, P.; Li, Z.; Duan, W.; Li, C. HO-1 induction in motor cortex and intestinal dysfunction in TDP-43 A315T transgenic mice. Brain Res. 2012, 1460, 88–95. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.G.; Wu, S.; Yi, J.; Xia, Y.; Jin, D.; Zhou, J.; Sun, J. Target Intestinal Microbiota to Alleviate Disease Progression in Amyotrophic Lateral Sclerosis. Clin. Ther. 2017, 39, 322–336. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Foidl, B.M.; Humpel, C. Chronic treatment with five vascular risk factors causes cerebral amyloid angiopathy but no Alzheimer pathology in C57BL6 mice. Brain Behav. Immun. 2019, 78, 52–64. [Google Scholar] [CrossRef] [PubMed]
- Puntener, U.; Booth, S.G.; Perry, V.H.; Teeling, J.L. Long-term impact of systemic bacterial infection on the cerebral vasculature and microglia. J. Neuroinflammation 2012, 9, 146. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matheoud, D.; Cannon, T.; Voisin, A.; Penttinen, A.M.; Ramet, L.; Fahmy, A.M.; Ducrot, C.; Laplante, A.; Bourque, M.J.; Zhu, L.; et al. Intestinal infection triggers Parkinson’s disease-like symptoms in Pink1(-/-) mice. Nature 2019, 571, 565–569. [Google Scholar] [CrossRef]
- Samieri, C.; Sonawane, A.R.; Lefevre-Arbogast, S.; Helmer, C.; Grodstein, F.; Glass, K. Using network science tools to identify novel diet patterns in prodromal dementia. Neurology 2020, 94, e2014–e2025. [Google Scholar] [CrossRef] [Green Version]
- La Rosa, F.; Clerici, M.; Ratto, D.; Occhinegro, A.; Licito, A.; Romeo, M.; Iorio, C.D.; Rossi, P. The Gut-Brain Axis in Alzheimer’s Disease and Omega-3. A Critical Overview of Clinical Trials. Nutrients 2018, 10. [Google Scholar] [CrossRef] [Green Version]
- Yamasaki, T.R.; Ono, K.; Ho, L.; Pasinetti, G.M. Gut Microbiome-Modified Polyphenolic Compounds Inhibit Alpha-Synuclein Seeding and Spreading in Alpha-Synucleinopathies. Front. Neurosci. 2020, 14, 398. [Google Scholar] [CrossRef]
