Phenotypes of Bronchopulmonary Dysplasia


1
Department of Pediatrics, Far Eastern Memorial Hospital, New Taipei City 22060, Taiwan
2
Department of Pediatrics, National Taiwan University Hospital, College of Medicine, National Taiwan University, Taipei 100225, Taiwan
3
Center for Developmental Biology & Regenerative Medicine, National Taiwan University, Taipei 100226, Taiwan
*
Author to whom correspondence should be addressed.
Int. J. Mol. Sci. 2020, 21(17), 6112; https://doi.org/10.3390/ijms21176112
Submission received: 2 July 2020
/
Revised: 20 August 2020
/
Accepted: 22 August 2020
/
Published: 25 August 2020
(This article belongs to the Special Issue Therapy Personalization in Pediatric Diseases: From Molecular Biology to Therapy)
Abstract
:Bronchopulmonary dysplasia (BPD) is the most common chronic morbidity in preterm infants. In the absence of effective interventions, BPD is currently a major therapeutic challenge. Several risk factors are known for this multifactorial disease that results in disrupted lung development. Inflammation plays an important role and leads to persistent airway and pulmonary vascular disease. Since corticosteroids are potent anti-inflammatory agents, postnatal corticosteroids have been used widely for BPD prevention and treatment. However, the clinical responses vary to a great degree across individuals, and steroid-related complications remain major concerns. Emerging studies on the molecular mechanism of lung alveolarization during inflammatory stress will elucidate the complicated pathway and help discover novel therapeutic targets. Moreover, with the advances in metabolomics, there are new opportunities to identify biomarkers for early diagnosis and prognosis prediction of BPD. Pharmacometabolomics is another novel field aiming to identify the metabolomic changes before and after a specific drug treatment. Through this “metabolic signature,” a more precise treatment may be developed, thereby avoiding unnecessary drug exposure in non-responders. In the future, more clinical, genetic, and translational studies would be required to improve the classification of BPD phenotypes and achieve individualized care to enhance the respiratory outcomes in preterm infants.
1. Introduction
In recent decades, owing to the advancements in obstetrical and neonatal care, such as antenatal corticosteroid [1] and exogenous surfactant therapy [2], the survival rate of premature infants has improved dramatically. The incidence of many prematurity-related complications, such as respiratory distress syndrome (RDS), intraventricular hemorrhage, and necrotizing enterocolitis (NEC), has shown a significant reduction. However, bronchopulmonary dysplasia (BPD) is still one of the most common complications in preterm infants [3,4]. Approximately 40–45% of extremely preterm neonates develop BPD [4,5]. This disease leads to persistent pulmonary dysfunction, such as chronic wheezing, frequent respiratory tract infection, and exercise intolerance in young adults [6].
Both the definition and characteristics of BPD have evolved over the past 50 years (Figure 1) [7,8,9,10,11]. In 1967, when BPD was first reported by Northway [7], the clinical picture for “classic BPD” was severe fibrosis. However, in the era of routine application of antenatal steroid, surfactant, and gentler mechanical ventilation strategy [12] in the neonatal intensive care unit (NICU), the “new BPD” was proposed in 2000 [9]. The definition of BPD is more comprehensive and based on the need for supplemental oxygen for at least 28 days and further categorization of the severity into none, mild, moderate, or severe according to O2 requirement at the postmenstrual age (PMA) of 36 weeks [9]. In terms of the pathogenesis, the concept of maturation arrest of alveologenesis and angiogenesis, further leading to more diffused inflammation and less scarring and fibrosis, has been focused on. [13]. This definition of BPD is a practical reference in clinical settings but does not provide much information on longer-term respiratory outcomes and does not consider BPD as a multifactorial disease. Moreover, new respiratory strategies that use a nasal cannula with different degrees of oxygen make some infants unclassifiable per the current definitions. The previous criteria also do not include infants who die of respiratory failure before the PMA of 36 weeks and the time point of BPD diagnosis, which is the most severe form of lethal BPD. To address these knowledge gaps, the workshop on BPD held by the National Institute of Child Health and Human Development (NICHD) in 2016 refined the definition of BPD according to the previous criteria and present clinical care [11]. An infant born at less than 32 weeks of gestation; having radiographic-confirmed persistent parenchymal lung disease; and at 36 weeks PMA requiring ventilator support for more than three consecutive days, either invasive or non-invasive (N-CPAP, NIPPV, nasal cannula, O2 hood), to maintain arterial oxygen saturation above 90%, would be diagnosed with BPD. This refinement considers newer modes of non-invasive respiratory support and uses grades I, II, and III instead of mild, moderate, or severe. Grade III would be the most severe form of BPD and III(A) is a new category that covers the early-death group (patients who die between 14 days of postnatal age and PMA of 36 weeks because of parenchymal lung disease and respiratory failure), which has been overlooked for a long time [11]. A multicenter assessment of 2677 preterm infants [14] has indicated that BPD severity categorization based on the mode of ventilatory support at 36 weeks of PMA best predicts early childhood morbidity, regardless of supplemental oxygen use.
BPD is a complex disease with poorly characterized classification; this is one of the reasons why definite treatments are still lacking. Multiple factors contribute to the pathogenesis of BPD, which include developmental arrest of the premature lung, infection, oxygen toxicity, and ventilation-induced lung injury, making it difficult to classify the disease into subgroups or the so-called clinical phenotypes. Different phenotypes are associated with different outcomes. Therefore, understanding the diversity of phenotypes may enable better risk stratification and formulation of targeted treatment guidelines [15,16]. Metabolomics is a promising field that involves study of the associations between changes in the metabolic pathways and human diseases, and may help uncover the underlying mechanisms of BPD as well as facilitate the identification of useful biomarkers in early diagnosis, treatment selection, and prognostic prediction [17,18].
Chronic inflammation and hyperoxia have been considered as the important mechanisms for BPD and a common pathway leads to different phenotypes. Corticosteroids have been commonly used to prevent or treat established BPD. However, there are debates about the right kind of steroid, and the right dosage with the right route at the right timing to get the most effective result and simultaneously attenuate the negative impact. Using steroids in preterm infants is challenging because of the variability in clinical effects and the inability to target the possible responders prior to the treatment [19]. Pharmacometabolomics may provide a solution to this clinical dilemma. By measuring the metabolites along with the treatment course, the drug response may be quantified more clearly.
In our review, we focus on the pathology-driven nomenclature of the multiple clinical phenotypes of BPD, updated research regarding the inflammatory pathway and biomarkers for early prediction of high-risk infants prone to developing BPD, and early initiation of preventive strategies. The recent scientific advances in the field of metabolomics and pharmacometabolomics also offer new opportunities to approach the complex pathophysiology of BPD. The ultimate goal would be to develop comprehensive and personalized care for these individuals to combat this devastating disease.
2. Respiratory Phenotypes
Based on the widely accepted diagnostic criteria, BPD is classified into mild, moderate, or severe types according to the respiratory support or the need for supplemental oxygen at PMA of 36 weeks for preterm infants [9]. However, this classification does not adequately represent the heterogeneous pathophysiology in an individual infant. BPD is now considered a clinical syndrome characterized by different mechanisms and involves different regions such as airways, alveoli, and vascular components that may have different impacts throughout life [10].
Wu et al. [15] performed a retrospective, single-center study of a referral cohort of preterm infants with severe BPD, aiming to define the frequency of three disease components and the clinical outcome at hospital discharge. A total of 73 of the 76 (96%) evaluated infants were classifiable and belonged to one or more of the phenotypic subgroups: up to 78% had parenchymal lung disease, 66% had pulmonary hypertension (PH), and 60% had large airway disease. The most common phenotypic presentation was the co-occurrence of all three disease components. PH and large airway disease, instead of moderate-severe parenchymal disease, stand a higher chance of having a worse composite outcome, including death prior to NICU discharge, tracheostomy, or pulmonary vasodilator use when discharged [15]. PH alone was associated with an increased risk of mortality [15]. Next, we will discuss the different phenotypes and the corresponding current treatments of BPD (Table 1).
2.1. Central Airways
Tracheomalacia, subglottic stenosis, bronchomalacia, and bronchial stenosis are the clinical manifestations of central airway diseases commonly seen in BPD [29]. The positive pressure ventilation may damage the highly susceptible preterm airway, leading to the development of malacia [20]. The prevalence of large airway malacia, such as tracheomalacia or bronchomalacia, was reported to be less than 2% among extremely preterm infants [21]. However, this prevalence may be underestimated because of the unawareness, lack of diagnostic criteria [36], and limited availability of bronchoscopic examination [37], and because the procedure was performed only in a small number of preterm infants [16,38]. To maintain a patent airway, infants with large airway disease were associated with a higher need for tracheostomy and home ventilation [15,21]. The symptoms of tracheomalacia usually improve gradually and resolve by age 2 [36]; however, the lifelong impact is not clearly known [16].
2.2. Small Airways
The small airways component of BPD, consisting of structural remodeling, bronchoconstriction, and hyperreactivity, manifests similarly to asthma [16,29]. Clinically, it may present with airway obstruction. The obstructive lung disease seen in BPD includes both a fixed component secondary to structural changes that respond poorly to standard asthma therapies and a reactive inflammatory component that responds fairly [16]. A study found that the survivors of BPD at school age present with low exhaled nitric oxide levels and poor response to β2-agonists during the evaluation for airflow limitation, which is quite different from the conditions of other typical asthmatic children. These data indicate that the pathogenesis of obstructive lung disease in patients with BPD might involve the structural changes in small airways instead of airway inflammation [39]. Of course, besides airway structural changes, airway inflammation also contributes to the development of BPD [40,41]. Recent studies have demonstrated that BPD is associated with a certain cytokine expression pattern, which suggests these cytokine profiles might contribute to the development of BPD in preterm infants [42]. However, it is unclear whether this cytokine profile was triggered by postnatal infection or it pre-existed in some preterm infants prenatally [43]. For example, maternal ureaplasma chorioamnionitis could trigger a pro-inflammatory state, which may contribute to the pathogenesis of developing BPD [44]. Studies in mice, rats, and rabbits have shown that blockers of inflammatory mediators, receptors, and signaling pathways improve BPD presentation [45]. However, using inflammatory modulators in treating BPD in infants has not been uniformly successful [46,47]. It is also unclear whether interventions such as corticosteroids that modify the inflammatory state alter long-term respiratory outcomes [22].
2.3. Distal Airspace and Vasculature
The “new” BPD focuses more on the aberrations in both alveolar and pulmonary vascular development and the distal component comprises decreased alveolarization, abnormal vascular remodeling, and impaired lymphatic function. The alveolar disease component results in impairment of gas exchange including poor oxygenation and hypercapnea [16]. The interruption of alveolarization by preterm birth leads to the development of fewer but larger alveoli and presents with reduced vital capacity, confirmed by lower carbon monoxide diffusion in BPD infants around the corrected age of 17.4 months [16,48]. Fortunately, owing to ongoing alveolar multiplication during postnatal life, most patients with BPD can be weaned off from ventilator support successfully [27,49,50,51,52]. However, many children and young adults with a history of preterm birth still present with exercise limitation and reduced pulmonary reserve, even with normal lung capacity [53,54,55].
Researchers nowadays pay more attention to the effects of prematurity on vascular development and focus more on pulmonary vascular disease in BPD [56]. The distal airspace has a complex interaction with the surrounding vasculature during lung growth and thus generates the concept of ‘vascular hypothesis’ of BPD, highlighting the contribution of the impaired lung vascular growth to the disordered development of distal airspace in the setting of preterm birth [57,58,59,60]. Pulmonary vascular disease (PVD), the vascular component of BPD, encompasses a spectrum of PVD phenotypes. Among them, PH has long been recognized as a strong contributor to poor survival in preterm infants with BPD [61]. However, the PVD spectrum is too wide, making it a challenge to define the presence of PH and the quantification of PVD under universally consented diagnostic standards [62]. This would be especially difficult in infants with “subclinical” PVD [56]. During intrauterine life, PH is necessary to support fetal circulation since the placenta is the main organ for gas exchange. Immediately after birth, a rapid and dramatic decrease in pulmonary vascular resistance occurs to direct the blood flow into the lungs of the newborn allowing for further ventilation and perfusion adaptation. Under certain circumstances, such as severe parenchymal lung disease, hypoxic-related intra-uterine growth restriction, oligohydramnios, prolonged premature rupture of membranes, or genetic predisposition [63,64,65,66], the vascular transition is delayed, and the pulmonary vascular resistance (PVR) does not decrease normally. It leads to extrapulmonary right-to-left shunting through the ductus arteriosus or across the foramen ovale, further resulting in profound cyanosis and hypoxemia [63,64,65]. Of note, the earlier we confirm the delayed pulmonary vascular transition by echocardiography [65], the higher the risk of developing BPD and pulmonary hypertension [65,67]. Based on retrospective studies, PH may occur in up to 17–43% of BPD affected preterm infants [68,69,70,71], and the mortality rate may be as high as 14–38% [61]. The affected infants often present with recurrent cyanotic episodes and have prolonged initial hospital admissions, require extra oxygen supplement or higher levels of respiratory support even after discharge compared with infants with BPD alone [72]. In terms of treatment strategy, the commonly used medications are the pulmonary vasodilators that deal with the reactive component of PH. In contrast, for the fixed component secondary to an underdeveloped pulmonary vascular bed, there is currently little that clinicians can do [16,25]. In the long run, children with chronic pulmonary vascular disease might have different degrees of exercise intolerance and persistent echocardiographic abnormalities along with cardiac remodeling and structural change into young adults [29]. Even among preterm infants of similar gestational ages, extreme phenotypic and severity variability in PVD exists and reflects in variable responses to vasodilators. Thus, researchers are trying to identify biomarkers for following PH longitudinally into further childhood [73].
