Targeting E3 Ubiquitin Ligases and Deubiquitinases in Ciliopathy and Cancer

 ,
,
Abstract
:1. Introduction
2. Roles of Cilia in Ciliopathy and Cancer
2.1. Intellectual Disability
2.2. Retinal Degeneration
2.3. Craniofacial Malformation
2.4. Laterality Disorders
2.5. Cystic Kidney Disease
2.6. Obesity
2.7. Scoliosis
2.8. Respiratory Distress
2.9. Infertility
2.10. Roles of Primary Cilia in Cancer
3. Roles of E3 Ubiquitin Ligases and DUBs in Ciliogenesis, Ciliopathy, and Cancer
3.1. CRL3KCTD17, USP8, and TCHP
3.2. MARCHF7, TRIM32, USP9X, and IQCB1
3.3. CYLD and MIB1
3.4. CRL2VHL
4. Future Directions
4.1. Identification of E3 Ubiquitin Ligases and DUBs Related to Cilia Assembly and Disassembly
4.2. Identification of E3 Ubiquitin Ligase and DUB Substrates
4.3. Identification of Drugs Targeting E3 Ubiquitin Ligases and DUBs
Supplementary Materials
Funding
Acknowledgments
Conflicts of Interest
References
- Anvarian, Z.; Mykytyn, K.; Mukhopadhyay, S.; Pedersen, L.B.; Christensen, S.T. Cellular signalling by primary cilia in development, organ function and disease. Nat. Reviews. Nephrol. 2019, 15, 199–219. [Google Scholar] [CrossRef] [PubMed]
- Malicki, J.J.; Johnson, C.A. The Cilium: Cellular Antenna and Central Processing Unit. Trends Cell Biol. 2017, 27, 126–140. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goto, H.; Inoko, A.; Inagaki, M. Cell cycle progression by the repression of primary cilia formation in proliferating cells. Cell. Mol. Life Sci. 2013, 70, 3893–3905. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Izawa, I.; Goto, H.; Kasahara, K.; Inagaki, M. Current topics of functional links between primary cilia and cell cycle. Cilia 2015, 4, 12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goto, H.; Inaba, H.; Inagaki, M. Mechanisms of ciliogenesis suppression in dividing cells. Cell. Mol. Life Sci. 2017, 74, 881–890. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nishimura, Y.; Kasahara, K.; Shiromizu, T.; Watanabe, M.; Inagaki, M. Primary cilia as signaling hubs in health and disease. Adv. Sci. 2019, 6, 1801138. [Google Scholar] [CrossRef] [Green Version]
- Valente, E.M.; Rosti, R.O.; Gibbs, E.; Gleeson, J.G. Primary cilia in neurodevelopmental disorders. Nat. Rev. Neurol. 2014, 10, 27–36. [Google Scholar] [CrossRef] [Green Version]
- Reiter, J.F.; Leroux, M.R. Genes and molecular pathways underpinning ciliopathies. Nat. Rev. Mol. Cell Biol. 2017, 18, 533–547. [Google Scholar] [CrossRef]
- Liu, H.; Kiseleva, A.A.; Golemis, E.A. Ciliary signalling in cancer. Nat. Rev. Cancer 2018, 18, 511–524. [Google Scholar] [CrossRef]
- Wang, L.; Dynlacht, B.D. The regulation of cilium assembly and disassembly in development and disease. Development 2018, 145. [Google Scholar] [CrossRef] [Green Version]
- Silverman, M.A.; Leroux, M.R. Intraflagellar transport and the generation of dynamic, structurally and functionally diverse cilia. Trends Cell Biol. 2009, 19, 306–316. [Google Scholar] [CrossRef] [PubMed]
- Loreng, T.D.; Smith, E.F. The Central Apparatus of Cilia and Eukaryotic Flagella. Cold Spring Harb. Perspect Biol. 2017, 9. [Google Scholar] [CrossRef] [PubMed]
- Ishikawa, T. Axoneme Structure from Motile Cilia. Cold Spring Harb. Perspect. Biol. 2017, 9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mitchison, H.M.; Valente, E.M. Motile and non-motile cilia in human pathology: From function to phenotypes. J. Pathol. 2017, 241, 294–309. [Google Scholar] [CrossRef] [PubMed]
- Reiter, J.F.; Blacque, O.E.; Leroux, M.R. The base of the cilium: Roles for transition fibres and the transition zone in ciliary formation, maintenance and compartmentalization. EMBO Rep. 2012, 13, 608–618. [Google Scholar] [CrossRef] [Green Version]
- Alieva, I.; Staub, C.; Uzbekova, S.; Uzbekov, R. A question of flagella origin for spermatids—Mother or daughter centriole? In Flagella and Cilia Types, Strucure and Functions; Uzbekov, R.E., Ed.; Nova Science Publishers, Inc.: New York, NY, USA, 2018; pp. 109–126. [Google Scholar]
- Al Jord, A.; Lemaître, A.I.; Delgehyr, N.; Faucourt, M.; Spassky, N.; Meunier, A. Centriole amplification by mother and daughter centrioles differs in multiciliated cells. Nature 2014, 516, 104–107. [Google Scholar] [CrossRef]
- Tucker, R.W.; Pardee, A.B.; Fujiwara, K. Centriole ciliation is related to quiescence and DNA synthesis in 3T3 cells. Cell 1979, 17, 527–535. [Google Scholar] [CrossRef]
- Tucker, R.W.; Scher, C.D.; Stiles, C.D. Centriole deciliation associated with the early response of 3T3 cells to growth factors but not to SV40. Cell 1979, 18, 1065–1072. [Google Scholar] [CrossRef]
- Rieder, C.L.; Jensen, C.G.; Jensen, L.C.W. The resorption of primary cilia during mitosis in a vertebrate (PtK1) cell line. J. Ultrastruct. Res. 1979, 68, 173–185. [Google Scholar] [CrossRef]
- Tsang, W.Y.; Dynlacht, B.D. CP110 and its network of partners coordinately regulate cilia assembly. Cilia 2013, 2, 9. [Google Scholar] [CrossRef] [Green Version]
- Yadav, S.P.; Sharma, N.K.; Liu, C.; Dong, L.; Li, T.; Swaroop, A. Centrosomal protein CP110 controls maturation of the mother centriole during cilia biogenesis. Developement 2016, 143, 1491–1501. [Google Scholar]
- Craft, J.M.; Harris, J.A.; Hyman, S.; Kner, P.; Lechtreck, K.F. Tubulin transport by IFT is upregulated during ciliary growth by a cilium-autonomous mechanism. J. Cell Biol. 2015, 208, 223–237. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Malicki, J.; Avidor-Reiss, T. From the cytoplasm into the cilium: Bon voyage. Organogenesis 2014, 10, 138–157. [Google Scholar] [CrossRef] [PubMed]
- Mirvis, M.; Stearns, T.; James Nelson, W. Cilium structure, assembly, and disassembly regulated by the cytoskeleton. Biochem. J. 2018, 475, 2329–2353. [Google Scholar] [CrossRef] [Green Version]
- Clague, M.J.; Heride, C.; Urbé, S. The demographics of the ubiquitin system. Trends Cell Biol. 2015, 25, 417–426. [Google Scholar] [CrossRef]
- Leznicki, P.; Kulathu, Y. Mechanisms of regulation and diversification of deubiquitylating enzyme function. J. Cell Sci. 2017, 130, 1997–2006. [Google Scholar] [CrossRef] [Green Version]
- Popovic, D.; Vucic, D.; Dikic, I. Ubiquitination in disease pathogenesis and treatment. Nat. Med. 2014, 20, 1242–1253. [Google Scholar] [CrossRef]
- Harrigan, J.A.; Jacq, X.; Martin, N.M.; Jackson, S.P. Deubiquitylating enzymes and drug discovery: Emerging opportunities. Nat. Rev. Drug Discov. 2018, 17, 57–78. [Google Scholar] [CrossRef]
- Senft, D.; Qi, J.; Ronai, Z.A. Ubiquitin ligases in oncogenic transformation and cancer therapy. Nat. Rev. Cancer 2018, 18, 69–88. [Google Scholar] [CrossRef]
- Shearer, R.F.; Saunders, D.N. Regulation of primary cilia formation by the ubiquitin-proteasome system. Biochem. Soc. Trans. 2016, 44, 1265–1271. [Google Scholar] [CrossRef]
