eATP/P2X7R Axis: An Orchestrated Pathway Triggering Inflammasome Activation in Muscle Diseases

 ,
, {kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. P2X7R in Skeletal Muscle Cells and Muscular Tissue at Steady State and Under Pathological Conditions
2.1. P2X7R Signaling in Muscle Cells
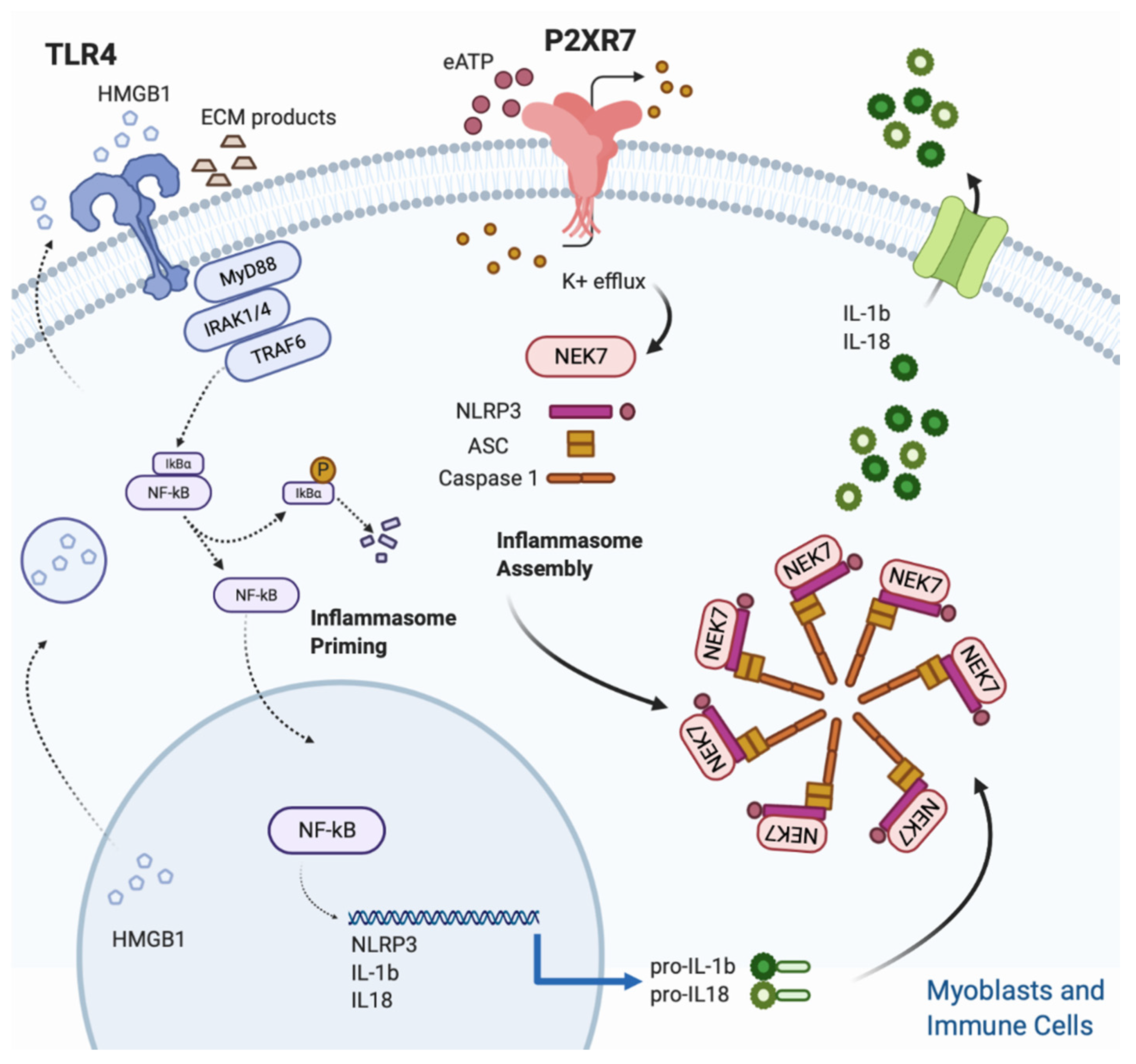
2.2. P2X7R Triggers the Inflammasome Signaling in Dystrophic Muscle Cells
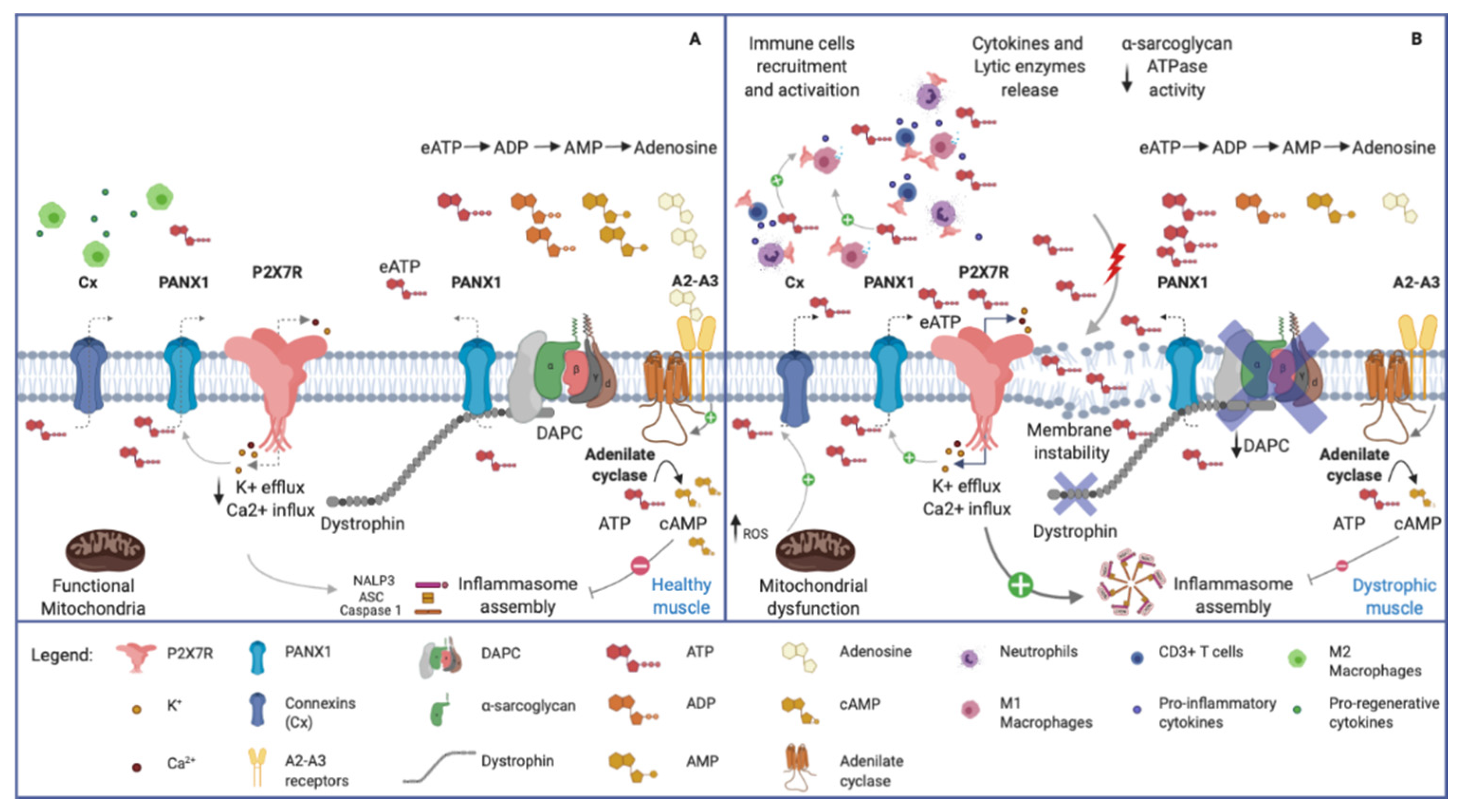
2.3. P2X7R in Dystrophic Skeletal Muscle
3. Other Regulators of P2X7R Signaling
3.1. Pannexins
3.2. Connexins
3.3. eATP/P2X7R Turning off Mechanisms in Skeletal Muscle
3.4. Adenosine and ADORA Receptors
4. Prospects, Challenges and Future Directions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Park, S.Y.; Kim, I.S. Engulfment signals and the phagocytic machinery for apoptotic cell clearance. Exp. Mol. Med. 2017, 49, e331. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Forrester, T.; Williams, C.A. Release of adenosine triphosphate from isolated adult heart cells in response to hypoxia. J. Physiol. 1977, 268, 371–390. [Google Scholar] [CrossRef] [PubMed]
- Kroemer, G.; Galluzzi, L.; Kepp, O.; Zitvogel, L. Immunogenic cell death in cancer therapy. Annu. Rev. Immunol. 2013, 31, 51–72. [Google Scholar] [CrossRef] [PubMed]
- Giuliani, A.L.; Sarti, A.C.; Di Virgilio, F. Extracellular nucleotides and nucleosides as signalling molecules. Immunol. Lett. 2018, 205, 16–24. [Google Scholar] [CrossRef] [PubMed]
- North, R.A. Molecular physiology of P2X receptors. Physiol. Rev. 2002, 82, 1013–1067. [Google Scholar] [CrossRef]
- North, R.A. P2X receptors. Philos. Trans. R. Soc. Lond B Biol. Sci. 2016, 371, 1700. [Google Scholar] [CrossRef]
- Karasawa, A.; Kawate, T. Structural basis for subtype-specific inhibition of the P2X7 receptor. Elife 2016, 5. [Google Scholar] [CrossRef]
- Harkat, M.; Peverini, L.; Cerdan, A.H.; Dunning, K.; Beudez, J.; Martz, A.; Calimet, N.; Specht, A.; Cecchini, M.; Chataigneau, T.; et al. On the permeation of large organic cations through the pore of ATP-gated P2X receptors. Proc. Natl. Acad Sci. USA 2017, 114, E3786–E3795. [Google Scholar] [CrossRef] [Green Version]
- Browne, L.E.; North, R.A. P2X receptor intermediate activation states have altered nucleotide selectivity. J. Neurosci. 2013, 33, 14801–14808. [Google Scholar] [CrossRef] [Green Version]
- Karasawa, A.; Michalski, K.; Mikhelzon, P.; Kawate, T. The P2X7 receptor forms a dye-permeable pore independent of its intracellular domain but dependent on membrane lipid composition. Elife 2017, 6. [Google Scholar] [CrossRef]
- Kaczmarek-Hajek, K.; Zhang, J.; Kopp, R.; Grosche, A.; Rissiek, B.; Saul, A.; Bruzzone, S.; Engel, T.; Jooss, T.; Krautloher, A.; et al. Re-evaluation of neuronal P2X7 expression using novel mouse models and a P2X7-specific nanobody. Elife 2018, 7. [Google Scholar] [CrossRef] [PubMed]
- Bruzzone, S.; Basile, G.; Chothi, M.P.; Nobbio, L.; Usai, C.; Jacchetti, E.; Schenone, A.; Guse, A.H.; Di Virgilio, F.; De Flora, A.; et al. Diadenosine homodinucleotide products of ADP-ribosyl cyclases behave as modulators of the purinergic receptor P2X7. J. Biol. Chem. 2010, 285, 21165–21174. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pelegrin, P.; Surprenant, A. Pannexin-1 couples to maitotoxin- and nigericin-induced interleukin-1beta release through a dye uptake-independent pathway. J. Biol. Chem. 2007, 282, 2386–2394. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baroja-Mazo, A.; Barbera-Cremades, M.; Pelegrin, P. The participation of plasma membrane hemichannels to purinergic signaling. Biochim. Biophys. Acta 2013, 1828, 79–93. [Google Scholar] [CrossRef] [Green Version]
- Di Virgilio, F.; Schmalzing, G.; Markwardt, F. The Elusive P2X7 Macropore. Trends Cell Biol. 2018, 28, 392–404. [Google Scholar] [CrossRef]
- Surprenant, A.; Rassendren, F.; Kawashima, E.; North, R.A.; Buell, G. The cytolytic P2Z receptor for extracellular ATP identified as a P2X receptor (P2X7). Science 1996, 272, 735–738. [Google Scholar] [CrossRef]
- Young, C.N.; Sinadinos, A.; Lefebvre, A.; Chan, P.; Arkle, S.; Vaudry, D.; Gorecki, D.C. A novel mechanism of autophagic cell death in dystrophic muscle regulated by P2RX7 receptor large-pore formation and HSP90. Autophagy 2015, 11, 113–130. [Google Scholar] [CrossRef]
