Genetic Variants in the FGB and FGG Genes Mapping in the Beta and Gamma Nodules of the Fibrinogen Molecule in Congenital Quantitative Fibrinogen Disorders Associated with a Thrombotic Phenotype



 , and
, and
Abstract
: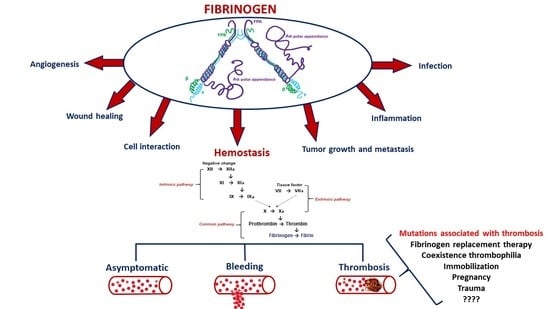
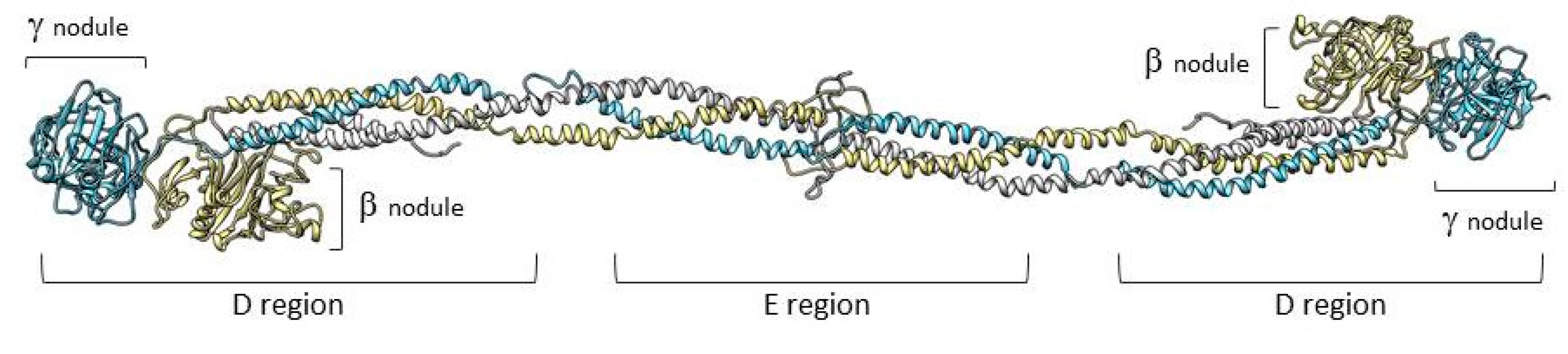
1. Structure and Function of Fibrinogen
2. Congenital Fibrinogen Disorders
2.1. Classification
2.2. Clinical Features
3. Pathogenesis and Risk Factors for Thrombosis in Congenital Quantitative Fibrinogen Disorders
4. Laboratory and Genetic Analysis of Congenital Quantitative Fibrinogen Disorders
4.1. Laboratory Analyses
4.2. Genetic Analyses
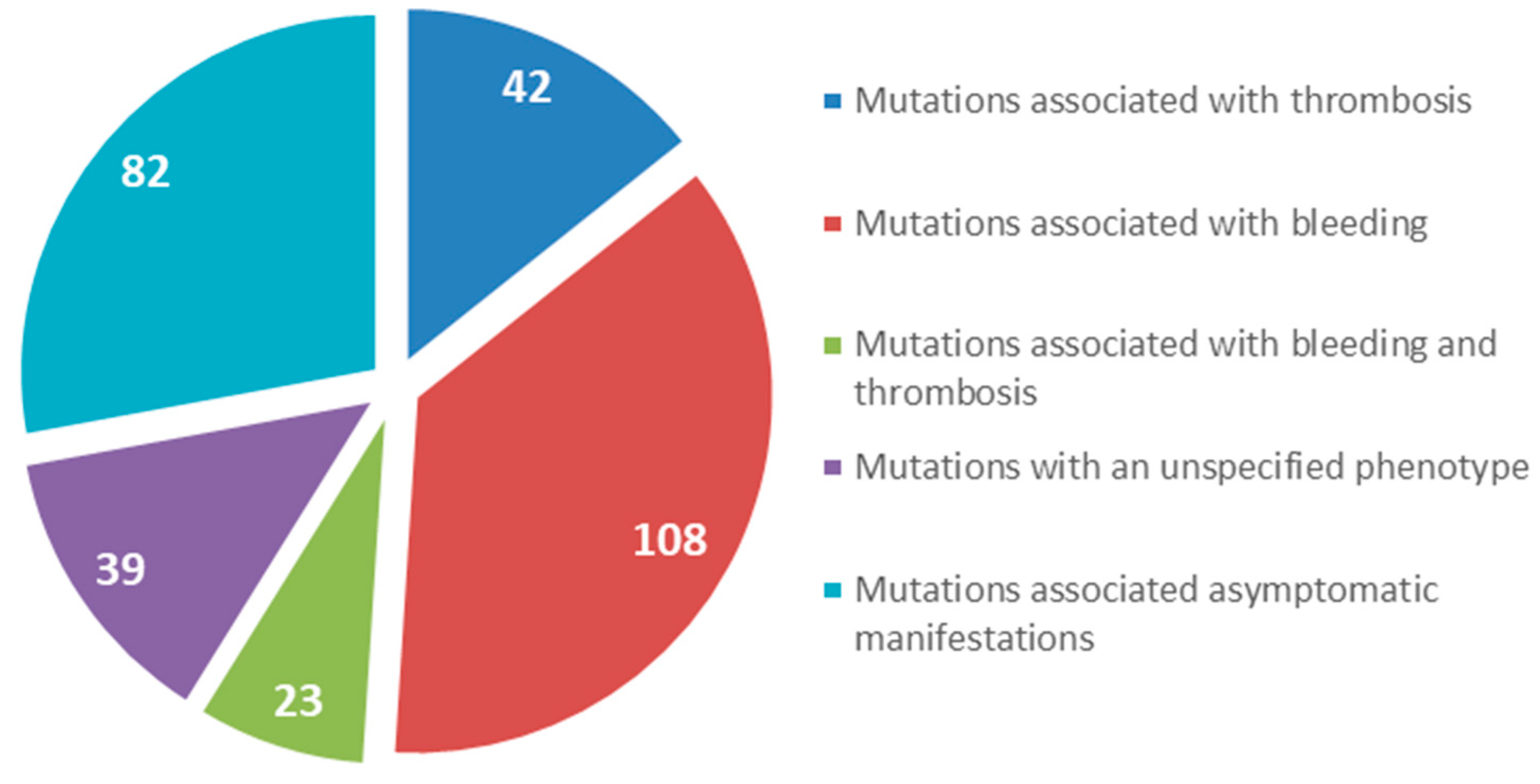
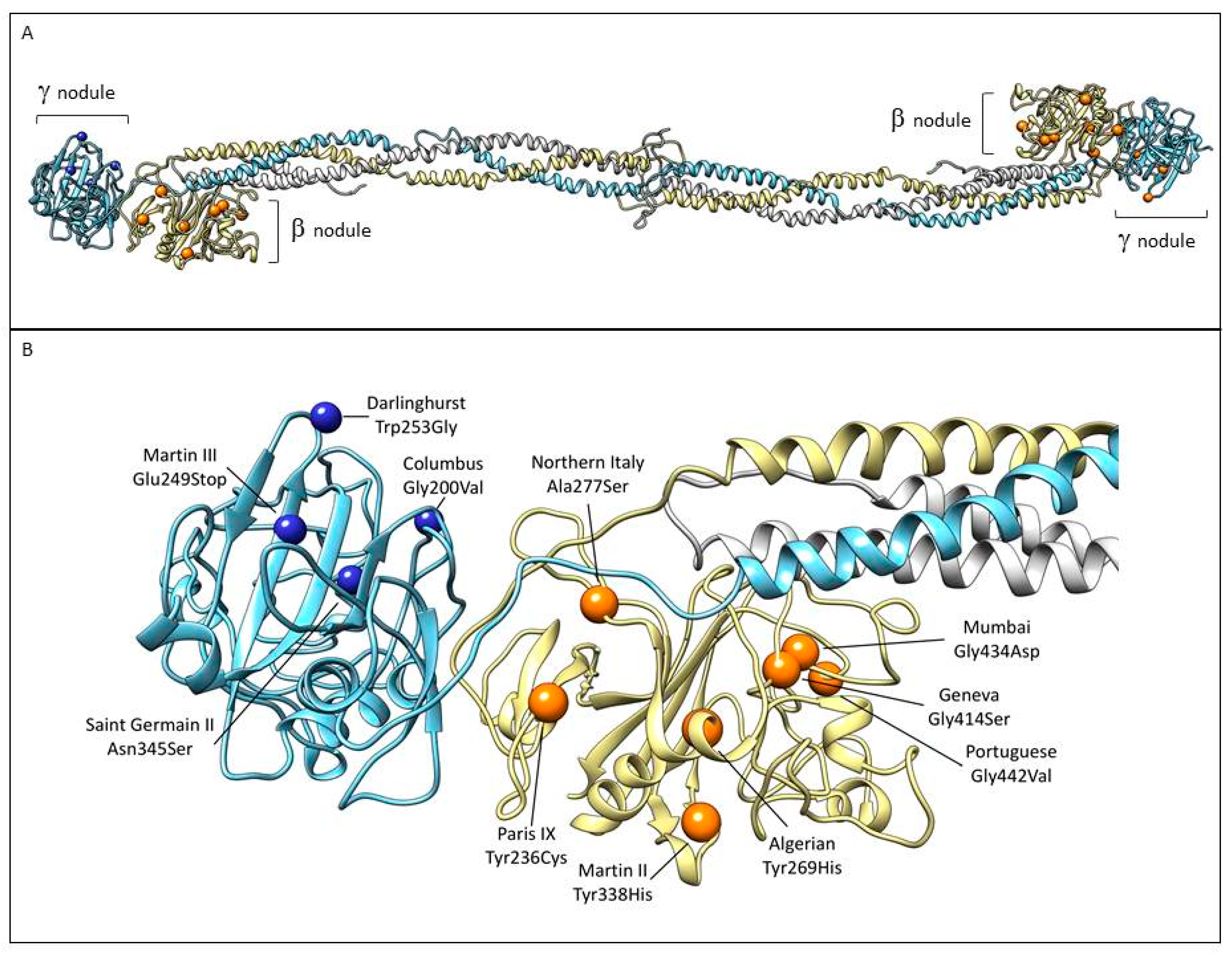
5. Case Reports of Mutations Located in the Beta and Gamma Nodules of Fibrinogen Bβ and γ Chains Associated with Thrombotic Complications
6. Mutations in the FGB Gene
6.1. Fibrinogen PARIS IX
6.2. Fibrinogen ALGERIAN
6.3. Fibrinogen NORTHERN ITALY
6.4. Fibrinogen MARTIN II
6.5. Fibrinogen GENEVA
6.6. Fibrinogen MUMBAI
6.7. Fibrinogen PORTUGUESE
7. Mutations in the FGG Gene
7.1. Fibrinogen COLUMBUS
7.2. Fibrinogen MARTIN III
7.3. Fibrinogen DARLINGHURST
7.4. Fibrinogen SAINT–GERMAIN II
8. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- De Moerloose, P.; Casini, A.; Neerman-Arbez, M. Congenital Fibrinogen Disorders: An Update. Semin. Thromb. Hemost. 2013, 39, 585–595. [Google Scholar] [PubMed] [Green Version]