- Hossain, D.; Tsang, W.Y. The role of ubiquitination in the regulation of primary cilia assembly and disassembly. Semin. Cell Dev. Biol. 2019, 93, 145–152. [Google Scholar] [CrossRef] [PubMed]
- Kasahara, K.; Kawakami, Y.; Kiyono, T.; Yonemura, S.; Kawamura, Y.; Era, S.; Matsuzaki, F.; Goshima, N.; Inagaki, M. Ubiquitin-proteasome system controls ciliogenesis at the initial step of axoneme extension. Nat. Commun. 2014, 5, 5081. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kasahara, K.; Aoki, H.; Kiyono, T.; Wang, S.; Kagiwada, H.; Yuge, M.; Tanaka, T.; Nishimura, Y.; Mizoguchi, A.; Goshima, N.; et al. EGF receptor kinase suppresses ciliogenesis through activation of USP8 deubiquitinase. Nat. Commun. 2018, 9, 758. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Toulis, V.; Marfany, G. By the Tips of Your Cilia: Ciliogenesis in the Retina and the Ubiquitin-Proteasome System. Adv. Exp. Med. Biol 2020, 1233, 303–310. [Google Scholar] [PubMed]
- Pazour, G.J.; Quarmby, L.; Smith, A.O.; Desai, P.B.; Schmidts, M. Cilia in cystic kidney and other diseases. Cell Signal. 2020, 69, 109519. [Google Scholar] [CrossRef] [PubMed]
- Hildebrandt, F.; Benzing, T.; Katsanis, N. Ciliopathies. N. Engl. J. Med. 2011, 364, 1533–1543. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Braun, D.A.; Hildebrandt, F. Ciliopathies. Cold Spring Harb. Perspect. Biol. 2017, 9. [Google Scholar] [CrossRef] [PubMed]
- Guo, J.; Higginbotham, H.; Li, J.; Nichols, J.; Hirt, J.; Ghukasyan, V.; Anton, E.S. Developmental disruptions underlying brain abnormalities in ciliopathies. Nat. Commun. 2015, 6, 7857. [Google Scholar] [CrossRef] [Green Version]
- Youn, Y.H.; Han, Y.G. Primary Cilia in Brain Development and Diseases. Am. J. Pathol. 2018, 188, 11–22. [Google Scholar] [CrossRef]
- Bujakowska, K.M.; Liu, Q.; Pierce, E.A. Photoreceptor Cilia and Retinal Ciliopathies. Cold Spring Harb. Perspect. Biol. 2017, 9. [Google Scholar] [CrossRef]
- Bachmann-Gagescu, R.; Neuhauss, S.C. The photoreceptor cilium and its diseases. Curr. Opin. Genet. Dev. 2019, 56, 22–33. [Google Scholar] [CrossRef] [PubMed]
- Schock, E.N.; Brugmann, S.A. Discovery, Diagnosis, and Etiology of Craniofacial Ciliopathies. Cold Spring Harb. Perspect. Biol. 2017, 9. [Google Scholar] [CrossRef]
- Cortés, C.R.; Metzis, V.; Wicking, C. Unmasking the ciliopathies: Craniofacial defects and the primary cilium. Wiley Interdiscip. Rev. Dev. Biol. 2015, 4, 637–653. [Google Scholar] [CrossRef] [PubMed]
- Dasgupta, A.; Amack, J.D. Cilia in vertebrate left-right patterning. Philos. Trans. R. Soc. Lond. B. Biol. Sci. 2016, 1710, 20150410. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grimes, D.T. Making and breaking symmetry in development, growth and disease. Development 2019, 146. [Google Scholar] [CrossRef] [Green Version]
- Ye, H.; Wang, X.; Constans, M.M.; Sussman, C.R.; Chebib, F.T.; Irazabal, M.V.; Young, W.F., Jr.; Harris, P.C.; Kirschner, L.S.; Torres, V.E. The regulatory 1α subunit of protein kinase A modulates renal cystogenesis. Am. J. Physiol. Ren. Physiol. 2017, 313, 677–686. [Google Scholar] [CrossRef] [Green Version]
- Cornec-Le Gall, E.; Alam, A.; Perrone, R.D. Autosomal dominant polycystic kidney disease. Lancet 2019, 393, 919–935. [Google Scholar] [CrossRef]
- Mariman, E.C.; Vink, R.G.; Roumans, N.J.; Bouwman, F.G.; Stumpel, C.T.; Aller, E.E.; van Baak, M.A.; Wang, P. The cilium: A cellular antenna with an influence on obesity risk. Br. J. Nutr. 2016, 116, 576–592. [Google Scholar] [CrossRef] [Green Version]
- Engle, S.E.; Bansal, R.; Antonellis, P.J.; Berbari, N.F. Cilia signaling and obesity. Semin. Cell Dev. Biol. 2020. Available online: https://www.sciencedirect.com/science/article/abs/pii/S1084952119301831 (accessed on 19 August 2020).
- Oliazadeh, N.; Gorman, K.F.; Eveleigh, R.; Bourque, G.; Moreau, A. Identification of Elongated Primary Cilia with Impaired Mechanotransduction in Idiopathic Scoliosis Patients. Sci. Rep. 2017, 7, 44260. [Google Scholar] [CrossRef] [Green Version]
- Qin, L.; Liu, W.; Cao, H.; Xiao, G. Molecular mechanosensors in osteocytes. Bone Res. 2020, 8, 23. [Google Scholar] [CrossRef]
- Sagel, S.D.; Davis, S.D.; Campisi, P.; Dell, S.D. Update of respiratory tract disease in children with primary ciliary dyskinesia. Proc. Am. Thorac. Soc. 2011, 8, 438–443. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mirra, V.; Werner, C.; Santamaria, F. Primary Ciliary Dyskinesia: An Update on Clinical Aspects, Genetics, Diagnosis, and Future Treatment Strategies. Front. Pediatr. 2017, 5, 135. [Google Scholar] [CrossRef] [PubMed]
- Sironen, A.; Shoemark, A.; Patel, M.; Loebinger, M.R.; Mitchison, H.M. Sperm defects in primary ciliary dyskinesia and related causes of male infertility. Cell. Mol. Life Sci. 2020, 77, 2029–2048. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Girardet, L.; Augière, C.; Asselin, M.-P.; Belleannée, C. Primary cilia: Biosensors of the male reproductive tract. Andrology 2019, 7, 588–602. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Juric-Sekhar, G.; Hevner, R.F. Malformations of Cerebral Cortex Development: Molecules and Mechanisms. Annu. Rev. Pathol. 2019, 14, 293–318. [Google Scholar] [CrossRef] [PubMed]
- Vecino, E.; Rodriguez, F.D.; Ruzafa, N.; Pereiro, X.; Sharma, S.C. Glia-neuron interactions in the mammalian retina. Prog. Retin. Eye Res. 2016, 51, 1–40. [Google Scholar] [CrossRef] [Green Version]
- May-Simera, H.; Nagel-Wolfrum, K.; Wolfrum, U. Cilia—The sensory antennae in the eye. Prog. Retin. Eye Res. 2017, 60, 144–180. [Google Scholar] [CrossRef]
- Jeong, J.; Mao, J.; Tenzen, T.; Kottmann, A.H.; McMahon, A.P. Hedgehog signaling in the neural crest cells regulates the patterning and growth of facial primordia. Genes Dev. 2004, 18, 937–951. [Google Scholar] [CrossRef] [Green Version]
- Tobin, J.L.; Di Franco, M.; Eichers, E.; May-Simera, H.; Garcia, M.; Yan, J.; Quinlan, R.; Justice, M.J.; Hennekam, R.C.; Briscoe, J.; et al. Inhibition of neural crest migration underlies craniofacial dysmorphology and Hirschsprung’s disease in Bardet-Biedl syndrome. Proc. Natl. Acad. Sci. USA 2008, 105, 6714–6719. [Google Scholar] [CrossRef] [Green Version]
- Brugmann, S.A.; Allen, N.C.; James, A.W.; Mekonnen, Z.; Madan, E.; Helms, J.A. A primary cilia-dependent etiology for midline facial disorders. Hum. Mol. Genet. 2010, 19, 1577–1592. [Google Scholar] [CrossRef] [Green Version]
- Hirokawa, N.; Tanaka, Y.; Okada, Y. Left-right determination: Involvement of molecular motor KIF3, cilia, and nodal flow. Cold Spring Harb. Perspect. Biol. 2009, 1, a000802. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kawasumi, A.; Nakamura, T.; Iwai, N.; Yashiro, K.; Saijoh, Y.; Belo, J.A.; Shiratori, H.; Hamada, H. Left-right asymmetry in the level of active Nodal protein produced in the node is translated into left-right asymmetry in the lateral plate of mouse embryos. Dev. Biol. 2011, 353, 321–330. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shiratori, H.; Hamada, H. TGFbeta signaling in establishing left-right asymmetry. Semin. Cell Dev. Biol. 2014, 32, 80–84. [Google Scholar] [CrossRef] [PubMed]
