Colonic Mucosal Microbiota and Association of Bacterial Taxa with the Expression of Host Antimicrobial Peptides in Pediatric Ulcerative Colitis

 , , and
, , and
Abstract
:1. Introduction
2. Results
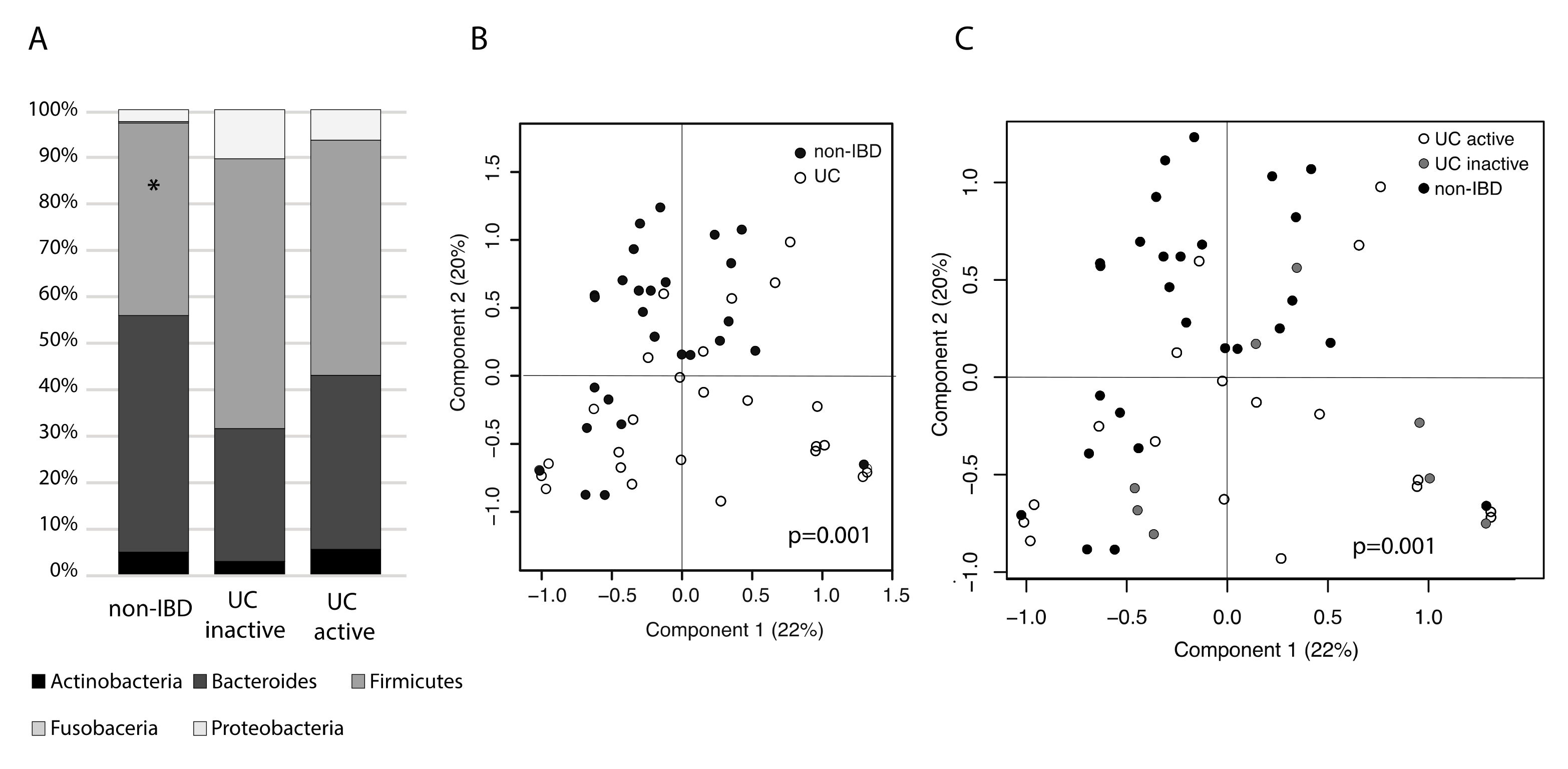
2.1. Study Cohort and Effect of Sampling on the Microbiota
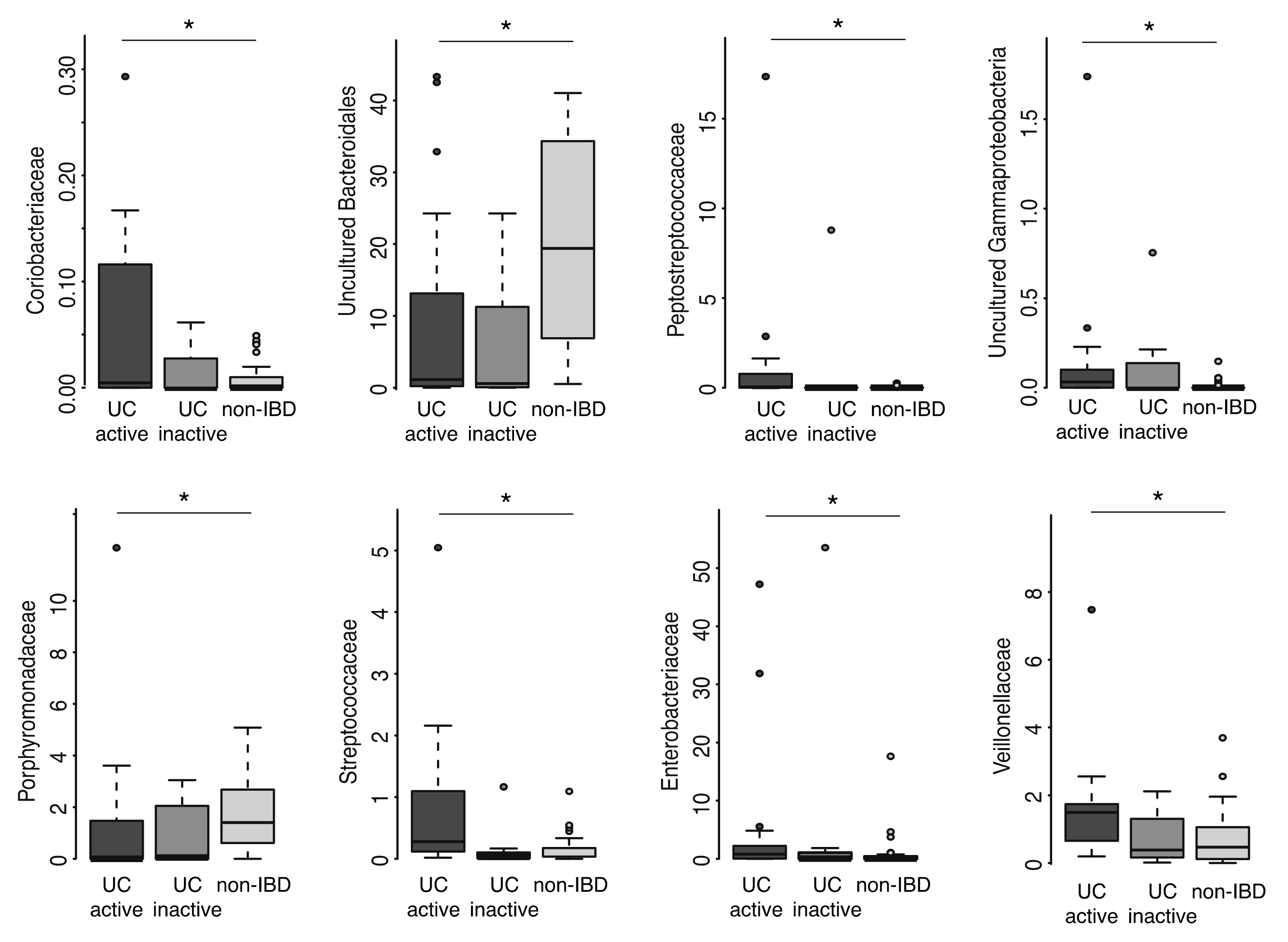
2.2. Mucosal Microbiota Associated with UC
2.3. Mucosal Gene exPression and Correlations with Microbial Taxa
3. Discussion
4. Materials and Methods
4.1. Patients
4.2. Isolation of Host RNA and Microbial DNA
4.3. 16.S rDNA Amplicon Sequencing
4.4. qRT-PCR of Mucosal Gene Expression
4.5. 16.S rDNA Amplicon Data Analysis
4.6. Statistical Analysis
4.7. Ethical Considerations
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| IBD | Inflammatory bowel diseases |
| UC | Ulcerative colitis |
| CD | Crohn’s disease |
| 16S rDNA | Small sub-unit ribosomal RNA gene |
| qRT-PCR | Quantitative reverse-transcriptase polymerase chain reaction |
| OTU | Operational taxonomic unit |
| PCoA | Principal co-ordinate analysis |
| FDR | False discovery rate |
References
- Burisch, J.; Katsanos, K.H.; Christodoulou, D.K.; Barros, L.; Magro, F.; Pedersen, N.; Kjeldsen, J.; Vegh, Z.; Lakatos, P.L.; Eriksson, C.; et al. Natural Disease Course of Ulcerative Colitis During the First Five Years of Follow-up in a European Population-based Inception Cohort-An Epi-IBD Study. J. Crohns Colitis 2019, 13, 198–208. [Google Scholar] [CrossRef] [PubMed]
- Alatab, S.; Sepanlou, S.G.; Ikuta, K.; Vahedi, H.; Bisignano, C.; Safiri, S.; Sadeghi, A.; Nixon, M.R.; Abdoli, A.; Abolhassani, H.; et al. The global, regional, and national burden of inflammatory bowel disease in 195 countries and territories, 1990–2017: A systematic analysis for the Global Burden of Disease Study 2017. Lancet Gastroenterol. Hepatol. 2020, 5, 17–30. [Google Scholar] [CrossRef] [Green Version]