3. History and Rationale for Using Postnatal Corticosteroids (PCS)
BPD is now considered a multifactorial disease and both prenatal and postnatal risk factors play a role in this disease. Exposure to an inflammatory environment has been one of the well-accepted theories regarding the development of BPD [74,75,76,77,78]. The exposure may begin prenatally. Preterm infants born to mothers with chorioamnionitis present with evident lung inflammation during postnatal days and are more prone to developing BPD [79]. Ureaplasma species, frequently colonized in the lower genitourinary tract of women, can invade the amniotic fluid to cause inflammation, making it the most common organism associated with chorioamnionitis. Antenatal exposure to Ureaplasma spp. induces pulmonary inflammation in preterm sheep, and evidence from other experimental data demonstrate a significant association between ureaplasma chorioamnionitis and BPD development in preterm infants [44]. Furthermore, intra-amniotic administration of potent pro-inflammatory stimuli, such as lipopolysaccharide (LPS) or Escherichia coli endotoxin in animal models seems to be harmful to the normal alveolar and vascular growth [80,81].
After birth, in addition to developmental arrest of the immature lung, the preterm infants are continuously exposed to inflammatory environments, including lung and systemic infection owing to immature immunity, invasive procedures, oxidative stress and free radicals generated from hyperoxia, and mechanical ventilation. The incidence of BPD increased among very-low birth-weight infants with a history of either early- or late-onset of sepsis [82,83]. By using newer and gentler ventilation methods, ventilator-associated lung injury has diminished, but inflammation remains the key basis of pathogenesis [84]. Antenatal steroid therapy reduces mortality and many prematurity-related morbidities, including RDS, intraventricular hemorrhage, and necrotizing enterocolitis, but BPD remains unsolved [85].
Since corticosteroids are powerful downregulators of inflammation, there is a long history of using corticosteroids postnatally to prevent and treat established BPD. Physiologically, steroid hormones are produced in the adrenal cortex. There are two main classes of corticosteroids—glucocorticoids and mineralocorticoids—involved in a wide range of metabolisms and the immune system during physiological stress. Cortisol, an endogenous glucocorticoid, is released in response to stress, and it aids in the metabolism of carbohydrate, fat, and protein, to increase blood sugar level and blood pressure, to control inflammation, and to suppress the immune system. However, fetal cortisol production only reaches significant levels after 30 weeks of gestation, and before 23 weeks of gestation, the production is very limited because of the immature hypothalamic-pituitary-adrenal axis [86,87,88]. As a result, the capacities of these extremely preterm infants to handle extrauterine stress, such as mechanical ventilation, painful procedures, infection, or noxious stimuli, are impaired. The imbalance between the immature adrenal gland function and the ongoing inflammatory process in preterm neonates has prompted studies investigating the effect of corticosteroids on the development of BPD for the past three decades.
There has been a long debate on the use of PCS. In the early 2000s, concerns about serious short- and long-term side effects such as neurodevelopmental impairment outweighed the possible beneficial effects of prevention or treatment of chronic lung disease, leading to recommendations against the routine use of systemic dexamethasone [89]. A small randomized controlled trial using low initial and total doses of dexamethasone after the first one week of life revealed shorter ventilator-dependent duration in preterm infants without obvious short-term morbidities [90], and there was no strong association with long-term complications [91]. This study reopened the debate regarding the use of corticosteroid therapy, especially in infants with multiple risk factors of BPD. Since both severe BPD and PCS are associated with impaired neurodevelopment, further evidence suggested that clinicians should balance the risks and benefits more carefully and consider the use of PCS when the risk of BPD is high (>50%) [92]. A European cohort in 2011 showed that PCS were prescribed for 13.9% of infants born between 24 and 29 weeks of gestation but the use of PCS in clinical practice varies widely across regions (3.1–49.4%) [93]. Some neonatal factors are associated with PCS prescription including low gestational age, small for gestational age, male sex, under mechanical ventilation, or receiving non-steroid anti-inflammatory drugs for patent ductus arteriosus (PDA). It may partly suggest that disease severity plays a role in, but cannot fully explain, the variation in PCS use. Despite all the concerns, the use of PCS has never been abandoned and further evaluation of the different kinds of corticosteroids and routes of administration may help to get the most out of steroid therapy [29].
3.1. Dexamethasone
Dexamethasone has been used in various studies owing to its two main characteristics, its potent glucocorticoid activity and long half-life [94]. Avery et al. published a controlled trial of dexamethasone in ventilator-dependent infants in 1985 and showed short-term improvement in lung function, especially a striking increase in compliance within 48–72 h after treatment, permitting rapid weaning from the ventilator [95]. To date, the debate on the use of dexamethasone is still ongoing. Recent Cochrane reviews by Doyle concluded that the benefit of corticosteroids, particularly dexamethasone, either given early (≤7 days) or late (>7 days), may not outweigh the adverse effects associated with this treatment [96,97]. Even though early corticosteroid treatment can improve pulmonary function rapidly, facilitate extubation, and reduce the risk of BPD and PDA, there are concerns, including acute complications, infection, gastrointestinal (GI) perforation, and also longer-term sequels such as growth inhibition and central nervous system (CNS) injury. Dexamethasone interferes with the hypothalamic-pituitary-adrenal axis, suppresses endogenous cortisol production, binds only to glucocorticoid receptors, and leads to apoptosis in the hippocampus [98]. Current consensus reserves the use of late corticosteroids for patients who are difficult to wean from the mechanical ventilator and to minimize both the dose and duration of any course of treatment [97].
3.2. Hydrocortisone
Since dexamethasone has a strong affinity for only the glucocorticoid receptor and long half-life, its clinical shortcomings are apparent. Researchers have turned to different kinds of PCS with different routes of administration. Preterm infants with lower cortisol levels in the first week of life have been linked to prolonged oxygen dependence and the development of BPD, possibly because of their inability to secrete adequate amounts of cortisol in response to stressful stimuli, resulting in damage to the vulnerable lung [99]. In addition, infants presenting with symptoms and signs of hypotension are sometimes treated with a stress dose of steroids. Thus, an individual patient data meta-analysis, including five eligible studies in extremely preterm infants, evaluating the efficacy of using early low-dose hydrocortisone as prophylaxis of adrenal insufficiency demonstrated that the therapy is beneficial for survival without BPD [100]. The most updated results for the use of hydrocortisone came from the PREMILOC trial [101]. They found that the rate of BPD-free survival at 36 weeks of PMA was increased significantly by prophylactic low-dose hydrocortisone (0.5 mg/kg every 12 h for 7 days and then 0.5 mg/kg daily for 3 days) [101]. Compared to a previous study that was stopped prematurely because of the increase in the frequency of GI perforation, the PREMILOC trial showed no increase in GI or other short-term complications, and the long-term neurodevelopmental outcomes were also similar, or even improved in extremely preterm infants [102,103]. In addition to the clinical prognosis, the PREMILOC study also evaluated the hydrocortisone effect on brain structure by assessing brain MRI at term equivalent of age and concluded that neither white matter brain damage nor overall moderate-to-severe brain lesions were statistically different among those who had early hydrocortisone exposure when adjusted for other neonatal variables [104]. The exposed infants should have a neurodevelopment assessment at preschool age to verify the safety of early hydrocortisone because re-evaluation at this age reflects a child’s general intellectual ability more accurately [105].
Regarding the timing for hydrocortisone intervention, a recent meta-analysis involving 12 studies using early (within the first week of life) and two using late hydrocortisone demonstrated that early systemic use significantly alleviated BPD at PMA of 36 weeks and also attenuated neurodevelopmental impairment. Some safety concerns remain, owing to a higher risk of intestinal perforation, especially among patients under NSAIDs treatment for patent ductus arteriosus at the same time. Owing to the limited number of studies using late hydrocortisone, there is no strong conclusion currently [106].
3.3. Inhaled Corticosteroids
Inhaled corticosteroids have been postulated to be an alternative strategy to decrease systemic absorption and the side effects. Owing to its strong topical effects, the incidence of BPD was lower among infants receiving early inhaled budesonide [107] but the effective delivery method for inhaled glucocorticoids in preterm infants is still under investigation.
In 2017, Shah et al. [108] presented this challenge in a Cochrane review, which found no net advantages of inhaled corticosteroids over systemic corticosteroids, neither in the primary outcome with the incidence of death or BPD at 36 weeks of PMA nor in the secondary outcomes as ventilator-dependent duration or length of admission course. In contrast, the adverse event profiles for inhaled compared with systemic steroids were similar. These results urge researchers to design a better delivery system for the inhaled steroids, ensuring selective delivery to the alveoli [108]. From animal studies, we have learned that aerosolized budesonide mostly remains in the airway, but with the help of surfactant, it distributes more evenly into distal alveoli, enhancing gas exchange and lowering lung inflammation [109,110,111,112,113]. In a meta-analysis involving two clinical trials [114], intra-tracheal administration of budesonide combined with surfactant at birth reduced BPD by 40% without a difference in mortality or other well-known adverse outcomes [41,115]. However, there are still concerns about using an inhaled corticosteroid in preterm infants because of their immature drug metabolic system. In children and adults, budesonide is kept in the lung tissue as budesonide esters for delayed glucocorticoid release, but in preterm animal models, this pharmacokinetic feature has not been definitely demonstrated [116,117,118,119]. The CYP3A enzymes in the liver metabolize and inactivate budesonide, which is absorbed systemically, further minimizing systemic exposures. However, these enzymes may be less developed in premature infants [119,120,121]. Studies in preterm sheep demonstrated that budesonide 0.25 mg/kg with surfactant is absorbed into the systemic circulation and alters hundreds of genes in the liver and brain [122]. A previous study also showed that the affinity of budesonide for the glucocorticoid receptor is about 10 times higher than that of dexamethasone [123]. The recent data on preterm lambs from a study by Hillman et al. searching for the lowest effective dose of budesonide for decreasing lung injury is consistent with the dose Yeh et al. used in their clinical studies, 0.25 mg/kg, though systemic effects should be monitored in infants [119]. Currently, a multicenter randomized controlled trial (Preventing Lung Disease Using Surfactant + Steroid, PLUSS) is ongoing to test whether early administration of intratracheal budesonide combined with surfactant (Curosurf) reduce the rate of BPD or death at 36 weeks of PMA compared with Curosurf alone. The study is recruiting 1060 extremely preterm infants, less than 28 weeks of gestation, born in 2017–2022 from centers in Australia, New Zealand, and Canada. It may provide more evidence for clinical practice.
To obtain the best clinical outcome, we need to know what kind of steroid to use with the right dose at the right time through the right route for the appropriate duration. Future studies should focus on the pathobiology at the molecular level; looking for practical biomarkers and the genetic-environment interplay will help to design clinical trials and elucidate the phenotypic responders.