- Marques, S.; Borges, A.C.; Silva, A.C.; Freitas, S.; Cordenonsi, M.; Belo, J.A. The activity of the Nodal antagonist Cerl-2 in the mouse node is required for correct L/R body axis. Genes Dev. 2004, 18, 2342–2347. [Google Scholar] [CrossRef] [Green Version]
- Nakamura, T.; Saito, D.; Kawasumi, A.; Shinohara, K.; Asai, Y.; Takaoka, K.; Dong, F.; Takamatsu, A.; Belo, J.A.; Mochizuki, A.; et al. Fluid flow and interlinked feedback loops establish left-right asymmetric decay of Cerl2 mRNA. Nat. Commun. 2012, 3, 1322. [Google Scholar] [CrossRef] [Green Version]
- Logan, M.; Pagan-Westphal, S.M.; Smith, D.M.; Paganessi, L.; Tabin, C.J. The transcription factor Pitx2 mediates situs-specific morphogenesis in response to left-right asymmetric signals. Cell 1998, 94, 307–317. [Google Scholar] [CrossRef] [Green Version]
- Saijoh, Y.; Adachi, H.; Sakuma, R.; Yeo, C.Y.; Yashiro, K.; Watanabe, M.; Hashiguchi, H.; Mochida, K.; Ohishi, S.; Kawabata, M.; et al. Left-right asymmetric expression of lefty2 and nodal is induced by a signaling pathway that includes the transcription factor FAST2. Mol. Cell 2000, 5, 35–47. [Google Scholar] [CrossRef]
- Shiratori, H.; Sakuma, R.; Watanabe, M.; Hashiguchi, H.; Mochida, K.; Sakai, Y.; Nishino, J.; Saijoh, Y.; Whitman, M.; Hamada, H. Two-step regulation of left-right asymmetric expression of Pitx2: Initiation by nodal signaling and maintenance by Nkx2. Mol. Cell 2001, 7, 137–149. [Google Scholar] [CrossRef]
- Botilde, Y.; Yoshiba, S.; Shinohara, K.; Hasegawa, T.; Nishimura, H.; Shiratori, H.; Hamada, H. Cluap1 localizes preferentially to the base and tip of cilia and is required for ciliogenesis in the mouse embryo. Dev. Biol. 2013, 381, 203–212. [Google Scholar] [CrossRef] [Green Version]
- Ware, S.M.; Aygun, M.G.; Hildebrandt, F. Spectrum of clinical diseases caused by disorders of primary cilia. Proc. Am. Thorac. Soc. 2011, 8, 444–450. [Google Scholar] [CrossRef] [Green Version]
- Oud, M.M.; Lamers, I.J.; Arts, H.H. Ciliopathies: Genetics in Pediatric Medicine. J. Pediatr. Genet. 2017, 6, 18–29. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matsui, T.; Bessho, Y. Left-right asymmetry in zebrafish. Cell. Mol. Life Sci. 2012, 69, 3069–3077. [Google Scholar] [CrossRef] [PubMed]
- Matsui, T.; Ishikawa, H.; Bessho, Y. Cell collectivity regulation within migrating cell cluster during Kupffer’s vesicle formation in zebrafish. Front. Cell Dev. Biol. 2015, 3, 27. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Devlin, L.A.; Sayer, J.A. Renal ciliopathies. Curr. Opin. Genet. Dev. 2019, 56, 49–60. [Google Scholar] [CrossRef]
- Mangolini, A.; de Stephanis, L.; Aguiari, G. Role of calcium in polycystic kidney disease: From signaling to pathology. World J. Nephrol 2016, 5, 76–83. [Google Scholar] [CrossRef]
- Avasthi, P.; Maser, R.L.; Tran, P.V. Primary Cilia in Cystic Kidney Disease. Results Probl. Cell Differ. 2017, 60, 281–321. [Google Scholar]
- Malekshahabi, T.; Khoshdel Rad, N.; Serra, A.L.; Moghadasali, R. Autosomal dominant polycystic kidney disease: Disrupted pathways and potential therapeutic interventions. J. Cell. Physiol. 2019, 234, 12451–12470. [Google Scholar] [CrossRef]
- Oh, E.C.; Vasanth, S.; Katsanis, N. Metabolic regulation and energy homeostasis through the primary Cilium. Cell Metab. 2015, 21, 21–31. [Google Scholar] [CrossRef] [Green Version]
- Vaisse, C.; Reiter, J.F.; Berbari, N.F. Cilia and Obesity. Cold Spring Harb. Perspect. Biol. 2017, 9, a028217. [Google Scholar] [CrossRef] [Green Version]
- Ernst, M.B.; Wunderlich, C.M.; Hess, S.; Paehler, M.; Mesaros, A.; Koralov, S.B.; Kleinridders, A.; Husch, A.; Munzberg, H.; Hampel, B.; et al. Enhanced Stat3 activation in POMC neurons provokes negative feedback inhibition of leptin and insulin signaling in obesity. J. Neurosci. 2009, 29, 11582–11593. [Google Scholar] [CrossRef] [Green Version]
- Mesaros, A.; Koralov, S.B.; Rother, E.; Wunderlich, F.T.; Ernst, M.B.; Barsh, G.S.; Rajewsky, K.; Bruning, J.C. Activation of Stat3 signaling in AgRP neurons promotes locomotor activity. Cell Metab. 2008, 7, 236–248. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Han, Y.M.; Kang, G.M.; Byun, K.; Ko, H.W.; Kim, J.; Shin, M.S.; Kim, H.K.; Gil, S.Y.; Yu, J.H.; Lee, B.; et al. Leptin-promoted cilia assembly is critical for normal energy balance. J. Clin. Investig. 2014, 124, 2193–2197. [Google Scholar] [CrossRef] [Green Version]
- Sebo, Z.L.; Rodeheffer, M.S. Assembling the adipose organ: Adipocyte lineage segregation and adipogenesis in vivo. Development 2019, 146. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marion, V.; Stoetzel, C.; Schlicht, D.; Messaddeq, N.; Koch, M.; Flori, E.; Danse, J.M.; Mandel, J.L.; Dollfus, H. Transient ciliogenesis involving Bardet-Biedl syndrome proteins is a fundamental characteristic of adipogenic differentiation. Proc. Natl. Acad. Sci. USA 2009, 106, 1820–1825. [Google Scholar] [CrossRef] [Green Version]
- Marion, V.; Mockel, A.; De Melo, C.; Obringer, C.; Claussmann, A.; Simon, A.; Messaddeq, N.; Durand, M.; Dupuis, L.; Loeffler, J.P.; et al. BBS-induced ciliary defect enhances adipogenesis, causing paradoxical higher-insulin sensitivity, glucose usage, and decreased inflammatory response. Cell Metab. 2012, 16, 363–377. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, D.; Shi, S.; Wang, H.; Liao, K. Growth arrest induces primary-cilium formation and sensitizes IGF-1-receptor signaling during differentiation induction of 3T3-L1 preadipocytes. J. Cell Sci. 2009, 122, 2760–2768. [Google Scholar] [CrossRef] [Green Version]
- Dalbay, M.T.; Thorpe, S.D.; Connelly, J.T.; Chapple, J.P.; Knight, M.M. Adipogenic Differentiation of hMSCs is Mediated by Recruitment of IGF-1r Onto the Primary Cilium Associated With Cilia Elongation. Stem Cells 2015, 33, 1952–1961. [Google Scholar] [CrossRef]
- Spasic, M.; Jacobs, C.R. Primary cilia: Cell and molecular mechanosensors directing whole tissue function. Semin. Cell Dev. Biol. 2017, 71, 42–52. [Google Scholar] [CrossRef]
- Uzbekov, R.E.; Maurel, D.B.; Aveline, P.C.; Pallu, S.; Benhamou, C.L.; Rochefort, G.Y. Centrosome fine ultrastructure of the osteocyte mechanosensitive primary cilium. Microsc. Microanal. 2012, 18, 1430–1441. [Google Scholar] [CrossRef] [PubMed]
- Takeuchi, K.; Kitano, M.; Ishinaga, H.; Kobayashi, M.; Ogawa, S.; Nakatani, K.; Masuda, S.; Nagao, M.; Fujisawa, T. Recent advances in primary ciliary dyskinesia. Auris Nasus Larynx 2016, 43, 229–236. [Google Scholar] [CrossRef] [PubMed]