- Tiscia, G.L.; Margaglione, M. Human Fibrinogen: Molecular and Genetic Aspects of Congenital Disorders. Int. J. Mol. Sci. 2018, 19, 1597. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cieśla, M.; Adamczyk, Z.; Barbasz, J.; Wasilewska, M. Mechanisms of fibrinogen adsorption at solid substrates at lower pH. Langmuir 2013, 29, 7005–7016. [Google Scholar] [CrossRef] [PubMed]
- Mosesson, M.W. Fibrinogen and fibrin structure and functions. J. Thromb. Haemost. 2005, 3, 1894–1904. [Google Scholar] [CrossRef] [PubMed]
- Simurda, T.; Caccia, S.; Asselta, R.; Zolkova, J.; Stasko, J.; Skornova, I.; Snahnicanova, Z.; Loderer, D.; Lasabova, Z.; Kubisz, P. Congenital hypofibrinogenemia associated with a novel heterozygous nonsense mutation in the globular C-terminal domain of the γ-chain (p.Glu275Stop). J. Thromb. Thrombolysis. 2019, 1–4. [Google Scholar] [CrossRef] [PubMed]
- Kollman, J.M.; Pandi, L.; Sawaya, M.R.; Riley, M.; Doolittle, R.F. Crystal structure of human fibrinogen. Biochemistry 2009, 4, 3877–3886. [Google Scholar] [CrossRef] [PubMed]
- Simurda, T.; Snahnicanova, Z.; Loderer, D.; Sokol, J.; Stasko, J.; Lasabova, Z.; Kubisz, P. Fibrinogen Martin: A Novel Mutation in FGB (Gln180Stop) Causing Congenital Afibrinogenemia. Semin. Thromb. Hemost. 2016, 42, 455–458. [Google Scholar]
- Litvinov, R.I.; Weisel, J.W. Not fibrin(ogen), but fibrinogen or fibrin. Blood 2015, 126, 1977–1978. [Google Scholar] [CrossRef]