4. Recent Advances in Hyperoxia Exposure and Inflammatory Pathway of BPD
The incidence of BPD remained the same [124,125,126] or even higher because the smallest preterm infants survived [5,127]. The most affected population is infants born less than 28 weeks of gestation [4,13,128,129]. Along with a population shift, the key pathological hallmark of BPD today is a changing histopathological picture with disrupted development of the alveolar airspaces of the lung [13,129]. There are several mediators impairing alveolarization that recent studies have focused on, including the oxidative stress-related genes, TGF-β signal pathway, inflammatory pathways, extracellular matrix modification or dysregulation, and non-coding RNA [130]. Among these, lung inflammation remains a key factor in the aberrated alveolarization of BPD; however, more studies are needed to understand the molecular mechanisms underlying the injurious effects on lung growth and the endogenous pathways invoked to preserve lung development during inflammation [131]. The inflammatory response is believed to be triggered antenatally by intrauterine stress or chorioamnionitis-related cytokine exposure and further increased postnatally by factors such as hyperoxia, ventilator-induced lung injury, systemic infections, or inadequate nutritional support [132]. It also explains why researchers use hyperoxia-exposed animal models to study the antioxidant responses or to understand lung development under oxidative stress. Immediately after birth, the infants are exposed to an oxygen-rich environment compared with the intrauterine life. For the extremely preterm babies, they often require even higher oxygen supplement during resuscitation and initial stabilization. Oxygen is a double-edged sword since the preterm infants do not have an adequate amount of endogenous antioxidants to deal with oxidant injury, further predisposing them to the development of BPD or NEC [133,134]. The influences on cell growth brought by hyperoxia are either from the accumulation of inflammatory mediators or direct insult from reactive oxygen species [135]. These reactive species cause damage to not only cellular macromolecules [136,137], but also at the nuclear or mitochondrial DNA level. Quantitative analysis of DNA demonstrated that both DNA strands broke and base damage occurred in the lungs of mice in a hyperoxia environment [138]. DNA strand breakage, despite a seemingly secondary effect during cell death in oxygen, occurs mainly in alveolar type 2 cells and relates to lung injury [138]. In addition to the direct DNA damage, epigenetic changes, such as acetylation of histone, also affects cell cycle regulation and are associated with hyperoxia-induced alveolar simplification [139]. Other cells in the lung are also affected by hyperoxia. The lung epithelium presented with markedly increased apoptotic cell numbers in the hyperoxia group and can be protected by Calcitonin gene-related peptide [135]. Fibroblasts help maintain alveolar and bronchial integrity, but following hyperoxia, significant morphological changes occur, with decreased proliferation and increased cell size, leading to a pro-fibrotic stage and resulting in abnormal repair in the lung [140].
The effect of antioxidants was demonstrated in a preclinical study of guinea pigs receiving glutathione additive parental nutrition; it seemed to have rescued the oxygen-related injury and prevented the loss of alveoli [141]. Certain genetic variants of antioxidant response genes, such as the single nucleotide polymorphism in the NFE2L2 gene or NQO1 gene, altered the susceptibility to BPD or the disease severity in preterm infants [142,143,144]. These effects were further examined using pharmacological induction of gene expression, but with variable results. For instance, the pharmacological induction of Nfe2l2 expression did not reverse the developmental arrest of alveolar growth after hyperoxia [145]. However, for the NQO1 gene, it increased genetic expression pharmacologically in mice and rescued hyperoxic lung injury [146]. The development of airways and pulmonary vasculature is a highly coordinated process during lung growth, and oxidative stress disturbs the interaction among them. The cGMP signaling is one of the key pathways regulating pulmonary vascular tone in neonates. After hyperoxia exposure, it also plays an important role in the pathogenesis of PH phenotype in BPD patients [147]. Superoxide dismutase (SOD) is a class of antioxidant enzymes that catalyzes the dismutation of the superoxide radical into oxygen and hydrogen peroxide. There are three different SOD isoforms in mammalian cells. Previous animal studies have found that different isoforms have different degrees of protection against hyperoxia-induced damage. SOD3 is highly expressed in the arterial wall and loss of SOD3 results in alveolar simplification, vascular remodeling, and pulmonary hypertension [148]. The studies above have emphasized the importance of optimal saturation targeting and broadened our understanding on the balance between oxidative stress and the lung antioxidant mechanism. These have also shed new light on the pathogenesis of BPD and might provide researchers with possible therapeutic targets in the future.
Several studies have focused on the molecular pathobiology involved in distal lung development during inflammatory stress. They found that the BPD preterm infants have higher levels of inflammatory cells in their tracheal aspirate following the elevation of IL-8 and IL-6 in the earliest phase of lung injury, which initiated a series of detrimental cascades and ended up in pulmonary microvascular edema, limiting epithelial growth factor expression and suppressing pulmonary development [149,150]. One of the cascades worthy of attention is the nuclear factor kappa B (NF-κB) signaling pathway. This NF-κB pathway is a key regulator in the release of cytokines and chemokines and can cause excessive inflammation resulting in lung diseases. Pharmacological inhibition of the NF-κB signaling pathway had an impact on lung alveolarization and pulmonary angiogenesis in newborn mouse pups [131]. The sophisticated interplay between the epithelium and the mesenchyme is crucial for normal lung development. Perinatal inflammation can damage the elastin assembly and blunt lung alveolarization by disrupting the NF-κB pathway in a mouse study [151]. The same study also demonstrated that the restoration of elastic fiber assembly in the developing lung is possible [151]. To test the possible targets for inflammatory modulation, several hyperoxia-based mice models have focused on the benefits of different interleukin (IL)-1 receptor antagonists, with or without combining other anti-inflammatory agents (such as protein C), to rescue the devastating effects on lung alveolarization and further examine other possible therapeutic targets, such as the Rac1 signaling pathway [152,153,154].
The adaptive immune system is also involved in inflammation-related lung pathology, probably because of their roles in tissue migration and immune surveillance. Several reports have demonstrated that T cell activation and alteration in lymphocyte subsets are related to several prematurity associated complications, such as BPD, NEC, and periventricular leukomalacia [42,155,156]. Umbilical cord blood collected from preterm infants present with lower levels of naïve CD8+ T cells and less degree of regulatory CD31 expression, which may explain why these small neonates are vulnerable to CD8+ T cell-mediated inflammation and have impaired T cell memory function [157]. Both chorioamnionitis and BPD are associated with pro-inflammatory conditions; therefore, it is not surprising that pro-inflammatory CD4+ non-regulatory T cell cytokines are increased. However, there is a decreased abundance of regulatory T-cells in the later disease, suggesting certain inflammatory changes in these chorioamnionitis-exposed and BPD infants [158]. In a preclinical study with a hyperoxia-based mouse model, anti-inflammatory CD11b+ mononuclear cells protected the developing lung from injury, suggesting that enhancing their functions may provide some benefits to prevent BPD [159].
Apart from the immune functions, trophic functions of macrophages are involved in many tissue development and physiological regulations as well [160,161]. Yolk sac-derived macrophages colonize the alveolar space and mature in the distal lung shortly after birth [162,163]. Myeloid-lineage macrophages are recruited to the lung under pathological conditions, including BPD, and provoke a localized inflammatory response. Macrophage activation inhibited the expression of multiple genes critical for physiological lung growth in vivo and reduced airway branching in vitro [164]. In assessing the causal roles of immune cells as mediators in lung organogenesis, it was found that a hyperoxia mouse model of BPD showed impaired lung recruitment of immune cells and that cells of the Csf1r-expressing monocyte/macrophage lineage contributed to stunted alveolarization [165]. Furthermore, hyperoxia-exposed animal models demonstrated a rapid accumulation of neutrophils in the airways right after the initiation of mechanical ventilation. The neutrophils become activated under postnatal inflammatory insults and adhere to the endothelium of the pulmonary vascular system, followed by decreased alveolarization and angiogenesis, thus leading to difficult consequences such as PH [78,166,167,168]. Administration of leukadherin-1 (LA1), a novel agonist of leucocyte surface integrins, enhances leukocyte adhesion to the vascular endothelium, prevents leukocyte transendothelial migration to the lung, and decreases macrophage influx to the injury site during hyperoxia exposure [169]. Other promising targets include the chemokine receptor 4 (CXCR4), which, together with the stromal-derived factor-1 (SDF-1), modulates the inflammatory response. Targeting CXCR4 with its antagonist alleviates lung injury with a decrease in macrophage and neutrophil counts in the bronchoalveolar lavage fluid [150].
The studies above highlight the role of inflammatory cells as pathogenic mediators and provide potential targets in the inflammatory signaling pathway to protect against injurious stimuli to lung alveolarization [130].
5. Screening and Prevention: Biomarkers and Omics
BPD is a multifactorial disease due to the exposure of an immature lung to complex interactions between genetic and environmental factors [170,171,172,173]. There are some commonly known risk factors for BPD, such as infection, hyperoxia, or ventilator-related lung injury, but not all exposed premature infants develop BPD [171]. Although there are methods to modify disease severity, there is no definite treatment and prevention. Early recognition of infants at risk and prevention using more personalized methods may be the only way to improve prognosis [174].
Although researchers are now working on BPD-related biomarkers, ranging from identifying infants at risk of disease development, early disease detection, understanding disease progression to effective and suitable intervention, to date, no single biomarker has been validated as clinically reliable. Metabolomics is an “omic” science involving the quantification and characterization of low-molecular-weight endogenous metabolites, reflecting instantaneous metabolic perturbation [175]. Considering the complicated mechanisms and different phenotypes of BPD, it would be more realistic to develop a panel of biomarkers, including those obtained using genomics, microbiomics, and metabolomics, to describe the metabolic profile. A recent review examined the evidence arising from metabolomics and its interaction with microbiomics and pharmacology [18]. The changes in metabolite composition represent the host response to a disease or during a pathophysiological state. Therefore, metabolomics can provide a unique fingerprint of a specific physiological interaction in an individual preterm infant.
Biomarkers can be identified in various body fluids, such as serum, cord blood, amniotic fluid, urine, tracheal aspirate, or bronchoalveolar lavage fluid. The metabolomics of BPD includes metabolites related to alterations in oxidative stress, lipid, and amino-acid metabolism. Baraldi et al. [176] used amniotic fluid to perform an untargeted metabolic analysis to investigate whether this approach could help identify infants prone to developing BPD. Twenty-one preterm neonates were analyzed according to their BPD status, and 10 of them developed BPD. Interestingly, the group with BPD was characterized by lower levels of 3β,16α-dihydroxyandrostenone sulfate, a metabolite of dehydroepiandrosterone sulfate. As mentioned earlier, one of the rationales for using corticosteroid is the observation of relative adrenal insufficiency in preterm infants. This metabolomic finding shows the association between developing BPD and adrenocortical insufficiency. This study suggests that the injury responsible for BPD may begin as early as intra-uterine life owing to prenatal oxidative stress exposure and metabolic dysregulations.
Pharmacometabolomics is a novel field of metabolomics aiming to identify changes in the metabolome before and after specific drug treatment. Analyzing the metabolomic change in conjunction with the drug response measurements and the clinical outcome will help scientists to elucidate the mechanisms of drug effect. Corticosteroids have shown promising effects in decreasing BPD but the unwanted side effects and highly heterogeneous drug responses pose an urgent need for a more individualized approach by identifying high-risk groups. Pharmacometabolomics may be a helpful tool to identify patients who might benefit from specific drug therapy [18]. Lewis et al. [19] performed the first pharmacometabolomic study in systemic-dexamethasone-treated BPD infants. They found that 11 metabolites in the blood and 15 in the urine differed significantly before and after steroid treatment. Among them, gluconic acid presented with the most significant change after steroid treatment, and the degree of urine gluconic acid change is related to drug response.
According to previous studies, gluconic acid, an oxidation product of glucose, has some distinct features that implicate inflammation. Fanos et al. [175] found that urine gluconates were associated with BPD development, taking four other urine metabolites into account. Another pilot study by Fattuoni et al. [177] also revealed that the only metabolite significantly upregulated in the cord blood of neonates born to mothers with chorioamnionitis was gluconic acid. Besides urine gluconic acid, the levels of uridine and mannitol also have a negative correlation with the degree of steroid response [19]. Conclusively, the changes in serum and urine metabolites indicate that dexamethasone therapy does have effects on the major metabolic pathways involved in the inflammatory cascade and oxidative stress [19].
6. Conclusions
To reduce the complications associated with BPD, we need a multi-pronged and more personalized approach. First, timely recognition of both prenatal and postnatal risk factors for the development of BPD may enable primary prevention at the optimal timing for infants at the highest risk. Second, screening for predictive biomarkers for BPD may allow for earlier treatment within the window of opportunity. Third, understanding the pathophysiology of the preterm pulmonary development by recognizing dysregulated pathways in different phenotypes, identifying biomarkers, and testing novel or more patient-specific treatments may help improve outcomes. For instance, subtyping the inflammation in preterm respiratory disease more precisely may help guide clinical decision to select a possible responder to PCS [16]. Finally, identifying the serious BPD phenotypes, such as pulmonary hypertension, which causes clinical deterioration, may help develop treatment guidelines and reduce the burden of disease.
Research on validated biomarkers and the development of accurate, rapid, and affordable point-of-care biomarker tests has been both challenging and promising. A panel of biomarkers could be applied in combination with clinical parameters to serve as predictors for later disease or long-term prognosis. To modify clinical outcomes, the ideal biomarkers should be expressed early in the postnatal days to enable physicians offer timely and targeted interventions. Future directions will emphasize the classification of BPD phenotypes using biomarkers and novel therapies tailored to the underlying pathophysiology. Trials targeting certain phenotypic pathophysiology are more patient-specific and more efficient than the traditional randomized controlled trials that enroll all at-risk neonates [73]. Taking the benchwork to the bedside would lead to “precision medicine,” which will provide better care to the at-risk premature infants.