- Horani, A.; Ferkol, T.W.; Dutcher, S.K.; Brody, S.L. Genetics and biology of primary ciliary dyskinesia. Paediatr. Respir. Rev. 2016, 18, 18–24. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pugacheva, E.N.; Jablonski, S.A.; Hartman, T.R.; Henske, E.P.; Golemis, E.A. HEF1-dependent Aurora A activation induces disassembly of the primary cilium. Cell 2007, 129, 1351–1363. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Otto, T.; Sicinski, P. Cell cycle proteins as promising targets in cancer therapy. Nat. Rev. Cancer 2017, 17, 93–115. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liang, Y.; Meng, D.; Zhu, B.; Pan, J. Mechanism of ciliary disassembly. Cell. Mol. Life Sci. 2016, 73, 1787–1802. [Google Scholar] [CrossRef] [PubMed]
- Korobeynikov, V.; Deneka, A.Y.; Golemis, E.A. Mechanisms for nonmitotic activation of Aurora-A at cilia. Biochem. Soc. Trans. 2017, 45, 37–49. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Plotnikova, O.V.; Nikonova, A.S.; Loskutov, Y.V.; Kozyulina, P.Y.; Pugacheva, E.N.; Golemis, E.A. Calmodulin activation of Aurora-A kinase (AURKA) is required during ciliary disassembly and in mitosis. Mol. Biol. Cell 2012, 23, 2658–2670. [Google Scholar] [CrossRef]
- Lee, K.H.; Johmura, Y.; Yu, L.R.; Park, J.E.; Gao, Y.; Bang, J.K.; Zhou, M.; Veenstra, T.D.; Yeon Kim, B.; Lee, K.S. Identification of a novel Wnt5a-CK1varepsilon-Dvl2-Plk1-mediated primary cilia disassembly pathway. EMBO J. 2012, 31, 3104–3117. [Google Scholar] [CrossRef]
- Plotnikova, O.V.; Seo, S.; Cottle, D.L.; Conduit, S.; Hakim, S.; Dyson, J.M.; Mitchell, C.A.; Smyth, I.M. INPP5E interacts with AURKA, linking phosphoinositide signaling to primary cilium stability. J. Cell Sci. 2015, 128, 364–372. [Google Scholar] [CrossRef] [Green Version]
- Inoko, A.; Matsuyama, M.; Goto, H.; Ohmuro-Matsuyama, Y.; Hayashi, Y.; Enomoto, M.; Ibi, M.; Urano, T.; Yonemura, S.; Kiyono, T.; et al. Trichoplein and Aurora A block aberrant primary cilia assembly in proliferating cells. J. Cell Biol. 2012, 197, 391–405. [Google Scholar] [CrossRef] [Green Version]
- Gabriel, E.; Wason, A.; Ramani, A.; Gooi, L.M.; Keller, P.; Pozniakovsky, A.; Poser, I.; Noack, F.; Telugu, N.S.; Calegari, F.; et al. CPAP promotes timely cilium disassembly to maintain neural progenitor pool. EMBO J. 2016, 35, 803–819. [Google Scholar] [CrossRef]
- Pazour, G.J.; Wilkerson, C.G.; Witman, G.B. A dynein light chain is essential for the retrograde particle movement of intraflagellar transport (IFT). J. Cell Biol. 1998, 141, 979–992. [Google Scholar] [CrossRef] [PubMed]
- Maskey, D.; Marlin, M.C.; Kim, S.; Kim, S.; Ong, E.C.; Li, G.; Tsiokas, L. Cell cycle-dependent ubiquitylation and destruction of NDE1 by CDK5-FBW7 regulates ciliary length. EMBO J. 2015, 34, 2424–2440. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nikonova, A.S.; Golemis, E.A. The tumor suppressor FBW7 controls ciliary length. EMBO J. 2015, 34, 2388–2390. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Inaba, H.; Goto, H.; Kasahara, K.; Kumamoto, K.; Yonemura, S.; Inoko, A.; Yamano, S.; Wanibuchi, H.; He, D.; Goshima, N.; et al. Ndel1 suppresses ciliogenesis in proliferating cells by regulating the trichoplein-Aurora A pathway. J. Cell Biol. 2016, 212, 409–423. [Google Scholar] [CrossRef] [Green Version]
- Fabbri, L.; Bost, F.; Mazure, N.M. Primary Cilium in Cancer Hallmarks. Int. J. Mol. Sci. 2019, 20, 1336. [Google Scholar] [CrossRef] [Green Version]
- Higgins, M.; Obaidi, I.; McMorrow, T. Primary cilia and their role in cancer. Oncol. Lett. 2019, 17, 3041–3047. [Google Scholar] [CrossRef]
- Peixoto, E.; Richard, S.; Pant, K.; Biswas, A.; Gradilone, S.A. The primary cilium: Its role as a tumor suppressor organelle. Biochem. Pharm. 2020, 175, 113906. [Google Scholar] [CrossRef] [PubMed]
- Urdiciain, A.; Erausquin, E.; Meléndez, B.; Rey, J.A.; Idoate, M.A.; Castresana, J.S. Tubastatin A, an inhibitor of HDAC6, enhances temozolomide-induced apoptosis and reverses the malignant phenotype of glioblastoma cells. Int. J. Oncol. 2019, 54, 1797–1808. [Google Scholar] [CrossRef]
- Chen, Q.; Li, J.; Yang, X.; Ma, J.; Gong, F.; Liu, Y. Prdx1 promotes the loss of primary cilia in esophageal squamous cell carcinoma. BMC Cancer 2020, 20, 372. [Google Scholar] [CrossRef]
- Rocha, C.; Papon, L.; Cacheux, W.; Marques Sousa, P.; Lascano, V.; Tort, O.; Giordano, T.; Vacher, S.; Lemmers, B.; Mariani, P.; et al. Tubulin glycylases are required for primary cilia, control of cell proliferation and tumor development in colon. EMBO J. 2014, 33, 2247–2260. [Google Scholar] [CrossRef] [Green Version]
- Gradilone, S.A.; Radtke, B.N.; Bogert, P.S.; Huang, B.Q.; Gajdos, G.B.; LaRusso, N.F. HDAC6 inhibition restores ciliary expression and decreases tumor growth. Cancer Res. 2013, 73, 2259–2270. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mansini, A.P.; Peixoto, E.; Thelen, K.M.; Gaspari, C.; Jin, S.; Gradilone, S.A. The cholangiocyte primary cilium in health and disease. Biochim. Biophys. Acta 2018, 1864, 1245–1253. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, T.; Nakazono, K.; Tokuda, M.; Mashima, Y.; Dynlacht, B.D.; Itoh, H. HDAC2 promotes loss of primary cilia in pancreatic ductal adenocarcinoma. EMBO Rep. 2017, 18, 334–343. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Esteban, M.A.; Harten, S.K.; Tran, M.G.; Maxwell, P.H. Formation of primary cilia in the renal epithelium is regulated by the von Hippel-Lindau tumor suppressor protein. J. Am. Soc. Nephrol. 2006, 17, 1801–1806. [Google Scholar] [CrossRef]
- Qie, Y.; Wang, L.; Du, E.; Chen, S.; Lu, C.; Ding, N.; Yang, K.; Xu, Y. TACC3 promotes prostate cancer cell proliferation and restrains primary cilium formation. Exp. Cell Res. 2020, 390, 111952. [Google Scholar] [CrossRef]
- Bhattacharya, R.; Kwon, J.; Ali, B.; Wang, E.; Patra, S.; Shridhar, V.; Mukherjee, P. Role of hedgehog signaling in ovarian cancer. Clin. Cancer Res. 2008, 14, 7659–7666. [Google Scholar] [CrossRef] [Green Version]
- Egeberg, D.L.; Lethan, M.; Manguso, R.; Schneider, L.; Awan, A.; Jorgensen, T.S.; Byskov, A.G.; Pedersen, L.B.; Christensen, S.T. Primary cilia and aberrant cell signaling in epithelial ovarian cancer. Cilia 2012, 1, 15. [Google Scholar] [CrossRef] [Green Version]
- Zingg, D.; Debbache, J.; Peña-Hernández, R.; Antunes, A.T.; Schaefer, S.M.; Cheng, P.F.; Zimmerli, D.; Haeusel, J.; Calçada, R.R.; Tuncer, E.; et al. EZH2-Mediated Primary Cilium Deconstruction Drives Metastatic Melanoma Formation. Cancer Cell 2018, 34, 69–84. [Google Scholar] [CrossRef]
- Xiang, W.; Guo, F.; Cheng, W.; Zhang, J.; Huang, J.; Wang, R.; Ma, Z.; Xu, K. HDAC6 inhibition suppresses chondrosarcoma by restoring the expression of primary cilia. Oncol. Rep. 2017, 38, 229–236. [Google Scholar] [CrossRef] [Green Version]
- Wong, S.Y.; Seol, A.D.; So, P.L.; Ermilov, A.N.; Bichakjian, C.K.; Epstein, E.H., Jr.; Dlugosz, A.A.; Reiter, J.F. Primary cilia can both mediate and suppress Hedgehog pathway-dependent tumorigenesis. Nat. Med. 2009, 15, 1055–1061. [Google Scholar] [CrossRef] [Green Version]