- Meng, Q.; Ying, Z.; Noble, E.; Zhao, Y.; Agrawal, R.; Mikhail, A.; Zhuang, Y.; Tyagi, E.; Zhang, Q.; Lee, J.H.; et al. Systems Nutrigenomics Reveals Brain Gene Networks Linking Metabolic and Brain Disorders. EBioMedicine 2016, 7, 157–166. [Google Scholar] [CrossRef] [Green Version]
- Raimondi, I.; Izzo, L.; Tunesi, M.; Comar, M.; Albani, D.; Giordano, C. Organ-On-A-Chip in vitro Models of the Brain and the Blood-Brain Barrier and Their Value to Study the Microbiota-Gut-Brain Axis in Neurodegeneration. Front. Bioeng. Biotechnol. 2019, 7, 435. [Google Scholar] [CrossRef]
- Boertien, J.M.; van der Zee, S.; Chrysou, A.; Gerritsen, M.J.J.; Jansonius, N.M.; Spikman, J.M.; van Laar, T.; Group, P.S. Study protocol of the DUtch PARkinson Cohort (DUPARC): A prospective, observational study of de novo Parkinson’s disease patients for the identification and validation of biomarkers for Parkinson’s disease subtypes, progression and pathophysiology. BMC Neurol. 2020, 20, 245. [Google Scholar] [CrossRef] [PubMed]
- Isaacson, R.S.; Ganzer, C.A.; Hristov, H.; Hackett, K.; Caesar, E.; Cohen, R.; Kachko, R.; Melendez-Cabrero, J.; Rahman, A.; Scheyer, O.; et al. The clinical practice of risk reduction for Alzheimer’s disease: A precision medicine approach. Alzheimer Dement. J. Alzheimer Assoc. 2018, 14, 1663–1673. [Google Scholar] [CrossRef] [PubMed]
- Hegelmaier, T.; Lebbing, M.; Duscha, A.; Tomaske, L.; Tonges, L.; Holm, J.B.; Bjorn Nielsen, H.; Gatermann, S.G.; Przuntek, H.; Haghikia, A. Interventional Influence of the Intestinal Microbiome Through Dietary Intervention and Bowel Cleansing Might Improve Motor Symptoms in Parkinson’s Disease. Cells 2020, 9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beilharz, J.E.; Kaakoush, N.O.; Maniam, J.; Morris, M.J. Cafeteria diet and probiotic therapy: Cross talk among memory, neuroplasticity, serotonin receptors and gut microbiota in the rat. Mol. Psychiatry 2018, 23, 351–361. [Google Scholar] [CrossRef]