- Burnstock, G.; Knight, G.E. The potential of P2X7 receptors as a therapeutic target, including inflammation and tumour progression. Purinergic Signal. 2018, 14, 1–18. [Google Scholar] [CrossRef] [Green Version]
- Volonte, C.; Apolloni, S.; Skaper, S.D.; Burnstock, G. P2X7 receptors: Channels, pores and more. CNS Neurol. Disord. Drug Targets 2012, 11, 705–721. [Google Scholar] [CrossRef]
- Monif, M.; Burnstock, G.; Williams, D.A. Microglia: Proliferation and activation driven by the P2X7 receptor. Int. J. Biochem. Cell Biol. 2010, 42, 1753–1756. [Google Scholar] [CrossRef]
- Massicot, F.; Hache, G.; David, L.; Chen, D.; Leuxe, C.; Garnier-Legrand, L.; Rat, P.; Laprevote, O.; Coudore, F. P2X7 Cell death receptor activation and mitochondrial impairment in oxaliplatin-induced apoptosis and neuronal injury: Cellular mechanisms and in vivo approach. PLoS ONE 2013, 8, e66830. [Google Scholar] [CrossRef]
- Nobbio, L.; Sturla, L.; Fiorese, F.; Usai, C.; Basile, G.; Moreschi, I.; Benvenuto, F.; Zocchi, E.; De Flora, A.; Schenone, A.; et al. P2X7-mediated increased intracellular calcium causes functional derangement in Schwann cells from rats with CMT1A neuropathy. J. Biol. Chem. 2009, 284, 23146–23158. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rissiek, B.; Haag, F.; Boyer, O.; Koch-Nolte, F.; Adriouch, S. P2X7 on mouse T cells: One channel, many functions. Front. Immunol. 2015, 6, 204. [Google Scholar] [CrossRef] [Green Version]
- Amoroso, F.; Capece, M.; Rotondo, A.; Cangelosi, D.; Ferracin, M.; Franceschini, A.; Raffaghello, L.; Pistoia, V.; Varesio, L.; Adinolfi, E. The P2X7 receptor is a key modulator of the PI3K/GSK3beta/VEGF signaling network: Evidence in experimental neuroblastoma. Oncogene 2015, 34, 5240–5251. [Google Scholar] [CrossRef] [PubMed]
- Orioli, E.; De Marchi, E.; Giuliani, A.L.; Adinolfi, E. P2X7 receptor orchestrates multiple signalling pathways triggering inflammation, autophagy and metabolic/trophic responses. Curr. Med. Chem. 2017, 24, 2261–2275. [Google Scholar] [CrossRef] [PubMed]
- Qiu, Y.; Li, W.H.; Zhang, H.Q.; Liu, Y.; Tian, X.X.; Fang, W.G. P2X7 mediates ATP-driven invasiveness in prostate cancer cells. PLoS ONE 2014, 9, e114371. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xia, J.; Yu, X.; Tang, L.; Li, G.; He, T. P2X7 receptor stimulates breast cancer cell invasion and migration via the AKT pathway. Oncol. Rep. 2015, 34, 103–110. [Google Scholar] [CrossRef] [Green Version]
- Murrell-Lagnado, R.D. Regulation of P2X purinergic receptor signaling by cholesterol. Curr. Top. Membr. 2017, 80, 211–232. [Google Scholar]
- Kim, J.E.; Kim, D.S.; Jin Ryu, H.; Il Kim, W.; Kim, M.J.; Won Kim, D.; Young Choi, S.; Kang, T.C. The effect of P2X7 receptor activation on nuclear factor-kappaB phosphorylation induced by status epilepticus in the rat hippocampus. Hippocampus 2013, 23, 500–514. [Google Scholar] [CrossRef]
- Cockcroft, S.; Gomperts, B.D. ATP induces nucleotide permeability in rat mast cells. Nature 1979, 279, 541–542. [Google Scholar] [CrossRef]
- Di Virgilio, F.; Bronte, V.; Collavo, D.; Zanovello, P. Responses of mouse lymphocytes to extracellular adenosine 5’-triphosphate (ATP). Lymphocytes with cytotoxic activity are resistant to the permeabilizing effects of ATP. J. Immunol. 1989, 143, 1955–1960. [Google Scholar] [PubMed]
- Di Virgilio, F.; Sarti, A.C.; Grassi, F. Modulation of innate and adaptive immunity by P2X ion channels. Curr. Opin. Immunol. 2018, 52, 51–59. [Google Scholar] [CrossRef] [PubMed]
- Bianchi, G.; Vuerich, M.; Pellegatti, P.; Marimpietri, D.; Emionite, L.; Marigo, I.; Bronte, V.; Di Virgilio, F.; Pistoia, V.; Raffaghello, L. ATP/P2X7 axis modulates myeloid-derived suppressor cell functions in neuroblastoma microenvironment. Cell Death Dis. 2014, 5, e1135. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Di Virgilio, F.; Dal Ben, D.; Sarti, A.C.; Giuliani, A.L.; Falzoni, S. The P2X7 Receptor in Infection and Inflammation. Immunity 2017, 47, 15–31. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Antonioli, L.; Fornai, M.; Blandizzi, C.; Pacher, P.; Hasko, G. Adenosine signaling and the immune system: When a lot could be too much. Immunol. Lett. 2018, 205, 9–15. [Google Scholar] [CrossRef]