- Han, Y.G.; Kim, H.J.; Dlugosz, A.A.; Ellison, D.W.; Gilbertson, R.J.; Alvarez-Buylla, A. Dual and opposing roles of primary cilia in medulloblastoma development. Nat. Med. 2009, 15, 1062–1065. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jeong, M.; Lee, E.W.; Seong, D.; Seo, J.; Kim, J.H.; Grootjans, S.; Kim, S.Y.; Vandenabeele, P.; Song, J. USP8 suppresses death receptor-mediated apoptosis by enhancing FLIP(L) stability. Oncogene 2017, 36, 458–470. [Google Scholar] [CrossRef] [PubMed]
- MacLeod, G.; Bozek, D.A.; Rajakulendran, N.; Monteiro, V.; Ahmadi, M.; Steinhart, Z.; Kushida, M.M.; Yu, H.; Coutinho, F.J.; Cavalli, F.M.G.; et al. Genome-Wide CRISPR-Cas9 Screens Expose Genetic Vulnerabilities and Mechanisms of Temozolomide Sensitivity in Glioblastoma Stem Cells. Cell Rep. 2019, 27, 971–986. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Das, A.; Qian, J.; Tsang, W.Y. USP9X counteracts differential ubiquitination of NPHP5 by MARCH7 and BBS11 to regulate ciliogenesis. PLoS Genet. 2017, 13, e1006791. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, J.; Meng, Y.; Zeng, J.; Zeng, B.; Jiang, X. Ubiquitin E3 Ligase MARCH7 promotes proliferation and invasion of cervical cancer cells through VAV2-RAC1-CDC42 pathway. Oncol. Lett. 2018, 16, 2312–2318. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kano, S.; Miyajima, N.; Fukuda, S.; Hatakeyama, S. Tripartite motif protein 32 facilitates cell growth and migration via degradation of Abl-interactor 2. Cancer Res. 2008, 68, 5572–5580. [Google Scholar] [CrossRef] [Green Version]
- Horn, E.J.; Albor, A.; Liu, Y.; El-Hizawi, S.; Vanderbeek, G.E.; Babcock, M.; Bowden, G.T.; Hennings, H.; Lozano, G.; Weinberg, W.C.; et al. RING protein Trim32 associated with skin carcinogenesis has anti-apoptotic and E3-ubiquitin ligase properties. Carcinogenesis 2004, 25, 157–167. [Google Scholar] [CrossRef]
- Reijnders, M.R.; Zachariadis, V.; Latour, B.; Jolly, L.; Mancini, G.M.; Pfundt, R.; Wu, K.M.; van Ravenswaaij-Arts, C.M.; Veenstra-Knol, H.E.; Anderlid, B.M.; et al. De Novo Loss-of-Function Mutations in USP9X Cause a Female-Specific Recognizable Syndrome with Developmental Delay and Congenital Malformations. Am. J. Hum. Genet. 2016, 98, 373–381. [Google Scholar] [CrossRef] [Green Version]
- Pérez-Mancera, P.A.; Rust, A.G.; van der Weyden, L.; Kristiansen, G.; Li, A.; Sarver, A.L.; Silverstein, K.A.T.; Grützmann, R.; Aust, D.; Rümmele, P.; et al. The deubiquitinase USP9X suppresses pancreatic ductal adenocarcinoma. Nature 2012, 486, 266–270. [Google Scholar] [CrossRef]
- Yang, Y.; Ran, J.; Liu, M.; Li, D.; Li, Y.; Shi, X.; Meng, D.; Pan, J.; Ou, G.; Aneja, R.; et al. CYLD mediates ciliogenesis in multiple organs by deubiquitinating Cep70 and inactivating HDAC6. Cell Res. 2014, 24, 1342–1353. [Google Scholar] [CrossRef] [Green Version]
- Douanne, T.; André-Grégoire, G.; Thys, A.; Trillet, K.; Gavard, J.; Bidère, N. CYLD Regulates Centriolar Satellites Proteostasis by Counteracting the E3 Ligase MIB1. Cell Rep. 2019, 27, 1657–1665. [Google Scholar] [CrossRef] [Green Version]
- Tse, W.K.F. Importance of deubiquitinases in zebrafish craniofacial development. Biochem. Biophys. Res. Commun. 2017, 487, 813–819. [Google Scholar] [CrossRef] [PubMed]
- Bignell, G.R.; Warren, W.; Seal, S.; Takahashi, M.; Rapley, E.; Barfoot, R.; Green, H.; Brown, C.; Biggs, P.J.; Lakhani, S.R.; et al. Identification of the familial cylindromatosis tumour-suppressor gene. Nat. Genet. 2000, 25, 160–165. [Google Scholar] [CrossRef]
- Wang, L.; Lee, K.; Malonis, R.; Sanchez, I.; Dynlacht, B.D. Tethering of an E3 ligase by PCM1 regulates the abundance of centrosomal KIAA0586/Talpid3 and promotes ciliogenesis. Elife 2016, 5, e12950. [Google Scholar] [CrossRef]
- Lei, Y.; Li, Z.; Qi, L.; Tong, S.; Li, B.; He, W.; Chen, M. The Prognostic Role of Ki-67/MIB-1 in Upper Urinary-Tract Urothelial Carcinomas: A Systematic Review and Meta-Analysis. J. Endourol. 2015, 29, 1302–1308. [Google Scholar] [CrossRef]
- Ang, S.O.; Chen, H.; Hirota, K.; Gordeuk, V.R.; Jelinek, J.; Guan, Y.; Liu, E.; Sergueeva, A.I.; Miasnikova, G.Y.; Mole, D.; et al. Disruption of oxygen homeostasis underlies congenital Chuvash polycythemia. Nat. Genet. 2002, 32, 614–621. [Google Scholar] [CrossRef] [PubMed]
- Schermer, B.; Ghenoiu, C.; Bartram, M.; Müller, R.U.; Kotsis, F.; Höhne, M.; Kühn, W.; Rapka, M.; Nitschke, R.; Zentgraf, H.; et al. The von Hippel-Lindau tumor suppressor protein controls ciliogenesis by orienting microtubule growth. J. Cell Biol. 2006, 175, 547–554. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaelin, W.G., Jr. The VHL Tumor Suppressor Gene: Insights into Oxygen Sensing and Cancer. Trans. Am. Clin. Clim. Assoc. 2017, 128, 298–307. [Google Scholar]
- Nishizawa, M.; Izawa, I.; Inoko, A.; Hayashi, Y.; Nagata, K.; Yokoyama, T.; Usukura, J.; Inagaki, M. Identification of trichoplein, a novel keratin filament-binding protein. J. Cell Sci. 2005, 118, 1081–1090. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ibi, M.; Zou, P.; Inoko, A.; Shiromizu, T.; Matsuyama, M.; Hayashi, Y.; Enomoto, M.; Mori, D.; Hirotsune, S.; Kiyono, T.; et al. Trichoplein controls microtubule anchoring at the centrosome by binding to Odf2 and ninein. J. Cell Sci. 2011, 124, 857–864. [Google Scholar] [CrossRef] [Green Version]
- Sasaki, S.; Shionoya, A.; Ishida, M.; Gambello, M.J.; Yingling, J.; Wynshaw-Boris, A.; Hirotsune, S. A LIS1/NUDEL/cytoplasmic dynein heavy chain complex in the developing and adult nervous system. Neuron 2000, 28, 681–696. [Google Scholar] [CrossRef] [Green Version]
- Niethammer, M.; Smith, D.S.; Ayala, R.; Peng, J.; Ko, J.; Lee, M.S.; Morabito, M.; Tsai, L.H. NUDEL is a novel Cdk5 substrate that associates with LIS1 and cytoplasmic dynein. Neuron 2000, 28, 697–711. [Google Scholar] [CrossRef] [Green Version]
- Barbelanne, M.; Song, J.; Ahmadzai, M.; Tsang, W.Y. Pathogenic NPHP5 mutations impair protein interaction with Cep290, a prerequisite for ciliogenesis. Hum. Mol. Genet. 2013, 22, 2482–2494. [Google Scholar] [CrossRef] [Green Version]
- Chiang, A.P.; Beck, J.S.; Yen, H.J.; Tayeh, M.K.; Scheetz, T.E.; Swiderski, R.E.; Nishimura, D.Y.; Braun, T.A.; Kim, K.Y.; Huang, J.; et al. Homozygosity mapping with SNP arrays identifies TRIM32, an E3 ubiquitin ligase, as a Bardet-Biedl syndrome gene (BBS11). Proc. Natl. Acad. Sci. USA 2006, 103, 6287–6292. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Otto, E.A.; Loeys, B.; Khanna, H.; Hellemans, J.; Sudbrak, R.; Fan, S.; Muerb, U.; O’Toole, J.F.; Helou, J.; Attanasio, M.; et al. Nephrocystin-5, a ciliary IQ domain protein, is mutated in Senior-Loken syndrome and interacts with RPGR and calmodulin. Nat. Genet. 2005, 37, 282–288. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Zhou, J. CYLD—a deubiquitylase that acts to fine-tune microtubule properties and functions. J. Cell Sci. 2016, 129, 2289–2295. [Google Scholar] [CrossRef] [Green Version]