- Rao, S.S.C.; Rehman, A.; Yu, S.; Andino, N.M. Brain fogginess, gas and bloating: A link between SIBO, probiotics and metabolic acidosis. Clin. Transl. Gastroenterol. 2018, 9, 162. [Google Scholar] [CrossRef]
- Vendrik, K.E.W.; Ooijevaar, R.E.; de Jong, P.R.C.; Laman, J.D.; van Oosten, B.W.; van Hilten, J.J.; Ducarmon, Q.R.; Keller, J.J.; Kuijper, E.J.; Contarino, M.F. Fecal Microbiota Transplantation in Neurological Disorders. Front. Cell. Infect. Microbiol. 2020, 10, 98. [Google Scholar] [CrossRef] [Green Version]
- Mandrioli, J.; Amedei, A.; Cammarota, G.; Niccolai, E.; Zucchi, E.; D’Amico, R.; Ricci, F.; Quaranta, G.; Spanu, T.; Masucci, L. FETR-ALS Study Protocol: A Randomized Clinical Trial of Fecal Microbiota Transplantation in Amyotrophic Lateral Sclerosis. Front. Neurol. 2019, 10, 1021. [Google Scholar] [CrossRef]




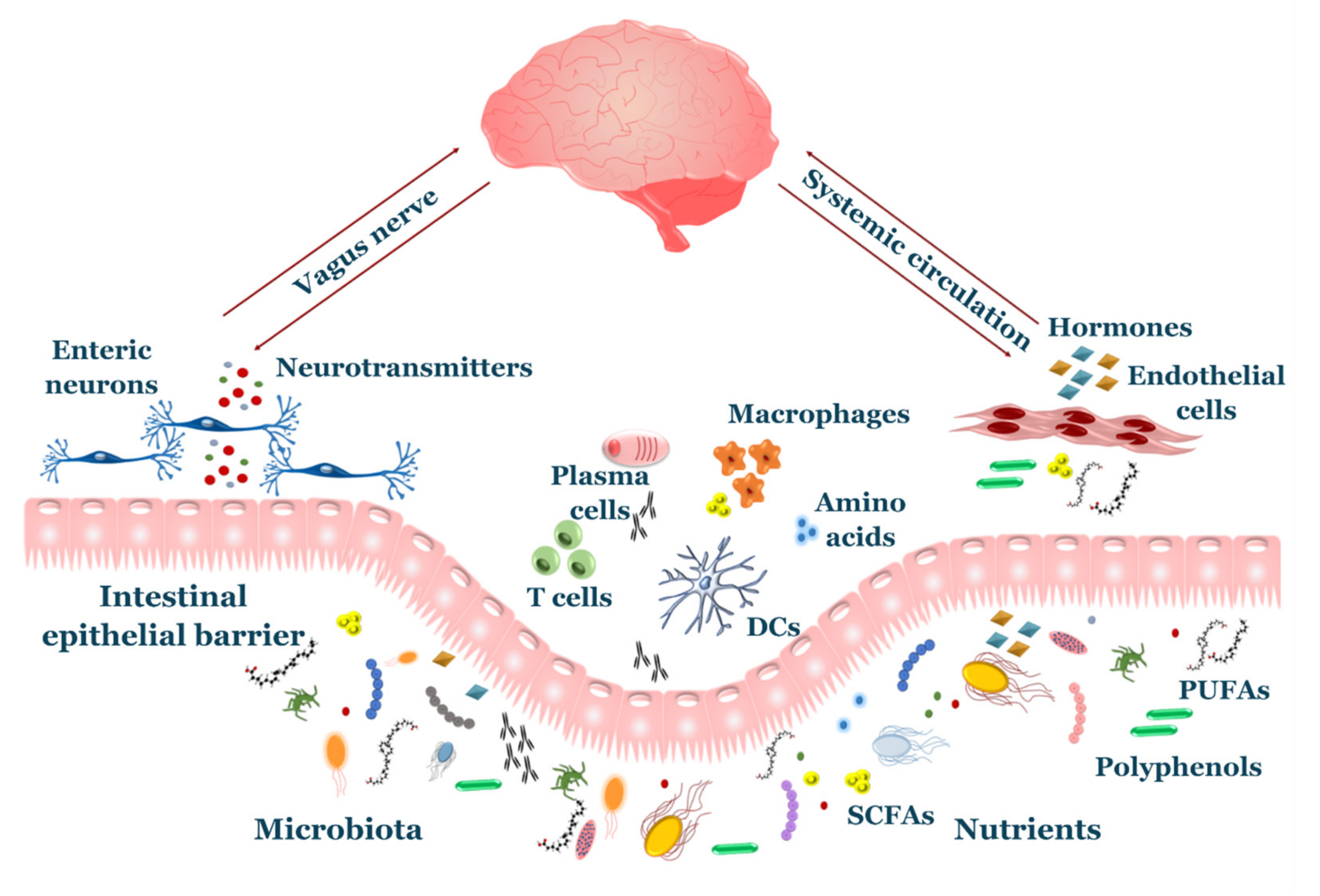{kind=link}
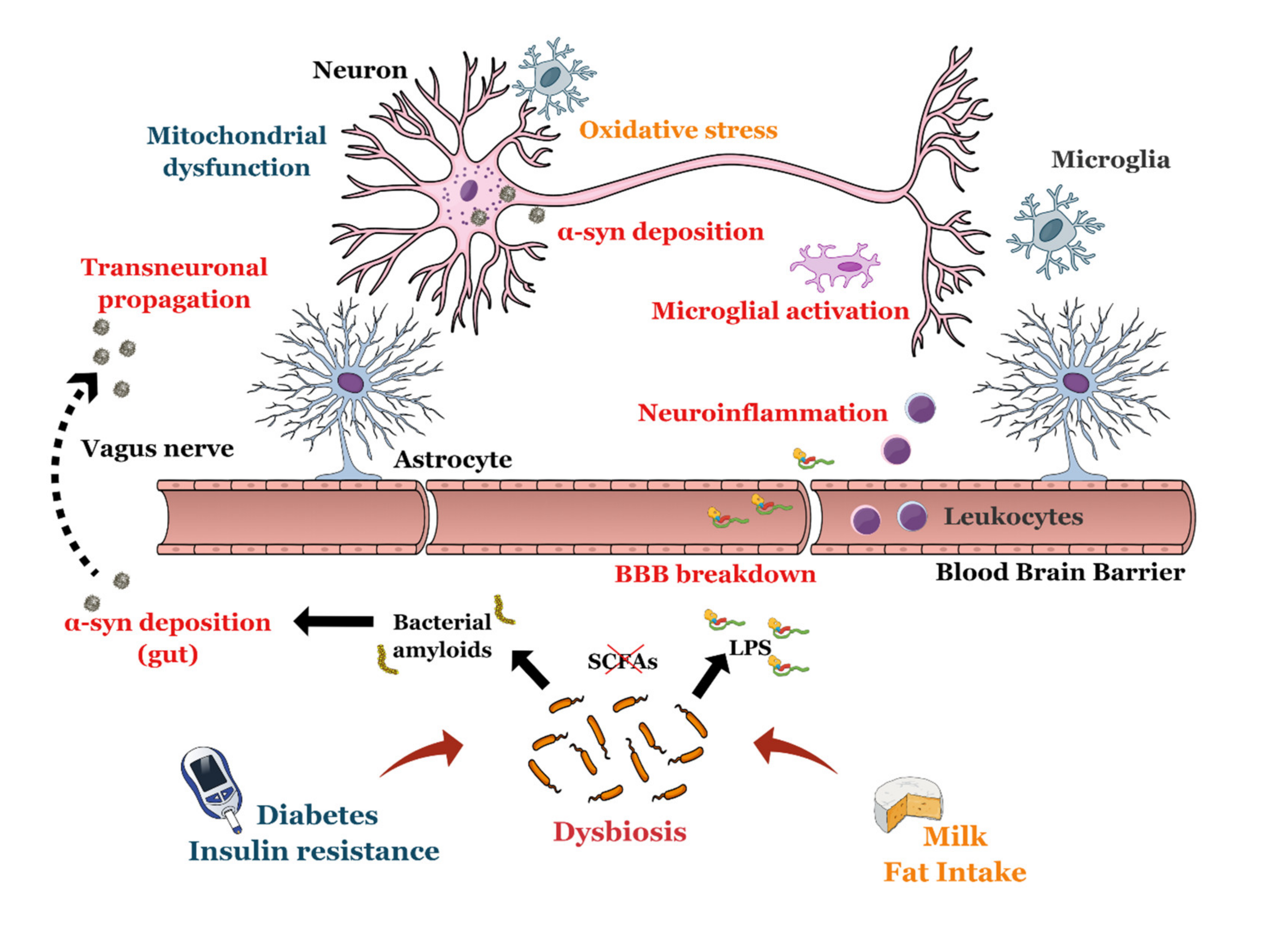{kind=link}
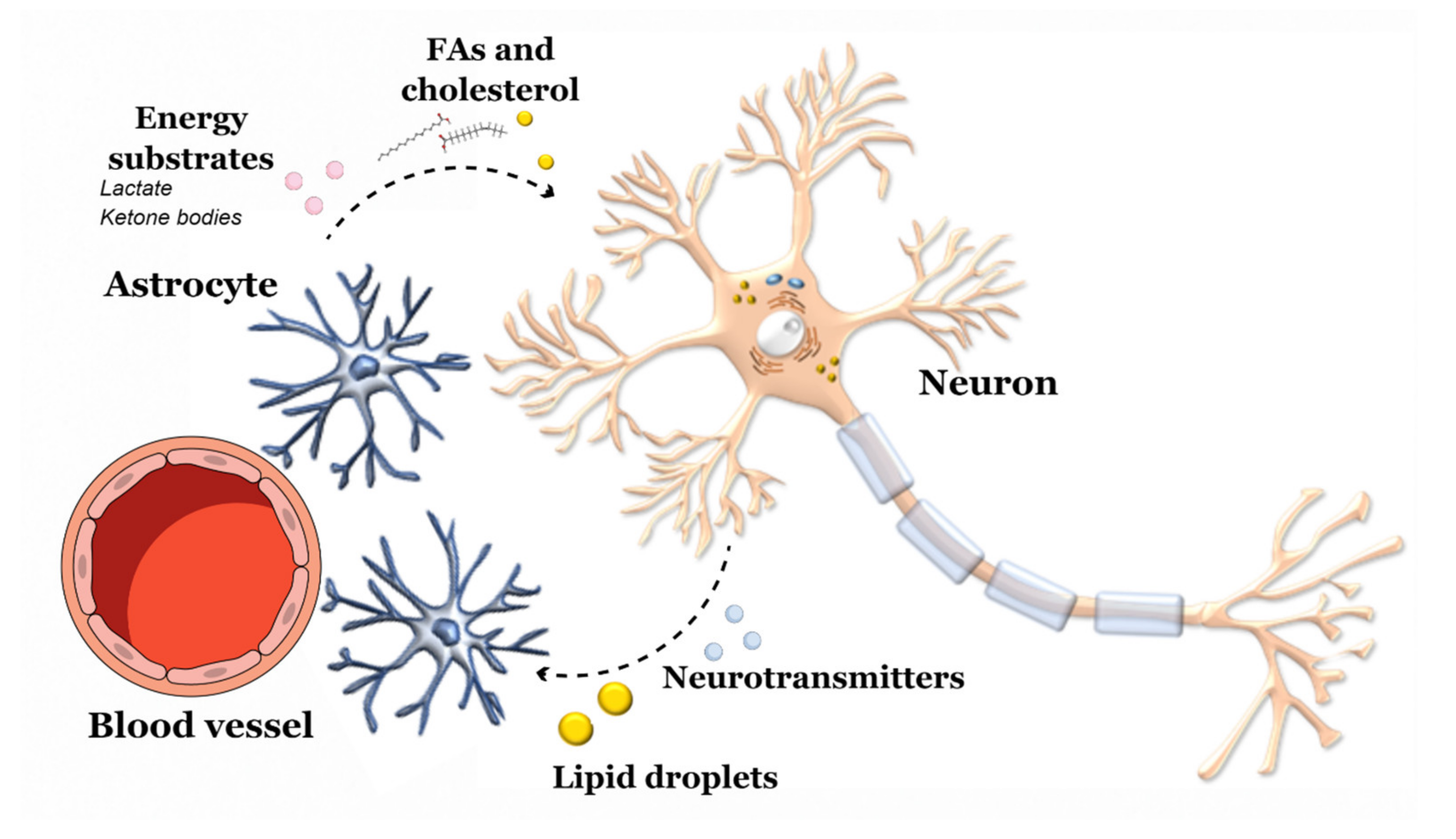{kind=link}
| Harmful | Protective | |
|---|---|---|
| Metabolism | ||
| Obesity | Human: risk in midlife. Mice: memory loss, Aβ deposition and gliosis. | |
| Diabetes | Human and mice: defects in insulin/IGF-1 pathway in brains. | |
| Dyslipidaemia | Human: risk in midlife. Mice: correlates with burden of Aβ pathology. | |
| MAFLD | Human: impaired cognition. Mice: AD-like pathology. | |
| Diet | ||
| Fat intake | Human: saturated fat. Mice: high-fat diet worsens severity in AD-mice. | Human and mice: ω-3 PUFAs protect neurons, reduce inflammation and vascular damage. |
| Antioxidants | Human and mice: counteract ROS production, lipid peroxidation, DNA damage. | |
| Dietary patterns | Human: MeD. Human and mice: caloric restriction. | |
| Microbiota | ||
| Composition | Opportunistic gram-negative bacteria | SCFA-producing bacteria. |