- Banachewicz, W.; Suplat, D.; Krzeminski, P.; Pomorski, P.; Baranska, J. P2 nucleotide receptors on C2C12 satellite cells. Purinergic Signal. 2005, 1, 249–257. [Google Scholar] [CrossRef] [Green Version]
- Martinello, T.; Baldoin, M.C.; Morbiato, L.; Paganin, M.; Tarricone, E.; Schiavo, G.; Bianchini, E.; Sandona, D.; Betto, R. Extracellular ATP signaling during differentiation of C2C12 skeletal muscle cells: Role in proliferation. Mol. Cell Biochem. 2011, 351, 183–196. [Google Scholar] [CrossRef]
- Araya, R.; Riquelme, M.A.; Brandan, E.; Saez, J.C. The formation of skeletal muscle myotubes requires functional membrane receptors activated by extracellular ATP. Brain Res. Brain Res. Rev. 2004, 47, 174–188. [Google Scholar] [CrossRef]
- Almada, A.E.; Wagers, A.J. Molecular circuitry of stem cell fate in skeletal muscle regeneration, ageing and disease. Nat. Rev. Mol. Cell Biol. 2016, 17, 267–279. [Google Scholar] [CrossRef] [Green Version]
- Zammit, P.S.; Golding, J.P.; Nagata, Y.; Hudon, V.; Partridge, T.A.; Beauchamp, J.R. Muscle satellite cells adopt divergent fates: A mechanism for self-renewal? J. Cell Biol. 2004, 166, 347–357. [Google Scholar] [CrossRef]
- Yin, H.; Price, F.; Rudnicki, M.A. Satellite cells and the muscle stem cell niche. Physiol. Rev. 2013, 93, 23–67. [Google Scholar] [CrossRef] [Green Version]
- Sinadinos, A.; Young, C.N.; Al-Khalidi, R.; Teti, A.; Kalinski, P.; Mohamad, S.; Floriot, L.; Henry, T.; Tozzi, G.; Jiang, T.; et al. P2RX7 purinoceptor: A therapeutic target for ameliorating the symptoms of duchenne muscular dystrophy. PLoS Med. 2015, 12, e1001888. [Google Scholar] [CrossRef] [PubMed]
- Sandona, D.; Gastaldello, S.; Martinello, T.; Betto, R. Characterization of the ATP-hydrolysing activity of alpha-sarcoglycan. Biochem. J. 2004, 381, 105–112. [Google Scholar] [CrossRef]
- Zimmermann, H. Extracellular metabolism of ATP and other nucleotides. Naunyn Schmiedebergs Arch. Pharmacol. 2000, 362, 299–309. [Google Scholar] [CrossRef] [PubMed]
- Bollen, M.; Gijsbers, R.; Ceulemans, H.; Stalmans, W.; Stefan, C. Nucleotide pyrophosphatases/phosphodiesterases on the move. Crit. Rev. Biochem. Mol. Biol. 2000, 35, 393–432. [Google Scholar] [CrossRef] [PubMed]
- Young, C.N.; Brutkowski, W.; Lien, C.F.; Arkle, S.; Lochmuller, H.; Zablocki, K.; Gorecki, D.C. P2X7 purinoceptor alterations in dystrophic mdx mouse muscles: Relationship to pathology and potential target for treatment. J. Cell Mol. Med. 2012, 16, 1026–1037. [Google Scholar] [CrossRef] [PubMed]
- Khakh, B.S.; North, R.A. P2X receptors as cell-surface ATP sensors in health and disease. Nature 2006, 442, 527–532. [Google Scholar] [CrossRef]
- Yeung, D.; Zablocki, K.; Lien, C.F.; Jiang, T.; Arkle, S.; Brutkowski, W.; Brown, J.; Lochmuller, H.; Simon, J.; Barnard, E.A.; et al. Increased susceptibility to ATP via alteration of P2X receptor function in dystrophic mdx mouse muscle cells. FASEB J. 2006, 20, 610–620. [Google Scholar] [CrossRef]
- Guerra, A.N.; Gavala, M.L.; Chung, H.S.; Bertics, P.J. Nucleotide receptor signalling and the generation of reactive oxygen species. Purinergic Signal. 2007, 3, 39–51. [Google Scholar] [CrossRef] [Green Version]
- Haslund-Vinding, J.; McBean, G.; Jaquet, V.; Vilhardt, F. NADPH oxidases in oxidant production by microglia: Activating receptors, pharmacology and association with disease. Br. J. Pharmacol. 2017, 174, 1733–1749. [Google Scholar] [CrossRef] [Green Version]
- Adinolfi, E.; Giuliani, A.L.; De Marchi, E.; Pegoraro, A.; Orioli, E.; Di Virgilio, F. The P2X7 receptor: A main player in inflammation. Biochem. Pharmacol. 2018, 151, 234–244. [Google Scholar] [CrossRef] [PubMed]
- Martinon, F.; Burns, K.; Tschopp, J. The inflammasome: A molecular platform triggering activation of inflammatory caspases and processing of proIL-beta. Mol. Cell 2002, 10, 417–426. [Google Scholar] [CrossRef]
- Dinarello, C.A. Immunological and inflammatory functions of the interleukin-1 family. Annu. Rev. Immunol. 2009, 27, 519–550. [Google Scholar] [CrossRef]
- Bryant, C.; Fitzgerald, K.A. Molecular mechanisms involved in inflammasome activation. Trends Cell Biol. 2009, 19, 455–464. [Google Scholar] [CrossRef] [PubMed]