- De Lange, K.M.; Moutsianas, L.; Lee, J.C.; Lamb, C.A.; Luo, Y.; Kennedy, N.A.; Jostins, L.; Rice, D.L.; Gutierrez-Achury, J.; Ji, S.; et al. Genome-wide association study implicates immune activation of multiple integrin genes in inflammatory bowel disease. Nat. Genet. 2017, 49, 256–261. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Howell, K.J.; Kraiczy, J.; Nayak, K.M.; Gasparetto, M.; Ross, A.; Lee, C.; Mak, T.N.; Koo, B.; Kumar, N.; Lawley, T.; et al. DNA Methylation and Transcription Patterns in Intestinal Epithelial Cells From Pediatric Patients with Inflammatory Bowel Diseases Differentiate Disease Subtypes and Associate with Outcome. Gastroenterology 2018, 154, 585–598. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kalliomaki, M.; Rajala, S.; Elamo, H.; Ashorn, M.; Ruuska, T. Increased expression of CXCL16, a bacterial scavenger receptor, in the colon of children with ulcerative colitis. J. Crohns Colitis 2014, 8, 1222–1226. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ostaff, M.J.; Stange, E.F.; Wehkamp, J. Antimicrobial peptides and gut microbiota in homeostasis and pathology. EMBO Mol. Med. 2013, 5, 1465–1483. [Google Scholar] [CrossRef] [PubMed]
- Stange, E.F.; Schroeder, B.O. Microbiota and mucosal defense in IBD: An update. Expert Rev. Gastroenterol. Hepatol. 2019, 13, 963–976. [Google Scholar] [CrossRef] [Green Version]
- Khor, B.; Gardet, A.; Xavier, R.J. Genetics and pathogenesis of inflammatory bowel disease. Nature 2011, 474, 307–317. [Google Scholar] [CrossRef] [Green Version]
- Manichanh, C.; Borruel, N.; Casellas, F.; Guarner, F. The gut microbiota in IBD. Nat. Rev. Gastroenterol. Hepatol. 2012, 9, 599–608. [Google Scholar] [CrossRef]
- Mukhopadhya, I.; Hansen, R.; El-Omar, E.M.; Hold, G.L. IBD-what role do Proteobacteria play? Nat. Rev. Gastroenterol. Hepatol. 2012, 9, 219–230. [Google Scholar] [CrossRef]
- Lloyd-Price, J.; Arze, C.; Ananthakrishnan, A.N.; Schirmer, M.; Avila-Pacheco, J.; Poon, T.W.; Andrews, E.; Ajami, N.J.; Bonham, K.S.; Brislawn, C.J.; et al. Multi-omics of the gut microbial ecosystem in inflammatory bowel diseases. Nature 2019, 569, 655–662. [Google Scholar] [CrossRef] [PubMed]
