Emerging Strategies Targeting Catabolic Muscle Stress Relief


1
School of Biomedical Sciences, University of Leeds, Leeds LS2 9JT, UK
2
Department of Experimental and Molecular Cardiology, TU Dresden, Heart Center Dresden, 01307 Dresden, Germany
3
Dresden Cardiovascular Research Institute and Core Laboratories GmbH, 01067 Dresden, Germany
4
Medical Faculty Mannheim, University of Heidelberg, 68167 Mannheim, Germany
5
Myomedix GmbH, Im Biengarten 36, 69151 Neckargemünd, Germany
*
Author to whom correspondence should be addressed.
Int. J. Mol. Sci. 2020, 21(13), 4681; https://doi.org/10.3390/ijms21134681
Submission received: 4 June 2020
/
Revised: 26 June 2020
/
Accepted: 29 June 2020
/
Published: 30 June 2020
(This article belongs to the Special Issue Muscle Atrophy: Discovery of Mechanisms and Potential Therapies)
{kind=link}
{kind=link}
{kind=link}
Abstract
:Skeletal muscle wasting represents a common trait in many conditions, including aging, cancer, heart failure, immobilization, and critical illness. Loss of muscle mass leads to impaired functional mobility and severely impedes the quality of life. At present, exercise training remains the only proven treatment for muscle atrophy, yet many patients are too ill, frail, bedridden, or neurologically impaired to perform physical exertion. The development of novel therapeutic strategies that can be applied to an in vivo context and attenuate secondary myopathies represents an unmet medical need. This review discusses recent progress in understanding the molecular pathways involved in regulating skeletal muscle wasting with a focus on pro-catabolic factors, in particular, the ubiquitin-proteasome system and its activating muscle-specific E3 ligase RING-finger protein 1 (MuRF1). Mechanistic progress has provided the opportunity to design experimental therapeutic concepts that may affect the ubiquitin-proteasome system and prevent subsequent muscle wasting, with novel advances made in regards to nutritional supplements, nuclear factor kappa-light-chain-enhancer of activated B cells (NFκB) inhibitors, myostatin antibodies, β2 adrenergic agonists, and small-molecules interfering with MuRF1, which all emerge as a novel in vivo treatment strategies for muscle wasting.
Keywords:
atrophy; diaphragm; disuse; mitochondria; MuRF1; MuRF2; skeletal muscle; ubiquitin; wasting1. Introduction
Skeletal muscle is the largest and one of the most dynamic organs in the human body, representing 30–40% of total body mass and containing up to 75% of the organism’s protein reserves [1]. Skeletal muscle is essential for life, supporting movement, respiration, thermoregulation, and metabolic homeostasis. As such, skeletal muscle is a highly adaptable organ that can sense and respond accordingly to match environmental cues. Skeletal muscle wasting (also referred to as atrophy) is a characteristic of several catabolic conditions, including aging (i.e., sarcopenia), starvation, and immobilization, but also many acute and chronic illnesses such as cancer, heart failure, sepsis, and diabetes [2]. Cachexia is inextricably linked to muscle atrophy in many illnesses, however, the former is defined as a complex multifactorial metabolic syndrome that is associated with a significant reduction in body mass underpinned by skeletal muscle loss (with or without fat mass loss), which is not fully reversible with nutritional aids [3]. Collectively, therefore, a loss of muscle mass leads to a decline in functional mobility, which contributes to poor quality of life and survival [4]. Accordingly, our current lack of direct treatments to rescue muscle wasting across millions of patients is a key concern, with exercise training the only established intervention [5]. However, given that many patients are too ill or bedridden to perform exertional exercise, the development of novel pharmacological strategies to inhibit muscle atrophy represents an important research avenue. In this context, targeted inhibition of procatabolic factors specifically activated under wasting conditions could offer the most beneficial treatment strategy for patients [6,7,8].
The current review, therefore, will address novel approaches that may permit modulation of procatabolic factors upregulated across various wasting conditions that impact muscle homeostasis. We will first introduce a major mechanism in the myofiber responsible for degrading proteins and thus, atrophy: the ubiquitin-proteasome system (UPS) and its muscle activating muscle-specific E3 ligase Muscle RING-finger protein 1 (MuRF1). We then discuss recent advances in pharmacological aids that have shown potential in vivo application for inhibiting both upstream and downstream control nodes in this system and thus limit muscle wasting across various catabolic conditions.
2. Pathways Modulating Muscle Atrophy
Atrophy can be defined as a cellular shrinkage of tissues and organs caused by loss of organelles, cytoplasm, and proteins [2]. Skeletal muscle atrophy, therefore, is underpinned by complex changes in the balance between rates of protein synthesis and protein degradation, with many wasting conditions characterized by increased rates of proteolysis and suppressed rates of protein synthesis. As detailed extensively elsewhere [2], the molecular signaling pathways that interact to control protein synthesis and degradation generally include the insulin-like growth factor 1 (IGF1)/protein kinase B (Akt)/mammalian target of rapamycin (mTOR)-forkhead box protein O (FoxO), TGF-β/myostatin/bone morphogenetic protein (BMP), nuclear factor kappa-light-chain-enhancer of activated B cells (NFκB), and glucocorticoid. These signaling pathways can interact to determine the terminal fate of many proteins, which include degradation by one of the four major proteolytic systems in the cell: ubiquitin-proteasome, autophagy-lysosome, calpain, and caspase [9]. The signaling pathways and proteolytic systems controlling muscle mass are tightly governed by upstream factors related to hormonal/cytokine, metabolic/nutrient, mechanical load, and neural activity, which helps to explain why sudden changes to our environment such as starvation, exercise, immobilization, or disease result in rapid alterations to muscle mass. This review will primarily focus on UPS-dependent degradation, and we refer the reader to other reviews detailing the additional proteolytic systems [2,10,11].
2.1. Proteasome-Dependent Degradation
Characterized by a large protein complex (2.5 MDa) called the 26S proteasome, the most important proteolytic pathway recognized to mediate muscle atrophy is the UPS [12,13,14]. This enzyme complex degrades structural and regulatory proteins selectively targeted following the covalent attachment of polyubiquitin chains to lysine residues (Figure 1). The UPS is composed of an active 20S catalytic core capped by regulatory 19S subunits that are responsible for unfolding proteins for entry [15]. A family of three enzymes exert a major control on the UPS, termed E1, E2, and E3. As shown in Figure 1, the E1 activating enzyme initially primes the ubiquitin protein in an ATP-consuming reaction, while the E2 conjugating enzyme passes ubiquitin to the E3 ligase enzyme that links ubiquitin to the target protein in a repeated process (termed polyubiquitination) for subsequent recognition and degradation by the 26S proteasome. While proteasome-dependent degradation is considered a main fate for many sarcomeric proteins, some early events in this process include activated calpains and caspases (i.e., caspase-3 and -8) initially disrupting sarcomere assembly (by targeting Z-disks to release major proteins such as titin, nebulin, filamin, troponin-T, and desmin) as well as cleaving actomyosin to form a 14 kDa actin fragment for subsequent UPS processing [10,16,17]. One major rate-limiting step considered in the UPS is the attachment of ubiquitin to target proteins via specific E3 ubiquitin ligases, which have attracted particular attention given their potential for therapeutic modulation not only for muscle atrophy but also in the regulation of inflammation and immunity [18].
2.2. Muscle-Specific E3 Ligases: A Rate-Limiting Step in the UPS
A major breakthrough in the muscle field was achieved when multiple studies identified a series of so-called “atrogenes” transcriptionally upregulated in a variety of wasting conditions (i.e., diabetes, fasting, uremia, immobilization, and denervation) but otherwise not generally expressed under basal conditions [19,20]. Two key atrogenes identified were the muscle-specific E3 ubiquitin-ligases MuRF1 and muscle atrophy F-box (MAFBx; also termed atrogin-1) [19,20]. Evidence is now conclusive that knockdown of either gene can attenuate muscle wasting in various catabolic conditions including denervation, hindlimb suspension, fasting, chronic kidney disease, and lung disease [21], although their role in sarcopenia remains controversial [22].
In particular, removal of many thick filaments of the sarcomere is achieved by MuRF1-dependent targeting of troponin I, myosin heavy chain, myosin-binding protein C, myosin light chain [23,24] and actin [25]. In contrast, fewer MAFbx-dependent targets have been reported, and these have been specific to regulatory proteins linked to protein synthesis (e.g., MyoD and eukaryotic translation initiation factor 3 subunit F). In addition, other key E3 ligases that can accelerate sarcomeric proteolysis include: tripartite motif-containing protein 32 (Trim 32, reported to target thin filaments such as actin, tropomyosin, and troponins but also Z band-located proteins such as alpha-actinin and desmin) [26], tumor necrosis factor receptor-associated factor 6 (TRAF6, reported to target thick filaments and regulatory proteins) [27], C terminus of HSC70-Interacting Protein (CHIP, which targets filament C via lysosomal-dependent degradation) [28], F-Box Protein 40 (Fbxo40, targeting anabolic-insulin signaling proteins such as Insulin receptor substrate 1) [29], as well as muscle ubiquitin ligase of SCF complex in atrophy-1 (MUSA1) and specific of muscle atrophy and regulated by transcription (SMART), whose targets remain unclear [30]. The activation of E3 ligase transcription is highly regulated by numerous signaling pathways and transcription factors that include IGF1/Akt/FoxO, myostatin/mothers against decapentaplegic homolog (Smad) 2-3, NFκB, and glucocorticoid/glucocorticoid receptor. As such, these pathways have yielded useful therapeutic targets for inhibiting muscle wasting [2].
3. Therapeutic Treatments to Inhibit Muscle Atrophy
Currently, the only proven treatment for muscle wasting is exercise training [5], yet many patients are simply too ill, frail, or bedridden to perform physical exertion. Other non-physical exercise approaches that may also yield beneficial effects include certain nutritional supplements (e.g., amino acids) or hormonal therapy (e.g., testosterone) [7]. Given the key role that elevated proteolysis plays in mediating muscle atrophy across various conditions [21], the development of novel pharmacological aids for in vivo administration that target this process would represent a significant breakthrough. Recent advances in new methodologies such as proteomics, metabolomics, and transcriptomics have been instrumental in shedding new light on the underlying molecular pathways involved in regulating skeletal muscle wasting. However, inhibition of certain proteolytic pathways (via pharmacological or genetic deletion) have proven ineffective or even detrimental: suppressing autophagy impairs clearance of damaged organelles and misfolded proteins which leads to a dystrophic phenotype [31,32,33], while overexpression of calpastatin (an endogenous calpain inhibitor) in models of disuse-induced atrophy (hindlimb suspension) and sepsis has failed to impact muscle mass [34,35]. Similarly, previous studies have shown that exogenous inhibition of caspase is effective in preserving muscle function but not mass [36,37]. In contrast, much attention has been focused on targeting regulatory steps in proteasome-dependent degradation, with some encouraging treatments achieved by targeting both upstream (e.g., cytokines, transcription factors) and downstream (e.g., E3 ligases, 26S proteasome) nodes in the UPS (Figure 1).