- Rawat, R.; Cohen, T.V.; Ampong, B.; Francia, D.; Henriques-Pons, A.; Hoffman, E.P.; Nagaraju, K. Inflammasome up-regulation and activation in dysferlin-deficient skeletal muscle. Am. J. Pathol. 2010, 176, 2891–2900. [Google Scholar] [CrossRef] [PubMed]
- Cohen, T.V.; Many, G.M.; Fleming, B.D.; Gnocchi, V.F.; Ghimbovschi, S.; Mosser, D.M.; Hoffman, E.P.; Partridge, T.A. Upregulated IL-1beta in dysferlin-deficient muscle attenuates regeneration by blunting the response to pro-inflammatory macrophages. Skelet. Muscle 2015, 5, 24. [Google Scholar] [CrossRef] [Green Version]
- Benny Klimek, M.E.; Sali, A.; Rayavarapu, S.; Van der Meulen, J.H.; Nagaraju, K. Effect of the IL-1 receptor antagonist kineret(r) on disease phenotype in mdx mice. PLoS ONE 2016, 11, e0155944. [Google Scholar] [CrossRef] [Green Version]
- Babelova, A.; Moreth, K.; Tsalastra-Greul, W.; Zeng-Brouwers, J.; Eickelberg, O.; Young, M.F.; Bruckner, P.; Pfeilschifter, J.; Schaefer, R.M.; Grone, H.J.; et al. Biglycan, a danger signal that activates the NLRP3 inflammasome via toll-like and P2X receptors. J. Biol. Chem. 2009, 284, 24035–24048. [Google Scholar] [CrossRef] [Green Version]
- Scaffidi, P.; Misteli, T.; Bianchi, M.E. Release of chromatin protein HMGB1 by necrotic cells triggers inflammation. Nature 2002, 418, 191–195. [Google Scholar] [CrossRef]
- De Mori, R.; Straino, S.; Di Carlo, A.; Mangoni, A.; Pompilio, G.; Palumbo, R.; Bianchi, M.E.; Capogrossi, M.C.; Germani, A. Multiple effects of high mobility group box protein 1 in skeletal muscle regeneration. Arter. Thromb. Vasc. Biol. 2007, 27, 2377–2383. [Google Scholar] [CrossRef] [Green Version]
- Sagheddu, R.; Chiappalupi, S.; Salvadori, L.; Riuzzi, F.; Donato, R.; Sorci, G. Targeting RAGE as a potential therapeutic approach to Duchenne muscular dystrophy. Hum. Mol. Genet. 2018, 27, 3734–3746. [Google Scholar] [CrossRef] [PubMed]
- Kelley, N.; Jeltema, D.; Duan, Y.; He, Y. The NLRP3 inflammasome: An overview of mechanisms of activation and regulation. Int. J. Mol. Sci. 2019, 20, 3328. [Google Scholar] [CrossRef] [Green Version]
- De Nardo, D. Toll-like receptors: Activation, signalling and transcriptional modulation. Cytokine 2015, 74, 181–189. [Google Scholar] [CrossRef] [PubMed]
- Shi, H.; Wang, Y.; Li, X.; Zhan, X.; Tang, M.; Fina, M.; Su, L.; Pratt, D.; Bu, C.H.; Hildebrand, S.; et al. NLRP3 activation and mitosis are mutually exclusive events coordinated by NEK7, a new inflammasome component. Nat. Immunol. 2016, 17, 250–258. [Google Scholar] [CrossRef] [PubMed]
- Munoz-Planillo, R.; Kuffa, P.; Martinez-Colon, G.; Smith, B.L.; Rajendiran, T.M.; Nunez, G. K(+) efflux is the common trigger of NLRP3 inflammasome activation by bacterial toxins and particulate matter. Immunity 2013, 38, 1142–1153. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Christgen, S.; Place, D.E.; Kanneganti, T.D. Toward targeting inflammasomes: Insights into their regulation and activation. Cell Res. 2020, 30, 315–327. [Google Scholar] [CrossRef] [Green Version]
- He, Y.; Zeng, M.Y.; Yang, D.; Motro, B.; Nunez, G. NEK7 is an essential mediator of NLRP3 activation downstream of potassium efflux. Nature 2016, 530, 354–357. [Google Scholar] [CrossRef] [Green Version]
- Mercuri, E.; Bonnemann, C.G.; Muntoni, F. Muscular dystrophies. Lancet 2019, 394, 2025–2038. [Google Scholar] [CrossRef]
- Birnkrant, D.J.; Bushby, K.; Bann, C.M.; Apkon, S.D.; Blackwell, A.; Colvin, M.K.; Cripe, L.; Herron, A.R.; Kennedy, A.; Kinnett, K.; et al. Diagnosis and management of Duchenne muscular dystrophy, part 3: Primary care, emergency management, psychosocial care, and transitions of care across the lifespan. Lancet Neurol. 2018, 17, 445–455. [Google Scholar] [CrossRef] [Green Version]
- Kirschner, J.; Lochmuller, H. Sarcoglycanopathies. Handb. Clin. Neurol. 2011, 101, 41–46. [Google Scholar]
- Petrilli, V.; Dostert, C.; Muruve, D.A.; Tschopp, J. The inflammasome: A danger sensing complex triggering innate immunity. Curr. Opin. Immunol. 2007, 19, 615–622. [Google Scholar] [CrossRef] [PubMed]
- Tidball, J.G.; Welc, S.S.; Wehling-Henricks, M. Immunobiology of inherited muscular dystrophies. Compr. Physiol. 2018, 8, 1313–1356. [Google Scholar] [PubMed]
- Wehling, M.; Spencer, M.J.; Tidball, J.G. A nitric oxide synthase transgene ameliorates muscular dystrophy in mdx mice. J. Cell Biol. 2001, 155, 123–131. [Google Scholar] [CrossRef] [PubMed]