| Mechanisms | Endotoxin exposure, IEB permeability, gut inflammation. | SCFAs supply to gut and brain |
| Gut-brain axis | Aβ deposition in the gut of animal models. Bacterial amyloids may aid in cross-seeding. Gut-to-brain transport after prolonged time. | |
| Gut microbiota-based Therapy | Improve metabolic markers. Efficacy on cognition is limited. | |
| Harmful | Protective | |
|---|---|---|
| Metabolism | ||
| Obesity | No relationship found | |
| Diabetes | Increase risk and clinical severity Mitochondrial dysfunction as a common mechanism | |
| Dyslipidaemia | Exacerbation of motor deficits in animal models. | Decrease risk and progression in humans |
| MAFLD | Unclear association DJ-1 dysfunction may be a potential common mechanism | |
| Diet | ||
| Fat intake | Uncertain association with saturated fats High-fat diet worsen motor symptoms and neuronal loss in mice. | Ω-3 PUFAs |
| Antioxidants | Uric acid decrease risk and progression | |
| Food | Milk (unknown mechanism). | Coffee (A2a receptor antagonist). |
| Dietary patterns | MeD Caloric restriction in humans and animals. | |
| Microbiota | ||
| Composition | Opportunistic gram-negative bacteria Lactobacillus, Bifidobacterium, Akkermansia | SCFA-producing bacteria. |
| Mechanisms | Endotoxemia, IEB permeability, gut inflammation. Sulfite production | SCFAs supply. |
| Gut-brain axis | α-syn deposition in the gut (humans and animals). Bacterial amyloids enhance α-syn aggregation. Gut-to-brain transport demonstrated. | |
| Gut microbiota-based Therapy | Probiotics improve constipation and metabolic markers. Potential to treat other non-motor symptoms (e.g., depression). | |
| Harmful | Protective | |
|---|---|---|
| Metabolism | ||
| Obesity | Decrease risk and progression Counteracts ALS hypermetabolism | |
| Diabetes | Mixed effects (depends on ethnicity) IGT observed in patients Different mechanisms form classic diabetes | |