Author Contributions
S.-H.W. wrote the paper; P.-N.T. did a critical revision of the manuscript and wrote the paper. All authors have read and agreed to the published version of the manuscript.
Funding
This research was supported by funding from National Taiwan University Hospital (NTUH.109-T15).
Conflicts of Interest
The authors declare no conflict of interest. The funders had no role in the design if the study; in the collection, analyses, or interpretation of data; in the writing of the manuscript, or in the decision to publish the results. The funders had no role in the design of the study; in the collection, analyses, or interpretation of data; in the writing of the manuscript, or in the decision to publish the results
Abbreviations
| BPD | Bronchopulmonary Dysplasia |
| CLD | Chronic Lung Disease |
| PDA | Patent ductus arteriosus |
| PCS | Postnatal Corticosteroids |
| PMA | Postmenstral Age |
References
- Liggins, G.C.; Howie, R.N. A controlled trial of antepartum glucocorticoid treatment for prevention of the respiratory distress syndrome in premature infants. Pediatrics 1972, 50, 515–525. [Google Scholar] [PubMed]
- Fujiwara, T.; Chida, S.; Watabe, Y.; Maeta, H.; Morita, T.; Abe, T. Artificial surfactant therapy in hyaline-membrane disease. Lancet 1980, 315, 55–59. [Google Scholar] [CrossRef]
- Horbar, J.D.; Edwards, E.M.; Greenberg, L.T.; Morrow, K.A.; Soll, R.F.; Buus-Frank, M.E.; Buzas, J.S. Variation in performance of neonatal intensive care units in the United States. JAMA Pediatr. 2017, 171, e164396. [Google Scholar] [CrossRef] [PubMed]
- Stoll, B.J.; Hansen, N.I.; Bell, E.F.; Walsh, M.C.; Carlo, W.A.; Shankaran, S.; Laptook, A.R.; Sánchez, P.J.; Van Meurs, K.P.; Wyckoff, M. Trends in care practices, morbidity, and mortality of extremely preterm neonates, 1993–2012. JAMA 2015, 314, 1039–1051. [Google Scholar] [CrossRef] [Green Version]
- Shah, P.; Sankaran, K.; Aziz, K.; Allen, A.; Seshia, M.; Ohlsson, A.; Lee, S. Outcomes of preterm infants <29 weeks gestation over 10-year period in Canada: A cause for concern? J. Perinatol. 2012, 32, 132–138. [Google Scholar] [PubMed] [Green Version]
- Maitre, N.L.; Ballard, R.A.; Ellenberg, J.H.; Davis, S.D.; Greenberg, J.M.; Hamvas, A.; Pryhuber, G.S. Respiratory consequences of prematurity: Evolution of a diagnosis and development of a comprehensive approach. J. Perinatol. 2015, 35, 313–321. [Google Scholar] [CrossRef] [Green Version]
- Northway, W.H., Jr.; Rosan, R.C.; Porter, D.Y. Pulmonary disease following respirator therapy of hyaline-membrane disease: Bronchopulmonary dysplasia. N. Engl. J. Med. 1967, 276, 357–368. [Google Scholar] [CrossRef]
- Shennan, A.T.; Dunn, M.S.; Ohlsson, A.; Lennox, K.; Hoskins, E.M. Abnormal pulmonary outcomes in premature infants: Prediction from oxygen requirement in the neonatal period. Pediatrics 1988, 82, 527–532. [Google Scholar]
- Ah, J.; Bancalari, E. Bronchopulmonary dysplasia. Am. J. Respir. Crit. Care Med. 2001, 163, 1723–1729. [Google Scholar]
- Walsh, M.C.; Yao, Q.; Gettner, P.; Hale, E.; Collins, M.; Hensman, A.; Everette, R.; Peters, N.; Miller, N.; Muran, G. Impact of a physiologic definition on bronchopulmonary dysplasia rates. Pediatrics 2004, 114, 1305–1311. [Google Scholar] [CrossRef]
- Higgins, R.D.; Jobe, A.H.; Koso-Thomas, M.; Bancalari, E.; Viscardi, R.M.; Hartert, T.V.; Ryan, R.M.; Kallapur, S.G.; Steinhorn, R.H.; Konduri, G.G. Bronchopulmonary dysplasia: Executive summary of a workshop. J. Pediatr. 2018, 197, 300–308. [Google Scholar] [CrossRef] [PubMed]
- Avery, M.E.; Tooley, W.H.; Keller, J.B.; Hurd, S.S.; Bryan, M.H.; Cotton, R.B.; Epstein, M.F.; Fitzhardinge, P.M.; Hansen, C.B.; Hansen, T.N. Is chronic lung disease in low birth weight infants preventable? A survey of eight centers. Pediatrics 1987, 79, 26–30. [Google Scholar] [PubMed]
- Jobe, A.H. The new bronchopulmonary dysplasia. Curr. Opin. Pediatr. 2011, 23, 167. [Google Scholar] [CrossRef] [Green Version]
- Jensen, E.A.; Dysart, K.; Gantz, M.G.; McDonald, S.; Bamat, N.A.; Keszler, M.; Kirpalani, H.; Laughon, M.M.; Poindexter, B.B.; Duncan, A.F. The diagnosis of bronchopulmonary dysplasia in very preterm infants. An evidence-based approach. Am. J. Respir. Crit. Care Med. 2019, 200, 751–759. [Google Scholar] [CrossRef] [PubMed]
- Wu, K.Y.; Jensen, E.A.; White, A.M.; Wang, Y.; Biko, D.M.; Nilan, K.; Fraga, M.V.; Mercer-Rosa, L.; Zhang, H.; Kirpalani, H. Characterization of disease phenotype in very preterm infants with severe bronchopulmonary dysplasia. Am. J. Respir. Crit. Care Med. 2020, 201, 1398–1406. [Google Scholar] [CrossRef] [PubMed]
- Collaco, J.M.; McGrath-Morrow, S.A. Respiratory phenotypes for preterm infants, children, and adults: Bronchopulmonary dysplasia and more. Ann. Am. Thorac. Soc. 2018, 15, 530–538. [Google Scholar] [CrossRef] [PubMed]
- Peterson, J.; Garges, S.; Giovanni, M.; McInnes, P.; Wang, L.; Schloss, J.A.; Bonazzi, V.; McEwen, J.E.; Wetterstrand, K.A.; Deal, C. The NIH human microbiome project. Genome Res. 2009, 19, 2317–2323. [Google Scholar] [PubMed] [Green Version]
- Piersigilli, F.; Bhandari, V. Metabolomics of bronchopulmonary dysplasia. Clin. Chim. Acta 2020, 500, 109–114. [Google Scholar] [CrossRef]
- Lewis, T.; Chalise, P.; Gauldin, C.; Truog, W. Pharmacometabolomics of respiratory phenotypic response to dexamethasone in preterm infants at risk for bronchopulmonary dysplasia. Clin. Transl. Sci. 2019, 12, 591–599. [Google Scholar] [CrossRef] [Green Version]
- Hysinger, E.B.; Panitch, H.B. Paediatric tracheomalacia. Paediatr. Respir. Rev. 2016, 17, 9–15. [Google Scholar] [CrossRef]
- Wai, K.C.; Keller, R.L.; Lusk, L.A.; Ballard, R.A.; Chan, D.K. Characteristics of extremely low gestational age newborns undergoing tracheotomy: A secondary analysis of the trial of late surfactant randomized clinical trial. JAMA Otolaryngol. Head Neck Surg. 2017, 143, 13–19. [Google Scholar] [CrossRef]
- Abman, S.H.; Collaco, J.M.; Shepherd, E.G.; Keszler, M.; Cuevas-Guaman, M.; Welty, S.E.; Truog, W.E.; McGrath-Morrow, S.A.; Moore, P.E.; Rhein, L.M. Interdisciplinary care of children with severe bronchopulmonary dysplasia. J. Pediatr. 2017, 181, 12–28. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abman, S.H.; Bancalari, E.; Jobe, A. The Evolution of Bronchopulmonary Dysplasia after 50 Years; American Thoracic Society: New York, NY, USA, 2017. [Google Scholar]
- Slaughter, J.L.; Stenger, M.R.; Reagan, P.B.; Jadcherla, S.R. Inhaled bronchodilator use for infants with bronchopulmonary dysplasia. J. Perinatol. 2015, 35, 61–66. [Google Scholar] [CrossRef] [Green Version]
- Abman, S.H.; Hansmann, G.; Archer, S.L.; Ivy, D.D.; Adatia, I.; Chung, W.K.; Hanna, B.D.; Rosenzweig, E.B.; Raj, J.U.; Cornfield, D. Pediatric pulmonary hypertension: Guidelines from the american heart association and american thoracic society. Circulation 2015, 132, 2037–2099. [Google Scholar] [CrossRef] [PubMed]
- Rupprecht, T.; Rupprecht, C.; Harms, D.; Sterlacci, W.; Vieth, M.; Seybold, K. Leukotriene receptor blockade as a life-saving treatment in severe bronchopulmonary dysplasia. Respiration 2014, 88, 285–290. [Google Scholar] [CrossRef] [PubMed]
- Thurlbeck, W.M. Postnatal human lung growth. Thorax 1982, 37, 564–571. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mourani, P.M.; Ivy, D.D.; Gao, D.; Abman, S.H. Pulmonary vascular effects of inhaled nitric oxide and oxygen tension in bronchopulmonary dysplasia. Am. J. Respir. Crit. Care Med. 2004, 170, 1006–1013. [Google Scholar] [CrossRef]
- Thebaud, B.; Goss, K.N.; Laughon, M.; Whitsett, J.A.; Abman, S.H.; Steinhorn, R.H.; Aschner, J.L.; Davis, P.G.; McGrath-Morrow, S.A.; Soll, R.F.; et al. Bronchopulmonary dysplasia. Nat. Rev. Dis. Primers 2019, 5, 78. [Google Scholar] [CrossRef] [PubMed]
- Mourani, P.M.; Abman, S.H. Pulmonary hypertension and vascular abnormalities in bronchopulmonary dysplasia. Clin. Perinatol. 2015, 42, 839–855. [Google Scholar] [CrossRef] [Green Version]
- Galiè, N.; Ghofrani, H.A.; Torbicki, A.; Barst, R.J.; Rubin, L.J.; Badesch, D.; Fleming, T.; Parpia, T.; Burgess, G.; Branzi, A. Sildenafil citrate therapy for pulmonary arterial hypertension. N. Engl. J. Med. 2005, 353, 2148–2157. [Google Scholar] [CrossRef] [Green Version]
- Mourani, P.M.; Sontag, M.K.; Ivy, D.D.; Abman, S.H. Effects of long-term sildenafil treatment for pulmonary hypertension in infants with chronic lung disease. J. Pediatr. 2009, 154, 379–384. [Google Scholar] [CrossRef] [Green Version]
- Sitbon, O.; Sattler, C.; Bertoletti, L.; Savale, L.; Cottin, V.; Jaïs, X.; De Groote, P.; Chaouat, A.; Chabannes, C.; Bergot, E. Initial dual oral combination therapy in pulmonary arterial hypertension. Eur. Respir. J. 2016, 47, 1727–1736. [Google Scholar] [CrossRef] [Green Version]
- Slaughter, J.L.; Stenger, M.R.; Reagan, P.B. Variation in the use of diuretic therapy for infants with bronchopulmonary dysplasia. Pediatrics 2013, 131, 716–723. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prabhu, V.G.; Keszler, M.; Dhanireddy, R. Pulmonary function changes after nebulised and intravenous frusemide in ventilated premature infants. Arch. Dis. Child. Fetal Neonatal Ed. 1997, 77, F32–F35. [Google Scholar] [CrossRef] [PubMed]