- Mosesson, M.W.; Siebenlist, K.R.; Meh, D.A. The structure and biological features of fibrinogen and fibrin. Ann. N. Y. Acad. Sci. 2001, 93, 11–30. [Google Scholar] [CrossRef]
- Platé, M.; Asselta, R.; Peyvandi, F.; Techini, M.L.; Duga, S. Molecular Characterization of the First Missense Mutation in the Fibrinogen Aalpha-chain Gene Identified in a Compound Heterozygous Afibrinogenemic Patient. Biochim. Biophys. Acta 2007, 1772, 781–787. [Google Scholar] [CrossRef] [Green Version]
- Brennan, S.O.; Fellowes, A.P.; George, P.M. Molecular Mechanisms of Hypo- And Afibrinogenemia. Ann. N. Y. Acad. Sci. 2001, 936, 91–100. [Google Scholar] [CrossRef]
- Asselta, R.; Duga, S.; Tenchini, M.L. The molecular basis of quantitative fibrinogen disorders. J. Thromb. Haemost. 2006, 4, 2115–2129. [Google Scholar] [CrossRef] [PubMed]
- Simurda, T.; Zolkova, J.; Kolkova, Z.; Loderer, D.; Dobrotova, M.; Skornova, I.; Brunclóková, M.; Grendar, M.; Lasabova, Z.; Stasko, J.; et al. Comparison of clinical phenotype with genetic and laboratory results in 31 patients with congenital dysfibrinogenemia in northern Slovakia. Int. J. Hematol. 2020, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Laurens, N.; Koolwijk, P.; de Maat, M.P.M. Fibrin Structure and Wound Healing. J. Thromb. Haemost. 2006, 4, 932–939. [Google Scholar] [CrossRef] [PubMed]
- Weisel, J.W.; Litvinov, R.I. Fibrin Formation, Structure and Properties. Subcell. Biochem. 2017, 82, 405–456. [Google Scholar] [PubMed] [Green Version]
