Amyloidogenic Propensities of Ribosomal S1 Proteins: Bioinformatics Screening and Experimental Checking

 , ,
, ,
Abstract
:1. Introduction
2. Results
2.1. Analysis of Amyloidogenic Propensities of Ribosomal S1 Proteins
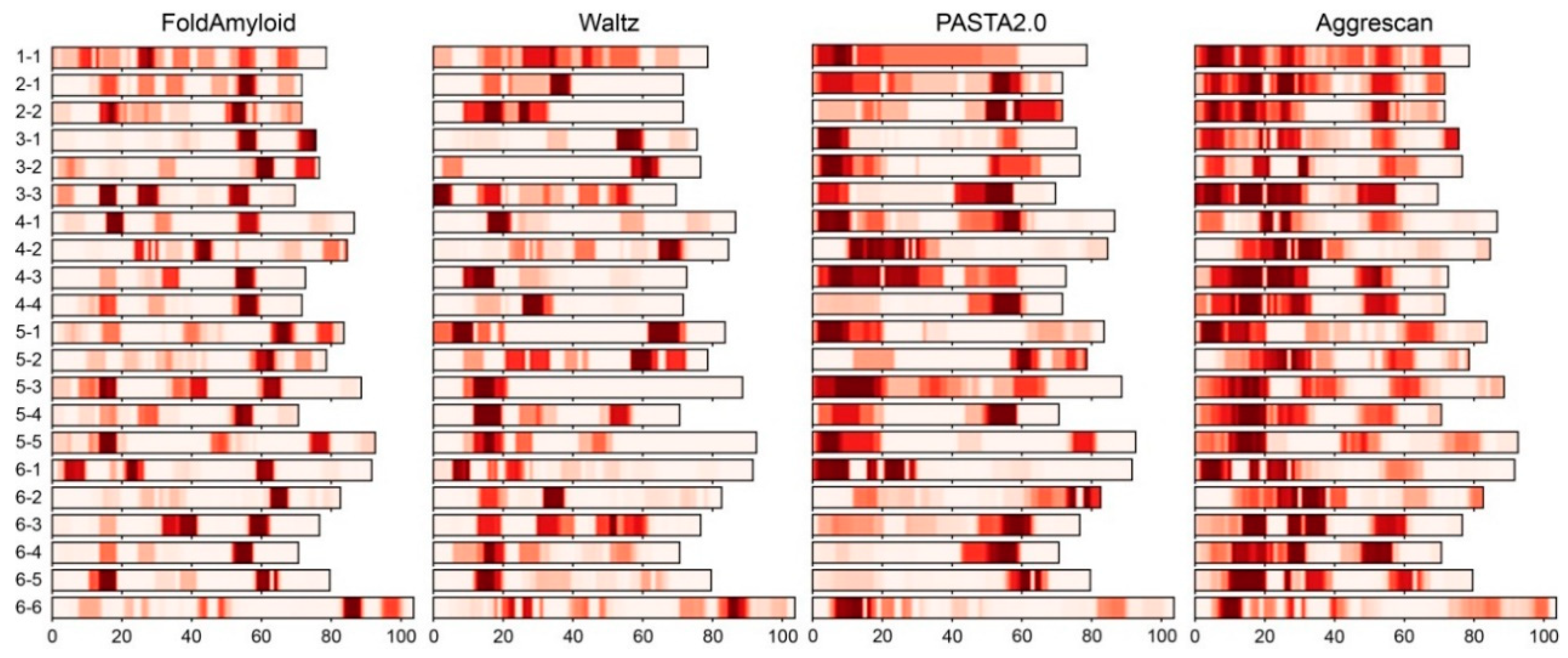
2.2. Analysis of Distribution of Amyloidogenic Regions in Ribosomal S1 Proteins
2.3. Correlation between Amyloidogenic Regions and Secondary Structures in S1 Proteins
2.4. Sequence Logos and Uniqueness of Amyloidogenic Regions in S1 Domain
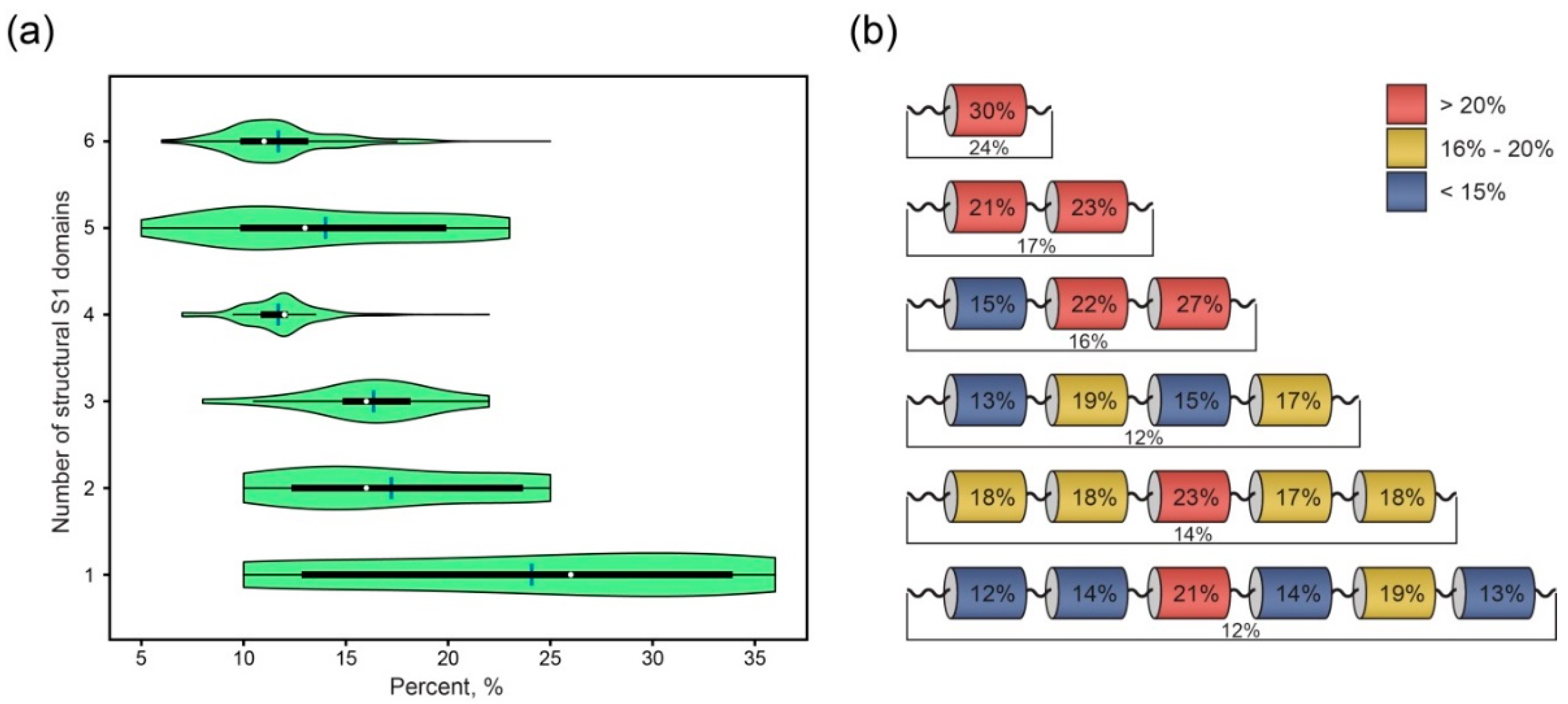
2.5. Amyloidogenic Propensities of Linkers between Structural S1 Domains
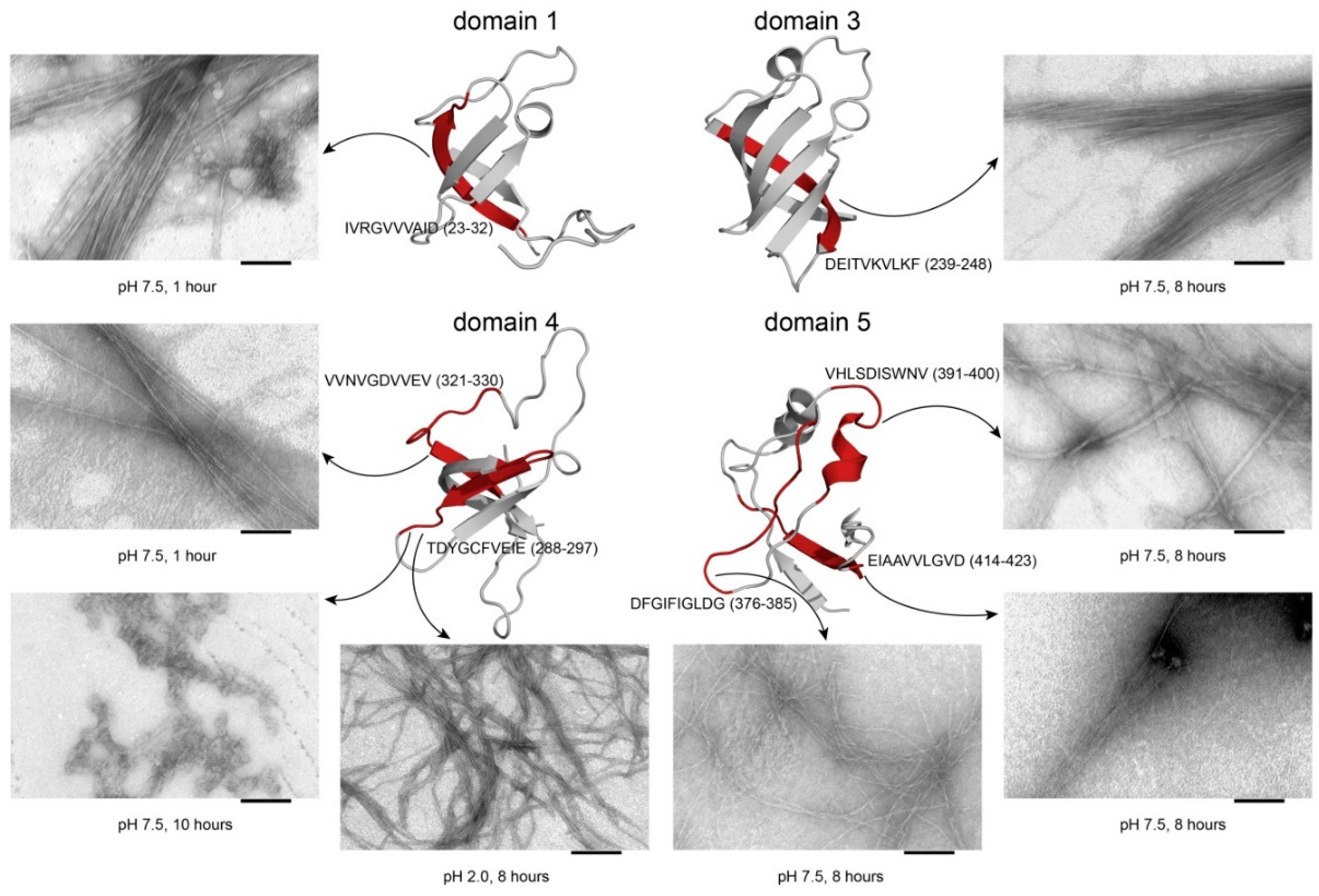
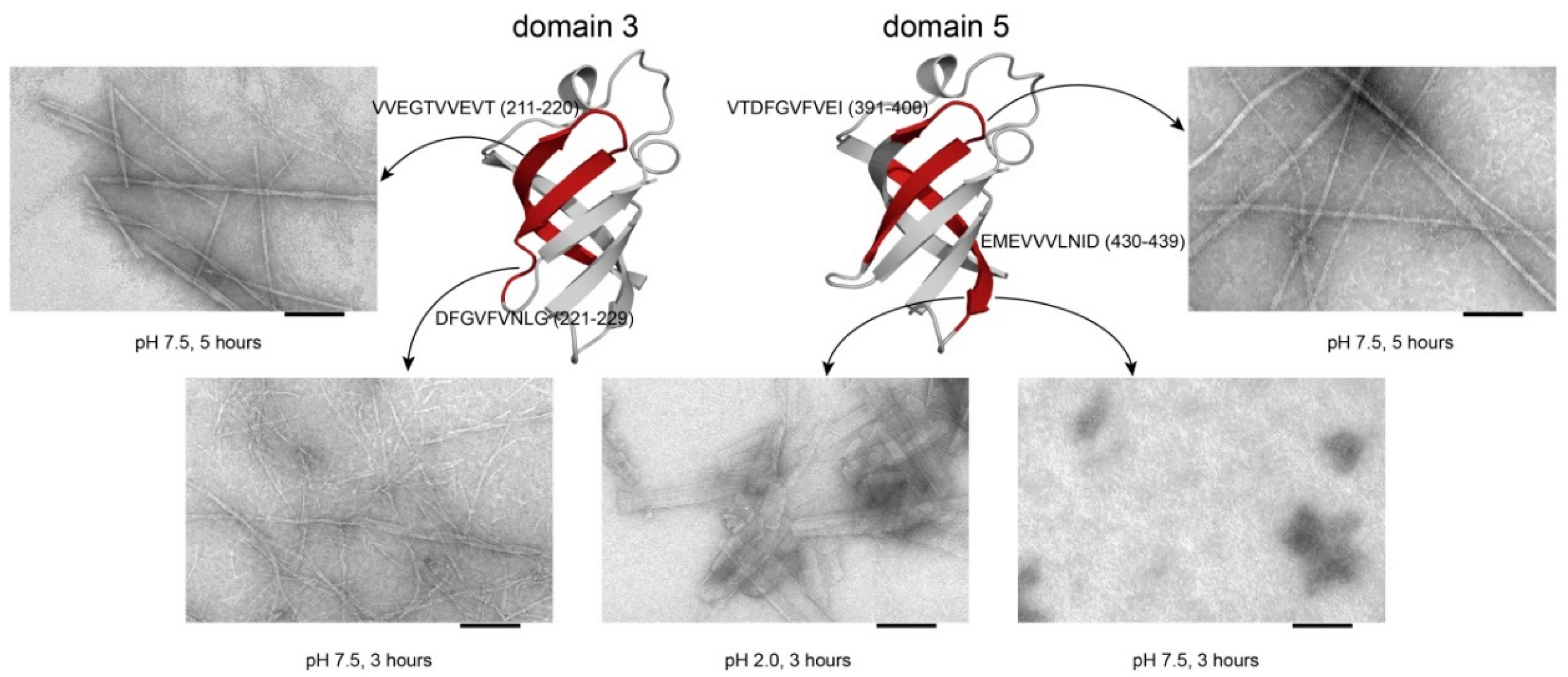
2.6. Analysis of Fluorescence of Thioflavin T and Data from Electron Microscopy
3. Discussion
4. Materials and Methods
4.1. Construction of Ribosomal S1 Protein Dataset
4.2. Prediction and Analysis of Amyloidogenic Regions
4.3. Alignment and Prediction of Secondary and Tertiary Structures
4.4. Realization
4.5. Chemical Synthesis of Peptides
4.6. ThT Fluorescence Assay
4.7. Electron Microscopy
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| OB-fold | oligosaccharide/oligonucleotide-binding fold |
| ThT | thioflavin T |
| DNA | deoxyribonucleic acid |
| RNA | ribonucleic acid |
| YB-1 | Y box-binding protein 1 |
| CSD | cold shock domain |
| a.a. | amino acid |
| β-barrel | beta barrel |
| α-helix | alpha helix |
| β-strand | beta strand |
| TEM | transmission electron microscopy |
| PADs | potential amyloidogenic determinants |
References
- Wower, I.K.; Zwieb, C.W.; Guven, S.A.; Wower, J. Binding and cross-linking of tmRNA to ribosomal protein S1, on and off the Escherichia coli ribosome. EMBO J. 2000, 19, 6612–6621. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Machulin, A.V.; Deryusheva, E.I.; Selivanova, O.M.; Galzitskaya, O.V. The number of domains in the ribosomal protein S1 as a hallmark of the phylogenetic grouping of bacteria. PLoS ONE 2019, 14, e0221370. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bycroft, M.; Hubbard, T.J.; Proctor, M.; Freund, S.M.; Murzin, A.G. The solution structure of the S1 RNA binding domain: A member of an ancient nucleic acid-binding fold. Cell 1997, 88, 235–242. [Google Scholar] [CrossRef] [Green Version]
- Amblar, M.; Barbas, A.; Gomez-Puertas, P.; Arraiano, C.M. The role of the S1 domain in exoribonucleolytic activity: Substrate specificity and multimerization. RNA 2007, 13, 317–327. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schubert, M.; Edge, R.E.; Lario, P.; Cook, M.A.; Strynadka, N.C.J.; Mackie, G.A.; McIntosh, L.P. Structural characterization of the RNase E S1 domain and identification of its oligonucleotide-binding and dimerization interfaces. J. Mol. Biol. 2004, 341, 37–54. [Google Scholar] [CrossRef] [PubMed]