- Tidball, J.G.; Villalta, S.A. Regulatory interactions between muscle and the immune system during muscle regeneration. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2010, 298, R1173–R1187. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burzyn, D.; Kuswanto, W.; Kolodin, D.; Shadrach, J.L.; Cerletti, M.; Jang, Y.; Sefik, E.; Tan, T.G.; Wagers, A.J.; Benoist, C.; et al. A special population of regulatory T cells potentiates muscle repair. Cell 2013, 155, 1282–1295. [Google Scholar] [CrossRef] [Green Version]
- Villalta, S.A.; Rosenthal, W.; Martinez, L.; Kaur, A.; Sparwasser, T.; Tidball, J.G.; Margeta, M.; Spencer, M.J.; Bluestone, J.A. Regulatory T cells suppress muscle inflammation and injury in muscular dystrophy. Sci. Transl. Med. 2014, 6, 258ra142. [Google Scholar] [CrossRef]
- Villalta, S.A.; Deng, B.; Rinaldi, C.; Wehling-Henricks, M.; Tidball, J.G. IFN-gamma promotes muscle damage in the mdx mouse model of Duchenne muscular dystrophy by suppressing M2 macrophage activation and inhibiting muscle cell proliferation. J. Immunol. 2011, 187, 5419–5428. [Google Scholar] [CrossRef] [Green Version]
- Gazzerro, E.; Baldassari, S.; Assereto, S.; Fruscione, F.; Pistorio, A.; Panicucci, C.; Volpi, S.; Perruzza, L.; Fiorillo, C.; Minetti, C.; et al. Enhancement of muscle T regulatory cells and improvement of muscular dystrophic process in mdx mice by blockade of extracellular ATP/P2X axis. Am. J. Pathol. 2015, 185, 3349–3360. [Google Scholar] [CrossRef] [Green Version]
- Ardissone, V.; Radaelli, E.; Zaratin, P.; Ardizzone, M.; Ladel, C.; Gattorno, M.; Martini, A.; Grassi, F.; Traggiai, E. Pharmacologic P2X purinergic receptor antagonism in the treatment of collagen-induced arthritis. Arthritis Rheum. 2011, 63, 3323–3332. [Google Scholar] [CrossRef]
- Vergani, A.; Fotino, C.; D’Addio, F.; Tezza, S.; Podetta, M.; Gatti, F.; Chin, M.; Bassi, R.; Molano, R.D.; Corradi, D.; et al. Effect of the purinergic inhibitor oxidized ATP in a model of islet allograft rejection. Diabetes 2013, 62, 1665–1675. [Google Scholar] [CrossRef] [Green Version]
- Schenk, U.; Frascoli, M.; Proietti, M.; Geffers, R.; Traggiai, E.; Buer, J.; Ricordi, C.; Westendorf, A.M.; Grassi, F. ATP inhibits the generation and function of regulatory T cells through the activation of purinergic P2X receptors. Sci. Signal. 2011, 4, ra12. [Google Scholar] [CrossRef] [PubMed]
- Frascoli, M.; Marcandalli, J.; Schenk, U.; Grassi, F. Purinergic P2X7 receptor drives T cell lineage choice and shapes peripheral gammadelta cells. J. Immunol. 2012, 189, 174–180. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sociali, G.; Visigalli, D.; Prukop, T.; Cervellini, I.; Mannino, E.; Venturi, C.; Bruzzone, S.; Sereda, M.W.; Schenone, A. Tolerability and efficacy study of P2X7 inhibition in experimental Charcot-Marie-Tooth type 1A (CMT1A) neuropathy. Neurobiol. Dis. 2016, 95, 145–157. [Google Scholar] [CrossRef] [PubMed]
- Al-Khalidi, R.; Panicucci, C.; Cox, P.; Chira, N.; Rog, J.; Young, C.N.J.; McGeehan, R.E.; Ambati, K.; Ambati, J.; Zablocki, K.; et al. Zidovudine ameliorates pathology in the mouse model of Duchenne muscular dystrophy via P2RX7 purinoceptor antagonism. Acta Neuropathol. Commun. 2018, 6, 27. [Google Scholar] [CrossRef]
- Fowler, B.J.; Gelfand, B.D.; Kim, Y.; Kerur, N.; Tarallo, V.; Hirano, Y.; Amarnath, S.; Fowler, D.H.; Radwan, M.; Young, M.T.; et al. Nucleoside reverse transcriptase inhibitors possess intrinsic anti-inflammatory activity. Science 2014, 346, 1000–1003. [Google Scholar] [CrossRef] [Green Version]
- Gazzerro, E.; Baratto, S.; Assereto, S.; Baldassari, S.; Panicucci, C.; Raffaghello, L.; Scudieri, P.; De Battista, D.; Fiorillo, C.; Volpi, S.; et al. The danger signal extracellular ATP is involved in the immunomediated damage of alpha-sarcoglycan-deficient muscular dystrophy. Am. J. Pathol. 2019, 189, 354–369. [Google Scholar] [CrossRef]
- Betto, R.; Senter, L.; Ceoldo, S.; Tarricone, E.; Biral, D.; Salviati, G. Ecto-ATPase activity of alpha-sarcoglycan (adhalin). J. Biol. Chem. 1999, 274, 7907–7912. [Google Scholar] [CrossRef] [Green Version]