- Vilar, R.; Fish, R.J.; Casini, A.; Neerman-Arbez, M. Fibrin(ogen) in human disease: Both friend and foe. Haematologica 2020, 105, 284–296. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Riedel, T.; Suttnar, J.; Brynda, E.; Houska, M.; Medved, L.; Dyr, J.E. Fibrinopeptides A and B release in the process of surface fibrin formation. Blood 2011, 117, 1700–1706. [Google Scholar] [CrossRef] [Green Version]
- Walton, B.L.; Byrnes, J.R.; Wolberg, A.S. Fibrinogen, red blood cells, and factor XIII in venous thrombosis. J. Thromb. Haemost. 2015, 13, 208–215. [Google Scholar] [CrossRef]
- Zuliani-Alvarez, L.; Midwood, K.S. Fibrinogen-Related Proteins in Tissue Repair: How a Unique Domain with a Common Structure Controls Diverse Aspects of Wound Healing. Adv. Wound Care 2015, 4, 273–285. [Google Scholar] [CrossRef] [Green Version]
- Seung-Jae, J. Mechanisms of Platelet Activation and Integrin αIIβ3. Korean Circ. J. 2012, 42, 295–301. [Google Scholar]
- Zafar, H.; Shang, Y.; Li, J.; David, G.A., III; Fernandez, J.P.; Molina, H.; Filizola, M.; Coller, B.S. αIIbβ3 binding to a fibrinogen fragment lacking the γ-chain dodecapeptide is activation dependent and EDTA inducible. Blood Adv. 2017, 1, 417–428. [Google Scholar] [CrossRef] [PubMed]
- Fuss, C.; Palmaz, J.C.; Sprague, E.A. Fibrinogen: Structure, function, ans surface interactions. J. Vasc. Interv. Radiol. 2001, 12, 677–682. [Google Scholar] [CrossRef]