3.1. Upstream Inhibition of UPS: Inflammatory Cytokines, Growth Factors, and Transcription Factors
During inflammatory diseases such as cancer, sepsis, or heart failure, an elevated and often chronic production of proinflammatory cytokines such as interleukin-1 (IL-1), interleukin-6 (IL-6), and tumor necrosis factor-alpha (TNFα) can act as upstream UPS triggers to induce atrophy [38]. Proinflammatory cytokines have been shown to modulate signaling via membrane-bound receptors activating a number of instrumental transcription factors that include NFκB, FoxO, and p38 MAPK, all of which are known to accelerate proteolysis involving MuRF1 (and MAFbx) activation [39,40,41,42]. Various studies have now shown that these effects can be inhibited by the use of novel antibodies (Figure 1). For example, the administration of MR16-1 antibody (anti-mouse IL-6 receptor antibody) in a mouse model of disuse-induced muscle atrophy (tail suspension) resulted in partial protection from muscle wasting alongside suppressed MuRF1 and MAFbx gene expression [42]. A similar study using RemicadeTM (an anti-TNFα drug that is clinically available) found that by interfering with TNFα activity, this drug reduced atrophy of dystrophic muscles by preventing NFκB translocation to the nucleus and, consequently, MuRF1 accumulation [40]. A link between cytokine-MuRF1 signaling has been confirmed in mice injected with exogenous TNFα [43], such that mice with genetic inactivation of MuRF1 were protected from force loss induced by TNFα treatment [43], which highlights an important role for MuRF1 in regulating myofibril contractile function as well as atrophy. In addition to cytokines, other secreted circulating factors known to induce muscle wasting include growth factors such as myostatin. Myostatin is a negative regulator of muscle mass and a member of the transforming growth factor-beta (TGF-β) superfamily, and acts by binding plasma membrane-located activin receptors type IIB and IIA (ActRIIB/A), which leads to subsequent phosphorylation of Smad2/3-Smad4 complex and inhibition of Akt signaling. An anti-myostatin antibody (ATA 842) administered for 4 weeks increased muscle mass in sarcopenic mice [44], while a soluble ActRIIB inhibitor rescued muscle wasting in cancer cachexia mice [45] (Figure 1).
Another member of the TGF-β superfamily, growth and differentiation factor 11 (GDF11), has been reported to work alongside myostatin to mediate muscle atrophy in vivo with MuRF1 involvement [46,47]. Treatment with GDF11 propeptide-Fc (a natural inhibitor of GDF11 derived from the GDF11 precursor protein) in dystrophic mdx mice (a model of Duchenne muscular dystrophy) improved muscle mass and strength but, to our knowledge, no data have been published on the effects of this treatment on MuRF1 expression [48]. Other approaches have targeted indirect myostatin inhibition by using small-molecule inhibitors (C188-9) to block the phosphotyrosyl peptide binding site and thus phosphorylation of Signal Transducer and Activator of Transcription 3 (Stat-3). Stat-3 is upregulated via IL-6-JAK signaling and can induce myostatin and MuRF1 expression via the transcription factor CCAAT-enhancer-binding protein delta (C/EBP-δ). Treatment with C188-9 suppressed MuRF1 expression alongside proteasome and caspase-3 activity, while preserving muscle mass in both cancer and diabetic mice [49].
Similar to myostatin and inflammatory cytokines, circulating glucocorticoids are also elevated under catabolic conditions and regulate atrophy via glucocorticoid receptor-dependent signaling that can increase MuRF1 expression (i.e., by direct or via FoxO signaling) [50]. However, treatment with the glucocorticoid receptor antagonist RU-38486 or RU 486 can suppress MuRF1, MAFbx, and proteasome levels in septic mice [51,52] (Figure 1). Since many catabolic conditions are associated with elevated oxidative stress that can directly promote proteasome-dependent degradation via MuRF1 and MAFBx expression, another approach has been to employ antioxidant treatments. Specifically, mitochondrial-targeted antioxidants have proven particularly effective in preventing muscle wasting in a range of conditions including mechanical ventilation, inflammation, denervation, and fasting [53,54,55] (Figure 1). However, given the important physiological and signaling roles played by reactive oxygen species (ROS), chronic blockade of these molecules could result in severe complications (e.g., cancer) [56].
An additional therapeutic target for muscle atrophy has been the selected inhibition of atrophic transcription factors such as NFκB and FoxO (Figure 1). Among these, the selective inhibitor IMD-0354 was developed in the late 90s to suppress NFκB [57]. However, despite promising results in vitro, administration of IMD-0354 during muscle unloading was unable to prevent MAFbx and MuRF1 accumulation as well as muscle wasting, despite preventing inhibitor of nuclear factor kappa-B kinase subunit beta (IκKβ) phosphorylation [58]. More recent experiments have been more encouraging, with a novel class of orally bioavailable NFκB inhibitors (termed edasalonexent and CAT-1041) able to inhibit muscle wasting in both murine and canine models of muscular dystrophy [46]. However, since multiple transcription factors control muscle mass, other studies have focused on inhibiting the FoxO family of transcription factors. Proteins belonging to this family of transcription factors, when not phosphorylated, translocate to the nucleus to increase MuRF1 and MAFbx transcription, and this can be controlled by Akt-dependent signaling [59,60]. Approaches targeting the Akt/FoxO signaling axis include administration of leucine [61,62] as well as its active metabolite Beta-Hydroxy b-methylbutyrate (HMB) [42,63,64,65,66]. Another candidate recently described in the literature is Matrine, an alkaloid found in the plant Sophorae flavescentes that showed strong antitumoral and anti-inflammatory activity [67]. This compound is approved by the China Food and Drug Administration for use in cachectic patients and was shown to attenuate MuRF1 mRNA expression and maintain fiber size via Akt/FoxO pathway in mice with cancer cachexia [67].
Another promising area has been the administration of the β2-adrenergic receptor (β2-AR) agonists, which can exert both pro-anabolic and anti-catabolic effects [68]. Conventional (e.g., formoterol) [69], as well as more novel β2-ARs such as 5-hydroxybenzothiazolone (5-HOB) [70] and espindolol/MT-102 [71,72], have shown benefits in promoting muscle growth and attenuating atrophy in experimental models of aging and cancer cachexia, possibly via NFκB/FoxO-dependent MuRF1 activation. However, the use of β2-AR can have adverse effects on cardiovascular function, which can have serious repercussions in many patients. Overall, while it seems that some viable treatments are available to inhibit multiple transcription factors and thus UPS activation, targeting a more central node where signaling networks converge, such as the ubiquitin-proteasome pathway per se, may be a more specific and thus beneficial approach.
3.2. Downstream Inhibition of UPS via the 26S Proteasome
As discussed earlier, muscle wasting often involves the degradation of polyubiquitinated proteins via the 26S proteasome [12]. Bortezomib (otherwise termed VelcadeTM or PS-341) is a selective boronic acid proteasome inhibitor approved by the United States Food and Drug Administration and used as a third-line treatment of multiple myeloma and mantle cell lymphoma [73]. Bortezomib functions by inhibiting the catalytic site of the proteasome complex without direct effects on ubiquitination or upstream activators [74]. Studies in murine models investigating the effects of bortezomib on muscle atrophy have produced mixed results showing either a significant reduction of muscle atrophy by up to 50% in the soleus muscle of denervated rats [75] or no effects in cancer mice [73]. Further experiments focused on the diaphragm have shown that bortezomib lowered proteasome activity and MAFBx/MuRF1 transcripts with normalized myosin protein levels and improved contractile function in heart failure rats [76], yet limited benefits were observed following acute mechanical ventilation-induced diaphragm atrophy [77,78]. Carfilzomib is a clinically approved irreversible selective proteasome inhibitor. Similar to bortezomib, this drug is employed as a second-line treatment for patients with multiple myeloma [79], with some evidence suggesting the efficacy of this drug to prevent muscle wasting and MuRF1 activity. For example, early treatment with Carfilzomib (2 mg/kg; 2 × per week) in mice with cancer-associated cachexia was effective in partly rescuing skeletal muscle wasting and, through the downregulation of angiotensin II, MuRF1 and MAFBx expression in skeletal muscle [80]. Other proteasome inhibitors tested include MG132, a reversible and cell-permeable proteasome inhibitor belonging to the class of synthetic peptide aldehydes. MG132 has been able to rescue muscle mass by ~50–75% alongside reducing the expression of both MuRF1 and MAFBx in mice following both limb immobilization [40,60] and cancer [81]. However, it is difficult to delineate the effects of MG132 on the proteasome per se, as this drug also inhibits the NFκB canonical pathway by preventing degradation of IκBα [60,81] as well as lysosomal proteases and calpains [40], with lack of clarity over benefits to muscle contractile function [82].
A major consideration for the treatment of proteasome inhibitors is that patients have shown dose-limiting toxicity, drug-resistance, and several adverse effects such as cardiac complications and even muscle weakness, which severely limit their application to the general population [26,83]. Overall, while proteasome-specific inhibitors have shown some benefits, there is a lack of consistency in positive outcomes, and it appears that maintaining proteasome-dependent degradation is essential for preserving cellular homeostasis [12]. As such, a more unique therapeutic approach that targets steps earlier in the UPS pathway, such as blocking the function of muscle-specific E3 ligases that are atrophy dependent, may be a more optimal approach with fewer side effects [6,11,26].
4. Targeted Small-Molecule Inhibition of the E3 Ligase MuRF1
There is a fast-growing field on how to target specific E3 ligases in different cellular contexts that were previously thought to be undruggable [84]. What evidence is there to support inhibiting one E3 ligase over another? Within the skeletal muscle context, there is good evidence favoring the MuRF1 protein over other E3 ligases. Based upon gene inactivation experiments, the deletion of MuRF1 is sufficient to provide partial protection from atrophy after denervation [19] and various other catabolic conditions [21]. In further support, MuRF1 is: (1) consistently upregulated in over 15 different settings of atrophy [21]; (2) central to catabolic rather than healthy basal conditions unlike other E3 ligases [85]; (3) able to regulate distinct pools of sarcomere-specific substrates that make up the majority of muscle bulk [86]; (4) muscle-specific which limits any adverse effects in other cell types [87,88,89]. As such, these data provide a strong rationale for developing effective in vivo drugs capable of suppressing MuRF1, but is this possible? In the next section, we discuss from a structural perspective the MuRF family and potential strategies for perturbing MuRF1 functions (i.e., properties that are specific to MuRF1 among the TRIM family) (Figure 2).
4.1. Structural Properties of the MuRFs
The muscle-specific RING finger proteins (MuRFs) are a group of three highly homologous proteins coded by different genes that are classified as part of the TRIpartite motif (TRIM) family, which include MuRF1 (TRIM 63) [90], MuRF2 (TRIM55) [91], and MuRF3 (TRIM64) [92,93]. MuRFs are expressed in skeletal and cardiac muscle and characterized by their RING finger, a unique MuRF-family-specific motif (MFC), a B-box, a coiled-coil (CC), and acidic-tail domains (Figure 2), which share high sequence identity apart from the N- and C-termini whose residue length differs considerably [92]. Like all TRIM family members, the RING motif is responsible for ubiquitination, B-box for protein binding, and CC domain for self-association.
As demonstrated in Figure 2, MuRF1′s RING domain was found in the N-terminal position with a conserved pattern of cysteine and histidine residues, which allowed attachment of ubiquitin to recognized substrates [94]. The central region, constituted by a B-box type II and a helical domain, is responsible for the interaction with different target proteins and is considered the functional unit of MuRF1 [94,95,96]. To our knowledge, while the full 3D structure of MuRF1 is yet to be reported, the B-box domain has been determined [97], which indicated MuRF1 self-associates and forms multimers. This self-multimerisation is presumably the reason why no structure of MuRF1 has been determined so far, as the structure of higher-order multimers would need to be resolved in the first place [97]. MuRF1 (and MuRF2) also presents a one signature-box (COS-box) sequence motif in its helical domain [95]. Evidence suggests that this domain plays a central role in protein recruitment by interacting with domains near the C-terminus of titin and so represents an interesting pharmacological target to limit protein activity [95,96,98]. In this regard, MuRF1 has received much attention for is its ability to bind at the M-line and interact with the large structural protein titin. Titin spans the entire sarcomere and is involved in regulating both sarcomeric structure and signaling [99]. While MuRF1 does not seem to target titin for degradation per se, this interaction may influence sarcomeric stability [100] and has recently been targeted by interfering small molecules to inhibit atrophy [82,101]. Beyond the M-line of the sarcomere, MuRF1 is also reported to locate at the Z-line, nucleus [102,103], and mitochondria [104]. While MuRF1 is best known for its involvement in sarcomeric protein degradation, more recently it has been recognized to have potential broad signaling effects that include regulation of protein synthesis, mitochondrial function, amino acid and carbohydrate metabolism, insulin homeostasis, apoptosis, and endoplasmic reticulum (ER) stress response [21,82,101,105] (Figure 3).