- Deryusheva, E.I.; Machulin, A.V.; Matyunin, M.A.; Galzitskaya, O.V. Investigation of the relationship between the S1 domain and its molecular functions derived from studies of the tertiary structure. Molecules 2019, 24, 3681. [Google Scholar] [CrossRef] [Green Version]
- Machulin, A.; Deryusheva, E.; Lobanov, M.; Galzitskaya, O. Repeats in S1 proteins: Flexibility and tendency for intrinsic disorder. Int. J. Mol. Sci. 2019, 20, 2377. [Google Scholar] [CrossRef] [Green Version]
- Deryusheva, E.I.; Machulin, A.V.; Selivanova, O.M.; Serdyuk, I.N. The S1 ribosomal protein family contains a unique conservative domain. Mol. Biol. 2010, 44, 642–647. [Google Scholar] [CrossRef]
- Sipe, J.D.; Cohen, A.S. Review: History of the amyloid fibril. J. Struct. Biol. 2000, 130, 88–98. [Google Scholar] [CrossRef] [Green Version]
- Selivanova, O.M.; Guryanov, S.G.; Enin, G.A.; Skabkin, M.A.; Ovchinnikov, L.P.; Serdyuk, I.N. YB-1 is capable of forming extended nanofibrils. Biochemistry 2010, 75, 115–120. [Google Scholar] [CrossRef]
- Guryanov, S.G.; Selivanova, O.M.; Nikulin, A.D.; Enin, G.A.; Melnik, B.S.; Kretov, D.A.; Serdyuk, I.N.; Ovchinnikov, L.P. Formation of amyloid-like fibrils by Y-box binding protein 1 (YB-1) is mediated by its cold shock domain and modulated by disordered terminal domains. PLoS ONE 2012, 7, e36969. [Google Scholar] [CrossRef] [Green Version]
- Galzitskaya, O.; Grishin, S.; Dzhus, U.; Selivanova, O.; Glyakina, A.; Deryusheva, E.; Machulin, A.; Suvorina, M.; Surin, A. Identification of amyloidogenic regions in S1 ribosomal proteins from Thermus thermophilus and Mycoplasma mobile. In Proceedings of the FEBS Open Bio, Krakow, Poland, 6–11 July 2019; p. 264. [Google Scholar]
- Grishin, S.Y.; Dzhus, U.F.; Selivanova, O.M.; Balobanov, V.A.; Surin, A.K.; Galzitskaya, O.V. Comparative Analysis of Aggregation of Thermus thermophilus Ribosomal Protein bS1 and Its Stable Fragment. Biochemistry 2020, 85, 344–354. [Google Scholar] [CrossRef] [PubMed]
- Martinez, K.A.; Kitko, R.D.; Mershon, J.P.; Adcox, H.E.; Malek, K.A.; Berkmen, M.B.; Slonczewski, J.L. Cytoplasmic pH response to acid stress in individual cells of Escherichia coli and Bacillus subtilis observed by fluorescence ratio imaging microscopy. Appl. Environ. Microbiol. 2012, 78, 3706–3714. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zilberstein, D.; Agmon, V.; Schuldiner, S.; Padan, E. Escherichia coli intracellular pH, membrane potential, and cell growth. J. Bacteriol. 1984, 158, 246–252. [Google Scholar] [CrossRef] [Green Version]