| Dyslipidaemia | Increased risk of disease | Decreased mortality Early switch to lipid metabolism in motor neurons and muscles may be an early compensatory mechanism |
| MAFLD | Hepatic steatosis frequent finding Unknown significance | |
| Diet | ||
| Fat intake | ω-3 PUFAs may exert a double-edge role | Associated with longer survival |
| Antioxidants | Lowers disease risk Intake correlates with higher functional scores Co-supply with ω-3 PUFAs may show synergic effects | |
| Dietary patterns | Human: MeD Mice: caloric restriction worsens disease severity | |
| Microbiota | ||
| Composition | Opportunistic gram-negative bacteria | SCFA-producing bacteria |
| Mechanisms | Endotoxemia, IEB permeability, gut inflammation Deletion of C9orf72 exaggerates systemic immune response | Nicotinamide and SCFAs levels |
| Gut-brain axis | TDP-43 deposition in the gut of animal models Gut-to-brain transport not assessed | |
| Gut microbiota-based Therapy | Mice: SCFAs alleviate motor symptoms | |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gentile, F.; Doneddu, P.E.; Riva, N.; Nobile-Orazio, E.; Quattrini, A. Diet, Microbiota and Brain Health: Unraveling the Network Intersecting Metabolism and Neurodegeneration. Int. J. Mol. Sci. 2020, 21, 7471. https://doi.org/10.3390/ijms21207471
Gentile F, Doneddu PE, Riva N, Nobile-Orazio E, Quattrini A. Diet, Microbiota and Brain Health: Unraveling the Network Intersecting Metabolism and Neurodegeneration. International Journal of Molecular Sciences. 2020; 21(20):7471. https://doi.org/10.3390/ijms21207471
Chicago/Turabian StyleGentile, Francesco, Pietro Emiliano Doneddu, Nilo Riva, Eduardo Nobile-Orazio, and Angelo Quattrini. 2020. "Diet, Microbiota and Brain Health: Unraveling the Network Intersecting Metabolism and Neurodegeneration" International Journal of Molecular Sciences 21, no. 20: 7471. https://doi.org/10.3390/ijms21207471