- Carden, K.A.; Boiselle, P.M.; Waltz, D.A.; Ernst, A. Tracheomalacia and tracheobronchomalacia in children and adults: An in-depth review. Chest 2005, 127, 984–1005. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mok, Q.; Negus, S.; McLaren, C.; Rajka, T.; Elliott, M.; Roebuck, D.; McHugh, K. Computed tomography versus bronchography in the diagnosis and management of tracheobronchomalacia in ventilator dependent infants. Arch. Dis. Child. Fetal Neonatal Ed. 2005, 90, F290–FF293. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hysinger, E.B.; Friedman, N.L.; Padula, M.A.; Shinohara, R.T.; Zhang, H.; Panitch, H.B.; Kawut, S.M. Tracheobronchomalacia is associated with increased morbidity in bronchopulmonary dysplasia. Ann. Am. Thorac. Soc. 2017, 14, 1428–1435. [Google Scholar] [CrossRef]
- Baraldi, E.; Bonetto, G.; Zacchello, F.; Filippone, M. Low exhaled nitric oxide in school-age children with bronchopulmonary dysplasia and airflow limitation. Am. J. Respir. Crit. Care Med. 2005, 171, 68–72. [Google Scholar] [CrossRef]
- Martinez, F.D. Early-life origins of chronic obstructive pulmonary disease. N. Engl. J. Med. 2016, 375, 871–878. [Google Scholar] [CrossRef] [Green Version]
- Yeh, T.F.; Chen, C.M.; Wu, S.Y.; Husan, Z.; Li, T.C.; Hsieh, W.S.; Tsai, C.H.; Lin, H.C. Intratracheal administration of budesonide/surfactant to prevent bronchopulmonary dysplasia. Am. J. Respir. Crit. Care Med. 2016, 193, 86–95. [Google Scholar] [CrossRef]
- Ambalavanan, N.; Carlo, W.A.; D’Angio, C.T.; McDonald, S.A.; Das, A.; Schendel, D.; Thorsen, P.; Higgins, R.D. Cytokines associated with bronchopulmonary dysplasia or death in extremely low birth weight infants. Pediatrics 2009, 123, 1132–1141. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McGrath-Morrow, S.A.; Lee, S.; Gibbs, K.; Lopez, A.; Collaco, J.M.; Neptune, E.; Soloski, M.J.; Scott, A.; D’Alessio, F. Immune response to intrapharyngeal LPS in neonatal and juvenile mice. Am. J. Respir. Cell Mol. Biol. 2015, 52, 323–331. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kallapur, S.G.; Kramer, B.W.; Jobe, A.H. Ureaplasma and BPD. In Seminars in Perinatology; Elsevier: Amsterdam, The Netherlands, 2013; pp. 94–101. [Google Scholar]
- Solaligue, D.E.S.; Rodríguez-Castillo, J.A.; Ahlbrecht, K.; Morty, R.E. Recent advances in our understanding of the mechanisms of late lung development and bronchopulmonary dysplasia. Am. J. Physiol. Lung Cell Mol. Physiol. 2017, 313, L1101–L1153. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.W.; Davis, J.M. Future applications of antioxidants in premature infants. Curr. Opin. Pediatr. 2011, 23, 161. [Google Scholar] [CrossRef] [Green Version]
- Savani, R.C. Modulators of inflammation in bronchopulmonary dysplasia. In Seminars in Perinatology; Elsevier: Amsterdam, The Netherlands, 2018; pp. 459–470. [Google Scholar]
- Chang, D.V.; Assaf, S.J.; Tiller, C.J.; Kisling, J.A.; Tepper, R.S. Membrane and capillary components of lung diffusion in infants with bronchopulmonary dysplasia. Am. J. Respir. Crit. Care Med. 2016, 193, 767–771. [Google Scholar] [CrossRef] [Green Version]
- Yeh, J.; McGrath-Morrow, S.A.; Collaco, J.M. Oxygen weaning after hospital discharge in children with bronchopulmonary dysplasia. Pediatr. Pulmonol. 2016, 51, 1206–1211. [Google Scholar] [CrossRef] [Green Version]
- Henningfeld, J.K.; Maletta, K.; Ren, B.; Richards, K.L.; Wegner, C.; D’Andrea, L.A. Liberation from home mechanical ventilation and decannulation in children. Pediatr. Pulmonol. 2016, 51, 838–849. [Google Scholar] [CrossRef]
- Saletti, A.; Stick, S.; Doherty, D.; Simmer, K. Home oxygen therapy after preterm birth in Western Australia. J. Paediatr. Child Health 2004, 40, 519–523. [Google Scholar] [CrossRef]
- Silva, D.; Hagan, R.; Sly, P. Home oxygen management of neonatal chronic lung disease in Western Australia. J. Paediatr. Child Health 1995, 31, 185–188. [Google Scholar] [CrossRef]
- Clemm, H.H.; Vollsæter, M.; Røksund, O.D.; Eide, G.E.; Markestad, T.; Halvorsen, T. Exercise capacity after extremely preterm birth. Development from adolescence to adulthood. Ann. Am. Thorac. Soc. 2014, 11, 537–545. [Google Scholar] [CrossRef]
- O’Donnell, D.E. Adult survivors of preterm birth. What spirometry conceals, exercise tests reveal. Ann. Am. Thorac. Soc. 2014, 11, 1606–1607. [Google Scholar] [CrossRef] [PubMed]
- MacLean, J.; DeHaan, K.; Fuhr, D.; Hariharan, S.; Kamstra, B.; Hendson, L.; Adatia, I.; Majaesic, C.; Lovering, A.; Thompson, R. Altered breathing mechanics and ventilatory response during exercise in children born extremely preterm. Thorax 2016, 71, 1012–1019. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Day, C.L.; Ryan, R.M. Bronchopulmonary dysplasia: New becomes old again! Pediatr. Res. 2017, 81, 210–213. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abman, S.H. Bronchopulmonary Dysplasia: “A vascular hypothesis”. Am. J. Respir. Crit. Care Med. 2001, 164, 1755–1756. [Google Scholar] [CrossRef] [PubMed]
- Bhatt, A.J.; Pryhuber, G.S.; Huyck, H.; Watkins, R.H.; Metlay, L.A.; Maniscalco, W.M. Disrupted pulmonary vasculature and decreased vascular endothelial growth factor, Flt-1, and TIE-2 in human infants dying with bronchopulmonary dysplasia. Am. J. Respir. Crit. Care Med. 2001, 164, 1971–1980. [Google Scholar] [CrossRef]
- Stenmark, K.R.; Abman, S.H. Lung vascular development: Implications for the pathogenesis of bronchopulmonary dysplasia. Annu. Rev. Physiol. 2005, 67, 623–661. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thébaud, B.; Abman, S.H. Bronchopulmonary dysplasia: Where have all the vessels gone? Roles of angiogenic growth factors in chronic lung disease. Am. J. Respir. Crit. Care Med. 2007, 175, 978–985. [Google Scholar] [CrossRef] [Green Version]
- Collaco, J.M.; Romer, L.H.; Stuart, B.D.; Coulson, J.D.; Everett, A.D.; Lawson, E.E.; Brenner, J.I.; Brown, A.T.; Nies, M.K.; Sekar, P. Frontiers in pulmonary hypertension in infants and children with bronchopulmonary dysplasia. Pediatr. Pulmonol. 2012, 47, 1042–1053. [Google Scholar] [CrossRef] [Green Version]
- Mourani, P.M.; Abman, S.H. Pulmonary vascular disease in bronchopulmonary dysplasia: Pulmonary hypertension and beyond. Curr. Opin. Pediatr. 2013, 25, 329–337. [Google Scholar] [CrossRef]
- Aikio, O.; Metsola, J.; Vuolteenaho, R.; Perhomaa, M.; Hallman, M. Transient defect in nitric oxide generation after rupture of fetal membranes and responsiveness to inhaled nitric oxide in very preterm infants with hypoxic respiratory failure. J. Pediatr. 2012, 161, 397–403. [Google Scholar] [CrossRef]
- Chandrasekharan, P.; Kozielski, R.; Kumar, V.H.; Rawat, M.; Manja, V.; Ma, C.; Lakshminrusimha, S. Early use of inhaled nitric oxide in preterm infants: Is there a rationale for selective approach? Am. J. Perinatol. 2017, 34, 428–440. [Google Scholar]
- Mirza, H.; Garcia, J.A.; Crawford, E.; Pepe, J.; Zussman, M.; Wadhawan, R.; Oh, W. Natural history of postnatal cardiopulmonary adaptation in infants born extremely preterm and risk for death or bronchopulmonary dysplasia. J. Pediatr. 2018, 198, 187–193. [Google Scholar] [CrossRef] [PubMed]
- Giesinger, R.E.; More, K.; Odame, J.; Jain, A.; Jankov, R.P.; McNamara, P.J. Controversies in the identification and management of acute pulmonary hypertension in preterm neonates. Pediatr. Res. 2017, 82, 901–914. [Google Scholar] [CrossRef] [PubMed]
- Berenz, A.; Vergales, J.E.; Swanson, J.R.; Sinkin, R.A. Evidence of early pulmonary hypertension is associated with increased mortality in very low birth weight infants. Am. J. Perinatol. 2017, 34, 801–807. [Google Scholar] [CrossRef]
- Kim, D.-H.; Kim, H.-S.; Choi, C.W.; Kim, E.-K.; Kim, B.I.; Choi, J.-H. Risk factors for pulmonary artery hypertension in preterm infants with moderate or severe bronchopulmonary dysplasia. Neonatology 2012, 101, 40–46. [Google Scholar] [CrossRef]
- An, H.S.; Bae, E.J.; Kim, G.B.; Kwon, B.S.; Beak, J.S.; Kim, E.K.; Kim, H.S.; Choi, J.-H.; Noh, C.I.; Yun, Y.S. Pulmonary hypertension in preterm infants with bronchopulmonary dysplasia. Korean Circ. J. 2010, 40, 131–136. [Google Scholar] [CrossRef] [Green Version]
- Khemani, E.; McElhinney, D.B.; Rhein, L.; Andrade, O.; Lacro, R.V.; Thomas, K.C.; Mullen, M.P. Pulmonary artery hypertension in formerly premature infants with bronchopulmonary dysplasia: Clinical features and outcomes in the surfactant era. Pediatrics 2007, 120, 1260–1269. [Google Scholar] [CrossRef] [PubMed]
- Slaughter, J.; Pakrashi, T.; Jones, D.; South, A.; Shah, T. Echocardiographic detection of pulmonary hypertension in extremely low birth weight infants with bronchopulmonary dysplasia requiring prolonged positive pressure ventilation. J. Perinatol. 2011, 31, 635–640. [Google Scholar] [CrossRef] [Green Version]
- Stuart, B.; Sekar, P.; Coulson, J.; Choi, S.; McGrath-Morrow, S.A.; Collaco, J. Health-care utilization and respiratory morbidities in preterm infants with pulmonary hypertension. J. Perinatol. 2013, 33, 543–547. [Google Scholar] [CrossRef]
- Aschner, J.L.; Gien, J.; Ambalavanan, N.; Kinsella, J.; Konduri, G.; Lakshminrusimha, S.; Saugstad, O.; Steinhorn, R.H. Challenges, priorities and novel therapies for hypoxemic respiratory failure and pulmonary hypertension in the neonate. J. Perinatol. 2016, 36, S32–S36. [Google Scholar] [CrossRef]
- Alvarez-Fuente, M.; Moreno, L.; Mitchell, J.A.; Reiss, I.K.; Lopez, P.; Elorza, D.; Duijts, L.; Avila-Alvarez, A.; Arruza, L.; Ramirez Orellana, M.; et al. Preventing bronchopulmonary dysplasia: New tools for an old challenge. Pediatr. Res. 2019, 85, 432–441. [Google Scholar] [CrossRef]
- Bhandari, V. Postnatal inflammation in the pathogenesis of bronchopulmonary dysplasia. Birth Defects Res. Part A Clin. Mol. Teratol. 2014, 100, 189–201. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wright, C.J.; Kirpalani, H. Targeting inflammation to prevent bronchopulmonary dysplasia: Can new insights be translated into therapies? Pediatrics 2011, 128, 111–126. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bry, K.; Lappalainen, U. Pathogenesis of bronchopulmonary dysplasia: The role of interleuken 1β in the regulation of inflammation-mediated pulmonary retinoic acid pathways in transgenic mice. In Seminars in Perinatology; Elsevier: Amsterdam, The Netherlands, 2006; pp. 121–128. [Google Scholar]