- Jostins, L.; Ripke, S.; Weersma, R.K.; Duerr, R.H.; McGovern, D.P.; Hui, K.Y.; Lee, J.C.; Schumm, L.P.; Sharma, Y.; Anderson, C.A.; et al. Host-microbe interactions have shaped the genetic architecture of inflammatory bowel disease. Nature 2012, 491, 119–124. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alipour, M.; Zaidi, D.; Valcheva, R.; Jovel, J.; Martinez, I.; Sergi, C.; Walter, J.; Mason, A.L.; Wong, G.K.; Dieleman, L.A.; et al. Mucosal Barrier Depletion and Loss of Bacterial Diversity are Primary Abnormalities in Paediatric Ulcerative Colitis. J. Crohns Colitis 2016, 10, 462–471. [Google Scholar] [CrossRef] [PubMed]
- Rajilić-Stojanović, M.S.F.; Guarner, F.; de Vos, W.M. Phylogenetic analysis of dysbiosis in ulcerative colitis during remission. Inflamm. Bowel Dis. 2013, 19, 481–488. [Google Scholar] [CrossRef] [PubMed]
- Martinez, C.; Antolin, M.; Santos, J.; Torrejon, A.; Casellas, F.; Borruel, N.; Francisco, G.; Juan-R, M. Unstable composition of the fecal microbiota in ulcerative colitis during clinical remission. Am. J. Gastroenterol. 2008, 103, 643–648. [Google Scholar] [CrossRef] [PubMed]
- Sokol, H.P.B.; Watterlot, L.; Lakhdari, O.; Bermúdez-Humarán, L.G.; Gratadoux, J.J.; Blugeon, S.; Bridonneau, C.; Furet, J.P.; Corthier, G.; Grangette, C.; et al. Faecalibacterium prausnitzii is an anti-inflammatory commensal bacterium identified by gut microbiota analysis of Crohn disease patients. Proc. Natl. Acad. Sci. USA 2008, 105, 16731–16736. [Google Scholar] [CrossRef] [Green Version]
- Gevers, D.; Kugathasan, S.; Denson, L.A.; Vazquez-Baeza, Y.; Van Treuren, W.; Ren, B.; Schwager, E.; Knights, D.; Song, S.J.; Yassour, M.; et al. The treatment-naive microbiome in new-onset Crohn’s disease. Cell Host Microbe 2014, 15, 382–392. [Google Scholar] [CrossRef] [Green Version]
- Schirmer, M.; Denson, L.; Vlamakis, H.; Franzosa, E.A.; Thomas, S.; Gotman, N.M.; Rufo, P.; Baker, S.S.; Sauer, C.; Markowitz, J.; et al. Compositional and Temporal Changes in the Gut Microbiome of Pediatric Ulcerative Colitis Patients are Linked to Disease Course. Cell Host Microbe 2018, 24, 600–610.e4. [Google Scholar] [CrossRef] [Green Version]
- Hyams, J.S.; Davis Thomas, S.; Gotman, N.; Haberman, Y.; Karns, R.; Schirmer, M.; Mo, A.; Mack, D.R.; Boyle, B.; Griffiths, A.M.; et al. Clinical and biological predictors of response to standardised paediatric colitis therapy (PROTECT): A multicentre inception cohort study. Lancet 2019, 393, 1708–1720. [Google Scholar] [CrossRef]