MuRF2 plays a central role in sarcomere assembly during development by transiently associating with titin and myosin [103]. Post-development, MuRF2 works in synergy with MuRF1 to mediate signal transduction in cardiomyocytes to prevent cardiac hypertrophy while stabilizing fast-fibers in skeletal muscle [82,103,106]. MuRF2 also interacts with the titin kinase in a stretch-dependent manner, regulating protein synthesis via the transcription factor serum response factor (SRF) [91]. Furthermore, recent evidence suggests that MuRF2 may act as an atrogene capable of regulating muscle wasting, as demonstrated in a mouse model of cardiac cachexia [106]. The final family member MuRF3 was the first of the MuRFs to be identified [93] and is required for skeletal myoblast differentiation, development of cellular microtubular networks and myogenesis [107,108,109]. Together with MuRF1, the MuRF3 protein localizes to the M-band and Z-disk, playing a key role in sarcomeric protein degradation [107]. MuRF3 KO mice have shown this ligase to be necessary for limiting cardiac abnormalities induced by heart failure [103] and diabetic cardiomyopathy [110]. Given similarities in sequence, structure, and functionality between MuRF1/2/3, some level of redundancy occurs, which has limited our understanding of the different roles played by each family member [108]. In addition, such close homology between MuRFs makes the development of inhibitors of this ligase family particularly challenging, with issues related to cross-reactivity [111] and potential lethality if multiple MuRFs are blocked simultaneously, as shown in mice with double gene knock-out of MuRF1/2 [112] or MuRF1/3 [108,109].
4.2. Developing Novel Small-Molecules to Inhibit MuRF1
Knowledge of MuRF1′s protein structure and comparison with other TRIM members suggests potential strategies for attenuating its function. These include interference with its coiled-coil region involved in protein recognition [112], its b-box domain involved in protein dimerization [92], or its MFC domain that is conserved between MuRFs but no other TRIMs [90]. Finally, direct targeting of the ubiquitin-transferring RING domain is a possible strategy. Another approach may be blocking MuRF1′s interaction with known targets that can influence protein turnover, such as the titin filament [113]. P013222 is a specific MuRF1 inhibitor developed over a decade ago, showing low toxicity [114]. In a cellular model of muscle atrophy (C2C12 cells treated with the synthetic glucocorticoid dexamethasone), this compound was able to inhibit MuRF1 autoubiquitination and, by stabilizing myosin heavy chain, to prevent its degradation in a dose-dependent manner (12.5–50 μM) [114]. Other approaches using adenoviral blockade to inhibit MuRF1 have shown similar results in myotubes stressed with dexamethasone [23,85]. To our knowledge, however, these inhibitors were never tested in vivo, thus limiting clinical translation and targeting inhibition of MuRF1′s catalytic RING domain, which seems necessary for maintaining muscle homeostasis [109]. Therefore, an alternative strategy might be to interfere with MuRF1’s recognition and binding domain towards targeted substrates, particularly those of the sarcomere, and leaving its RING finger unperturbed, as recently described [82,101]. This approach may have the advantage of maintaining the basal activity of MuRF1’s catalytic N-terminal RING domain while limiting possible risks of myopathy (e.g., genetic inactivation of MuRF1 and these can result in cardiac septum ruptures and accumulation of protein aggregates) [109].
One small molecule identified and characterized so far that targets MuRF1 coiled-coil region, as demonstrated in a series of multiple mouse models of muscle wasting, appears to have low toxicity, while functionally having the ability to interfere with MuRF1 target-recognition [82,101] (Figure 2). Intriguingly, in two mechanistically distinct murine models of cardiac cachexia, the administration of this small molecule mixed with chow (over 6–10 weeks) was able to reduce skeletal muscle MuRF1 expression and proteasome activity, while fiber contractile dysfunction and atrophy were attenuated [82,101]. We should stress that these studies are recent and still ongoing, and mostly derived from a single small molecule [82,101]. Clearly, they will require follow-up in different animal models and expanded toxicology. However, one of the most striking findings from these experiments using this specific small molecule was its ability to rescue force loss in stressed mice, which may be regulated by changes to both sarcomeric and metabolic proteins [82,101].
Expression of sarcomeric proteins that are important for force generation, such as actin and telethonin, were also normalized by treatment using this specific small molecule [82,101]. We have also started exploratory proteomics studies to identify potential underlying pathways affected by this identified small molecule that may regulate muscle function. This approach has revealed that proteins participating in protein synthesis, apoptosis, and mitochondrial ATP synthesis were protected under stress when treated with this specific small molecule [82,101]. Consistent with this, we have found treatment with this small molecule can partially rescue muscle mitochondrial morphology and function (e.g., porin content, citrate synthase activity, mitochondrial complex I activity, and mitochondrial ribosomal Protein S5) [82], which are possibly in line with earlier yeast two-hybrid studies that showed that MuRF1 can preferentially target mitochondrial in addition to sarcomeric proteins [115], and implicate possible localization within mitochondria as recently reported in cardiac muscle [116]. Furthermore, genetically engineered mice have shown that MuRF1 can negatively regulate pyruvate dehydrogenase (PDH) function, insulin sensitivity, and hepatic glycogen stores [117], raising the intriguing proposition that MuRF1 could play a role in driving insulin-resistance associated with muscle wasting [118]. Overall, these data may support previous evidence that MuRF1 possesses broad signaling effects within both the myofiber and whole-organism (Figure 3).
It should be noted that this identified small molecule can also affect MuRF2 protein expression [82]. The construct used for screening had high homology between MuRF1 and MuRF2 since both MuRFs share very high homology in their MFC-Bbox-cc segments (Figure 2). Also, the genetic knockdown of MuRF1 has been shown to affect MuRF2 [106,119]. Finally, recent experiments have confirmed that mice with a genetic deletion of MuRF2 do not suffer muscle wasting in cardiac cachexia [106]. Considering the central coiled-coil fragment was used to develop the inhibitor, and this domain remains highly conserved between MuRF1 and MuRF2, further in vivo and in vitro experiments are warranted to clarify the specificity of this identified small molecule [82,101]. In addition, future work is required to determine the metabolism, bioavailability, and pharmacokinetics of this small molecule as well as optimal treatment durations, doses, and potential adverse effects. While the development of simultaneous MuRF1/2/3 inhibitors remains an intriguing possibility, careful consideration is required as parallel genetic knockdown of multiple MuRFs can induce severe cardiac failure despite skeletal muscle hypertrophy [112,116].
4.3. Other Novel Approaches to Inhibit MuRF1 Function
Rather than direct inhibition, other emerging studies have touted alternative approaches to target MuRF1 blockade. For example, MuRF1 was recently shown to be post-translationally regulated by attachment of small ubiquitin-like modifier (SUMO)-1 via the E2/E3 ligases UBC9 and PIASγ/4 at the coiled-coil domain [120], which may represent another pharmacological target to modulate MuRF1. Another ubiquitin-like protein is neuronal precursor cell-expressed developmentally downregulated protein 8 (NEDD8). This protein shares a higher amino acid homology (60%) with ubiquitin compared to SUMO (about 20%) and is ubiquitously expressed in all cell types with the highest expression observed in the heart and skeletal muscle [121]. Under catabolic conditions, NEDD8 has been proposed to function as a Ub substitute in order to prevent depletion of the ubiquitin pool, with recent proteome-wide high-throughput screens showing NEDD8 is a MuRF1 interacting partner [122]. Treatment of C2C12 cells exposed to the glucocorticoid dexamethasone with the neddylation inhibitor MLN4924 has shown promising effects, limiting myosin heavy chain loss by suppressing MuRF1 expression [122]. As such, further studies are clearly warranted to fully assess the efficacy of MLN4924 and the impact that NEDD8 inhibition may have to rescue skeletal muscle function and mass in vivo. Furthermore, PROteolysis TArgeting Chimeras (PROTACs) is a promising and appealing technology able to regulate protein function by degrading target proteins instead of inhibiting them. More specifically, these hetero-bifunctional molecules recruit an E3 ubiquitin ligase to a given substrate protein resulting in its targeted degradation [123]. This approach, that resembles a chemical knockdown strategy, shows more sensitivity to drug-resistant targets and a greater chance to regulate non-enzymatic functions [124]. As such, implementing this technology to target specific interactions of MuRF1 with known targets could be one promising strategy to combat muscle wasting. In addition, others have identified novel MuRF1-E2 networks [125], suggesting that targeting the E2 ligases interacting with MuRF1 could offer a new therapeutic opportunity towards inhibiting atrophy.
In contrast, other groups have also focused on small-molecule therapeutics using non-biased approaches without any predefined targets based upon mRNA signatures in atrophy [126]. These studies have resulted in anabolic signaling pathways being primarily modulated as a mean to inhibit subsequent proteolysis, with ursolic acid and tomatidine identified as novel small-molecules that suppress MuRF1 expression and muscle wasting in aging, starvation and disuse atrophy, by blocking expression of activating transcription factor 4 (ATF4)-Gadd45a/MEKK4 kinase complex activation [126]. While these experimental developments for MuRF1 inhibition are exciting, we should note that other approaches in the past have been able to reduce MuRF1 levels in line with improved muscle mass and function. These include, for example, nutritional supplements (e.g., leucine; see Figure 1) [61,62,98] as well as aerobic exercise training [127,128].
5. Conclusions
Skeletal muscle wasting represents a common trait in aging and many clinical settings, including cancer, heart failure, and critical illness, characterized by a shift towards a procatabolic state. During the past few years, an increasing number of experimental approaches have succeeded to attenuate muscle wasting in animal models, which have targeted procatabolic factors as a new emerging paradigm to support muscle function. In particular, strategies that have been able to suppress the activity of the main cellular pathway responsible for proteolysis (i.e., the UPS), and the upstream E3 ligase MuRF1 using small-molecules, have proven highly beneficial in the attenuation of muscle atrophy. However, an optimal approach to reduce muscle atrophy is likely via the combination of various therapies tailored to the patient’s current condition, which could include exercise training, leucine supplementation, β2-ARs, and small molecule inhibitors of MuRF1. This holistic approach is probably the most powerful stimuli for inhibiting a variety of procatabolic factors and thus muscle atrophy. Next, large animal models and ultimately, human patients will be required to validate many of these novel experimental concepts.
Funding
TSB supported by MRC UK (MR/S025472/1) and Heart Research UK (TRP 16/19).
Acknowledgments
Figures created using BioRender.com.
Conflicts of Interest
The authors declare no conflict of interest.