- Speer, C.P. Inflammation and bronchopulmonary dysplasia: A continuing story. In Seminars in Fetal and Neonatal Medicine; Elsevier: Amsterdam, The Netherlands, 2006; pp. 354–362. [Google Scholar]
- Watterberg, K.L.; Demers, L.M.; Scott, S.M.; Murphy, S. Chorioamnionitis and early lung inflammation in infants in whom bronchopulmonary dysplasia develops. Pediatrics 1996, 97, 210–215. [Google Scholar] [PubMed]
- Ueda, K.; Cho, K.; Matsuda, T.; Okajima, S.; Uchida, M.; Kobayashi, Y.; Minakami, H.; Kobayashi, K. A rat model for arrest of alveolarization induced by antenatal endotoxin administration. Pediatr. Res. 2006, 59, 396–400. [Google Scholar] [CrossRef] [Green Version]
- Willet, K.E.; Jobe, A.H.; Ikegami, M.; Newnham, J.; Brennan, S.; Sly, P.D. Antenatal endotoxin and glucocorticoid effects on lung morphometry in preterm lambs. Pediatr. Res. 2000, 48, 782–788. [Google Scholar] [CrossRef] [PubMed]
- Fanaroff, A.A.; Korones, S.B.; Wright, L.L.; Verter, J.; Poland, R.L.; Bauer, C.R.; Tyson, J.E.; Philips, J.B., III; Edwards, W.; Lucey, J.F. Incidence, presenting features, risk factors and significance of late onset septicemia in very low birth weight infants. Pediatr. Infect. Dis. J. 1998, 17, 593–598. [Google Scholar] [CrossRef]
- Klinger, G.; Levy, I.; Sirota, L.; Boyko, V.; Lerner-Geva, L.; Reichman, B.; Network, I.N. Outcome of early-onset sepsis in a national cohort of very low birth weight infants. Pediatrics 2010, 125, e736–e740. [Google Scholar] [CrossRef]
- Baud, O.; Watterberg, K.L. Prophylactic postnatal corticosteroids: Early hydrocortisone. In Seminars in Fetal and Neonatal Medicine; Elsevier: Amsterdam, The Netherlands, 2019. [Google Scholar]
- Roberts, D.; Brown, J.; Medley, N.; Dalziel, S.R. Antenatal corticosteroids for accelerating fetal lung maturation for women at risk of preterm birth. Cochrane Database Syst. Rev. 2017, 3, CD004454. [Google Scholar] [CrossRef]
- Donaldson, A.; Nicolini, U.; Symes, E.K.; Rodeck, C.H.; Tannirandorn, Y. Changes in concentrations of cortisol, dehydroepiandrosterone sulphate and progesterone in fetal and maternal serum during pregnancy. Clin. Endocrinol. 1991, 35, 447–451. [Google Scholar] [CrossRef]
- Economides, D.; Nicolaides, K.; Linton, E.; Perry, L.; Chard, T. Plasma cortisol and adrenocorticotropin in appropriate and small for gestational age fetuses. Fetal. Diagn. Ther. 1988, 3, 158–164. [Google Scholar] [CrossRef] [PubMed]
- Bagnoli, F.; Mori, A.; Fommei, C.; Coriolani, G.; Badii, S.; Tomasini, B. ACTH and cortisol cord plasma concentrations in preterm and term infants. J. Perinatol. 2013, 33, 520–524. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jefferies, A.L. Postnatal corticosteroids to treat or prevent chronic lung disease in preterm infants. Paediatr. Child Health 2002, 7, 20–28. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Doyle, L.W.; Davis, P.G.; Morley, C.J.; McPhee, A.; Carlin, J.B. Low-dose dexamethasone facilitates extubation among chronically ventilator-dependent infants: A multicenter, international, randomized, controlled trial. Pediatrics 2006, 117, 75–83. [Google Scholar] [CrossRef]
- Doyle, L.W.; Davis, P.G.; Morley, C.J.; McPhee, A.; Carlin, J.B. Outcome at 2 years of age of infants from the DART study: A multicenter, international, randomized, controlled trial of low-dose dexamethasonef. Pediatrics 2007, 119, 716–721. [Google Scholar] [CrossRef] [Green Version]
- Doyle, L.W.; Halliday, H.L.; Ehrenkranz, R.A.; Davis, P.G.; Sinclair, J.C. An update on the impact of postnatal systemic corticosteroids on mortality and cerebral palsy in preterm infants: Effect modification by risk of bronchopulmonary dysplasia. J. Pediatr. 2014, 165, 1258–1260. [Google Scholar] [CrossRef]
- Nuytten, A.; Behal, H.; Duhamel, A.; Jarreau, P.-H.; Mazela, J.; Milligan, D.; Gortner, L.; Piedvache, A.; Zeitlin, J.; Truffert, P. Evidence-based neonatal unit practices and determinants of postnatal corticosteroid-use in preterm births below 30 weeks GA in Europe. A population-based cohort study. PLoS ONE 2017, 12, e0170234. [Google Scholar] [CrossRef] [Green Version]
- Mammel, M.; Johnson, D.; Green, T.; Thompson, T. Controlled trial of dexamethasone therapy in infants with bronchopulmonary dysplasia. Lancet 1983, 321, 1356–1358. [Google Scholar] [CrossRef]
- Avery, G.B.; Fletcher, A.B.; Kaplan, M.; Brudno, D.S. Controlled trial of dexamethasone in respirator-dependent infants with bronchopulmonary dysplasia. Pediatrics 1985, 75, 106–111. [Google Scholar]
- Doyle, L.W.; Cheong, J.L.; Ehrenkranz, R.A.; Halliday, H.L. Early (<8 days) systemic postnatal corticosteroids for prevention of bronchopulmonary dysplasia in preterm infants. Cochrane Database Syst. Rev. 2017, 10, CD001146. [Google Scholar]
- Doyle, L.W.; Cheong, J.L.; Ehrenkranz, R.A.; Halliday, H.L. Late (>7 days) systemic postnatal corticosteroids for prevention of bronchopulmonary dysplasia in preterm infants. Cochrane Database Syst. Rev. 2017, 10, CD001145. [Google Scholar] [CrossRef] [PubMed]
- Crochemore, C.; Lu, J.; Wu, Y.; Liposits, Z.; Sousa, N.; Holsboer, F.; Almeida, O. Direct targeting of hippocampal neurons for apoptosis by glucocorticoids is reversible by mineralocorticoid receptor activation. Mol. Psychiatry 2005, 10, 790–798. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Watterberg, K.L.; Scott, S.M. Evidence of early adrenal insufficiency in babies who develop bronchopulmonary dysplasia. Pediatrics 1995, 95, 120–125. [Google Scholar] [PubMed]
- Shaffer, M.L.; Baud, O.; Lacaze-Masmonteil, T.; Peltoniemi, O.M.; Bonsante, F.; Watterberg, K.L. Effect of prophylaxis for early adrenal insufficiency using low-dose hydrocortisone in very preterm infants: An individual patient data meta-analysis. J. Pediatr. 2019, 207, 136–142. [Google Scholar] [CrossRef] [Green Version]
- Baud, O.; Maury, L.; Lebail, F.; Ramful, D.; El Moussawi, F.; Nicaise, C.; Zupan-Simunek, V.; Coursol, A.; Beuchée, A.; Bolot, P. Effect of early low-dose hydrocortisone on survival without bronchopulmonary dysplasia in extremely preterm infants (PREMILOC): A double-blind, placebo-controlled, multicentre, randomised trial. Lancet 2016, 387, 1827–1836. [Google Scholar] [CrossRef]
- Baud, O.; Trousson, C.; Biran, V.; Leroy, E.; Mohamed, D.; Alberti, C.; Group, P.T. Association between early low-dose hydrocortisone therapy in extremely preterm neonates and neurodevelopmental outcomes at 2 years of age. JAMA 2017, 317, 1329–1337. [Google Scholar] [CrossRef]
- Baud, O.; Trousson, C.; Biran, V.; Leroy, E.; Mohamed, D.; Alberti, C.; Group, P.T. Two-year neurodevelopmental outcomes of extremely preterm infants treated with early hydrocortisone: Treatment effect according to gestational age at birth. Arch. Dis. Child. Fetal Neonatal Ed. 2019, 104, F30–F35. [Google Scholar] [CrossRef]
- Alison, M.; Tilea, B.; Toumazi, A.; Biran, V.; Mohamed, D.; Alberti, C.; Bourmaud, A.; Baud, O. Prophylactic hydrocortisone in extremely preterm infants and brain MRI abnormality. Arch. Dis. Child. Fetal Neonatal Ed. 2020, 105, 520–525. [Google Scholar] [CrossRef]
- Deary, I.J.; Johnson, W. Intelligence and education: Causal perceptions drive analytic processes and therefore conclusions. Int. J. Epidemiol. 2010, 39, 1362–1369. [Google Scholar] [CrossRef]
- Morris, I.P.; Goel, N.; Chakraborty, M. Efficacy and safety of systemic hydrocortisone for the prevention of bronchopulmonary dysplasia in preterm infants: A systematic review and meta-analysis. Eur. J. Pediatr. 2019, 178, 1171–1184. [Google Scholar] [CrossRef] [Green Version]
- Bassler, D.; Plavka, R.; Shinwell, E.S.; Hallman, M.; Jarreau, P.-H.; Carnielli, V.; Van den Anker, J.N.; Meisner, C.; Engel, C.; Schwab, M. Early inhaled budesonide for the prevention of bronchopulmonary dysplasia. N. Engl. J. Med. 2015, 373, 1497–1506. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shah, S.S.; Ohlsson, A.; Halliday, H.L.; Shah, V.S. Inhaled versus systemic corticosteroids for the treatment of bronchopulmonary dysplasia in ventilated very low birth weight preterm infants. Cochrane Database Syst. Rev. 2017, 10, CD002057. [Google Scholar] [CrossRef] [PubMed]
- Fajardo, C.; Levin, D.; Garcia, M.; Abrams, D.; Adamson, I. Surfactant versus saline as a vehicle for corticosteroid delivery to the lungs of ventilated rabbits. Pediatr. Res. 1998, 43, 542–547. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, C.-M.; Fang, C.-L.; Chang, C.-H. Surfactant and corticosteroid effects on lung function in a rat model of acute lung injury. Crit. Care Med. 2001, 29, 2169–2175. [Google Scholar] [CrossRef] [PubMed]
- Nimmo, A.J.; Carstairs, J.R.; Patole, S.K.; Whitehall, J.; Davidson, K.; Vink, R. Intratracheal administration of glucocorticoids using surfactant as a vehicle. Clin. Exp. Pharm. Physiol. 2002, 29, 661–665. [Google Scholar] [CrossRef]
- Huang, L.-T.; Yeh, T.-F.; Kuo, Y.-L.; Chen, P.-C.; Chen, C.-M. Effect of surfactant and budesonide on the pulmonary distribution of fluorescent dye in mice. Pediatr. Neonatol. 2015, 56, 19–24. [Google Scholar] [CrossRef] [Green Version]
- Chen, C.-M.; Chang, C.-H.; Chao, C.-H.; Wang, M.-H.; Yeh, T.-F. Biophysical and chemical stability of surfactant/budesonide and the pulmonary distribution following intra-tracheal administration. Drug Deliv. 2019, 26, 604–611. [Google Scholar] [CrossRef] [Green Version]