- Kattula, S.; Byrnes, J.R.; Wolberg, A.S. Fibrinogen and Fibrin in Hemostasis and Thrombosis. Arter. Thromb. Vasc. Biol. 2017, 37, 13–21. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Casini, A.; de Moerloose, P.; Neerman-Arbez, M. Clinical Features and Management of Congenital Fibrinogen Deficiencies. Semin. Thromb. Hemost. 2016, 42, 366–374. [Google Scholar] [PubMed]
- Casini, A.; Brungs, T.; Lavenu-Bombled, C.; Vilar, R.; Neerman-Arbez, M.; de Moerloose, P. Genetics, diagnosis and clinical features of congenital hypodysfibrinogenemia: A systematic literature review and report of a novel mutation. J. Thromb. Haemost. 2017, 15, 876–888. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Simurda, T.; Zolkova, J.; Snahnicanova, Z.; Loderer, D.; Skornova, I.; Sokol, J.; Hudecek, J.; Stasko, J.; Lasabova, Z.; Kubisz, P. Identification of Two Novel Fibrinogen Bβ Chain Mutations in Two Slovak Families with Quantitative Fibrinogen Disorders. Int. J. Mol. Sci. 2017, 19, 100. [Google Scholar] [CrossRef] [Green Version]
- Casini, A.; Undas, A.; Palla, R.; Thachil, J.; de Moerloose, P. For the Subcommittee on Factor XIII and Fibrinogen. Diagnosis and Classification of Congenital Fibrinogen Disorders: Communication From the SSC of the ISTH. J. Thromb. Haemost. 2018, 16, 1887–1890. [Google Scholar] [CrossRef]
- Report on the WFH Annual Global Survey 2017. Available online: http://www1.wfh.org/publications/files/pdf-1714.pdf (accessed on 30 December 2019).