Abbreviations
| 5-HOB | 5-hydroxybenzothiazolone |
| β2-AR | β2-adrenergic receptor |
| ActRIIB/A | activin receptors type IIB and IIA |
| Akt | protein kinase B |
| ATF4 | activating transcription factor 4 |
| BMP | TGF-β/myostatin/bone morphogenetic protein |
| C/EBP-δ | CCAAT-enhancer binding protein delta |
| CC | coiled coil |
| CHIP | C terminus of HSC70-interacting protein |
| COS-box | C-terminal subgroup one signature |
| Fbxo40 | F-box protein 40 |
| FoxO | Forkhead box protein O |
| GDF11 | growth and differentiation factor 11 |
| HMB | Beta-Hydroxy b-methylbutyrate |
| IGF1 | insulin-like growth gactor 1 |
| IkBa | kappa light polypeptide gene enhancer in B-cells inhibitor, alpha |
| IkKb | inhibitor of nuclear factor kappa-B kinase subunit beta |
| IL-1 | Interleukin-1 |
| IL-6 | Interleukin-6 |
| MAFBx | muscle atrophy F-box |
| MFC | MuRF-family-specific motif |
| mTOR | mammalian target of rapamycin |
| MuRF1 | muscle-specific E3 ligase RING-finger protein 1 |
| MuRF2 | muscle-specific E3 ligase RING-finger protein 2 |
| MuRF3 | muscle-specific E3 ligase RING-finger protein 3 |
| MuRFs | muscle-specific RING finger proteins |
| MUSA1 | muscle ubiquitin ligase of SCF complex in atrophy-1 |
| NFkB | nuclear factor kappa-light-chain-enhancer of activated B cells |
| PDH | pyruvate dehydrogenase |
| PROTACs | PROteolysis TArgeting Chimeras |
| RING | really interesting new gene |
| ROS | reactive oxygen species |
| Smad | mothers against decapentaplegic homolog |
| SMART | specific of muscle atrophy and regulated by transcription |
| SRF | serum response factor |
| Stat-3 | signal transducer and activator of transcription 3 |
| SUMO | small ubiquitin-like modifier |
| TGF-beta | transforming growth factor-beta |
| TNF alpha | tumor necrosis factor alpha |
| TRAF6 | tumor necrosis factor receptor (TNFR)-associated factor 6 |
| TRIM | TRIpartite motif |
| Trim 32 | tripartite motif-containing protein 32 |
| UPS | ubiquitin proteasome system |
References
- Frontera, W.R.; Ochala, J. Skeletal Muscle: A Brief Review of Structure and Function. Behav. Genet. 2015, 45, 183–195. [Google Scholar] [CrossRef]
- Bonaldo, P.; Sandri, M. Cellular and molecular mechanisms of muscle atrophy. DMM Dis. Model. Mech. 2013, 6, 25–39. [Google Scholar] [CrossRef] [Green Version]
- Argilés, J.M.; Busquets, S.; Stemmler, B.; López-Soriano, F.J. Cachexia and sarcopenia: Mechanisms and potential targets for intervention. Curr. Opin. Pharmacol. 2015, 22, 100–106. [Google Scholar] [CrossRef] [PubMed]
- Powers, S.K.; Lynch, G.S.; Murphy, K.T.; Reid, M.B.; Zijdewind, I. Disease-Induced Skeletal Muscle Atrophy and Fatigue. Med. Sci. Sport Exerc. 2016, 48, 2307–2319. [Google Scholar] [CrossRef] [PubMed]
- Bowen, T.S.; Schuler, G.; Adams, V. Skeletal muscle wasting in cachexia and sarcopenia: Molecular pathophysiology and impact of exercise training. J. Cachexia Sarcopenia Muscle 2015, 6, 197–207. [Google Scholar] [CrossRef] [PubMed]
- Braun, T.; Gautel, M. Transcriptional mechanisms regulating skeletal muscle differentiation, growth and homeostasis. Nat. Rev. Mol. Cell Biol. 2011, 12, 349–361. [Google Scholar] [CrossRef] [PubMed]
- Cohen, S.; Nathan, J.A.; Goldberg, A.L. Muscle wasting in disease: Molecular mechanisms and promising therapies. Nat. Rev. Drug Discov. 2014, 14, 58–74. [Google Scholar] [CrossRef]
- Wang, X.H.; Mitch, W.E. Mechanisms of muscle wasting in chronic kidney disease. Nat. Rev. Nephrol. 2014, 10, 504–516. [Google Scholar] [CrossRef] [Green Version]
- Bell, R.A.V.; Al-Khalaf, M.; Megeney, L.A. The beneficial role of proteolysis in skeletal muscle growth and stress adaptation. Skelet. Muscle 2016, 6, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Pandurangan, M.; Hwang, I. The role of calpain in skeletal muscle. Anim. Cells Syst. 2012, 16, 431–437. [Google Scholar] [CrossRef] [Green Version]
- Sandri, M. Protein breakdown in muscle wasting: Role of autophagy-lysosome and ubiquitin-proteasome. Int. J. Biochem. Cell Biol. 2013, 45, 2121–2129. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lecker, S.H.; Goldberg, A.L.; Mitch, W.E. Protein degradation by the ubiquitin-proteasome pathway in normal and disease states. J. Am. Soc. Nephrol. 2006, 17, 1807–1819. [Google Scholar] [CrossRef] [PubMed]
- Reid, M.B.; Judge, A.R.; Bodine, S.C. CrossTalk opposing view: The dominant mechanism causing disuse muscle atrophy is proteolysis. J. Physiol. 2014, 592, 5345–5347. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murton, A.J.; Constantin, D.; Greenhaff, P.L. The involvement of the ubiquitin proteasome system in human skeletal muscle remodelling and atrophy. Biochim. Biophys. Acta Mol. Basis Dis. 2008, 1782, 730–743. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumari, N.; Lee, K.K.; Jha, S. Targeting the Ubiquitin Proteasome System in Cancer. In Neoplasm; InTech: London, UK, 2018. [Google Scholar] [CrossRef] [Green Version]
- Bartoli, M.; Richard, I. Calpains in muscle wasting. Int. J. Biochem. Cell Biol. 2005, 37, 2115–2133. [Google Scholar] [CrossRef]
- Du, J.; Wang, X.; Miereles, C.; Bailey, J.L.; Debigaré, R.; Zheng, B.; Price, S.R.; Mitch, W.E. Activation of caspase-3 is an initial step triggering accelerated muscle proteolysis in catabolic conditions. J. Clin. Investig. 2004, 113, 115–123. [Google Scholar] [CrossRef]
- Bulatov, E.; Zagidullin, A.A.; Valiullina, A.; Sayarova, R.; Rizvanov, A.A. Small Molecule Modulators of RING-Type E3 Ligases: MDM and Cullin Families as Targets. Front. Pharmacol. 2018, 9, 1–8. [Google Scholar] [CrossRef]
- Bodine, S.C.; Latres, E.; Baumhueter, S.; Lai, V.K.M.; Nunez, L.; Clarke, B.A.; Poueymirou, W.T.; Panaro, F.J.; Na, E.; Dharmarajan, K.; et al. Identification of ubiquitin ligases required for skeletal Muscle Atrophy. Science 2001, 294, 1704–1708. [Google Scholar] [CrossRef]
- Gomes, M.D.; Lecker, S.H.; Jagoe, R.T.; Navon, A.; Goldberg, A.L. Expressed During Muscle Atrophy. Structure 2001, 1–6. [Google Scholar]
- Bodine, S.C.; Baehr, L.M. Skeletal muscle atrophy and the E3 ubiquitin ligases MuRF1 and MAFbx/atrogin-1. Am. J. Physiol. Endocrinol. Metab. 2014, 307, E469–E484. [Google Scholar] [CrossRef] [Green Version]
- Larsson, L.; Degens, H.; Li, M.; Salviati, L.; Lee, Y.I.; Thompson, W.; Kirkland, J.L.; Sandri, M. Sarcopenia: Aging-related loss of muscle mass and function. Physiol. Rev. 2019, 99, 427–511. [Google Scholar] [CrossRef]
- Clarke, B.A.; Drujan, D.; Willis, M.S.; Murphy, L.O.; Corpina, R.A.; Burova, E.; Rakhilin, S.V.; Stitt, T.N.; Patterson, C.; Latres, E.; et al. The E3 Ligase MuRF1 Degrades Myosin Heavy Chain Protein in Dexamethasone-Treated Skeletal Muscle. Cell. Metab. 2007, 6, 376–385. [Google Scholar] [CrossRef] [Green Version]
- Cohen, S.; Brault, J.J.; Gygi, S.P.; Glass, D.J.; Valenzuela, D.M.; Gartner, C.; Latres, E.; Goldberg, A.L. During muscle atrophy, thick, but not thin, filament components are degraded by MuRF1-dependent ubiquitylation. J. Cell Biol. 2009, 185, 1083–1095. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Polge, C.; Heng, A.-E.; Jarzaguet, M.; Ventadour, S.; Claustre, A.; Combaret, L.; Bechet, D.; Matondo, M.; Uttenweiler, S.; Monsarrat, B.; et al. Muscle actin is polyubiquitinylated in vitro and in vivo and targeted for breakdown by the E3 ligase MuRF1. FASEB J. 2011, 25, 3790–3802. [Google Scholar] [CrossRef]
- Cohen, S.; Zhai, B.; Gygi, S.P.; Goldberg, A.L. Ubiquitylation by Trim32 causes coupled loss of desmin, Z-bands, and thin filaments in muscle atrophy. J. Cell Biol. 2012, 198, 575–589. [Google Scholar] [CrossRef] [PubMed]
- Paul, P.K.; Gupta, S.K.; Bhatnagar, S.; Panguluri, S.K.; Darnay, B.G.; Choi, Y.; Kumar, A. Targeted ablation of TRAF6 inhibits skeletal muscle wasting in mice. J. Cell Biol. 2010, 191, 1395–1411. [Google Scholar] [CrossRef] [Green Version]
- Arndt, V.; Dick, N.; Tawo, R.; Dreiseidler, M.; Wenzel, D.; Hesse, M.; Fürst, D.O.; Saftig, P.; Saint, R.; Fleischmann, B.K.; et al. Chaperone-Assisted Selective Autophagy Is Essential for Muscle Maintenance. Curr. Biol. 2010, 20, 143–148. [Google Scholar] [CrossRef] [PubMed]
- Shi, J.; Luo, L.; Eash, J.; Ibebunjo, C.; Glass, D.J. The SCF-Fbxo40 complex induces IRS1 ubiquitination in skeletal muscle, limiting IGF1 signaling. Dev. Cell. 2011, 21, 835–847. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Milan, G.; Romanello, V.; Pescatore, F.; Armani, A.; Paik, J.-H.; Frasson, L.; Seydel, A.; Zhao, J.; Abraham, R.; Goldberg, A.L.; et al. Regulation of autophagy and the ubiquitin-proteasome system by the FoxO transcriptional network during muscle atrophy. Nat. Commun. 2015, 6, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Masiero, E.; Agatea, L.; Mammucari, C.; Blaauw, B.; Loro, E.; Komatsu, M.; Metzger, D.; Reggiani, C.; Schiaffino, S.; Sandri, M. Autophagy Is Required to Maintain Muscle Mass. Cell Metab. 2009, 10, 507–515. [Google Scholar] [CrossRef]