- Moriarty, G.M.; Olson, M.P.; Atieh, T.B.; Janowska, M.K.; Khare, S.D.; Baum, J. A pH-dependent switch promotes β-synuclein fibril formation via glutamate residues. J. Biol. Chem. 2017, 292, 16368–16379. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Esbjörner, E.K.; Chan, F.; Rees, E.; Erdelyi, M.; Luheshi, L.M.; Bertoncini, C.W.; Kaminski, C.F.; Dobson, C.M.; Kaminski Schierle, G.S. Direct observations of amyloid β Self-assembly in live cells provide insights into differences in the kinetics of Aβ(1-40) and Aβ(1-42) aggregation. Chem. Biol. 2014, 21, 732–742. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iannuzzi, C.; Borriello, M.; Portaccio, M.; Irace, G.; Sirangelo, I. Insights into Insulin Fibril Assembly at Physiological and Acidic pH and Related Amyloid Intrinsic Fluorescence. Int. J. Mol. Sci. 2017, 18, 2551. [Google Scholar] [CrossRef] [Green Version]
- Duval, M.; Korepanov, A.; Fuchsbauer, O.; Fechter, P.; Haller, A.; Fabbretti, A.; Choulier, L.; Micura, R.; Klaholz, B.P.; Romby, P.; et al. Escherichia coli ribosomal protein S1 unfolds structured mRNAs onto the ribosome for active translation initiation. PLoS Biol. 2013, 11, e1001731. [Google Scholar] [CrossRef] [Green Version]
- Miyawaki, S.; Uemura, Y.; Hongo, K.; Kawata, Y.; Mizobata, T. Acid-denatured small heat shock protein HdeA from Escherichia coli forms reversible fibrils with an atypical secondary structure. J. Biol. Chem. 2019, 294, 1590–1601. [Google Scholar] [CrossRef] [Green Version]
- Bezsonov, E.E.; Groenning, M.; Galzitskaya, O.V.; Gorkovskii, A.A.; Semisotnov, G.V.; Selyakh, I.O.; Ziganshin, R.H.; Rekstina, V.V.; Kudryashova, I.B.; Kuznetsov, S.A.; et al. Amyloidogenic peptides of yeast cell wall glucantransferase Bgl2p as a model for the investigation of its pH-dependent fibril formation. Prion 2013, 7, 175–184. [Google Scholar] [CrossRef] [Green Version]
- Garbuzynskiy, S.O.; Lobanov, M.Y.; Galzitskaya, O.V. FoldAmyloid: A method of prediction of amyloidogenic regions from protein sequence. Bioinformatics 2010, 26, 326–332. [Google Scholar] [CrossRef] [PubMed]
- Oliveberg, M. Waltz, an exciting new move in amyloid prediction. Nat. Methods 2010, 7, 187–188. [Google Scholar] [CrossRef] [PubMed]
- Walsh, I.; Seno, F.; Tosatto, S.C.E.; Trovato, A. PASTA 2.0: An improved server for protein aggregation prediction. Nucleic Acids Res. 2014, 42, W301–W307. [Google Scholar] [CrossRef]
- Conchillo-Solé, O.; de Groot, N.S.; Avilés, F.X.; Vendrell, J.; Daura, X.; Ventura, S. AGGRESCAN: A server for the prediction and evaluation of “hot spots” of aggregation in polypeptides. BMC Bioinform. 2007, 8, 65. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Drozdetskiy, A.; Cole, C.; Procter, J.; Barton, G.J. JPred4: A protein secondary structure prediction server. Nucleic Acids Res. 2015, 43, W389–W394. [Google Scholar] [CrossRef]