- Mohamad, N.S.; Sinadinos, A.; Gorecki, D.C.; Zioupos, P.; Tong, J. Impact of P2RX7 ablation on the morphological, mechanical and tissue properties of bones in a murine model of duchenne muscular dystrophy. J. Biomech. 2016, 49, 3444–3451. [Google Scholar] [CrossRef] [Green Version]
- Saez, J.C.; Berthoud, V.M.; Branes, M.C.; Martinez, A.D.; Beyer, E.C. Plasma membrane channels formed by connexins: Their regulation and functions. Physiol. Rev. 2003, 83, 1359–1400. [Google Scholar] [CrossRef] [Green Version]
- Bruzzone, S.; Guida, L.; Zocchi, E.; Franco, L.; De Flora, A. Connexin 43 hemi channels mediate Ca2+-regulated transmembrane NAD+ fluxes in intact cells. FASEB J. 2001, 15, 10–12. [Google Scholar] [CrossRef]
- Navis, K.E.; Fan, C.Y.; Trang, T.; Thompson, R.J.; Derksen, D.J. Pannexin 1 channels as a therapeutic target: Structure, inhibition, and outlook. ACS Chem. Neurosci. 2020, 11, 2163–2172. [Google Scholar] [CrossRef] [PubMed]
- Cea, L.A.; Riquelme, M.A.; Cisterna, B.A.; Puebla, C.; Vega, J.L.; Rovegno, M.; Saez, J.C. Connexin- and pannexin-based channels in normal skeletal muscles and their possible role in muscle atrophy. J. Membr. Biol. 2012, 245, 423–436. [Google Scholar] [CrossRef] [PubMed]
- Osorio-Fuentealba, C.; Contreras-Ferrat, A.E.; Altamirano, F.; Espinosa, A.; Li, Q.; Niu, W.; Lavandero, S.; Klip, A.; Jaimovich, E. Electrical stimuli release ATP to increase GLUT4 translocation and glucose uptake via PI3Kgamma-Akt-AS160 in skeletal muscle cells. Diabetes 2013, 62, 1519–1526. [Google Scholar] [CrossRef] [Green Version]
- Linden, J.; Koch-Nolte, F.; Dahl, G. Purine release, metabolism, and signaling in the inflammatory response. Annu. Rev. Immunol. 2019, 37, 325–347. [Google Scholar] [CrossRef] [PubMed]
- Langlois, S.; Xiang, X.; Young, K.; Cowan, B.J.; Penuela, S.; Cowan, K.N. Pannexin 1 and pannexin 3 channels regulate skeletal muscle myoblast proliferation and differentiation. J. Biol. Chem. 2014, 289, 30717–30731. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pham, T.L.; St-Pierre, M.E.; Ravel-Chapuis, A.; Parks, T.E.C.; Langlois, S.; Penuela, S.; Jasmin, B.J.; Cowan, K.N. Expression of Pannexin 1 and Pannexin 3 during skeletal muscle development, regeneration, and Duchenne muscular dystrophy. J. Cell Physiol. 2018, 233, 7057–7070. [Google Scholar] [CrossRef] [PubMed]
- Cea, L.A.; Riquelme, M.A.; Vargas, A.A.; Urrutia, C.; Saez, J.C. Pannexin 1 channels in skeletal muscles. Front. Physiol. 2014, 5, 139. [Google Scholar] [CrossRef] [Green Version]
- Riquelme, M.A.; Cea, L.A.; Vega, J.L.; Boric, M.P.; Monyer, H.; Bennett, M.V.; Frank, M.; Willecke, K.; Saez, J.C. The ATP required for potentiation of skeletal muscle contraction is released via pannexin hemichannels. Neuropharmacology 2013, 75, 594–603. [Google Scholar] [CrossRef]
- Jorquera, G.; Altamirano, F.; Contreras-Ferrat, A.; Almarza, G.; Buvinic, S.; Jacquemond, V.; Jaimovich, E.; Casas, M. Cav1.1 controls frequency-dependent events regulating adult skeletal muscle plasticity. J. Cell Sci. 2013, 126, 1189–1198. [Google Scholar] [CrossRef] [Green Version]
- Pillon, N.J.; Li, Y.E.; Fink, L.N.; Brozinick, J.T.; Nikolayev, A.; Kuo, M.S.; Bilan, P.J.; Klip, A. Nucleotides released from palmitate-challenged muscle cells through pannexin-3 attract monocytes. Diabetes 2014, 63, 3815–3826. [Google Scholar] [CrossRef] [Green Version]
- Diaz-Vegas, A.; Campos, C.A.; Contreras-Ferrat, A.; Casas, M.; Buvinic, S.; Jaimovich, E.; Espinosa, A. ROS production via P2Y1-PKC-NOX2 is triggered by extracellular atp after electrical stimulation of skeletal muscle cells. PLoS ONE 2015, 10, e0129882. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Plotkin, L.I.; Davis, H.M.; Cisterna, B.A.; Saez, J.C. Connexins and pannexins in bone and skeletal muscle. Curr. Osteoporos. Rep. 2017, 15, 326–334. [Google Scholar] [CrossRef] [PubMed]
- Bao, L.; Locovei, S.; Dahl, G. Pannexin membrane channels are mechanosensitive conduits for ATP. FEBS Lett. 2004, 572, 65–68. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pelegrin, P.; Surprenant, A. Pannexin-1 mediates large pore formation and interleukin-1beta release by the ATP-gated P2X7 receptor. EMBO J. 2006, 25, 5071–5082. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Locovei, S.; Scemes, E.; Qiu, F.; Spray, D.C.; Dahl, G. Pannexin1 is part of the pore forming unit of the P2X (7) receptor death complex. FEBS Lett. 2007, 581, 483–488. [Google Scholar] [CrossRef] [Green Version]