- Jokl, E.J.; Blanco, G. Disrupted autophagy undermines skeletal muscle adaptation and integrity. Mamm. Genome 2016, 27, 525–537. [Google Scholar] [CrossRef]
- Bilodeau, P.A.; Coyne, E.S.; Wing, S.S. The ubiquitin proteasome system in atrophying skeletal muscle: Roles and regulation. Am. J. Physiol. Cell Physiol. 2016, 311, C392–C403. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Salazar, J.J.; Michele, D.E.; Brooks, S.V. Inhibition of calpain prevents muscle weakness and disruption of sarcomere structure during hindlimb suspension. J. Appl. Physiol. 2010, 108, 120–127. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Supinski, G.S.; Wang, L.; Song, X.H.; Moylan, J.S.; Callahan, L.A. Muscle-specific calpastatin overexpression prevents diaphragm weakness in cecal ligation puncture-induced sepsis. J. Appl. Physiol. 2014, 117, 921–929. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Supinski, G.S.; Callahan, L.A. Caspase activation contributes to endotoxin-induced diaphragm weakness. J. Appl. Physiol. 2006, 100, 1770–1777. [Google Scholar] [CrossRef]
- Stratos, I.; Li, Z.; Rotter, R.; Herlyn, P.; Mittlmeier, T.; Vollmar, B. Inhibition of caspase mediated apoptosis restores muscle function after crush injury in rat skeletal muscle. Apoptosis 2012, 17, 269–277. [Google Scholar] [CrossRef] [Green Version]
- Rea, I.M.; Gibson, D.S.; McGilligan, V.; McNerlan, S.E.; Denis Alexander, H.; Ross, O.A. Age and age-related diseases: Role of inflammation triggers and cytokines. Front. Immunol. 2018, 9, 1–28. [Google Scholar] [CrossRef]
- Jin, B.; Li, Y.-P. Curcumin prevents lipopolysaccharide-induced atrogin-1/MAFbx upregulation and muscle mass loss. J. Cell Biochem. 2007, 100, 960–969. [Google Scholar] [CrossRef] [Green Version]
- Caron, A.Z.; Haroun, S.; Leblanc, E.; Trensz, F.; Guindi, C.; Amrani, A.; Grenier, G. The proteasome inhibitor MG132 reduces immobilization-induced skeletal muscle atrophy in mice. BMC Musculoskelet. Disord. 2011, 12, 185. [Google Scholar] [CrossRef] [Green Version]
- Pérez-Baos, S.; Prieto-Potin, I.; Román-Blas, J.A.; Sánchez-Pernaute, O.; Largo, R.; Herrero-Beaumont, G. Mediators and patterns of muscle loss in chronic systemic inflammation. Front. Physiol. 2018, 9, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Yakabe, M.; Ogawa, S.; Ota, H.; Iijima, K.; Eto, M.; Ouchi, Y.; Akishita, M. Inhibition of interleukin-6 decreases atrogene expression and ameliorates tail suspension-induced skeletal muscle atrophy. PLoS ONE 2018, 13, e20592. [Google Scholar] [CrossRef] [PubMed]
- Adams, V.; Mangner, N.; Gasch, A.; Krohne, C.; Gielen, S.; Hirner, S.; Thierse, H.-J.; Witt, C.C.; Linke, A.; Schuler, G.; et al. Induction of MuRF1 Is Essential for TNF-α-Induced Loss of Muscle Function in Mice. J. Mol. Biol. 2008, 384, 48–59. [Google Scholar] [CrossRef] [PubMed]
- Camporez, J.-P.G.; Petersen, M.C.; Abudukadier, A.; Moreira, G.V.; Jurczak, M.J.; Friedman, G.; Haqq, C.M.; Petersen, K.F.; Shulman, G.I. Anti-myostatin antibody increases muscle mass and strength and improves insulin sensitivity in old mice. Proc. Natl. Acad. Sci USA 2016, 113, 2212–2217. [Google Scholar] [CrossRef] [Green Version]
- Zhou, X.; Wang, J.L.; Lu, J.; Song, Y.; Kwak, K.S.; Jiao, Q.; Rosenfeld, R.; Chen, Q.; Boone, T.; Simonet, W.S.; et al. Reversal of cancer cachexia and muscle wasting by ActRIIB antagonism leads to prolonged survival. Cell 2010, 142, 531–543. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hammers, D.W.; Sleeper, M.M.; Forbes, S.C.; Coker, C.C.; Jirousek, M.R.; Zimmer, M.; Walter, G.A.; Sweeney, H.L. Disease-modifying effects of orally bioavailable NF-κB inhibitors in dystrophin-deficient muscle. JCI Insight 2016, 1. [Google Scholar] [CrossRef] [Green Version]
- Zimmers, T.A.; Jiang, Y.; Wang, M.; Liang, T.W.; Rupert, J.E.; Au, E.; Marino, F.E.; Couch, M.E.; Koniaris, L.G. Exogenous GDF11 induces cardiac and skeletal muscle dysfunction and wasting. Basic Res. Cardiol. 2017, 112, 48. [Google Scholar] [CrossRef]
- Jin, Q.; Qiao, C.; Li, J.; Xiao, B.; Li, J.; Xiao, X. A GDF11/myostatin inhibitor, GDF11 propeptide-Fc, increases skeletal muscle mass and improves muscle strength in dystrophic mdx mice. Skelet. Muscle. 2019, 9, 1–16. [Google Scholar] [CrossRef]
- Zhang, L.; Pan, J.; Dong, Y.; Tweardy, D.J.; Dong, Y.; Garibotto, G.; Mitch, W.E. Stat3 Activation Links a C/EBPδ to Myostatin Pathway to Stimulate Loss of Muscle Mass. Cell Metab. 2013, 18, 368–379. [Google Scholar] [CrossRef] [Green Version]
- Schakman, O.; Kalista, S.; Barbé, C.; Loumaye, A.; Thissen, J.P. Glucocorticoid-induced skeletal muscle atrophy. Int. J. Biochem. Cell Biol. 2013, 45, 2163–2172. [Google Scholar] [CrossRef]
- Frost, R.A.; Nystrom, G.J.; Jefferson, L.S.; Lang, C.H. Hormone, cytokine, and nutritional regulation of sepsis-induced increases in atrogin-1 and MuRF1 in skeletal muscle. Am. J. Physiol. Endocrinol. Metab. 2007, 292. [Google Scholar] [CrossRef] [Green Version]
- Tiao, G.; Fagan, J.; Roegner, V.; Lieberman, M.; Wang, J.J.; Fischer, J.E.; Hasselgren, P.O. Energy-ubiquitin-dependent muscle proteolysis during sepsis in rats is regulated by glucocorticoids. J. Clin. Investig. 1996, 97, 339–348. [Google Scholar] [CrossRef] [Green Version]
- Qiu, J.; Fang, Q.; Xu, T.; Wu, C.; Xu, L.; Wang, L.; Yang, X.; Yu, S.; Zhang, Q.; Ding, F.; et al. Mechanistic role of reactive oxygen species and therapeutic potential of antioxidants in denervation-or fasting-induced skeletal muscle atrophy. Front. Physiol. 2018, 9, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Powers, S.K.; Hudson, M.B.; Nelson, W.; Talbert, E.E.; Min, K.; Szeto, H.H.; Kavazis, A.N.; Smuder, A.J. Mitochondria-targeted antioxidants protect against mechanical ventilation-induced diaphragm weakness*. Crit. Care Med. 2011, 39, 1749–1759. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abrigo, J.; Rivera, J.C.; Simon, F.; Cabrera, D.; Cabello-Verrugio, C. Transforming growth factor type beta (TGF-β) requires reactive oxygen species to induce skeletal muscle atrophy. Cell Signal 2016, 28, 366–376. [Google Scholar] [CrossRef] [PubMed]
- Patel, R.; Rinker, L.; Peng, J.; Chilian, W.M. Reactive Oxygen Species: The Good and the Bad. In Reactive Oxygen Species (ROS) in Living Cells; InTech: London, UK, 2018. [Google Scholar] [CrossRef] [Green Version]
- Rothwarf, D.M.; Karin, M. The NF-kappa B activation pathway: A paradigm in information transfer from membrane to nucleus. Sci. STKE 1999, 1999. [Google Scholar] [CrossRef]
- Belova, S.P.; Shenkman, B.S.; Kostrominova, T.Y.; Nemirovskaya, T.L. Paradoxical effect of IKKβ inhibition on the expression of E3 ubiquitin ligases and unloading-induced skeletal muscle atrophy. Physiol. Rep. 2017, 5, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Shimizu, H.; Langenbacher, A.D.; Huang, J.; Wang, K.; Otto, G.W.; Geisler, R.; Wang, Y.; Chen, J.-N. The calcineurin-FoxO-MuRF1 signaling pathway regulates myofibril integrity in cardiomyocytes. eLife 2017, 6, 1–19. [Google Scholar] [CrossRef] [PubMed]
- Jamart, C.; Raymackers, J.M.; An, G.L.; Deldicque, L.; Francaux, M. Prevention of muscle disuse atrophy by MG132 proteasome inhibitor. Muscle Nerve 2011, 43, 708–715. [Google Scholar] [CrossRef]
- Baptista, I.L.; Silvestre, J.G.; Silva, W.J.; Labeit, S.; Moriscot, A.S. FoxO3a suppression and VPS34 activity are essential to anti-atrophic effects of leucine in skeletal muscle. Cell Tissue Res. 2017, 369, 381–394. [Google Scholar] [CrossRef]
- Baptista, I.L.; Leal, M.L.; Artioli, G.G.; Aoki, M.S.; Fiamoncini, J.; Turri, A.O.; Curi, R.; Miyabara, E.H.; Moriscot, A. Leucine attenuates skeletal muscle wasting via inhibition of ubiquitin ligases. Muscle Nerve 2010, 41, 800–808. [Google Scholar] [CrossRef]
- Smith, H.J.; Mukerji, P.; Tisdale, M.J. Attenuation of proteasome-induced proteolysis in skeletal muscle by β-hydroxy-β-methylbutyrate in cancer-induced muscle loss. Cancer Res. 2005, 65, 277–283. [Google Scholar]
- Hao, Y.; Jackson, J.R.; Wang, Y.; Edens, N.; Pereira, S.L.; Alway, S.E. β-Hydroxy-β-methylbutyrate reduces myonuclear apoptosis during recovery from hind limb suspension-induced muscle fiber atrophy in aged rats. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2011, 301. [Google Scholar] [CrossRef] [Green Version]
- Noh, K.K.; Chung, K.W.; Choi, Y.J.; Park, M.H.; Jang, E.J.; Park, C.H.; Yoon, C.; Kim, N.D.; Kim, M.K.; Chung, H.Y. β-hydroxy β-methylbutyrate improves dexamethasone-induced muscle atrophy by modulating the muscle degradation pathway in SD rat. PLoS ONE 2014, 9, e102947. [Google Scholar] [CrossRef]