- Shiryev, S.A.; Papadopoulos, J.S.; Schaffer, A.A.; Agarwala, R. Improved BLAST searches using longer words for protein seeding. Bioinformatics 2007, 23, 2949–2951. [Google Scholar] [CrossRef] [Green Version]
- Timchenko, A.A.; Shiryaev, V.M.; Fedorova, Y.Y.; Kihara, H.; Kimura, K.; Willumeit, R.; Garamus, V.M.; Selivanova, O.M. Conformation of Thermus thermophilus ribosomal protein S1 in solution at different ionic strengths. Biophysics 2007, 52, 162–167. [Google Scholar] [CrossRef]
- Stefani, M.; Rigacci, S. Protein folding and aggregation into amyloid: The interference by natural phenolic compounds. Int. J. Mol. Sci. 2013, 14, 12411–12457. [Google Scholar] [CrossRef] [Green Version]
- Fedyukina, D.V.; Jennaro, T.S.; Cavagnero, S. Charge segregation and low hydrophobicity are key features of ribosomal proteins from different organisms. J. Biol. Chem. 2014, 289, 6740–6750. [Google Scholar] [CrossRef] [Green Version]
- Zhou, H.X.; Pang, X. Electrostatic interactions in protein structure, folding, binding, and condensation. Chem. Rev. 2018, 118, 1691–1741. [Google Scholar] [CrossRef]
- Uversky, V. Amyloidogenesis of natively unfolded proteins. Curr. Alzheimer Res. 2008, 5, 260–287. [Google Scholar] [CrossRef] [PubMed]
- Galzitskaya, O.V.; Lobanov, M.Y. Proteome-scale understanding of relationship between homo-repeat enrichments and protein aggregation properties. PLoS ONE 2018, 13, e0206941. [Google Scholar] [CrossRef]
- Borgia, A.; Kemplen, K.R.; Borgia, M.B.; Soranno, A.; Shammas, S.; Wunderlich, B.; Nettels, D.; Best, R.B.; Clarke, J.; Schuler, B. Transient misfolding dominates multidomain protein folding. Nat. Commun. 2015, 6, 8861. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wright, C.F.; Teichmann, S.A.; Clarke, J.; Dobson, C.M. The importance of sequence diversity in the aggregation and evolution of proteins. Nature 2005, 438, 878–881. [Google Scholar] [CrossRef]
- An, L.; Fitzpatrick, D.; Harrison, P.M. Emergence and evolution of yeast prion and prion-like proteins. BMC Evol. Biol. 2016, 16, 24. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nizhnikov, A.A.; Antonets, K.S.; Bondarev, S.A.; Inge-Vechtomov, S.G.; Derkatch, I.L. Prions, amyloids, and RNA: Pieces of a puzzle. Prion 2016, 10, 182–206. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, C.-S.; Wang, C.Y.-H.; Chen, B.P.-W.; He, R.-Y.; Liu, G.C.-H.; Wang, C.-H.; Chen, W.; Chern, Y.; Huang, J.J.-T. The influence of pathological mutations and proline substitutions in TDP-43 glycine-rich peptides on its amyloid properties and cellular toxicity. PLoS ONE 2014, 9, e103644. [Google Scholar] [CrossRef] [Green Version]
- Reinke, A.A.; Abulwerdi, G.A.; Gestwicki, J.E. Quantifying prefibrillar amyloids in vitro by using a “thioflavin- like” spectroscopic method. ChemBioChem 2010, 11, 1889–1895. [Google Scholar] [CrossRef]