- Farooqi, I.S. Genetic and hereditary aspects of childhood obesity. Best Pract. Res. Clin. Endocrinol. Metab. 2005, 19, 359–374. [Google Scholar] [CrossRef]
- Stephen, L.A.; Tawamie, H.; Davis, G.M.; Tebbe, L.; Nürnberg, P.; Nürnberg, G.; Thiele, H.; Thoenes, M.; Boltshauser, E.; Uebe, S.; et al. TALPID3 controls centrosome and cell polarity and the human ortholog KIAA0586 is mutated in Joubert syndrome (JBTS23). Elife 2015, 4, e08077. [Google Scholar] [CrossRef]
- Kamura, T.; Koepp, D.M.; Conrad, M.N.; Skowyra, D.; Moreland, R.J.; Iliopoulos, O.; Lane, W.S.; Kaelin, W.G., Jr.; Elledge, S.J.; Conaway, R.C.; et al. Rbx1, a component of the VHL tumor suppressor complex and SCF ubiquitin ligase. Science 1999, 284, 657–661. [Google Scholar] [CrossRef]
- Kuehn, E.W.; Walz, G.; Benzing, T. Von hippel-lindau: A tumor suppressor links microtubules to ciliogenesis and cancer development. Cancer Res. 2007, 67, 4537–4540. [Google Scholar] [CrossRef] [Green Version]
- Ohh, M.; Park, C.W.; Ivan, M.; Hoffman, M.A.; Kim, T.-Y.; Huang, L.E.; Pavletich, N.; Chau, V.; Kaelin, W.G. Ubiquitination of hypoxia-inducible factor requires direct binding to the β-domain of the von Hippel–Lindau protein. Nat. Cell Biol. 2000, 2, 423–427. [Google Scholar] [CrossRef]
- Hayashi, Y.; Yokota, A.; Harada, H.; Huang, G. Hypoxia/pseudohypoxia-mediated activation of hypoxia-inducible factor-1α in cancer. Cancer Sci. 2019, 110, 1510–1517. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, K.; Diener, D.R.; Rosenbaum, J.L. The ubiquitin conjugation system is involved in the disassembly of cilia and flagella. J. Cell Biol. 2009, 186, 601–613. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- D’Angiolella, V.; Donato, V.; Vijayakumar, S.; Saraf, A.; Florens, L.; Washburn, M.P.; Dynlacht, B.; Pagano, M. SCF(Cyclin F) controls centrosome homeostasis and mitotic fidelity through CP110 degradation. Nature 2010, 466, 138–142. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mick, D.U.; Rodrigues, R.B.; Leib, R.D.; Adams, C.M.; Chien, A.S.; Gygi, S.P.; Nachury, M.V. Proteomics of Primary Cilia by Proximity Labeling. Dev. Cell 2015, 35, 497–512. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shearer, R.F.; Frikstad, K.M.; McKenna, J.; McCloy, R.A.; Deng, N.; Burgess, A.; Stokke, T.; Patzke, S.; Saunders, D.N. The E3 ubiquitin ligase UBR5 regulates centriolar satellite stability and primary cilia. Mol. Biol. Cell 2018, 29, 1542–1554. [Google Scholar] [CrossRef] [PubMed]
- Massa, F.; Tammaro, R.; Prado, M.A.; Cesana, M.; Lee, B.H.; Finley, D.; Franco, B.; Morleo, M. The deubiquitinating enzyme Usp14 controls ciliogenesis and Hedgehog signaling. Hum. Mol. Genet. 2019, 28, 764–777. [Google Scholar] [CrossRef]
- Li, J.; D’Angiolella, V.; Seeley, E.S.; Kim, S.; Kobayashi, T.; Fu, W.; Campos, E.I.; Pagano, M.; Dynlacht, B.D. USP33 regulates centrosome biogenesis via deubiquitination of the centriolar protein CP110. Nature 2013, 495, 255–259. [Google Scholar] [CrossRef] [Green Version]
- Wheway, G.; Schmidts, M.; Mans, D.A.; Szymanska, K.; Nguyen, T.T.; Racher, H.; Phelps, I.G.; Toedt, G.; Kennedy, J.; Wunderlich, K.A.; et al. An siRNA-based functional genomics screen for the identification of regulators of ciliogenesis and ciliopathy genes. Nat. Cell Biol. 2015, 17, 1074–1087. [Google Scholar] [CrossRef]
- Kim, J.H.; Ki, S.M.; Joung, J.G.; Scott, E.; Heynen-Genel, S.; Aza-Blanc, P.; Kwon, C.H.; Kim, J.; Gleeson, J.G.; Lee, J.E. Genome-wide screen identifies novel machineries required for both ciliogenesis and cell cycle arrest upon serum starvation. Biochim. Biophys. Acta 2016, 1863, 1307–1318. [Google Scholar] [CrossRef] [Green Version]
- Kohli, P.; Höhne, M.; Jüngst, C.; Bertsch, S.; Ebert, L.K.; Schauss, A.C.; Benzing, T.; Rinschen, M.M.; Schermer, B. The ciliary membrane-associated proteome reveals actin-binding proteins as key components of cilia. EMBO Rep. 2017, 18, 1521–1535. [Google Scholar] [CrossRef] [PubMed]
- Lundberg, E.; Borner, G.H.H. Spatial proteomics: A powerful discovery tool for cell biology. Nat. Rev. Mol. Cell Biol. 2019, 20, 285–302. [Google Scholar] [CrossRef] [PubMed]
- Kliza, K.; Husnjak, K. Resolving the Complexity of Ubiquitin Networks. Front. Mol. Biosci. 2020, 7, 21. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Poulsen, J.W.; Madsen, C.T.; Young, C.; Kelstrup, C.D.; Grell, H.C.; Henriksen, P.; Juhl-Jensen, L.; Nielsen, M.L. Comprehensive profiling of proteome changes upon sequential deletion of deubiquitylating enzymes. J. Proteom. 2012, 75, 3886–3897. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Lu, D.; Gao, J.; Zhu, H.; Zhou, Y.; Gao, D.; Zhou, H. Identification of a USP9X Substrate NFX1-123 by SILAC-Based Quantitative Proteomics. J. Proteome Res. 2019, 18, 2654–2665. [Google Scholar] [CrossRef] [PubMed]
- Gomez-Ferreria, M.A.; Bashkurov, M.; Mullin, M.; Gingras, A.C.; Pelletier, L. CEP192 interacts physically and functionally with the K63-deubiquitinase CYLD to promote mitotic spindle assembly. Cell Cycle 2012, 11, 3555–3558. [Google Scholar] [CrossRef] [Green Version]
- Nakayasu, E.S.; Sydor, M.A.; Brown, R.N.; Sontag, R.L.; Sobreira, T.J.P.; Slysz, G.W.; Humphrys, D.R.; Skarina, T.; Onoprienko, O.; Di Leo, R.; et al. Identification of Salmonella Typhimurium Deubiquitinase SseL Substrates by Immunoaffinity Enrichment and Quantitative Proteomic Analysis. J. Proteome Res. 2015, 14, 4029–4038. [Google Scholar] [CrossRef] [Green Version]
- Iconomou, M.; Saunders, D.N. Systematic approaches to identify E3 ligase substrates. Biochem. J. 2016, 473, 4083–4101. [Google Scholar] [CrossRef] [Green Version]
- O’Connor, H.F.; Huibregtse, J.M. Enzyme-substrate relationships in the ubiquitin system: Approaches for identifying substrates of ubiquitin ligases. Cell. Mol. Life Sci. 2017, 74, 3363–3375. [Google Scholar] [CrossRef]
- Mattern, M.; Sutherland, J.; Kadimisetty, K.; Barrio, R.; Rodriguez, M.S. Using Ubiquitin Binders to Decipher the Ubiquitin Code. Trends Biochem. Sci. 2019, 44, 599–615. [Google Scholar] [CrossRef]
- Huang, Q.; Zhang, X. Emerging Roles and Research Tools of Atypical Ubiquitination. Proteomics 2020, 20, 1900100. [Google Scholar] [CrossRef] [PubMed]
- Dammermann, A.; Merdes, A. Assembly of centrosomal proteins and microtubule organization depends on PCM-1. J. Cell Biol. 2002, 159, 255–266. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shinde, S.R.; Nager, A.R.; Nachury, M.V. Lysine63-linked ubiquitin chains earmark GPCRs for BBSome-mediated removal from cilia. bioRxiv 2020. Available online: https://www.biorxiv.org/content/10.1101/2020.03.04.977090v1 (accessed on 17 August 2020).