- Virta, L.; Auvinen, A.; Helenius, H.; Huovinen, P.; Kolho, K.L. Association of repeated exposure to antibiotics with the development of pediatric Crohn’s disease—A nationwide, register-based finnish case-control study. Am. J. Epidemiol. 2012, 175, 775–784. [Google Scholar] [CrossRef] [Green Version]
- Kolho, K.L.; Korpela, K.; Jaakkola, T.; Pichai, M.V.; Zoetendal, E.G.; Salonen, A.; de Vos, W.M. Fecal Microbiota in Pediatric Inflammatory Bowel Disease and Its Relation to Inflammation. Am. J. Gastroenterol. 2015, 110, 921–930. [Google Scholar] [CrossRef] [PubMed]
- Hviid, A.; Svanstrom, H.; Frisch, M. Antibiotic use and inflammatory bowel diseases in childhood. Gut 2011, 60, 49–54. [Google Scholar] [CrossRef] [PubMed]
- Cheng, J.; Ringel-Kulka, T.; Heikamp-de Jong, I.; Ringel, Y.; Carroll, I.; de Vos, W.M.; Salojärvi, A.; Satokari, R. Discordant temporal development of bacterial phyla and the emergence of core in the fecal microbiota of young children. ISME J. 2016, 10, 1002–1014. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shen, Y.; Giardino Torchia, M.L.; Lawson, G.W.; Karp, C.L.; Ashwell, J.D.; Mazmanian, S.K. Outer membrane vesicles of a human commensal mediate immune regulation and disease protection. Cell Host Microbe 2012, 12, 509–520. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Waidmann, M.; Bechtold, O.; Frick, J.-S.; Lehr, H.-A.; Schubert, S.; Dobrindt, U.; Loeffler, J.; Bohn, E.; Autenrieth, I.B. Bacteroides vulgatus protects against escherichia coli-induced colitis in gnotobiotic interleukin-2-deficient mice. Gastroenterology 2003, 125, 162–177. [Google Scholar] [CrossRef]
- Brown, E.M.; Ke, X.; Hitchcock, D.; Jeanfavre, S.; Avila-Pacheco, J.; Nakata, T.; Arthur, T.D.; Fornelos, N.; Heim, C.; Franzosa, E.A.; et al. Bacteroides-Derived Sphingolipids Are Critical for Maintaining Intestinal Homeostasis and Symbiosis. Cell Host Microbe 2019, 25, 668–680.e7. [Google Scholar] [CrossRef]