- Selivanova, O.M.; Surin, A.K.; Ryzhykau, Y.L.; Glyakina, A.V.; Suvorina, M.Y.; Kuklin, A.I.; Rogachevsky, V.V.; Galzitskaya, O.V. To be fibrils or to be nanofilms? Oligomers are building blocks for fibril and nanofilm formation of fragments of Aβ peptide. Langmuir 2018, 34, 2332–2343. [Google Scholar] [CrossRef]
- Trovato, A.; Seno, F.; Tosatto, S.C.E. The PASTA server for protein aggregation prediction. Protein Eng. Des. Sel. 2007, 20, 521–523. [Google Scholar] [CrossRef] [Green Version]
- Maurer-Stroh, S.; Debulpaep, M.; Kuemmerer, N.; de la Paz, M.L.; Martins, I.C.; Reumers, J.; Morris, K.L.; Copland, A.; Serpell, L.; Serrano, L.; et al. Exploring the sequence determinants of amyloid structure using position-specific scoring matrices. Nat. Methods 2010, 7, 237–242. [Google Scholar] [CrossRef] [PubMed]
- Madeira, F.; Park, Y.M.; Lee, J.; Buso, N.; Gur, T.; Madhusoodanan, N.; Basutkar, P.; Tivey, A.R.N.; Potter, S.C.; Finn, R.D.; et al. The EMBL-EBI search and sequence analysis tools APIs in 2019. Nucleic Acids Res. 2019, 47, W636–W641. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, D.E.; Chivian, D.; Baker, D. Protein structure prediction and analysis using the Robetta server. Nucleic Acids Res. 2004, 32, W526–W531. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Crooks, G.E.; Hon, G.; Chandonia, J.-M.; Brenner, S.E. WebLogo: A sequence logo generator. Genome Res. 2004, 14, 1188–1190. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hunter, J.D. Matplotlib: A 2D graphics environment. Comput. Sci. Eng. 2007, 9, 90–95. [Google Scholar] [CrossRef]
- Wilkins, M.R.; Lindskog, I.; Gasteiger, E.; Bairoch, A.; Sanchez, J.-C.; Hochstrasser, D.F.; Appel, R.D. Detailed peptide characterization using PEPTIDEMASS—A World-Wide-Web-accessible tool. Electrophoresis 1997, 18, 403–408. [Google Scholar] [CrossRef] [PubMed]
- Beyermann, M.; Henklein, P.; Klose, A.; Sohr, R.; Bienert, M. Effect of tertiary amine on the carbodiimide-mediated peptide synthesis. Int. J. Pept. Protein Res. 2009, 37, 252–256. [Google Scholar] [CrossRef]
- Atherton, E.; Sheppard, R.C. Solid Phase Peptide Synthesis: A Practical Approach; IRL Press: Oxford, UK, 1989; ISBN 0199630666. [Google Scholar]
- Fields, G.B.; Noble, R.L. Solid phase peptide synthesis utilizing 9-fluorenylmethoxycarbonyl amino acids. Int. J. Pept. Protein Res. 1990, 35, 161–214. [Google Scholar] [CrossRef]
- Chan, W.C.; White, P.D. Basic Procedures. In Fmoc Solid Phase Peptide Synthesis: A Practical Approach; Oxford University Press: Oxford, UK, 2000. [Google Scholar]