- Holeček, M. Beta-hydroxy-beta-methylbutyrate supplementation and skeletal muscle in healthy and muscle-wasting conditions. J. Cachexia Sarcopenia Muscle 2017, 8, 529–541. [Google Scholar] [CrossRef]
- Chen, L.; Chen, L.; Wan, L.; Huo, Y.; Huang, J.; Li, J.; Lu, J.; Xin, B.; Yang, Q.; Guo, C. Matrine improves skeletal muscle atrophy by inhibiting E3 ubiquitin ligases and activating the Akt/mTOR/FoxO3α signaling pathway in C2C12 myotubes and mice. Oncol. Rep. 2019, 42, 479–494. [Google Scholar] [CrossRef]
- Lynch, G.S.; Ryall, J.G. Role of β-adrenoceptor signaling in skeletal muscle: Implications for muscle wasting and disease. Physiol. Rev. 2008, 88, 729–767. [Google Scholar] [CrossRef] [Green Version]
- Salazar-Degracia, A.; Busquets, S.; Argilés, J.M.; Bargalló-Gispert, N.; López-Soriano, F.J.; Barreiro, E. Effects of the beta 2 agonist formoterol on atrophy signaling, autophagy, and muscle phenotype in respiratory and limb muscles of rats with cancer-induced cachexia. Biochimie 2018, 149, 79–91. [Google Scholar] [CrossRef] [Green Version]
- Koziczak-Holbro, M.; Rigel, D.F.; Dumotier, B.; Sykes, D.A.; Tsao, J.; Nguyen, N.-H.; Bosch, J.; Jourdain, M.; Flotte, L.; Adachi, Y.; et al. Pharmacological characterization of a novel 5-hydroxybenzothiazolone-derived b2-adrenoceptor agonist with functional selectivity for anabolic effects on skeletal muscle resulting in a wider cardiovascular safety window in preclinical studiess. J. Pharmacol. Exp. Ther. 2019, 369, 188–199. [Google Scholar] [CrossRef] [Green Version]
- Pötsch, M.S.; Ishida, J.; Palus, S.; Tschirner, A.; Von Haehling, S.; Doehner, W.; Anker, S.D.; Springer, J. MT-102 prevents tissue wasting and improves survival in a rat model of severe cancer cachexia. J. Cachexia Sarcopenia Muscle 2020, 11, 594–605. [Google Scholar] [CrossRef]
- Pötsch, M.S.; Tschirner, A.; Palus, S.; Von Haehling, S.; Doehner, W.; Beadle, J.; Coats, A.J.S.; Anker, S.D.; Springer, J. The anabolic catabolic transforming agent (ACTA) espindolol increases muscle mass and decreases fat mass in old rats. J. Cachexia Sarcopenia Muscle 2014, 5, 149–158. [Google Scholar] [CrossRef]
- Penna, F.; Bonetto, A.; Aversa, Z.; Minero, V.G.; Fanelli, F.R.; Costelli, P.; Muscaritoli, M. Effect of the specific proteasome inhibitor bortezomib on cancer-related muscle wasting. J. Cachexia Sarcopenia Muscle 2016, 345–354. [Google Scholar] [CrossRef]
- Thibaudeau, T.A.; Smith, D.M. A practical review of proteasome pharmacology. Pharmacol. Rev. 2019, 71, 170–197. [Google Scholar] [CrossRef] [Green Version]
- Beehler, B.C.; Sleph, P.G.; Benmassaoud, L.; Grover, G.J. Reduction of Skeletal Muscle Atrophy by a Proteasome Inhibitor in a Rat Model of Denervation. Exp. Biol. Med. 2006, 231, 335–341. [Google Scholar] [CrossRef] [PubMed]
- Van Hees, H.W.H.; Li, Y.-P.; Ottenheijm, C.A.C.; Jin, B.; Pigmans, C.J.C.; Linkels, M.; Dekhuijzen, P.N.R.; Heunks, L.M.A. Proteasome inhibition improves diaphragm function in congestive heart failure rats Hieronymus. Am. J. Physiol. Lung Cell Mol. Physiol. 2008, 294. [Google Scholar] [CrossRef]
- Agten, A.; Maes, K.; Thomas, D.; Cielen, N.; Van Hees, H.W.; Dekhuijzen, R.P.; Decramer, M.; Gayan-Ramirez, G. Bortezomib partially protects the rat diaphragm from ventilator-induced diaphragm dysfunction. Crit. Care Med. 2012, 40, 2449–2455. [Google Scholar] [CrossRef]
- Smuder, A.J.; Nelson, W.B.; Hudson, M.B.; Kavazis, A.N.; Powers, S.K. Inhibition of the ubiquitin-proteasome pathway does not protect against ventilator-induced accelerated proteolysis or atrophy in the diaphragm. Anesthesiology 2014, 121, 115–126. [Google Scholar] [CrossRef] [Green Version]
- Hungria, V.; Crusoé, E.D.Q.; Bittencourt, R.I.; Maiolino, A.; Magalhães, R.J.P.; Sobrinho, J.D.N.; Pinto, J.V.; Fortes, R.C.; Moreira, E.D.S.; Tanaka, P.Y. New Proteasome Inhibitors in the Treatment of Multiple Myeloma. Hematol. Transfus. Cell Ther. 2019, 41, 76–83. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Li, C.; Peng, X.; Kang, Q.; Deng, D.; Zhang, L.; Zheng, Y.; Wang, C.; Qiao, Z.; Guo, D.; et al. Combined Treatment of Carfilzomib and Z-VAD-Fmk Inhibits Skeletal Proteolysis and Apoptosis and Ameliorates Cancer Cachexia. Med. Oncol. 2015, 32. [Google Scholar] [CrossRef]
- Zhang, L.; Tang, H.; Kou, Y.; Li, R.; Zheng, Y.; Wang, Q.; Zhou, X.; Jin, L. MG132-mediated inhibition of the ubiquitin-proteasome pathway ameliorates cancer cachexia. J. Cancer Res. Clin. Oncol. 2013, 139, 1105–1115. [Google Scholar] [CrossRef]
- Adams, V.; Bowen, T.S.; Werner, S.; Barthel, P.; Amberger, C.; Konzer, A.; Graumann, J.; Sehr, P.; Lewis, J.; Provaznik, J.; et al. Small-molecule-mediated chemical knock-down of MuRF1/MuRF2 and attenuation of diaphragm dysfunction in chronic heart failure. J. Cachexia Sarcopenia Muscle 2019, 10, 1102–1115. [Google Scholar] [CrossRef]
- Guglielmi, V.; Nowis, D.; Tinelli, M.; Malatesta, M.; Paoli, L.; Marini, M.; Manganotti, P.; Sadowski, R.; Wilczyński, G.M.; Meneghini, V.; et al. Bortezomib-induced muscle toxicity in multiple myeloma. J. Neuropathol. Exp. Neurol. 2017, 76, 620–630. [Google Scholar] [CrossRef] [PubMed]
- Galdeano, C. Drugging the undruggable: Targeting challenging E3 ligases for personalized medicine. Future Med. Chem. 2017, 9, 347–350. [Google Scholar] [CrossRef] [PubMed]
- Castillero, E.; Alamdari, N.; Lecker, S.H.; Hasselgren, P.O. Suppression of atrogin-1 and MuRF1 prevents dexamethasone-induced atrophy of cultured myotubes. Metabolism 2013, 62, 1495–1502. [Google Scholar] [CrossRef] [PubMed]
- Lyon, R.C.; Lange, S.; Sheikh, F. Breaking down protein degradation mechanisms in cardiac muscle. Trends Mol. Med. 2013, 19, 239–249. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kudryashova, E.; Wu, J.; Havton, L.A.; Spencer, M.J. Deficiency of the E3 ubiquitin ligase TRIM32 in mice leads to a myopathy with a neurogenic component. Hum. Mol. Genet. 2009, 18, 1353–1367. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Wang, Z.; De La Torre, R.; Barling, A.; Tsujikawa, T.; Hornick, N.; Hanifin, J.; Simpson, E.; Wang, Y.; Swanzey, E.; et al. Trim32 Deficiency Enhances Th2 Immunity and Predisposes to Features of Atopic Dermatitis. J. Investig. Dermatol. 2017, 137, 359–366. [Google Scholar] [CrossRef] [Green Version]
- Wang, M.; Luo, W.; Zhang, Y.; Yang, R.; Li, X.; Guo, Y.; Zhang, C.; Yang, R.; Gao, W.-Q. Trim32 suppresses cerebellar development and tumorigenesis by degrading Gli1/sonic hedgehog signaling. Cell Death Differ. 2020, 27, 1286–1299. [Google Scholar] [CrossRef]
- Centner, T.; Yano, J.; Kimura, E.; McElhinny, A.S.; Pelin, K.; Witt, C.C.; Bang, M.-L.; Trombitas, K.; Granzier, H.; Gregorio, C.C.; et al. Identification of muscle specific ring finger proteins as potential regulators of the titin kinase domain. J. Mol. Biol. 2001, 306, 717–726. [Google Scholar] [CrossRef] [Green Version]
- Pizon, V.; Iakovenko, A.; Van Der Ven, P.F.M.; Kelly, R.; Fatu, C.; Fürst, D.O.; Karsenti, E.; Gautel, M. Transient association of titin and myosin with microtubules in nascent myofibrils directed by the MURF2 RING-finger protein. J. Cell Sci. 2002, 115, 4469–4482. [Google Scholar] [CrossRef] [Green Version]
- Mayans, O.; Labeit, S. MuRFs Specialized Members of the TRIM/RBCC Family with Roles in the Regulation of the Trophic State of Muscle and Its Metabolism; Landes Bioscience: Austin, TX, USA; Springer: Berlin, Germany, 2012; pp. 119–130. [Google Scholar]
- Spencer, J.A.; Eliazer, S.; Ilaria, R.L.; Richardson, J.A.; Olson, E.N. Regulation of microtubule dynamics and myogenic differentiation by MURF, a striated muscle RING-finger protein. J. Cell Biol. 2000, 150, 771–784. [Google Scholar] [CrossRef] [Green Version]
- Esposito, D.; Koliopoulos, M.G.; Rittinger, K. Structural determinants of TRIM protein function. Biochem. Soc. Trans. 2017, 45, 183–191. [Google Scholar] [CrossRef] [PubMed]
- Stevens, M.; Franke, B.; Skorupka, K.A.; Cafiso, D.S.; Pornillos, O.; Mayans, O.; Norman, D.G. Exploration of the TRIM Fold of MuRF1 Using EPR Reveals a Canonical Antiparallel Structure and Extended COS-Box. J. Mol. Biol. 2019, 431, 2900–2909. [Google Scholar] [CrossRef]
- Franke, B.; Gasch, A.; Rodriguez, D.; Chami, M.; Khan, M.M.; Rudolf, R.; Bibby, J.; Hanashima, A.; Bogomolovas, J.; Von Castelmur, E.; et al. Molecular basis for the fold organization and sarcomeric targeting of the muscle atrogin muRF1. Open Biol. 2014, 4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mrosek, M.; Meier, S.; Ucurum-Fotiadis, Z.; Von Castelmur, E.; Hedbom, E.; Lustig, A.; Grzesiek, S.; Labeit, D.; Labeit, S.; Mayans, O. Structural Analysis of B-Box 2 from MuRF1: Identification of a Novel Self-Association Pattern in a RING-like Fold. Biochemistry 2008, 47, 10722–10730. [Google Scholar] [CrossRef] [PubMed]