- Venkataraman, R.; Kamaluddeen, M.; Hasan, S.U.; Robertson, H.L.; Lodha, A. Intratracheal administration of budesonide-surfactant in prevention of bronchopulmonary dysplasia in very low birth weight infants: A systematic review and meta-analysis. Pediatr. Pulmonol. 2017, 52, 968–975. [Google Scholar] [CrossRef]
- Yeh, T.F.; Lin, H.C.; Chang, C.H.; Wu, T.S.; Su, B.H.; Li, T.C.; Pyati, S.; Tsai, C.H. Early intratracheal instillation of budesonide using surfactant as a vehicle to prevent chronic lung disease in preterm infants: A pilot study. Pediatrics 2008, 121, e1310–e1318. [Google Scholar] [CrossRef]
- Brattsand, R.; Miller-Larsson, A. The role of intracellular esterification in budesonide once-daily dosing and airway selectivity. Clin. Ther. 2003, 25, C28–C41. [Google Scholar] [CrossRef]
- Van Den Brink, K.I.M.; Boorsma, M.; Staal-van den Brekel, A.J.; Edsbäcker, S.; Wouters, E.F.; Thorsson, L. Evidence of the in vivo esterification of budesonide in human airways. Br. J. Clin. Pharm. 2008, 66, 27–35. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roberts, J.K.; Stockmann, C.; Dahl, M.J.; Albertine, K.H.; Egan, E.; Lin, Z.; Reilly, C.A.; Ballard, P.L.; Ballard, R.A.; Ward, R.M. Pharmacokinetics of budesonide administered with surfactant in premature lambs: Implications for neonatal clinical trials. Curr. Clin. Pharm. 2016, 11, 53–61. [Google Scholar] [CrossRef] [PubMed]
- Hillman, N.H.; Abugisisa, L.; Royse, E.; Fee, E.; Kemp, M.W.; Kramer, B.W.; Schmidt, A.F.; Salomone, F.; Clarke, M.W.; Musk, G.C.; et al. Dose of budesonide with surfactant affects lung and systemic inflammation after normal and injurious ventilation in preterm lambs. Pediatr. Res. 2020. [Google Scholar] [CrossRef] [PubMed]
- Alcorn, J.; McNamara, P.J. Ontogeny of hepatic and renal systemic clearance pathways in infants part I. Clin. Pharm. 2002, 41, 959–998. [Google Scholar] [CrossRef] [PubMed]
- Moore, C.D.; Roberts, J.K.; Orton, C.R.; Murai, T.; Fidler, T.P.; Reilly, C.A.; Ward, R.M.; Yost, G.S. Metabolic pathways of inhaled glucocorticoids by the CYP3A enzymes. Drug Metab. Dispos. 2013, 41, 379–389. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hillman, N.H.; Kothe, T.B.; Schmidt, A.F.; Kemp, M.W.; Royse, E.; Fee, E.; Salomone, F.; Clarke, M.W.; Musk, G.C.; Jobe, A.H. Surfactant plus budesonide decreases lung and systemic responses to injurious ventilation in preterm sheep. Am. J. Physiol. Lung Cell Mol. Physiol. 2020, 318, L41–L48. [Google Scholar] [CrossRef]
- Esmailpour, N.; Högger, P.; Rohdewald, P. Binding kinetics of budesonide to the human glucocorticoid receptor. Eur. J. Pharm. Sci. 1998, 6, 219–223. [Google Scholar] [CrossRef]
- Fanaroff, A.A.; Stoll, B.J.; Wright, L.L.; Carlo, W.A.; Ehrenkranz, R.A.; Stark, A.R.; Bauer, C.R.; Donovan, E.F.; Korones, S.B.; Laptook, A.R. Trends in neonatal morbidity and mortality for very low birthweight infants. Am. J. Obs. Gynecol. 2007, 196, 147.e1–147.e8. [Google Scholar] [CrossRef]
- Smith, V.C.; Zupancic, J.A.; McCormick, M.C.; Croen, L.A.; Greene, J.; Escobar, G.J.; Richardson, D.K. Trends in severe bronchopulmonary dysplasia rates between 1994 and 2002. J. Pediatr. 2005, 146, 469–473. [Google Scholar] [CrossRef]
- Berger, T.M.; Bachmann, I.I.; Adams, M.; Schubiger, G. Impact of improved survival of very low-birth-weight infants on incidence and severity of bronchopulmonary dysplasia. Neonatology 2004, 86, 124. [Google Scholar] [CrossRef]
- Parker, R.A.; Lindstrom, D.P.; Cotton, R.B. Improved survival accounts for most, but not all, of the increase in bronchopulmonary dysplasia. Pediatrics 1992, 90, 663–668. [Google Scholar] [PubMed]
- Alvira, C.M.; Morty, R.E. Can we understand the pathobiology of bronchopulmonary dysplasia? J. Pediatr. 2017, 190, 27–37. [Google Scholar] [CrossRef] [PubMed]
- Jobe, A. What is BPD in 2012 and what will BPD become? Early Hum. Dev. 2012, 88, S27. [Google Scholar] [CrossRef]
- Morty, R.E. Recent advances in the pathogenesis of BPD. In Seminars in Perinatology; Elsevier: Amsterdam, The Netherlands, 2018; pp. 404–412. [Google Scholar]
- Hou, Y.; Liu, M.; Husted, C.; Chen, C.; Thiagarajan, K.; Johns, J.L.; Rao, S.P.; Alvira, C.M. Activation of the nuclear factor-κB pathway during postnatal lung inflammation preserves alveolarization by suppressing macrophage inflammatory protein-2. Am. J. Physiol. Lung Cell Mol. Physiol. 2015, 309, L593–L604. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jobe, A.J. The new BPD: An arrest of lung development. Pediatr. Res. 1999, 46, 641. [Google Scholar] [CrossRef] [Green Version]
- Abdel Ghany, E.A.G.; Alsharany, W.; Ali, A.A.; Youness, E.R.; Hussein, J.S. Anti-oxidant profiles and markers of oxidative stress in preterm neonates. Paediatr. Int. Child Health 2016, 36, 134–140. [Google Scholar] [CrossRef]
- Mohamed, I.; Elremaly, W.; Rouleau, T.; Lavoie, J.-C. Oxygen and parenteral nutrition two main oxidants for extremely preterm infants:‘It all adds up’. J. Neonatal Perinat. Med. 2015, 8, 189–197. [Google Scholar] [CrossRef]
- Fu, H.; Zhang, T.; Huang, R.; Yang, Z.; Liu, C.; Li, M.; Fang, F.; Xu, F. Calcitonin gene-related peptide protects type II alveolar epithelial cells from hyperoxia-induced DNA damage and cell death. Exp. Ther. Med. 2017, 13, 1279–1284. [Google Scholar] [CrossRef] [Green Version]
- Cochrane, C.G. Cellular injury by oxidants. In Molecular Aspects of Inflammation; Springer: Berlin/Heidelberg, Germany, 1991; pp. 177–188. [Google Scholar]
- Halliwell, B.; Aruoma, O.I. DNA damage by oxygen-derived species Its mechanism and measurement in mammalian systems. FEBS Lett. 1991, 281, 9–19. [Google Scholar] [CrossRef] [Green Version]
- Barker, G.F.; Manzo, N.D.; Cotich, K.L.; Shone, R.K.; Waxman, A.B. DNA damage induced by hyperoxia: Quantitation and correlation with lung injury. Am. J. Respir. Cell Mol. Biol. 2006, 35, 277–288. [Google Scholar] [CrossRef]
- Londhe, V.A.; Sundar, I.K.; Lopez, B.; Maisonet, T.M.; Yu, Y.; Aghai, Z.H.; Rahman, I. Hyperoxia impairs alveolar formation and induces senescence through decreased histone deacetylase activity and up-regulation of p21 in neonatal mouse lung. Pediatr. Res. 2011, 69, 371–377. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- You, K.; Parikh, P.; Khandalavala, K.; Wicher, S.A.; Manlove, L.; Yang, B.; Roesler, A.; Roos, B.B.; Teske, J.J.; Britt, R.D., Jr. Moderate hyperoxia induces senescence in developing human lung fibroblasts. Am. J. Physiol. Lung Cell Mol. Physiol. 2019, 317, L525–L536. [Google Scholar] [CrossRef] [PubMed]
- Elremaly, W.; Mohamed, I.; Rouleau, T.; Lavoie, J.-C. Adding glutathione to parenteral nutrition prevents alveolar loss in newborn Guinea pig. Free Radic. Biol. Med. 2015, 87, 274–281. [Google Scholar] [CrossRef] [PubMed]
- Sampath, V.; Garland, J.S.; Helbling, D.; Dimmock, D.; Mulrooney, N.P.; Simpson, P.M.; Murray, J.C.; Dagle, J.M. Antioxidant response genes sequence variants and BPD susceptibility in VLBW infants. Pediatr. Res. 2015, 77, 477–483. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cho, H.-Y.; van Houten, B.; Wang, X.; Miller-DeGraff, L.; Fostel, J.; Gladwell, W.; Perrow, L.; Panduri, V.; Kobzik, L.; Yamamoto, M. Targeted deletion of nrf2 impairs lung development and oxidant injury in neonatal mice. Antioxid. Redox Signal. 2012, 17, 1066–1082. [Google Scholar] [CrossRef] [Green Version]
- Gavrili, S.; Zachaki, S.; Daraki, A.; Polycarpou, E.; Manola, K.; Stavropoulou, C.; Sambani, C.; Baroutis, G. Association of C609T-inborn polymorphism of NAD (P) H: Quinone oxidoreductase 1 with the risk of bronchopulmonary dysplasia in preterm neonates. Am. J. Perinatol. 2016, 33, 535–539. [Google Scholar] [CrossRef]
- McGrath-Morrow, S.A.; Lauer, T.; Collaco, J.M.; Lopez, A.; Malhotra, D.; Alekseyev, Y.O.; Neptune, E.; Wise, R.; Biswal, S. Transcriptional responses of neonatal mouse lung to hyperoxia by Nrf2 status. Cytokine 2014, 65, 4–9. [Google Scholar] [CrossRef] [Green Version]
- Maturu, P.; Wei-Liang, Y.; Jiang, W.; Wang, L.; Lingappan, K.; Barrios, R.; Liang, Y.; Moorthy, B.; Couroucli, X.I. Newborn mice lacking the gene for Cyp1a1 are more susceptible to oxygen-mediated lung injury, and are rescued by postnatal β-naphthoflavone administration: Implications for bronchopulmonary dysplasia in premature infants. Toxicol. Sci. 2017, 157, 260–271. [Google Scholar] [CrossRef]
- Gupta, A.; Perez, M.; Lee, K.J.; Taylor, J.M.; Farrow, K.N. SOD2 activity is not impacted by hyperoxia in murine neonatal pulmonary artery smooth muscle cells and mice. Int. J. Mol. Sci. 2015, 16, 6373–6390. [Google Scholar] [CrossRef] [Green Version]
- Delaney, C.; Wright, R.H.; Tang, J.-R.; Woods, C.; Villegas, L.; Sherlock, L.; Savani, R.C.; Abman, S.H.; Nozik-Grayck, E. Lack of EC-SOD worsens alveolar and vascular development in a neonatal mouse model of bleomycin-induced bronchopulmonary dysplasia and pulmonary hypertension. Pediatr. Res. 2015, 78, 634–640. [Google Scholar] [CrossRef] [Green Version]
- Munshi, U.K.; Niu, J.O.; Siddiq, M.M.; Parton, L.A. Elevation of interleukin-8 and interleukin-6 precedes the influx of neutrophils in tracheal aspirates from preterm infants who develop bronchopulmonary dysplasia. Pediatr. Pulmonol. 1997, 24, 331–336. [Google Scholar] [CrossRef]