- Hiippala, K.; Kainulainen, V.; Kalliomaki, M.; Arkkila, P.; Satokari, R. Mucosal Prevalence and Interactions with the Epithelium Indicate Commensalism of Sutterella spp. Front Microbiol. 2016, 7, 1706. [Google Scholar] [CrossRef] [Green Version]
- Rigottier-Gois, L. Dysbiosis in inflammatory bowel diseases: The oxygen hypothesis. ISME J. 2013, 7, 1256–1261. [Google Scholar] [CrossRef]
- Bor, B.; Bedree, J.K.; Shi, W.; McLean, J.S.; He, X. Saccharibacteria (TM7) in the Human Oral Microbiome. J. Dent. Res. 2019, 98, 500–509. [Google Scholar] [CrossRef]
- He, X.; McLean, J.S.; Edlund, A.; Yooseph, S.; Hall, A.P.; Liu, S.Y.; Dorrestein, P.C.; Esquenazi, E.; Hunter, R.C.; Cheng, G.; et al. Cultivation of a human-associated TM7 phylotype reveals a reduced genome and epibiotic parasitic lifestyle. Proc. Natl. Acad. Sci. USA 2015, 112, 244–249. [Google Scholar] [CrossRef] [Green Version]
- Planell, N.; Lozano, J.J.; Mora-Buch, R.; Masamunt, M.C.; Jimeno, M.; Ordas, I.; Esteller, M.; Ricart, E.; Piqué, J.M.; Panés, J.; et al. Transcriptional analysis of the intestinal mucosa of patients with ulcerative colitis in remission reveals lasting epithelial cell alterations. Gut 2013, 62, 967–976. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Plaza-Diaz, J.; Gomez-Llorente, C.; Fontana, L.; Gil, A. Modulation of immunity and inflammatory gene expression in the gut, in inflammatory diseases of the gut and in the liver by probiotics. World J. Gastroenterol. 2014, 20, 15632–15649. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Zhang, W.; Wang, S.; Liu, H.; Zhang, D.; Wang, Y.; Ji, H. Swine-Derived Probiotic Lactobacillus plantarum Modulates Porcine Intestinal Endogenous Host Defense Peptide Synthesis Through TLR2/MAPK/AP-1 Signaling Pathway. Front Immunol. 2019, 10, 2691. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kobayashi, K.; Honme, Y.; Sashihara, T. Lactobacillus delbrueckii subsp. bulgaricus 2038 and Streptococcus thermophilus 1131 Induce the Expression of the REG3 Family in the Small Intestine of Mice via the Stimulation of Dendritic Cells and Type 3 Innate Lymphoid Cells. Nutrients 2019, 11, 2998. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hrdý, J.; Alard, J.; Couturier-Maillard, A.; Boulard, O.; Boutillier, D.; Delacre, M.; Carmen Lapadatescu, A.C. Lactobacillus reuteri 5454 and Bifidobacterium animalis ssp. lactis 5764 improve colitis while differentially impacting dendritic cells maturation and antimicrobial responses. Sci. Rep. 2020, 10, 5345. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schroeder, K.W.; Tremaine, W.J.; Ilstrup, D.M. Coated oral 5-aminosalicylic acid therapy for mildly to moderately active ulcerative colitis. A randomized study. N. Engl. J. Med. 1987, 317, 1625–1629. [Google Scholar] [CrossRef] [PubMed]
- Kalliomaki, M.; Satokari, R.; Lahteenoja, H.; Vahamiko, S.; Gronlund, J.; Routi, T.; Salminen, S. Expression of microbiota, Toll-like receptors, and their regulators in the small intestinal mucosa in celiac disease. J. Pediatr. Gastroenterol. Nutr. 2012, 54, 727–732. [Google Scholar] [CrossRef]
- Van den Bogert, B.; de Vos, W.M.; Zoetendal, E.G.; Kleerebezem, M. Microarray analysis and barcoded pyrosequencing provide consistent microbial profiles depending on the source of human intestinal samples. Appl. Environ. Microbiol. 2011, 77, 2071–2080. [Google Scholar] [CrossRef] [Green Version]
- Caporaso, J.G.; Kuczynski, J.; Stombaugh, J.; Bittinger, K.; Bushman, F.D.; Costello, E.K.; Fierer, N.; Peña, A.G.; Goodrich, J.K.; Gordon, J.I.; et al. QIIME allows analysis of high-throughput community sequencing data. Nat. Methods 2010, 7, 335–336. [Google Scholar] [CrossRef] [Green Version]
- Edgar, R.C. Search and clustering orders of magnitude faster than BLAST. Bioinformatics 2010, 26, 2460–2461. [Google Scholar] [CrossRef] [Green Version]
- Haas, B.J.; Gevers, D.; Earl, A.M.; Feldgarden, M.; Ward, D.V.; Giannoukos, G.; Ciulla, D.; Tabbaa, D.; Highlander, S.K.; Sodergren, E.; et al. Chimeric 16S rRNA sequence formation and detection in Sanger and 454-pyrosequenced PCR amplicons. Genome Res. 2011, 21, 494–504. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Korpela, K. mare: Microbiota Analysis in R Easily. R Package version 1.0. 2016. Available online: https://github.com/katrikorpela/mare (accessed on 21 August 2020).