- Palla, R.; Peyvandi, F.; Shapiro, A.D. Rare Bleeding Disorders: Diagnosis and Treatment. Blood 2015, 125, 2052–2061. [Google Scholar] [CrossRef]
- Simurda, T.; Stanciakova, L.; Stasko, J.; Dobrotova, M.; Kubisz, P. Yes or No for Secondary Prophylaxis in Afibrinogenemia? Blood Coagul. Fibrinolysis 2015, 26, 978–980. [Google Scholar] [CrossRef]
- Paraboschi, E.M.; Duga, S.; Asselta, R. Fibrinogen as a Pleiotropic Protein Causing Human Diseases: The Mutational Burden of Aα, Bβ, and γ Chains. Int. J. Mol. Sci. 2017, 18, 2711. [Google Scholar] [CrossRef] [Green Version]
- Naz, A.; Biswas, A.; Khan, T.N.; Goodeve, A.; Ahmed, N.; Saqlain, N.; Ahmed, S.; Din Ujjan, I.; Shamsi, T.S.; Oldenburg, J. Identification of novel mutations in congenital afibrinogenemia patients and molecular modeling of missense mutations in Pakistani population. Thromb. J. 2017, 15, 24. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Simurda, T.; Casini, A.; Stasko, J.; Hudecek, J.; Skornova, I.; Vilar, R.; Neerman-Arbez, N.; Kubisz, P. Perioperative Management of a Severe Congenital Hypofibrinogenemia With Thrombotic Phenotype. Thromb. Res. 2020, 188, 1–4. [Google Scholar] [CrossRef] [PubMed]
- Abolghasemi, H.; Shahverdi, E. Umbilical bleeding: A presenting feature for congenital afibrinogenemia. Blood Coagul. Fibrinolysis 2015, 26, 834–835. [Google Scholar] [CrossRef] [PubMed]
- Casini, A.; de Moerloose, P. Can the phenotype of inherited fibrinogen disorders be predicted? Haemophilia 2016, 22, 667–675. [Google Scholar] [CrossRef]
- Stanciakova, L.; Kubisz, P.; Dobrotova, M.; Stasko, J. Congenital Afibrinogenemia: From Etiopathogenesis to Challenging Clinical Management. Expert. Rev. Hematol. 2016, 9, 639–648. [Google Scholar] [CrossRef]
- Yılmaz, A.K.; Yaman, Y.; Isguder, R.; Cartı, O.; Demirag, B.; Agin, H.; Ozek, G.; Gunes Tatlı, B.; Albudak, E.; Berksoy, E. Spontaneous epidural and subdural hematoma in a child with afibrinogenemia and postoperative management. Blood Coagul. Fibrinolysis 2014, 25, 398–400. [Google Scholar]
- Neerman-Arbez, M.; Casini, A. Clinical Consequences and Molecular Bases of Low Fibrinogen Levels. Int. J. Mol. Sci. 2018, 19, 192. [Google Scholar] [CrossRef] [Green Version]
- Ruiz-Saez, A. Occurrence of thrombosis in rare bleeding disorders. Semin. Thromb. Hemost. 2013, 39, 684–692. [Google Scholar] [CrossRef]
- Patra, S.; Reddy, B.; Nagesh, C.M. Recurrent myocardial infarction in a case of congenital afibrinogenemia. Heart Views 2014, 15, 16–18. [Google Scholar] [CrossRef]
- De Moerloose, P.; Boehlen, F.; Neerman-Arbez, M. Fibrinogen and the risk of thrombosis. Semin. Thromb. Hemost. 2010, 36, 7–17. [Google Scholar] [CrossRef]
- Fuchs, R.J.; Levin, J.; Tadel, M.; Merritt, W. Perioperative coagulation management in a patient with afibrinogenaemia undergoing liver transplantation. Liver Transpl. 2007, 13, 752–756. [Google Scholar] [CrossRef] [PubMed]
- Asselta, R.; Platé, M.; Robusto, M.; Borhany, M.; Guella, I.; Soldà, G.; Afrasiabi, A.; Menegatti, M.; Shamsi, T.; Pyvandi, F.; et al. Clinical and molecular characterisation of 21 patients affected by quantitative fibrinogen deficiency. Thromb. Haemost. 2016, 113, 567–576. [Google Scholar] [CrossRef] [PubMed]