- Drummond, S.; Ramachandran, S.; Torres, E.; Huang, J.; Hehre, D.; Suguihara, C.; Young, K.C. CXCR4 blockade attenuates hyperoxia-induced lung injury in neonatal rats. Neonatology 2015, 107, 304–311. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Benjamin, J.T.; van der Meer, R.; Im, A.M.; Plosa, E.J.; Zaynagetdinov, R.; Burman, A.; Havrilla, M.E.; Gleaves, L.A.; Polosukhin, V.V.; Deutsch, G.H. Epithelial-derived inflammation disrupts elastin assembly and alters saccular stage lung development. Am. J. Pathol. 2016, 186, 1786–1800. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rudloff, I.; Cho, S.X.; Bui, C.B.; McLean, C.; Veldman, A.; Berger, P.J.; Nold, M.F.; Nold-Petry, C.A. Refining anti-inflammatory therapy strategies for bronchopulmonary dysplasia. J. Cell. Mol. Med. 2017, 21, 1128–1138. [Google Scholar] [CrossRef] [Green Version]
- Royce, S.G.; Nold, M.F.; Bui, C.; Donovan, C.; Lam, M.; Lamanna, E.; Rudloff, I.; Bourke, J.E.; Nold-Petry, C.A. Airway remodeling and hyperreactivity in a model of bronchopulmonary dysplasia and their modulation by IL-1 receptor antagonist. Am. J. Respir. Cell Mol. Biol. 2016, 55, 858–868. [Google Scholar] [CrossRef]
- Hummler, J.K.; Dapaah-Siakwan, F.; Vaidya, R.; Zambrano, R.; Luo, S.; Chen, S.; Kerr, N.; de Rivero Vaccari, J.P.; Keane, R.W.; Dietrich, W.D. Inhibition of Rac1 signaling downregulates inflammasome activation and attenuates lung injury in neonatal rats exposed to hyperoxia. Neonatology 2017, 111, 280–288. [Google Scholar] [CrossRef]
- Ballabh, P.; Simm, M.; Kumari, J.; Krauss, A.N.; Jain, A.; Auld, P.A.; Cunningham-Rundles, S. Lymphocyte subpopulations in bronchopulmonary dysplasia. Am. J. Perinatol. 2003, 20, 465–476. [Google Scholar]
- Duggan, P.; Maalouf, E.; Watts, T.; Sullivan, M.; Counsell, S.; Allsop, J.; Al-Nakib, L.; Rutherford, M.; Battin, M.; Roberts, I. Intrauterine T-cell activation and increased proinflammatory cytokine concentrations in preterm infants with cerebral lesions. Lancet 2001, 358, 1699–1700. [Google Scholar] [CrossRef]
- Scheible, K.M.; Emo, J.; Yang, H.; Holden-Wiltse, J.; Straw, A.; Huyck, H.; Misra, S.; Topham, D.J.; Ryan, R.M.; Reynolds, A.M. Developmentally determined reduction in CD31 during gestation is associated with CD8+ T cell effector differentiation in preterm infants. Clin. Immunol. 2015, 161, 65–74. [Google Scholar] [CrossRef] [Green Version]
- Misra, R.S.; Shah, S.; Fowell, D.J.; Wang, H.; Scheible, K.; Misra, S.K.; Huyck, H.; Wyman, C.P.; Ryan, R.M.; Reynolds, A.M. Preterm cord blood CD4+ T cells exhibit increased IL-6 production in chorioamnionitis and decreased CD4+ T cells in bronchopulmonary dysplasia. Hum. Immunol. 2015, 76, 329–338. [Google Scholar] [CrossRef] [Green Version]
- Eldredge, L.C.; Treuting, P.M.; Manicone, A.M.; Ziegler, S.F.; Parks, W.C.; McGuire, J.K. CD11b+ mononuclear cells mitigate hyperoxia-induced lung injury in neonatal mice. Am. J. Respir. Cell Mol. Biol. 2016, 54, 273–283. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wynn, T.A.; Chawla, A.; Pollard, J.W. Macrophage biology in development, homeostasis and disease. Nature 2013, 496, 445–455. [Google Scholar] [CrossRef] [PubMed]
- Pollard, J.W. Trophic macrophages in development and disease. Nat. Rev. Immunol. 2009, 9, 259–270. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guilliams, M.; De Kleer, I.; Henri, S.; Post, S.; Vanhoutte, L.; De Prijck, S.; Deswarte, K.; Malissen, B.; Hammad, H.; Lambrecht, B.N. Alveolar macrophages develop from fetal monocytes that differentiate into long-lived cells in the first week of life via GM-CSF. J. Exp. Med. 2013, 210, 1977–1992. [Google Scholar] [CrossRef] [Green Version]
- Jones, C.V.; Williams, T.M.; Walker, K.A.; Dickinson, H.; Sakkal, S.; Rumballe, B.A.; Little, M.H.; Jenkin, G.; Ricardo, S.D. M2 macrophage polarisation is associated with alveolar formation during postnatal lung development. Respir. Res. 2013, 14, 41. [Google Scholar] [CrossRef] [Green Version]
- Blackwell, T.S.; Hipps, A.N.; Yamamoto, Y.; Han, W.; Barham, W.J.; Ostrowski, M.C.; Yull, F.E.; Prince, L.S. NF-κB signaling in fetal lung macrophages disrupts airway morphogenesis. J. Immunol. 2011, 187, 2740–2747. [Google Scholar] [CrossRef] [Green Version]
- Kalymbetova, T.V.; Selvakumar, B.; Rodríguez-Castillo, J.A.; Gunjak, M.; Malainou, C.; Heindl, M.R.; Moiseenko, A.; Chao, C.M.; Vadász, I.; Mayer, K. Resident alveolar macrophages are master regulators of arrested alveolarization in experimental bronchopulmonary dysplasia. J. Pathol. 2018, 245, 153–159. [Google Scholar] [CrossRef]
- Bland, R.D.; Mokres, L.M.; Ertsey, R.; Jacobson, B.E.; Jiang, S.; Rabinovitch, M.; Xu, L.; Shinwell, E.S.; Zhang, F.; Beasley, M.A. Mechanical ventilation with 40% oxygen reduces pulmonary expression of genes that regulate lung development and impairs alveolar septation in newborn mice. Am. J. Physiol. Lung Cell Mol. Physiol. 2007, 293, L1099–L1110. [Google Scholar] [CrossRef]
- Hilgendorff, A.; Parai, K.; Ertsey, R.; Juliana Rey-Parra, G.; Thébaud, B.; Tamosiuniene, R.; Jain, N.; Navarro, E.F.; Starcher, B.C.; Nicolls, M.R. Neonatal mice genetically modified to express the elastase inhibitor elafin are protected against the adverse effects of mechanical ventilation on lung growth. Am. J. Physiol. Lung Cell Mol. Physiol. 2012, 303, L215–L227. [Google Scholar] [CrossRef]
- Ogden, B.E.; Murphy, S.A.; Saunders, G.C.; Pathak, D.; Johnson, J.D. Neonatal lung neutrophils and elastase/proteinase inhibitor imbalance. Am. Rev. Respir. Dis. 1984, 130, 817–821. [Google Scholar]
- Jagarapu, J.; Kelchtermans, J.; Rong, M.; Chen, S.; Hehre, D.; Hummler, S.; Faridi, M.H.; Gupta, V.; Wu, S. Efficacy of leukadherin-1 in the prevention of hyperoxia-induced lung injury in neonatal rats. Am. J. Respir. Cell Mol. Biol. 2015, 53, 793–801. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bhandari, A.; Bhandari, V. Pitfalls, problems, and progress in bronchopulmonary dysplasia. Pediatrics 2009, 123, 1562–1573. [Google Scholar] [CrossRef] [PubMed]
- Bhandari, V.; Bizzarro, M.J.; Shetty, A.; Zhong, X.; Page, G.P.; Zhang, H.; Ment, L.R.; Gruen, J.R. Familial and genetic susceptibility to major neonatal morbidities in preterm twins. Pediatrics 2006, 117, 1901–1906. [Google Scholar] [CrossRef] [PubMed]
- Lavoie, P.M.; Pham, C.; Jang, K.L. Heritability of bronchopulmonary dysplasia, defined according to the consensus statement of the national institutes of health. Pediatrics 2008, 122, 479–485. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bhandari, V.; Gruen, J.R. The genetics of bronchopulmonary dysplasia. In Seminars in Perinatology; Elsevier: Amsterdam, The Netherlands, 2006; pp. 185–191. [Google Scholar]
- Bhandari, A.; Bhandari, V. Biomarkers in bronchopulmonary dysplasia. Paediatr. Respir. Rev. 2013, 14, 173–179. [Google Scholar] [CrossRef]
- Fanos, V.; Cristina Pintus, M.; Lussu, M.; Atzori, L.; Noto, A.; Stronati, M.; Guimaraes, H.; Marcialis, M.A.; Rocha, G.; Moretti, C. Urinary metabolomics of bronchopulmonary dysplasia (BPD): Preliminary data at birth suggest it is a congenital disease. J. Matern. Fetal. Neonatal Med. 2014, 27 (Suppl. S2), 39–45. [Google Scholar] [CrossRef]
- Baraldi, E.; Giordano, G.; Stocchero, M.; Moschino, L.; Zaramella, P.; Tran, M.R.; Carraro, S.; Romero, R.; Gervasi, M.T. Untargeted metabolomic analysis of amniotic fluid in the prediction of preterm delivery and bronchopulmonary dysplasia. PLoS ONE 2016, 11, e0164211. [Google Scholar] [CrossRef]
- Fattuoni, C.; Pietrasanta, C.; Pugni, L.; Ronchi, A.; Palmas, F.; Barberini, L.; Dessì, A.; Pintus, R.; Fanos, V.; Noto, A. Urinary metabolomic analysis to identify preterm neonates exposed to histological chorioamnionitis: A pilot study. PLoS ONE 2017, 12, e0189120. [Google Scholar] [CrossRef]
Figure 1.
Bronchopulmonary dysplasia (BPD) definition and evolution. PMA: postmenstrual age. NHLBI: National heart, Lung, and Blood Institute.NICHD: National Institute of Child Health and Human Development. Pediatrics [1]. Lancet [2]. N. Engl. J. Med. [7]. Pediatrics [8]. Am. J. Respir. Crit. Care Med. [9]. Pediatrics [10]. J. Pediatr [11]. Pediatrics [12].
Figure 1.
Bronchopulmonary dysplasia (BPD) definition and evolution. PMA: postmenstrual age. NHLBI: National heart, Lung, and Blood Institute.NICHD: National Institute of Child Health and Human Development. Pediatrics [1]. Lancet [2]. N. Engl. J. Med. [7]. Pediatrics [8]. Am. J. Respir. Crit. Care Med. [9]. Pediatrics [10]. J. Pediatr [11]. Pediatrics [12].


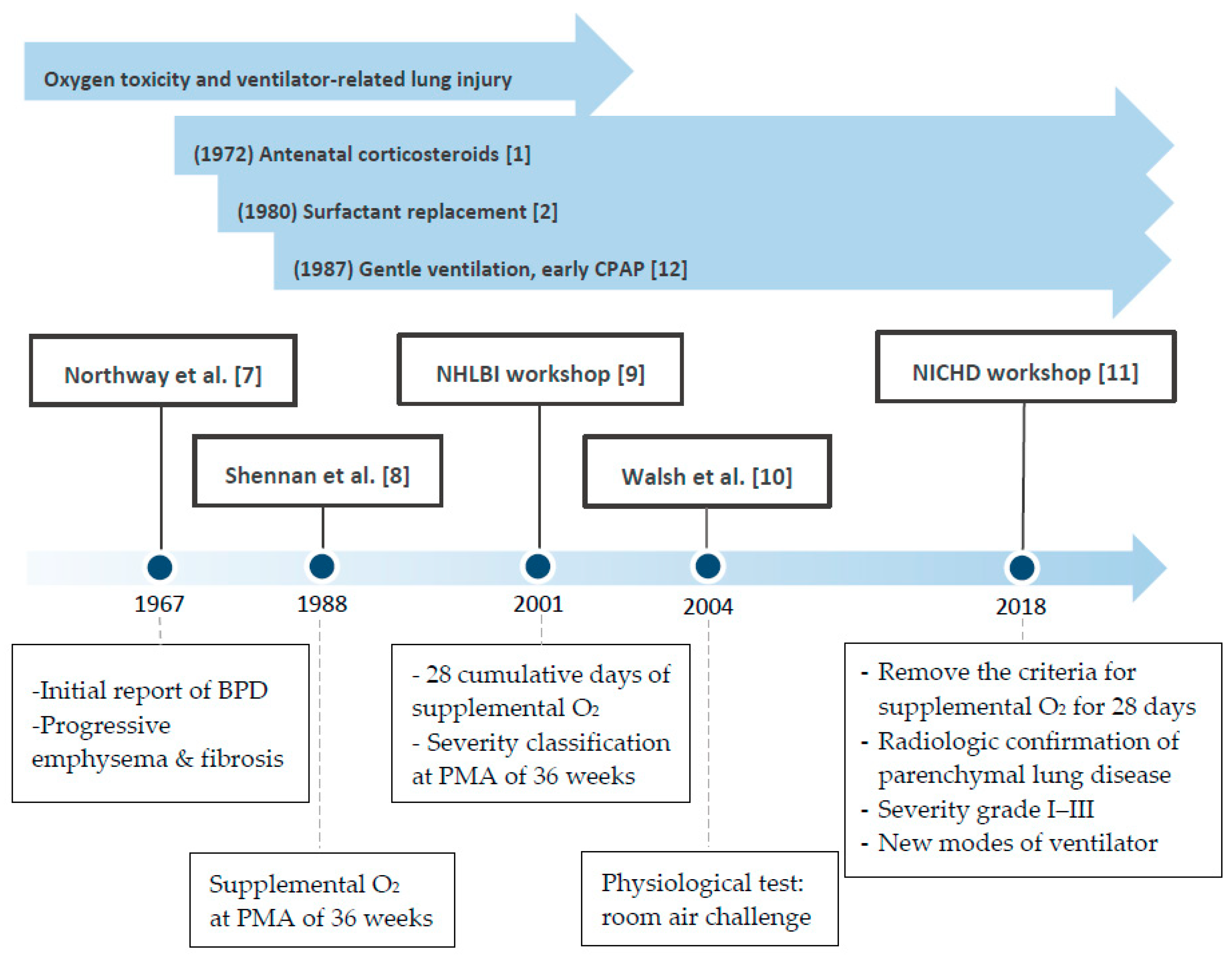{kind=link}
Table 1.
Phenotypes of BPD and current therapeutic practice.
| Phenotypes | Current Therapeutic Practice | |
|---|---|---|
| Central airways | Tracheostomy, home ventilator [20,21,22] | |
| Small airways | Structural change | Poor response to standard asthma treatment [16,22] |
| Airway inflammation | Bronchodilators [23,24] Systemic or inhaled corticosteroids [22,25] Leukotriene receptor antagonists [26] | |
| Distal airspace and vasculature | Interruption of alveolarization | Postnatal alveolar multiplication [27] |
| Pulmonary vascular disease | Inhaled Nitric Oxide [22,28,29] Oxygen therapy [25,29] Pulmonary vasodilators (Sildenafil, Bosentan) [25,30,31,32,33] Diuretics [22,34,35] | |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Wang, S.-H.; Tsao, P.-N. Phenotypes of Bronchopulmonary Dysplasia. Int. J. Mol. Sci. 2020, 21, 6112. https://doi.org/10.3390/ijms21176112
AMA Style
Wang S-H, Tsao P-N. Phenotypes of Bronchopulmonary Dysplasia. International Journal of Molecular Sciences. 2020; 21(17):6112. https://doi.org/10.3390/ijms21176112
Chicago/Turabian StyleWang, Shih-Hsin, and Po-Nien Tsao. 2020. "Phenotypes of Bronchopulmonary Dysplasia" International Journal of Molecular Sciences 21, no. 17: 6112. https://doi.org/10.3390/ijms21176112
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.