- Bhagwat, S.R.; Hajela, K.; Kumar, A. Proteolysis to Identify Protease Substrates: Cleave to Decipher. Proteomics 2018, 18, e1800011. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.Q.; Baker, D.; Ovaa, H. Small molecules that target the ubiquitin system. Biochem Soc. Trans. 2020, 48, 479–497. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ran, X.; Gestwicki, J.E. Inhibitors of protein–protein interactions (PPIs): An analysis of scaffold choices and buried surface area. Curr. Opin. Chem. Biol. 2018, 44, 75–86. [Google Scholar] [CrossRef]
- Andrei, S.A.; Sijbesma, E.; Hann, M.; Davis, J.; O’Mahony, G.; Perry, M.W.D.; Karawajczyk, A.; Eickhoff, J.; Brunsveld, L.; Doveston, R.G.; et al. Stabilization of protein-protein interactions in drug discovery. Expert Opin. Drug Discov. 2017, 12, 925–940. [Google Scholar] [CrossRef] [Green Version]
- Huang, X.; Dixit, V.M. Drugging the undruggables: Exploring the ubiquitin system for drug development. Cell Res. 2016, 26, 484–498. [Google Scholar] [CrossRef]
- Wu, L.; Grigoryan, A.V.; Li, Y.; Hao, B.; Pagano, M.; Cardozo, T.J. Specific small molecule inhibitors of Skp2-mediated p27 degradation. Chem. Biol. 2012, 19, 1515–1524. [Google Scholar] [CrossRef] [Green Version]
- Ganoth, D.; Bornstein, G.; Ko, T.K.; Larsen, B.; Tyers, M.; Pagano, M.; Hershko, A. The cell-cycle regulatory protein Cks1 is required for SCF(Skp2)-mediated ubiquitinylation of p27. Nat. Cell Biol. 2001, 3, 321–324. [Google Scholar] [CrossRef]
- Spruck, C.; Strohmaier, H.; Watson, M.; Smith, A.P.; Ryan, A.; Krek, T.W.; Reed, S.I. A CDK-independent function of mammalian Cks1: Targeting of SCF(Skp2) to the CDK inhibitor p27Kip1. Mol. Cell 2001, 7, 639–650. [Google Scholar] [CrossRef]
- Das, T.; Shin, S.C.; Song, E.J.; Kim, E.E. Regulation of Deubiquitinating Enzymes by Post-Translational Modifications. Int. J. Mol. Sci. 2020, 21, 4028. [Google Scholar] [CrossRef] [PubMed]
- Das, T.; Kim, E.E.; Song, E.J. Phosphorylation of USP15 and USP4 Regulates Localization and Spliceosomal Deubiquitination. J. Mol. Biol 2019, 431, 3900–3912. [Google Scholar] [CrossRef]
- Yamanaka, S.; Sato, Y.; Oikawa, D.; Goto, E.; Fukai, S.; Tokunaga, F.; Takahashi, H.; Sawasaki, T. Subquinocin, a small molecule inhibitor of CYLD and USP-family deubiquitinating enzymes, promotes NF-κB signaling. Biochem. Biophys. Res. Commun. 2020, 524, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Dufner, A.; Knobeloch, K.P. Ubiquitin-specific protease 8 (USP8/UBPy): A prototypic multidomain deubiquitinating enzyme with pleiotropic functions. Biochem. Soc. Trans. 2019, 47, 1867–1879. [Google Scholar] [CrossRef]
- Murtaza, M.; Jolly, L.A.; Gecz, J.; Wood, S.A. La FAM fatale: USP9X in development and disease. Cell. Mol. Life Sci. 2015, 72, 2075–2089. [Google Scholar] [CrossRef] [PubMed] [Green Version]


{kind=link}
| Ciliopathy Phenotype | Role of Cilia in the Phenotype | References |
|---|---|---|
| Intellectual disability | Dysfunction of cilia in radial glial progenitors impairs the proliferation, migration, and differentiation, resulting in the disruption of cerebral cortical development and intellectual disability. | [39,40] |
| Retinal degeneration | Mutation of genes related to the structure and function of axoneme in photoreceptor cells impair protein (e.g., rhodopsin) transport along the axoneme, resulting in retinal degeneration | [41,42] |
| Craniofacial malformation | Dysfunction of cilia in cranial neural crest cells impairs the epithelial-mesenchymal transition and the formation of facial prominences, causing craniofacial malformation such as cleft lip/palate | [43,44] |
| Laterality disorders | Dysfunction of cilia in ventral node fails to break left-right symmetry, left or right-side morphogenesis, causing laterality disorders, such as situs inversus and heterotaxy. | [45,46] |
| Cystic kidney disease | Dysfunction of cilia in renal tubular cells fails to detect fluid flow, increase Ca2+ concentration, and suppress protein kinase A, causing renal cystogenesis through dysregulated proliferation, apoptosis, and cell polarity. | [47,48] |
| Obesity | Dysfunction of cilia in hypothalamic neurons and adipocyte progenitor cess fails to suppress appetite and regulate appropriate differentiation to adipocytes, respectively, causing obesity. | [49,50] |
| Scoliosis | Primary cilia of osteoblasts are abnormally elongated and dysfunctional in mechanotransduction, which may impair loading-induced bone adaptation and cause scoliosis | [51,52] |