- Nouet, J.; Himelman, E.; Lahey, K.C.; Zhao, Q.; Fraidenraich, D. Connexin-43 reduction prevents muscle defects in a mouse model of manifesting Duchenne muscular dystrophy female carriers. Sci. Rep. 2020, 10, 5683. [Google Scholar] [CrossRef] [Green Version]
- Araya, R.; Eckardt, D.; Riquelme, M.A.; Willecke, K.; Saez, J.C. Presence and importance of connexin43 during myogenesis. Cell Commun. Adhes. 2003, 10, 451–456. [Google Scholar] [CrossRef]
- Cea, L.A.; Bevilacqua, J.A.; Arriagada, C.; Cardenas, A.M.; Bigot, A.; Mouly, V.; Saez, J.C.; Caviedes, P. The absence of dysferlin induces the expression of functional connexin-based hemichannels in human myotubes. BMC Cell Biol. 2016, 17 (Suppl. 1), 15. [Google Scholar] [CrossRef] [Green Version]
- Cea, L.A.; Puebla, C.; Cisterna, B.A.; Escamilla, R.; Vargas, A.A.; Frank, M.; Martinez-Montero, P.; Prior, C.; Molano, J.; Esteban-Rodriguez, I.; et al. Fast skeletal myofibers of mdx mouse, model of Duchenne muscular dystrophy, express connexin hemichannels that lead to apoptosis. Cell Mol. Life Sci. 2016, 73, 2583–2599. [Google Scholar] [CrossRef]
- Wang, N.; De Bock, M.; Decrock, E.; Bol, M.; Gadicherla, A.; Vinken, M.; Rogiers, V.; Bukauskas, F.F.; Bultynck, G.; Leybaert, L. Paracrine signaling through plasma membrane hemichannels. Biochim. Biophys. Acta 2013, 1828, 35–50. [Google Scholar] [CrossRef]
- Wang, X.; Qin, W.; Xu, X.; Xiong, Y.; Zhang, Y.; Zhang, H.; Sun, B. Endotoxin-induced autocrine ATP signaling inhibits neutrophil chemotaxis through enhancing myosin light chain phosphorylation. Proc. Natl. Acad. Sci. USA 2017, 114, 4483–4488. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eltzschig, H.K.; Eckle, T.; Mager, A.; Kuper, N.; Karcher, C.; Weissmuller, T.; Boengler, K.; Schulz, R.; Robson, S.C.; Colgan, S.P. ATP release from activated neutrophils occurs via connexin 43 and modulates adenosine-dependent endothelial cell function. Circ. Res. 2006, 99, 1100–1108. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gu, B.J.; Wiley, J.S. Rapid ATP-induced release of matrix metalloproteinase 9 is mediated by the P2X7 receptor. Blood 2006, 107, 4946–4953. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Young, C.N.J.; Chira, N.; Rog, J.; Al-Khalidi, R.; Benard, M.; Galas, L.; Chan, P.; Vaudry, D.; Zablocki, K.; Gorecki, D.C. Sustained activation of P2X7 induces MMP-2-evoked cleavage and functional purinoceptor inhibition. J. Mol. Cell Biol. 2018, 10, 229–242. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cui, N.; Hu, M.; Khalil, R.A. Biochemical and biological attributes of matrix metalloproteinases. Prog. Mol. Biol. Transl. Sci. 2017, 147, 1–73. [Google Scholar]
- Young, C.N.J.; Gorecki, D.C. P2RX7 purinoceptor as a therapeutic target-the second coming? Front. Chem. 2018, 6, 248. [Google Scholar] [CrossRef] [Green Version]
- Bours, M.J.; Swennen, E.L.; Di Virgilio, F.; Cronstein, B.N.; Dagnelie, P.C. Adenosine 5’-triphosphate and adenosine as endogenous signaling molecules in immunity and inflammation. Pharmacol. Ther. 2006, 112, 358–404. [Google Scholar] [CrossRef]
- De Torre-Minguela, C.; Mesa Del Castillo, P.; Pelegrin, P. The NLRP3 and pyrin inflammasomes: Implications in the pathophysiology of autoinflammatory diseases. Front. Immunol. 2017, 8, 43. [Google Scholar] [CrossRef] [Green Version]
- Csoka, B.; Selmeczy, Z.; Koscso, B.; Nemeth, Z.H.; Pacher, P.; Murray, P.J.; Kepka-Lenhart, D.; Morris, S.M., Jr.; Gause, W.C.; Leibovich, S.J.; et al. Adenosine promotes alternative macrophage activation via A2A and A2B receptors. FASEB J. 2012, 26, 376–386. [Google Scholar] [CrossRef] [Green Version]
- Wilson, J.M.; Ross, W.G.; Agbai, O.N.; Frazier, R.; Figler, R.A.; Rieger, J.; Linden, J.; Ernst, P.B. The A2B adenosine receptor impairs the maturation and immunogenicity of dendritic cells. J. Immunol. 2009, 182, 4616–4623. [Google Scholar] [CrossRef] [Green Version]
- Ehrentraut, H.; Westrich, J.A.; Eltzschig, H.K.; Clambey, E.T. Adora2b adenosine receptor engagement enhances regulatory T cell abundance during endotoxin-induced pulmonary inflammation. PLoS ONE 2012, 7, e32416. [Google Scholar] [CrossRef] [PubMed]