- Pereira, M.G.; Baptista, I.L.; Carlassara, E.O.C.; Moriscot, A.S.; Aoki, M.S.; Miyabara, E.H. Leucine supplementation improves skeletal muscle regeneration after cryolesion in rats. PLoS ONE 2014, 9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Granzier, H.L.; Labeit, S. The Giant Protein Titin: A Major Player in Myocardial Mechanics, Signaling, and Disease. Circ. Res. 2004, 94, 284–295. [Google Scholar] [CrossRef] [Green Version]
- Kedar, V.; McDonough, H.; Arya, R.; Li, H.H.; Rockman, H.A.; Patterson, C. Muscle-specific RING finger 1 is a bona fide ubiquitin ligase that degrades cardiac troponin I. Proc. Natl. Acad. Sci. USA 2004, 101, 18135–18140. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bowen, T.S.; Adams, V.; Werner, S.; Fischer, T.; Vinke, P.; Brogger, M.N.; Mangner, N.; Linke, A.; Sehr, P.; Lewis, J.; et al. Small-molecule inhibition of MuRF1 attenuates skeletal muscle atrophy and dysfunction in cardiac cachexia. J. Cachexia Sarcopenia Muscle 2017, 8, 939–953. [Google Scholar] [CrossRef] [PubMed]
- Perera, S.; Mankoo, B.; Gautel, M. Developmental regulation of MURF E3 ubiquitin ligases in skeletal muscle. J. Muscle Res. Cell Motil. 2012, 33, 107–122. [Google Scholar] [CrossRef] [Green Version]
- Lodka, D.; Pahuja, A.; Geers-Knörr, C.; Scheibe, R.J.; Nowak, M.; Hamati, J.; Köhncke, C.; Purfürst, B.; Kanashova, T.; Schmidt, S.; et al. Muscle RING-finger 2 and 3 maintain striated-muscle structure and function. J. Cachexia Sarcopenia Muscle 2016, 7, 165–180. [Google Scholar] [CrossRef] [Green Version]
- Mattox, T.A.; Young, M.E.; Rubel, C.E.; Spaniel, C.; Rodriguez, J.E.; Grevengoed, T.J.; Gautel, M.; Xu, Z.; Anderson, E.J.; Willis, M.S. MuRF1 activity is present in cardiac mitochondria and regulates reactive oxygen species production in vivo. J. Bioenerg. Biomembr. 2014, 46, 173–187. [Google Scholar] [CrossRef] [Green Version]
- Hwee, D.T.; Baehr, L.M.; Philp, A.; Baar, K.; Bodine, S.C. Maintenance of muscle mass and load-induced growth in Muscle RING Finger 1 null mice with age. Aging Cell 2014, 13, 92–101. [Google Scholar] [CrossRef]
- Nguyen, T.; Bowen, T.S.; Augstein, A.; Schauer, A.; Gasch, A.; Linke, A.; Labeit, S.; Adams, V. Expression of MuRF1 or MuRF2 is essential for the induction of skeletal muscle atrophy and dysfunction in a murine pulmonary hypertension model. Skelet. Muscle. 2020, 10, 12. [Google Scholar] [CrossRef]
- Olive, M.; Abdul-Hussein, S.; Oldfors, A.; González-Costello, J.; Van Der Ven, P.F.; Fürst, D.O.; González, L.; Moreno, D.; Torrejón-Escribano, B.; Alió, J.; et al. New cardiac and skeletal protein aggregate myopathy associated with combined MuRF1 and MuRF3 mutations. Hum. Mol. Genet. 2015, 24, 3638–3650. [Google Scholar] [CrossRef]
- Fielitz, J.; Kim, M.-S.; Shelton, J.M.; Latif, S.; Spencer, J.A.; Glass, D.J.; Richardson, J.A.; Bassel-Duby, R.; Olson, E.N. Myosin accumulation and striated muscle myopathy result from the loss of muscle RING finger 1 and 3. J. Clin. Investig. 2007, 117, 2486–2495. [Google Scholar] [CrossRef]
- Fielitz, J.; Van Rooij, E.; Spencer, J.A.; Shelton, J.M.; Latif, S.; Van Der Nagel, R.; Bezprozvannaya, S.; De Windt, L.; Richardson, J.A.; Bassel-Duby, R.; et al. Loss of muscle-specific RING-finger 3 predisposes the heart to cardiac rupture after myocardial infarction. Proc. Natl. Acad. Sci. USA 2007, 104, 4377–4382. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quintana, M.T.; He, J.; Sullivan, J.; Grevengoed, T.J.; Schisler, J.; Han, Y.; Hill, J.A.; Yates, C.; Stansfield, E.W.; Mapanga, R.F.; et al. Muscle ring finger-3 protects against diabetic cardiomyopathy induced by a high fat diet. BMC Endocr. Disord. 2015, 15, e36. [Google Scholar] [CrossRef] [Green Version]
- Edelmann, M.J.; Nicholson, B.; Kessler, B.M. Pharmacological targets in the ubiquitin system offer new ways of treating cancer, neurodegenerative disorders and infectious diseases. Expert Rev. Mol. Med. 2011, 13, 1–17. [Google Scholar] [CrossRef] [Green Version]
- Witt, C.C.; Witt, S.H.; Lerche, S.; Labeit, D.; Back, W.; Labeit, S. Cooperative control of striated muscle mass and metabolism by MuRF1 and MuRF2. EMBO J. 2008, 27, 350–360. [Google Scholar] [CrossRef] [Green Version]
- Wexler, B.C. Alloxan-induced diabetes in young vs old sprague-dawley rats. Exp. Gerontol. 1981, 16, 47–58. [Google Scholar] [CrossRef]
- Eddins, M.J.; Marblestone, J.G.; Kumar, K.G.S.; Leach, C.A.; Sterner, D.E.; Mattern, M.R.; Nicholson, B. Targeting the Ubiquitin E3 Ligase MuRF1 to Inhibit Muscle Atrophy. Cell Biochem. Biophys. 2011, 60, 113–118. [Google Scholar] [CrossRef] [PubMed]
- Witt, S.H.; Granzier, H.; Witt, C.C.; Labeit, S. MURF-1 and MURF-2 target a specific subset of myofibrillar proteins redundantly: Towards understanding MURF-dependent muscle ubiquitination. J. Mol. Biol. 2005, 350, 713–722. [Google Scholar] [CrossRef]
- Willis, M.S.; Wadosky, K.; Rodriguez, J.E.; Schisler, J.; Lockyer, P.; Hilliard, E.G.; Glass, D.J.; Patterson, C. Muscle ring finger 1 and muscle ring finger 2 are necessary but functionally redundant during developmental cardiac growth and regulate E2F1-mediated gene expression in vivo. Cell Biochem. Funct. 2014, 32, 39–50. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hirner, S.; Krohne, C.; Schuster, A.; Hoffmann, S.; Witt, S.; Erber, R.; Sticht, C.; Gasch, A.; Labeit, S.; Labeit, D. MuRF1-dependent Regulation of Systemic Carbohydrate Metabolism as Revealed from Transgenic Mouse Studies. J. Mol. Biol. 2008, 379, 666–677. [Google Scholar] [CrossRef]
- Crossland, H.; Skirrow, S.; Puthucheary, Z.A.; Constantin-Teodosiu, D.; Greenhaff, P.L. The impact of immobilisation and inflammation on the regulation of muscle mass and insulin resistance: Different routes to similar end-points. J. Physiol. 2019, 597, 1259–1270. [Google Scholar] [CrossRef] [Green Version]
- Silvestre, J.; Baptista, I.L.; Silva, W.; Cruz, A.; Silva, M.; Miyabara, E.H.; Labeit, S.; Moriscot, A. The e3 ligase murf2 plays a key role in the functional capacity of skeletal muscle fibroblasts. Braz. J. Med. Biol. Res. 2019, 52, 1–10. [Google Scholar] [CrossRef]
- Heras, G.; Namuduri, A.V.; Traini, L.; Shevchenko, G.; Falk, A.; Lind, S.B.; Jia, M.; Tian, G.; Gastaldello, S.; Mi, J. Muscle RING-finger protein-1 (MuRF1) functions and cellular localization are regulated by SUMO1 post-translational modification. J. Mol. Cell Biol. 2019, 11, 356–370. [Google Scholar] [CrossRef] [Green Version]
- Li, J.; Johnson, J.A.; Su, H. Ubiquitin and Ubiquitin-like Proteins in Cardiac Disease and Protection. Curr. Drug Targets 2018, 19, 989–1002. [Google Scholar] [CrossRef]
- Nowak, M.; Suenkel, B.; Porras, P.; Migotti, R.; Schmidt, F.; Kny, M.; Zhu, X.; Wanker, E.; Dittmar, G.; Fielitz, J.; et al. DCAF8, a Novel MuRF1 Interaction Partner, Promotes Muscle Atrophy. J. Cell Sci. 2019, 132. [Google Scholar] [CrossRef] [Green Version]
- Smith, B.; Wang, S.; Jaime-Figueroa, S.; Harbin, A.; Wang, J.; Hamman, B.D.; Crews, C.M. Differential PROTAC Substrate Specificity Dictated by Orientation of Recruited E3 Ligase. Nat. Commun. 2019, 10, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Sun, X.; Gao, H.; Yang, Y.; He, M.; Wu, Y.; Song, Y.; Tong, Y.; Rao, Y. Protacs: Great Opportunities for Academia and Industry. Signal Transduct. Targeted Ther. 2019, 4. [Google Scholar] [CrossRef] [Green Version]
- Polge, C.; Cabantous, S.; Deval, C.; Claustre, A.; Hauvette, A.; Bouchenot, C.; Aniort, J.; Béchet, D.; Combaret, L.; Attaix, D.; et al. A muscle-specific MuRF1-E2 network requires stabilization of MuRF1-E2 complexes by telethonin, a newly identified substrate. J. Cachexia Sarcopenia Muscle 2018, 9, 129–145. [Google Scholar] [CrossRef] [PubMed]
- Ebert, S.M.; Dierdorff, J.M.; Meyerholz, D.K.; Bullard, S.A.; Al-Zougbi, A.; Delau, A.D.; Tomcheck, K.C.; Skopec, Z.P.; Marcotte, G.R.; Bodine, S.C.; et al. An investigation of p53 in skeletal muscle aging. J. Appl. Physiol. 2019, 127, 1075–1084. [Google Scholar] [CrossRef] [PubMed]
- Mangner, N.; Bowen, T.S.; Werner, S.; Fischer, T.; Kullnick, Y.; Oberbach, A.; Linke, A.; Steil, L.; Schuler, G.; Adams, V. Exercise Training Prevents Diaphragm Contractile Dysfunction in Heart Failure. Med. Sci. Sport Exerc. 2016, 48, 2118–2124. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gielen, S.; Sandri, M.; Kozarez, I.; Kratzsch, J.; Teupser, D.; Thiery, J.; Erbs, S.; Mangner, N.; Lenk, K.; Hambrecht, R.; et al. Exercise Training Attenuates MuRF-1 Expression in the Skeletal Muscle of Patients With Chronic Heart Failure Independent of Age: The Randomized Leipzig Exercise Intervention in Chronic Heart Failure and Aging Catabolism Study. Circulation 2012, 125, 2716–2727. [Google Scholar] [CrossRef] [Green Version]
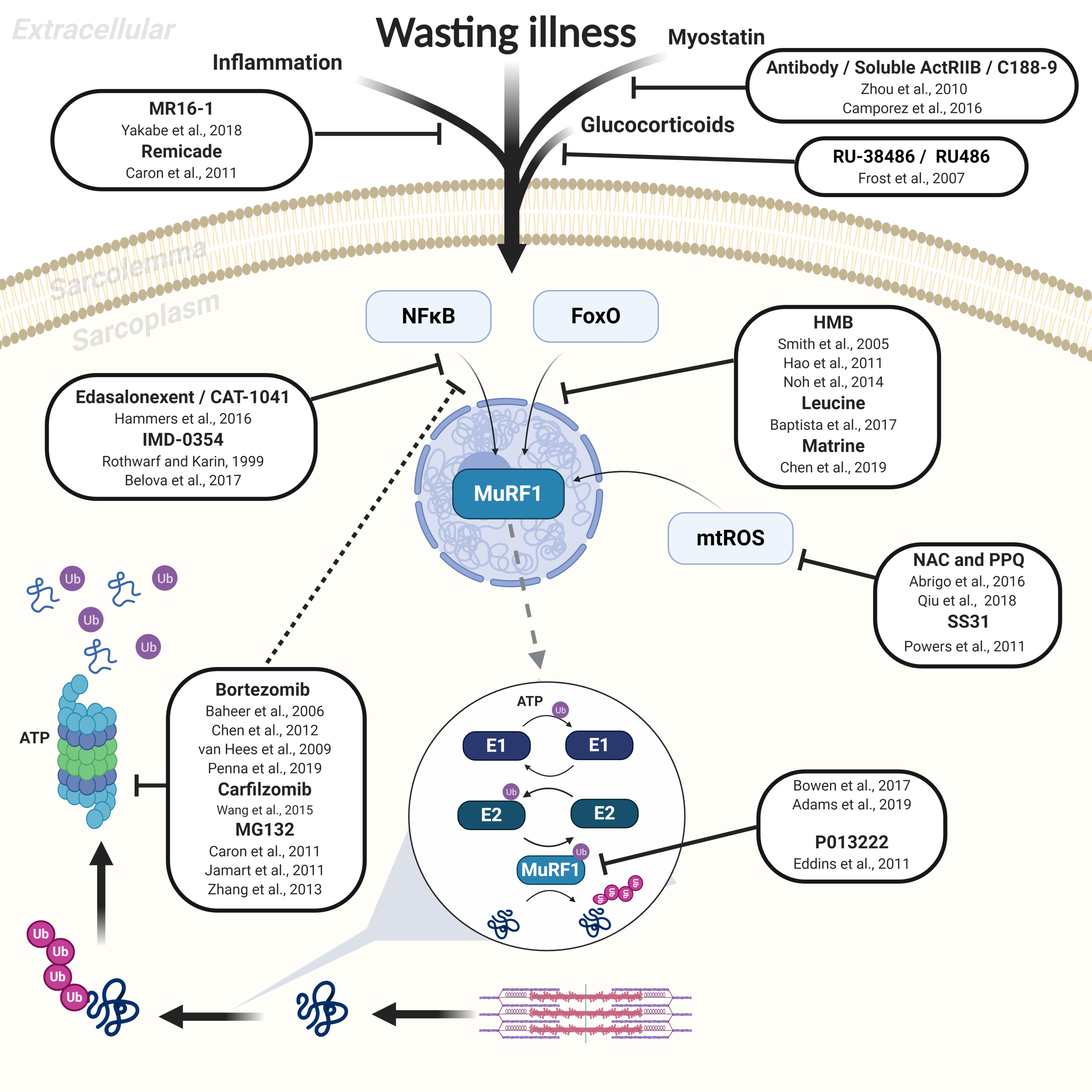
Figure 1.