- Olivera, E.; Marchetto, R.; Jubilut, G.N.; Paiva, A.C.M.; Nakaie, C.R. Correlation between rate of coupling reaction and swelling of resin beads: Influence of solvents, peptide sequence, chaotropic salt and acylation methods. In Chemistry, Structure and Biology, Proceedings of the 12th American Peptide Symposium, Cambridge, MA, USA, 16–21 June 1991; Smith, J., Rivier, J., Eds.; ESCOM: Leiden, The Netherlands, 1992; pp. 569–570. [Google Scholar]
- Stewart, J.M.; Klis, W.A. Polystyrene-based solid phase peptide synthesis: The sate of the art. In Innovations and Perspectives in Solid Phase Synthesis, Proceedings of the 1st International Symposium, Oxford, UK, August 19–September 2 1989; Epton, R., Ed.; SPCC Ltd.: Birmingham, UK, 1990; pp. 1–10. [Google Scholar]
- Zhang, L.; Goldammer, C.; Henkel, B.; Zühl, F.; Panhaus, G.; Jung, C.; Bayer, E. “Magic mixture”, a powerful solvent system for solid-phase synthesis of “difficult sequences.”. In Innovations and Perspectives in Solid Phase Synthesis, Proceedings of the 3rd International Symposium, Oxford, UK, August 31–September 4 1993; Epton, R., Ed.; Mayflower Worldwide Ltd.: Birmingham, UK, 1994; pp. 711–716. [Google Scholar]
- Selivanova, O.M.; Suvorina, M.Y.; Dovidchenko, N.V.; Eliseeva, I.A.; Surin, A.K.; Finkelstein, A.V.; Schmatchenko, V.V.; Galzitskaya, O.V. How to determine the size of folding nuclei of protofibrils from the concentration dependence of the rate and lag-time of Aggregation. II. Experimental application for insulin and LysPro insulin: Aggregation morphology, kinetics, and sizes of nuclei. J. Phys. Chem. B 2014, 118, 1198–1206. [Google Scholar] [CrossRef]
- Dovidchenko, N.V.; Glyakina, A.V.; Selivanova, O.M.; Grigorashvili, E.I.; Suvorina, M.Y.; Dzhus, U.F.; Mikhailina, A.O.; Shiliaev, N.G.; Marchenkov, V.V.; Surin, A.K.; et al. One of the possible mechanisms of amyloid fibrils formation based on the sizes of primary and secondary folding nuclei of Aβ40 and Aβ42. J. Struct. Biol. 2016, 194, 404–414. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Amount of Structural S1 Domains | FoldAmyloid | Waltz | PASTA 2.0 | AggreScan | ||||
|---|---|---|---|---|---|---|---|---|
| % Amyloidogenic Regions | Number of Sequences | % Amyloidogenic Regions | Number of Sequences | % Amyloidogenic Regions | Number of Sequences | % Amyloidogenic Regions | Number of Sequences | |
| 1S1 | 24 ± 10 | 12 | 15 ± 13 | 7 | 29 ± 19 | 12 | 43 ± 13 | 12 |
| 2S1 | 17 ± 6 | 9 | 9 ± 6 | 6 | 16 ± 7 | 9 | 39 ± 10 | 9 |
| 3S1 | 16 ± 3 | 26 | 5 ± 3 | 26 | 10 ± 4 | 26 | 27 ± 4 | 26 |
| 4S1 | 12 ± 2 | 460 | 6 ± 2 | 458 | 13 ± 4 | 460 | 28 ± 6 | 459 |
| 5S1 | 14 ± 5 | 16 | 5 ± 3 | 15 | 10 ± 4 | 16 | 31 ± 9 | 15 |
| 6S1 | 12 ± 3 | 851 | 4 ± 3 | 816 | 8 ± 3 | 851 | 29 ± 5 | 848 |
| Number of Structural S1 Domains | FoldAmyloid | Waltz | PASTA2.0 | Aggrescan | ||||