- Da Silva, J.L.G.; Passos, D.F.; Bernardes, V.M.; Leal, D.B.R. ATP and adenosine: Role in the immunopathogenesis of rheumatoid arthritis. Immunol. Lett. 2019, 214, 55–64. [Google Scholar] [CrossRef] [PubMed]
- Garrido, W.; Jara, C.; Torres, A.; Suarez, R.; Cappelli, C.; Oyarzun, C.; Quezada, C.; San Martin, R. Blockade of the adenosine A3 receptor attenuates caspase 1 activation in renal tubule epithelial cells and decreases interleukins IL-1beta and IL-18 in diabetic rats. Int. J. Mol. Sci. 2019, 20, 4531. [Google Scholar] [CrossRef] [Green Version]
- Lynge, J.; Hellsten, Y. Distribution of adenosine A1, A2A and A2B receptors in human skeletal muscle. Acta Physiol. Scand. 2000, 169, 283–290. [Google Scholar] [CrossRef] [PubMed]
- Lynge, J.; Schulte, G.; Nordsborg, N.; Fredholm, B.B.; Hellsten, Y. Adenosine A 2B receptors modulate cAMP levels and induce CREB but not ERK1/2 and p38 phosphorylation in rat skeletal muscle cells. Biochem. Biophys. Res. Commun. 2003, 307, 180–187. [Google Scholar] [CrossRef]
- Dobson, J.G., Jr.; Rubio, R.; Berne, R.M. Role of adenine nucleotides, adenosine, and inorganic phosphate in the regulation of skeletal muscle blood flow. Circ. Res. 1971, 29, 375–384. [Google Scholar] [CrossRef]
- Vergauwen, L.; Hespel, P.; Richter, E.A. Adenosine receptors mediate synergistic stimulation of glucose uptake and transport by insulin and by contractions in rat skeletal muscle. J. Clin. Investig. 1994, 93, 974–981. [Google Scholar] [CrossRef] [Green Version]
- Soslow, J.H.; Markham, L.W.; Burnette, W.B.; Galindo, C.L.; Feoktistov, I.; Raucci, F.J., Jr.; Damon, B.M.; Sawyer, D.B.; Ryzhov, S. Increased number of circulating CD8/CD26 T cells in the blood of duchenne muscular dystrophy patients is associated with augmented binding of adenosine deaminase and higher muscular strength scores. Front. Pharmacol. 2017, 8, 914. [Google Scholar] [CrossRef] [Green Version]
- Latroche, C.; Weiss-Gayet, M.; Muller, L.; Gitiaux, C.; Leblanc, P.; Liot, S.; Ben-Larbi, S.; Abou-Khalil, R.; Verger, N.; Bardot, P.; et al. Coupling between myogenesis and angiogenesis during skeletal muscle regeneration is stimulated by restorative macrophages. Stem Cell Rep. 2017, 9, 2018–2033. [Google Scholar] [CrossRef] [Green Version]
- Dort, J.; Fabre, P.; Molina, T.; Dumont, N.A. Macrophages are key regulators of stem cells during skeletal muscle regeneration and diseases. Stem Cells Int. 2019, 2019, 4761427. [Google Scholar] [CrossRef]
- Koch-Nolte, F.; Eichhoff, A.; Pinto-Espinoza, C.; Schwarz, N.; Schafer, T.; Menzel, S.; Haag, F.; Demeules, M.; Gonde, H.; Adriouch, S. Novel biologics targeting the P2X7 ion channel. Curr. Opin. Pharmacol. 2019, 47, 110–118. [Google Scholar] [CrossRef] [PubMed]
- Danquah, W.; Meyer-Schwesinger, C.; Rissiek, B.; Pinto, C.; Serracant-Prat, A.; Amadi, M.; Iacenda, D.; Knop, J.H.; Hammel, A.; Bergmann, P.; et al. Nanobodies that block gating of the P2X7 ion channel ameliorate inflammation. Sci. Transl. Med. 2016, 8, 366ra162. [Google Scholar] [CrossRef] [PubMed]
- Zahid, A.; Li, B.; Kombe, A.J.K.; Jin, T.; Tao, J. Pharmacological inhibitors of the NLRP3 inflammasome. Front. Immunol. 2019, 10, 2538. [Google Scholar] [CrossRef] [PubMed] [Green Version]



© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Panicucci, C.; Raffaghello, L.; Bruzzone, S.; Baratto, S.; Principi, E.; Minetti, C.; Gazzerro, E.; Bruno, C. eATP/P2X7R Axis: An Orchestrated Pathway Triggering Inflammasome Activation in Muscle Diseases. Int. J. Mol. Sci. 2020, 21, 5963. https://doi.org/10.3390/ijms21175963
Panicucci C, Raffaghello L, Bruzzone S, Baratto S, Principi E, Minetti C, Gazzerro E, Bruno C. eATP/P2X7R Axis: An Orchestrated Pathway Triggering Inflammasome Activation in Muscle Diseases. International Journal of Molecular Sciences. 2020; 21(17):5963. https://doi.org/10.3390/ijms21175963
Chicago/Turabian StylePanicucci, Chiara, Lizzia Raffaghello, Santina Bruzzone, Serena Baratto, Elisa Principi, Carlo Minetti, Elisabetta Gazzerro, and Claudio Bruno. 2020. "eATP/P2X7R Axis: An Orchestrated Pathway Triggering Inflammasome Activation in Muscle Diseases" International Journal of Molecular Sciences 21, no. 17: 5963. https://doi.org/10.3390/ijms21175963