Overview of proteasome-dependent protein degradation via the muscle-specific E3 ligase MuRF1 as induced in various wasting illness, with recognized pharmacological inhibitors also presented. The most important proteolytic pathway to mediate muscle atrophy is the ATP-dependent ubiquitin-proteasome system, whereby structural and regulatory proteins are degraded by the 26S proteasome following polyubiquitination that involves activation of E1, E2, and E3 enzymes. MuRF1 (an E3 ligase) plays a fundamental role in tagging proteins with ubiquitin during catabolic conditions, which is activated by various stimuli and signaling pathways that include NFκB and FoxO. A selection of reported pharmacological inhibitors of MuRF1-dependent muscle atrophy are presented along with the study reference. Solid lines indicate a direct effect on the target, while dotted lines indicate a secondary indirect effect. See the main text for full details.
Figure 1.
Overview of proteasome-dependent protein degradation via the muscle-specific E3 ligase MuRF1 as induced in various wasting illness, with recognized pharmacological inhibitors also presented. The most important proteolytic pathway to mediate muscle atrophy is the ATP-dependent ubiquitin-proteasome system, whereby structural and regulatory proteins are degraded by the 26S proteasome following polyubiquitination that involves activation of E1, E2, and E3 enzymes. MuRF1 (an E3 ligase) plays a fundamental role in tagging proteins with ubiquitin during catabolic conditions, which is activated by various stimuli and signaling pathways that include NFκB and FoxO. A selection of reported pharmacological inhibitors of MuRF1-dependent muscle atrophy are presented along with the study reference. Solid lines indicate a direct effect on the target, while dotted lines indicate a secondary indirect effect. See the main text for full details.

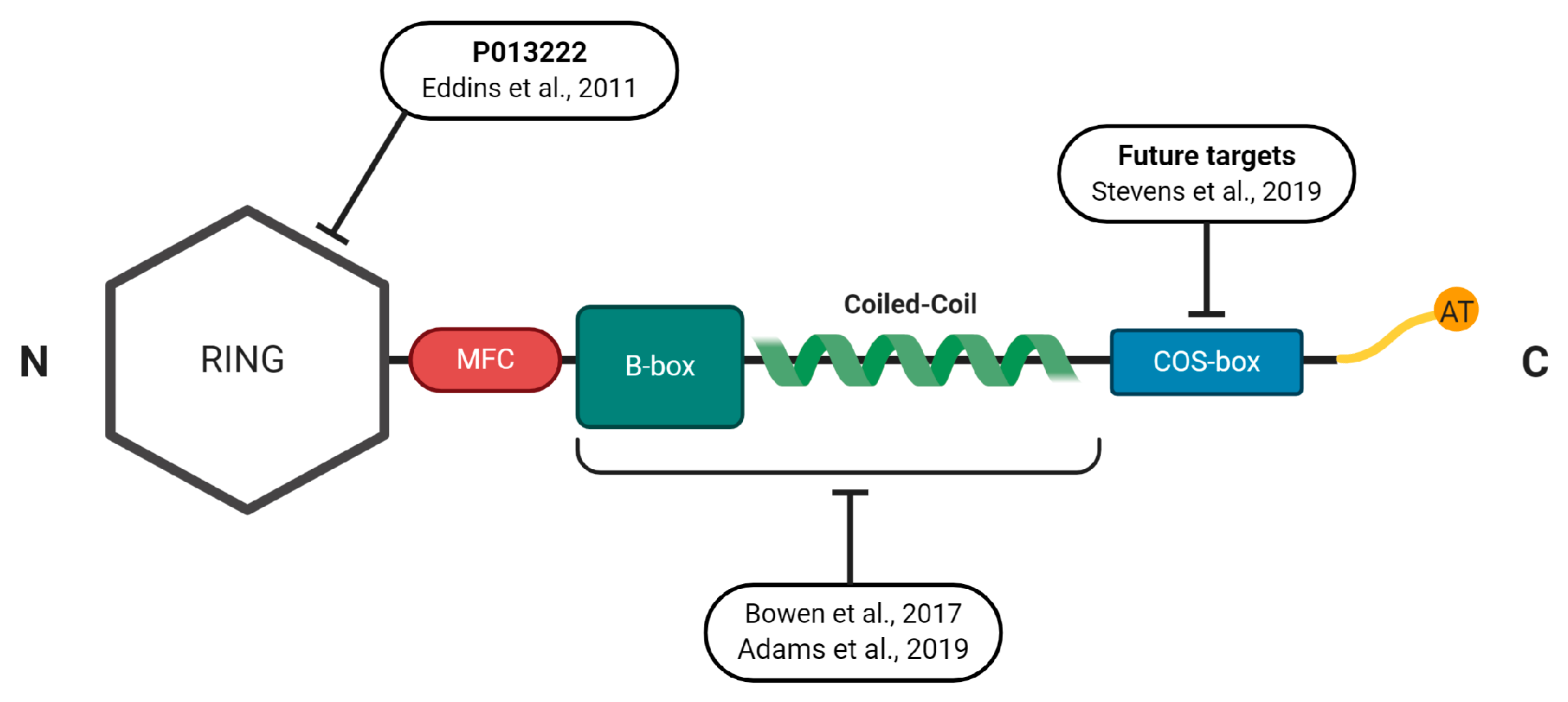
Figure 2.
Schematic representation of MuRF1′s protein structure indicating the regions that have been, or could be, exploited for small molecule inhibition alongside the relevant study reference. See the main text for full details.
Figure 2.
Schematic representation of MuRF1′s protein structure indicating the regions that have been, or could be, exploited for small molecule inhibition alongside the relevant study reference. See the main text for full details.

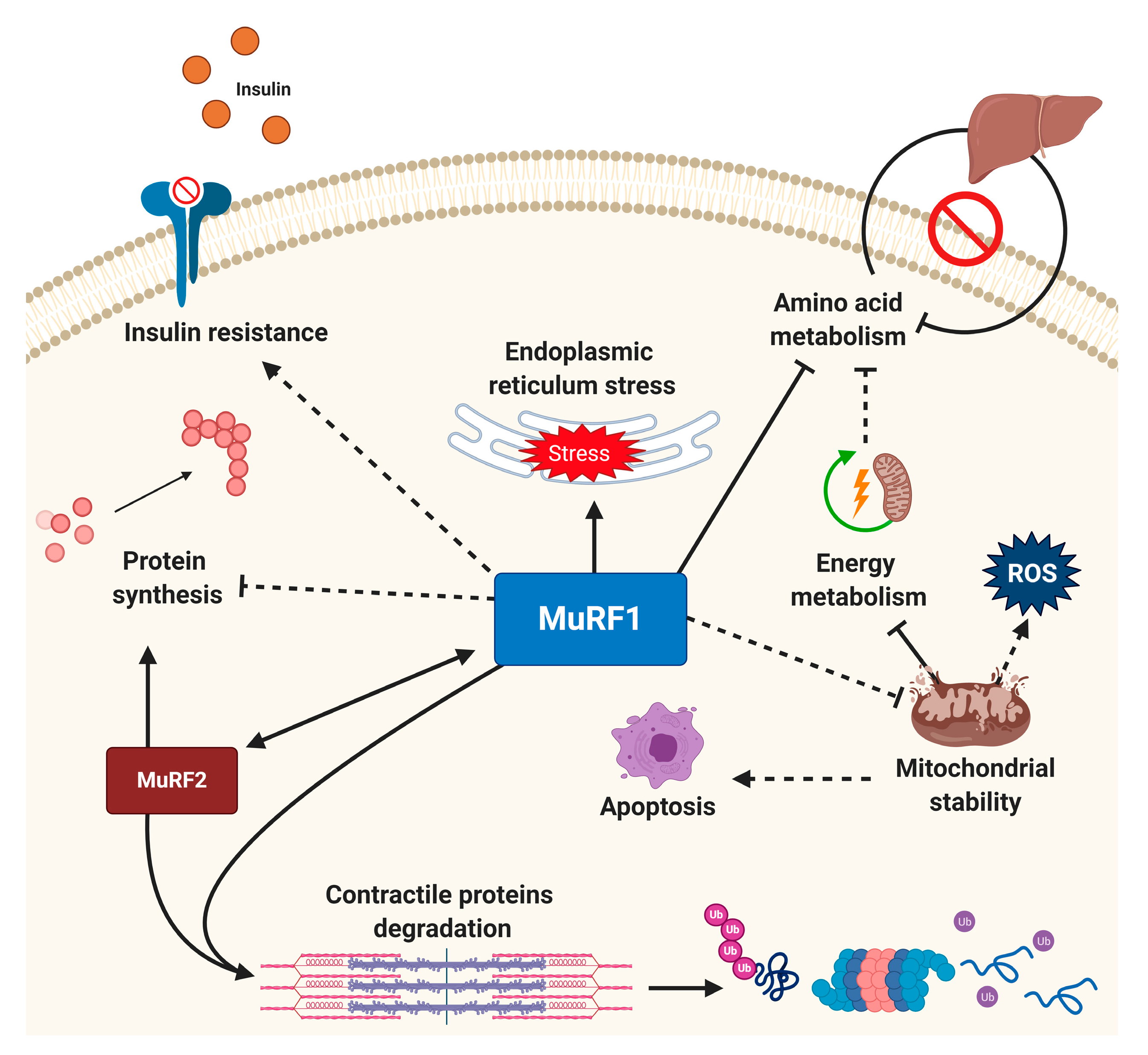
Figure 3.
MuRF1 as a potential central node for regulating cellular homeostasis. While MuRF1 is known to play a primary role in sarcomeric protein degradation, recent evidence indicates it may govern multiple cellular processes that include mitochondrial health, apoptosis, insulin resistance, endoplasmic reticulum stress response, amino acids, and carbohydrate metabolism. MuRF1 also works in synergy with MuRF2 to mediate muscle wasting. Solid lines indicate mechanisms with strong supporting evidence for direct effects MuRF1 effects, with recently emerging mechanisms that require further validation represented by dotted lines (direct or indirect). See the main text for full details.
Figure 3.
MuRF1 as a potential central node for regulating cellular homeostasis. While MuRF1 is known to play a primary role in sarcomeric protein degradation, recent evidence indicates it may govern multiple cellular processes that include mitochondrial health, apoptosis, insulin resistance, endoplasmic reticulum stress response, amino acids, and carbohydrate metabolism. MuRF1 also works in synergy with MuRF2 to mediate muscle wasting. Solid lines indicate mechanisms with strong supporting evidence for direct effects MuRF1 effects, with recently emerging mechanisms that require further validation represented by dotted lines (direct or indirect). See the main text for full details.

© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Scalabrin, M.; Adams, V.; Labeit, S.; Bowen, T.S. Emerging Strategies Targeting Catabolic Muscle Stress Relief. Int. J. Mol. Sci. 2020, 21, 4681. https://doi.org/10.3390/ijms21134681
AMA Style
Scalabrin M, Adams V, Labeit S, Bowen TS. Emerging Strategies Targeting Catabolic Muscle Stress Relief. International Journal of Molecular Sciences. 2020; 21(13):4681. https://doi.org/10.3390/ijms21134681
Chicago/Turabian StyleScalabrin, Mattia, Volker Adams, Siegfried Labeit, and T. Scott Bowen. 2020. "Emerging Strategies Targeting Catabolic Muscle Stress Relief" International Journal of Molecular Sciences 21, no. 13: 4681. https://doi.org/10.3390/ijms21134681
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.