|---|---|---|---|---|---|---|---|---|
| % Amyloidogenic Regions | Number of Sequences | % Amyloidogenic Regions | Number of Sequences | % Amyloidogenic Regions | Number of Sequences | % Amyloidogenic Regions | Number of Sequences | |
| 1-1 | 30 ± 15 | 12 | 30 ± 23 | 5 | 46 ± 30 | 12 | 61 ± 21 | 12 |
| 2-1 | 21 ± 9 | 8 | 14 ± 10 | 5 | 28 ± 17 | 8 | 47±13 | 9 |
| 2-2 | 23 ± 8 | 9 | 15 ± 8 | 5 | 31 ± 13 | 5 | 54 ± 17 | 9 |
| 3-1 | 15 ± 4 | 25 | 13 ± 4 | 23 | 14 ± 9 | 20 | 35 ± 13 | 26 |
| 3-2 | 22 ± 8 | 26 | 15 ± 5 | 5 | 22 ± 10 | 8 | 24 ± 10 | 26 |
| 3-3 | 27 ± 6 | 26 | 13 ± 6 | 10 | 33 ± 10 | 22 | 56 ± 7 | 26 |
| 4-1 | 13 ± 6 | 453 | 12 ± 4 | 140 | 25 ± 12 | 252 | 28 ± 16 | 459 |
| 4-2 | 19 ± 6 | 459 | 13 ± 5 | 57 | 24 ± 14 | 294 | 36 ± 9 | 459 |
| 4-3 | 15 ± 4 | 456 | 14 ± 3 | 343 | 42 ± 20 | 355 | 49 ± 6 | 459 |
| 4-4 | 17 ± 5 | 460 | 14 ± 6 | 387 | 23 ± 9 | 331 | 46 ± 7 | 459 |
| 5-1 | 18 ± 7 | 15 | 16 ± 9 | 4 | 28 ± 11 | 11 | 38 ± 15 | 15 |
| 5-2 | 18 ± 9 | 15 | 12 ± 4 | 9 | 17 ± 8 | 4 | 38 ± 10 | 15 |
| 5-3 | 23 ± 11 | 15 | 14 ± 3 | 4 | 31 ± 16 | 13 | 43 ± 14 | 15 |
| 5-4 | 17 ± 7 | 16 | 13 ± 8 | 9 | 17 ± 5 | 7 | 42 ± 10 | 15 |
| 5-5 | 18 ± 8 | 16 | 11 ± 7 | 7 | 27 ± 12 | 8 | 38 ± 12 | 15 |
| 6-1 | 12 ± 6 | 637 | 11 ± 5 | 581 | 32 ± 8 | 715 | 35 ± 11 | 847 |
| 6-2 | 14 ± 8 | 847 | 14 ± 6 | 346 | 20 ± 11 | 159 | 38 ± 11 | 848 |
| 6-3 | 21 ± 5 | 851 | 13 ± 7 | 166 | 27 ± 16 | 329 | 43 ± 7 | 848 |
| 6-4 | 14 ± 6 | 850 | 13 ± 7 | 166 | 20 ± 7 | 457 | 43 ± 11 | 848 |
| 6-5 | 19 ± 6 | 848 | 13 ± 6 | 239 | 15 ± 8 | 266 | 39 ± 9 | 848 |
| 6-6 | 13 ± 6 | 524 | 13 ± 7 | 176 | 19 ± 10 | 310 | 25 ± 13 | 809 |
| Number of S1 Structural Domains | Position of the Amyloidogenic Regions in S1 Domains (a.a.)* | |||
|---|---|---|---|---|
| 10–15 | 25–30 | 55–60 | 65–70 | |
| 1-1 | VXRY | (F/K)(G/C)(E/Y)(L/Y) | (V/E)KVL | L(L/V)(L/V)SFK |
| 2-1 | G(V/A/L)X(V/A) | (F/R)G(F/V)YP | EV(K/X)VL | R/G(L/V)(V/Y)LS |
| 2-2 | G(A/Y)EV(R/V)(L/Y) | (G/A)(L/F)(V/L)(H/P) | (V/E)(V/L)X(F/V)KV | (L/V)(S/H)X(K/R) |
| 3-1 | (E/H)(F/V)(F/L)(I/V) | (Q/M)(L/V)ILS | ||
| 3-2 | LRGFIP | (L/I)(P/T)(V/L)(K/A)FL | (K/R)LVLS | |
| 3-3 | (F/Y)G(V/A)F(I/V) | GLLHIS | (V/L)K(V/A)(M/L)(I/V) | |
| 4-1 | (E/Q)(V/A)L(V/L)D | V(I/L)(P/T)(S/L)R | (V/I/L)(E/D)(V/A/L)LV | LSK(K/R) |
| 4-2 | L(I/V)(V/L)X(D/E) | (M/L)V(D/E)(M/T/H)R | (N/R)(V/E) (I/V)LSRR | |
| 4-3 | FGAF(I/V)D | (E/Q)(I/L)S(W/H/Y)(K/E) | (V/I)(T/E/K)V(E/K) (I/V)L | |
| 4-4 | FG(V/A)F(I/V) | LVH(I/V)S | (V/I/L)XVK(V/I)(I/L) | |
| 5-1 | VXVDI | (G/A)X(V/I/L)PL | DX(V/I/L)X(V/A/L)(Y/Q)V | GX(I/Y)(L/Y)LSR |
| 5-2 | (I/V)X (G/V)X(I/V) | G(I/V)R(G/A)F(M/L) | (V/L)E(F/V)K(I/V)(I/L) | (N/R)(V/I)(V/I)(L/Y)LSR |
| 5-3 | (F/Y)G(V/A)F(I/V) | (E/R)(I/L)S(W/H/Y)(K/R) | VX(V/A)XV(I/L) | |