{kind=link}
{kind=link}
| UC Active | UC Inactive | Control (HC) | |
|---|---|---|---|
| Number of patients | n = 18 | n = 8 | n = 27 |
| Age (range) | 13 (5–17) | 14 (10–16) | 8 (3–15) |
| Biopsy collected | |||
| ascending | 8 | 3 | 2 |
| cecum | 4 | 4 | 17 |
| descending | 6 | 1 | 8 |
| Mayo score | |||
| 0 | - | 8 | 27 |
| 1 | 11 | - | - |
| 2 | 5 | - | - |
| 3 | 2 | - | - |
| Diagnosis or reason for diagnoctic colonoscopy | |||
| Pancolitis | 16 | 7 | - |
| Left-sided colitis | 2 | 1 | - |
| Diabetes mellitus | - | - | 2 |
| Diarrhea | - | - | 7 |
| Asthma | - | - | 1 |
| Abdominal pain | - | - | 8 |
| Hematochezia | - | - | 9 |
| Medication | |||
| None | 7 | 0 | 24 |
| 5-ASA | 11 | 8 | - |
| Prednisolone | 1 | 0 | - |
| Azathioprine | 4 | 3 | - |
| Metotrexate | - | 1 | - |
| Insulin | - | - | 2 |
| Budesonide | - | - | 1 |
| Taxon | Fold Change in UC as Compared to HC | p-Value | q-Value |
|---|---|---|---|
| Uncultured Bacteroidetes | 0.58 | 0.003 | 0.06 |
| Uncultured Erysipelotrichia | 7.27 | 0.021 | 0.17 |
| Uncultured Negativicutes | 0.61 | 0.005 | 0.06 |
| Veillonellaceae | 2.93 | 0.013 | 0.13 |
| Sutterellaceae | 4.35 | 2.96E-07 | 1.15E-05 |
| Fold Change in Gene Expression | Associations between Gene-Expression and Microbiota | |||
|---|---|---|---|---|
| Gene Expression | UC/HC | UC Active/ UC Inactive | Negative | Positive |
| CXCL16 | 1.31 * | 1.56 * | Lactobacillaceae | Veillonellaceae |
| CXCR6 | ns. | ns. | - | Sutterellaceae, Veillonellaceae |
| DEFB1 | 0.76 * | 0.34 *** | Enterobacteriaceae | - |
| DEFB103B | ns. | ns. | - | - |
| DEFB4A | ns. | ns. | Uncultured Betaproteobacteria | - |
| IL8 | 68.19 *** | 38.36 *** | - | Sutterellaceae, Veillonellaceae |
| LCN2 | 6.32 ** | 7.59 ** | - | Enterobacteriaceae |
| MUC2 | ns. | ns. | - | Desulfovibrionaceae |
| REGIIIg | ns. | ns. | Peptostreptococcaceae | - |
| RETNLB | ns. | ns. | - | - |
| S100A8 | 18.78 ** | 29.30 * | Lactobacillaceae | Actinomycetaceae |
| S100A9 | 8.58 * | 17.12 ** | Lactobacillaceae | Actinomycetaceae |
| TFF3 | ns. | ns. | - | - |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jalanka, J.; Cheng, J.; Hiippala, K.; Ritari, J.; Salojärvi, J.; Ruuska, T.; Kalliomäki, M.; Satokari, R. Colonic Mucosal Microbiota and Association of Bacterial Taxa with the Expression of Host Antimicrobial Peptides in Pediatric Ulcerative Colitis. Int. J. Mol. Sci. 2020, 21, 6044. https://doi.org/10.3390/ijms21176044
Jalanka J, Cheng J, Hiippala K, Ritari J, Salojärvi J, Ruuska T, Kalliomäki M, Satokari R. Colonic Mucosal Microbiota and Association of Bacterial Taxa with the Expression of Host Antimicrobial Peptides in Pediatric Ulcerative Colitis. International Journal of Molecular Sciences. 2020; 21(17):6044. https://doi.org/10.3390/ijms21176044
Chicago/Turabian StyleJalanka, Jonna, Jing Cheng, Kaisa Hiippala, Jarmo Ritari, Jarkko Salojärvi, Tarja Ruuska, Marko Kalliomäki, and Reetta Satokari. 2020. "Colonic Mucosal Microbiota and Association of Bacterial Taxa with the Expression of Host Antimicrobial Peptides in Pediatric Ulcerative Colitis" International Journal of Molecular Sciences 21, no. 17: 6044. https://doi.org/10.3390/ijms21176044