| Respiratory distress | Mutations of genes affecting dynein arm, radial spoke, central apparatus or multiciliation impair the structure and/or function of motile cilia of epithelial cells lining most of the respiratory tract, resulting in mucus obstruction and respiratory failure. | [53,54] |
| Infertility | Impairment of sperm tail, which has microtubule arrangement similar to that of motile cilia, cause sperm immotility and male infertility. Dysfunction of motile and primary cilia at the reproductive tract also causes both male and female infertility. | [55,56] |
| Cancer Cell | The Role of PC in the Cancer | References |
|---|---|---|
| Glioblastoma | Inhibition of HDAC6 restores the loss of PC and suppressed the proliferation | [110] |
| Esophageal squamous cell carcinoma | KD of PRDX1 restores the loss of PC and suppressed the proliferation | [111] |
| Colon cancer | Knockout of TTLL3 causes the loss of PC and promotes tumorigenesis in colon | [112] |
| Cholangiocarcinoma | The number of PC is frequently reduced. Inhibition of HDAC6 restores the loss of PC and suppressed the proliferation | [113,114] |
| Pancreatic ductal adenocarcinoma | Inhibition of HDAC2 in Panc1 induces ciliogenesis and suppressed the proliferation | [115] |
| Clear cell renal carcinoma | PC is lost by inactivation of VHL tumor suppressor | [116] |
| Prostate cancer | KD of TACC3 restores the loss of PC and suppressed the proliferation | [117] |
| Epithelial ovarian cancer | The number of PC is reduced, which is associated with centrosomal localization of AURKA. KD of AURKA restores the loss of PC and suppressed the oncogenic hedgehog signaling | [118,119] |
| Melanoma | Deconstruction of PC is sufficient to drive metastatic formation | [120] |
| Chondrosarcoma | Inhibition of HDAC6 restores the loss of PC and suppressed the proliferation | [121] |
| Medulloblastoma, basal cell carcinoma | - | - |
| with GOF mutation of SMO | PC increase transcriptional activator and stimulate proliferation | [122,123] |
| with GOF mutation of GLI2 | PC increase transcriptional suppressor and inhibit proliferation | [122,123] |
| E3 Ligase or DUB | The Role in Ciliogenesis (Substrate) | The Role in Ciliopathy and/or Cancer | References |
|---|---|---|---|
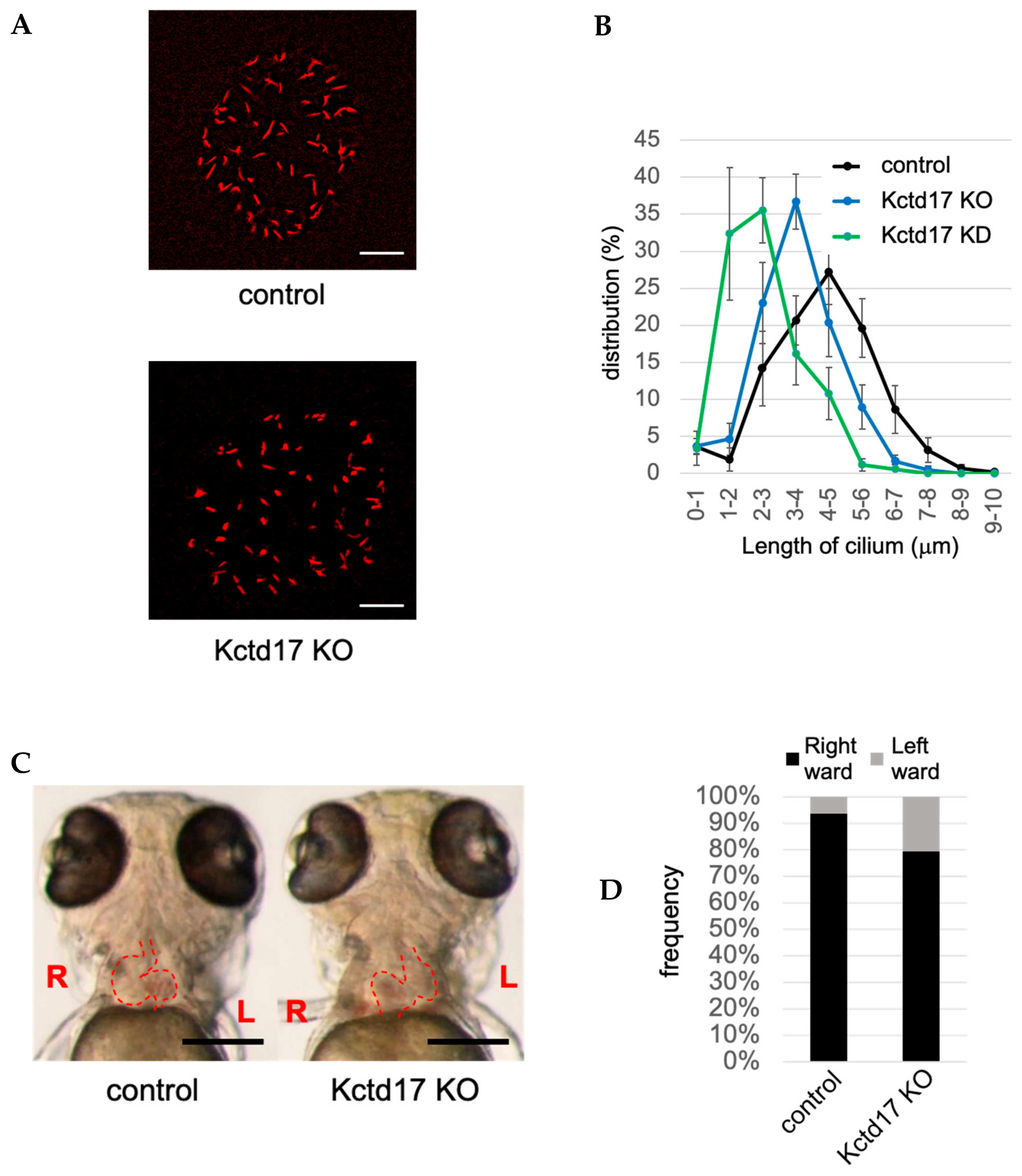
| CRL3-KCTD17 | inhibited by KD of KCTD17 (TCHP) | KO of KCTD17 causes situs inversus in zebrafish | [33], this paper (Figure 1) |
| USP8 | stimulated by KD of USP8 (TCHP) | KO of USP8 causes cystic kidney in zebrafish | [34] |
| USP8 is highly expressed and oncogenic in melanoma | [124] | ||
| Inhibition of USP8 suppresses the proliferation of glioblastoma stem cells | [125] | ||
| MARCHF7 | inhibited by OE of MARCHF7 (IQCB1) | MARCHF7 promotes proliferation and invasion of cervical cancer cells | [126,127] |
| TRIM32 | inhibited by OE of TRIM32 (IQCB1) | TRIM32 is a causative gene of BBS (BBS11) | [126,127] |
| TRIM32 is oncogenic in head and neck squamous cell carcinoma and skin cancer | [128,129] | ||
| USP9X | inhibited by KD of USP9X (IQCB1) | LOF mutations in USP9X cause phenotypes related to ciliopathy | [126,130] |
| USP9X is a major tumor suppressor gene in pancreatic ductal adenocarcinoma | [131] | ||
| CYLD | inhibited by KD/KO of CYLD (CEP70, MIB1) | KO/KD of CYLD causes ciliopathy-related phenotype in mouse and zebrafish | [132,133,134] |
| LOF mutation in CYLD cause skin cancer (familial cylindromatosis) | [135] | ||
| MIB1 | inhibited by OE of MIB1 (PCM1, KIAA0586) | MIB1 is oncogenic in upper urinary-tract urothelial carcinomas | [133,136,137] |
| CRL2-VHL | stimulated by OE of VHL (HIF1A) | KO of VHL causes cystic kidney | [138,139] |
| VHL is tumor-suppressive in renal cancers | [140] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shiromizu, T.; Yuge, M.; Kasahara, K.; Yamakawa, D.; Matsui, T.; Bessho, Y.; Inagaki, M.; Nishimura, Y. Targeting E3 Ubiquitin Ligases and Deubiquitinases in Ciliopathy and Cancer. Int. J. Mol. Sci. 2020, 21, 5962. https://doi.org/10.3390/ijms21175962
Shiromizu T, Yuge M, Kasahara K, Yamakawa D, Matsui T, Bessho Y, Inagaki M, Nishimura Y. Targeting E3 Ubiquitin Ligases and Deubiquitinases in Ciliopathy and Cancer. International Journal of Molecular Sciences. 2020; 21(17):5962. https://doi.org/10.3390/ijms21175962
Chicago/Turabian StyleShiromizu, Takashi, Mizuki Yuge, Kousuke Kasahara, Daishi Yamakawa, Takaaki Matsui, Yasumasa Bessho, Masaki Inagaki, and Yuhei Nishimura. 2020. "Targeting E3 Ubiquitin Ligases and Deubiquitinases in Ciliopathy and Cancer" International Journal of Molecular Sciences 21, no. 17: 5962. https://doi.org/10.3390/ijms21175962