| 5-4 | (F/Y)GAF(I/V)E | (E/D)GL(I/V)H(I/V) | VE(V/A)X (V/I)/L | |
| 5-5 | EGXF(I/V)(E/A) | (E/D)GL(I/V)H(I/V) | EXXV(I/L) | |
| 6-1 | (G/A)XV(I/V) | (V/A)(L/Y)(V/I)(D/N)(A/Y) | DX(V/I)XV(A/V)(L/I/V) | |
| 6-2 | GX(I/V) | GETV(D/E) | EFK(V/I)(I/L)K | |
| 6-3 | YG(A/V)D | LH(I/V)TD(M/I/L)(A/S)W(K/R) | (V/I)X(V/A)K(V/I) (L/I) (K/R) | |
| 6-4 | YG(A/C)FV | EGL(V/Y)H(V/I) | (V/E)V(E/D)(V/A)(M/V)(V/I)L | |
| 6-5 | FG(V/I/L)F(I/V) | D(I/L)SW | (V/I/L)X(A/Y)V(V/I)L | |
| 6-6 | VXGX(I/V) | VEAK(V/I/L) | RX(V/I/L)XLS(V/I)(K/R) | |
| Sequences, Company | Localization in S1 Sequence, Domain (D), Species | Peptides Mw * | Peptides pI ** |
|---|---|---|---|
| Peptide 1, VVEGTVVEVT (V10T)T, 1 | (211–220 a.a), D3, T. thermophilus | 1031.2 | 3.5 |
| Peptide 2, DFGVFVNLG (D9G)T, 2 | (221–229 a.a.), D3, T. thermophilus | 967.1 | 3.8 |
| Peptide 3, VTDFGVFVEI (V10I)T, 1 | (391–400 a.a.), D5, T. thermophilus | 1125.3 | 3.5 |
| Peptide 4, EMEVVVLNID (E10D)T, 2 | (430–439), D5, T. thermophilus | 1160.3 | 3.4 |
| Peptide 1, IVRGVVVAID (I10D)E, 3 | (23–32 a.a.), D1, E. coli | 1040.3 | 6.3 |
| Peptide 2, DEITVKVLKF (D10F)E, 3 | (239–248 a.a.), D3, E. coli | 1191.4 | 6.3 |
| Peptide 3, TDYGCFVEIE (T10E)E, 3 | (288–297 a.a.), D4, E. coli | 1175.3 | 3.4 |
| Peptide 4, VVNVGDVVEV (V10V)E, 3 | (321–330 a.a.), D4, E. coli | 1028.2 | 3.5 |
| Peptide 5, DFGIFIGLDG (D10G)E, 3 | (376–385 a.a.), D5, E. coli | 1053.2 | 3.5 |
| Peptide 6, VHLSDISWNV (V10NV)E,3 | (391–400 a.a.), D5, E. coli | 1169.3 | 5.5 |
| Peptide 7, EIAAVVLQVD (E10D)E, 3 | (414–423 a.a.), D5, E. coli | 1055.6 | 3.5 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Grishin, S.Y.; Deryusheva, E.I.; Machulin, A.V.; Selivanova, O.M.; Glyakina, A.V.; Gorbunova, E.Y.; Mustaeva, L.G.; Azev, V.N.; Rekstina, V.V.; Kalebina, T.S.; et al. Amyloidogenic Propensities of Ribosomal S1 Proteins: Bioinformatics Screening and Experimental Checking. Int. J. Mol. Sci. 2020, 21, 5199. https://doi.org/10.3390/ijms21155199
Grishin SY, Deryusheva EI, Machulin AV, Selivanova OM, Glyakina AV, Gorbunova EY, Mustaeva LG, Azev VN, Rekstina VV, Kalebina TS, et al. Amyloidogenic Propensities of Ribosomal S1 Proteins: Bioinformatics Screening and Experimental Checking. International Journal of Molecular Sciences. 2020; 21(15):5199. https://doi.org/10.3390/ijms21155199
Chicago/Turabian StyleGrishin, Sergei Y., Evgeniya I. Deryusheva, Andrey V. Machulin, Olga M. Selivanova, Anna V. Glyakina, Elena Y. Gorbunova, Leila G. Mustaeva, Vyacheslav N. Azev, Valentina V. Rekstina, Tatyana S. Kalebina, and et al. 2020. "Amyloidogenic Propensities of Ribosomal S1 Proteins: Bioinformatics Screening and Experimental Checking" International Journal of Molecular Sciences 21, no. 15: 5199. https://doi.org/10.3390/ijms21155199