- Korte, W.; Poon, M.C.; Iorio, A.; Makris, M. Thrombosis in Inherited Fibrinogen Disorders. Transfus. Med. Hemother. 2017, 44, 70–76. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ozdemir, M.A.; Isik, B.; Patiroglu, T.; Karakukcu, M.; Mutlu, F.T.; Yilmaz, E.; Unal, E. A case of congenital afibrinogenemia complicated with thromboembolic events that required repeated amputations. Blood Coagul. Fibrinolysis 2015, 26, 354–356. [Google Scholar] [CrossRef] [PubMed]
- Mosesson, M.W. Update on Antithrombin I (Fibrin). Thromb. Haemost. 2007, 98, 105–108. [Google Scholar] [CrossRef] [Green Version]
- Asselta, R.; Robusto, M.; Platé, M.; Santoro, C.; Peyvandi, F.; Duga, S. Molecular characterization of 7 patients affected by dys- or hypo-dysfibrinogenemia: Identification of a novel mutation in the fibrinogen Bbeta chain causing a gain of glycosylation. Thromb. Res. 2015, 136, 168–174. [Google Scholar] [CrossRef]
- Casini, A.; Lukowski, S.; Quntard, V.L.; Crutu, A.; Zak, M.; Regazzoni, S.; de Moerloose, P.; Neerman-Arbez, M. FGB mutations leading to congenital quantitative fibrinogen deficiencies: An update and report of four novel mutations. Thromb. Res. 2014, 13, 868–874. [Google Scholar] [CrossRef]
- Rottenstreich, A.; Lask, A.; Schliamser, L.; Zivelin, A.; Seligsohn, U.; Kalish, Y. Thromboembolic Events in Patients With Severe Inherited Fibrinogen Deficiency. J. Thromb. Thrombolysis 2016, 42, 261–266. [Google Scholar] [CrossRef]
- Simurda, T.; Kubisz, P.; Dobrotova, M.; Necas, L.; Stasko, J. Perioperative Coagulation Management in a Patient with Congenital Afibrinogenemia during Revision Total Hip Arthroplasty. Semin. Thromb. Hemost. 2016, 42, 689–692. [Google Scholar] [CrossRef] [Green Version]
- Previtali, E.; Bucciarelli, P.; Passamonti, S.M.; Martinelli, I. Risk factors for venous and arterial thrombosis. High Speed Blood Transfus. Equip. 2010, 9, 120–138. [Google Scholar]
- Peyvandi, F.; Palla, R.; Menegatti, M.; Siboni, S.M.; Halimeh, S.; Faeser, B.; Pergantou, H.; Platokouki, H.; Giangrande, P.; Peerlinck, K.; et al. Coagulation factor activity and clinical bleeding severity in rare bleeding disorders: results from the European Network of Rare Bleeding Disorders. J. Thromb. Haemost. 2012, 10, 615–621. [Google Scholar] [CrossRef]
- Casini, A.; Blondon, M.; Lebreton, A.; Koegel, J.; Tintillier, V.; De Maistre, E.; Gautier, P.; Biron, C.; Neerman-Arbez, M.; De Moerloose, P. Natural history of patients with congenital dysfibrinogenemia. Blood 2015, 125, 553–561. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Collet, J.; Woodhead, J.; Soria, J.; Soria, C.; Mirshahi, M.; Caen, J.; Weisel, J. Fibrinogen Dusart: electron microscopy of molecules, fibers and clots, and viscoelastic properties of clots. Biophys. J. 1996, 70, 500–510. [Google Scholar] [CrossRef] [Green Version]
- Meh, D.A.; Mosesson, M.W.; Siebenlist, K.R.; Simpson-Haidaris, P.J.; Brennan, S.O.; Diorio, J.P.; Thompson, K.; Di Minno, G. Fibrinogen naples I (B beta A68T) nonsubstrate thrombin-binding capacities. Thromb. Res. 2001, 103, 63–73. [Google Scholar] [CrossRef]
- De Moerloose, P.; Casini, A.; Neerman-Arbez, M. Laboratory and Genetic Investigation of Mutations Accounting for Congenital Fibrinogen Disorders. Semin. Thromb. Hemost. 2016, 42, 356–365. [Google Scholar] [CrossRef] [PubMed]
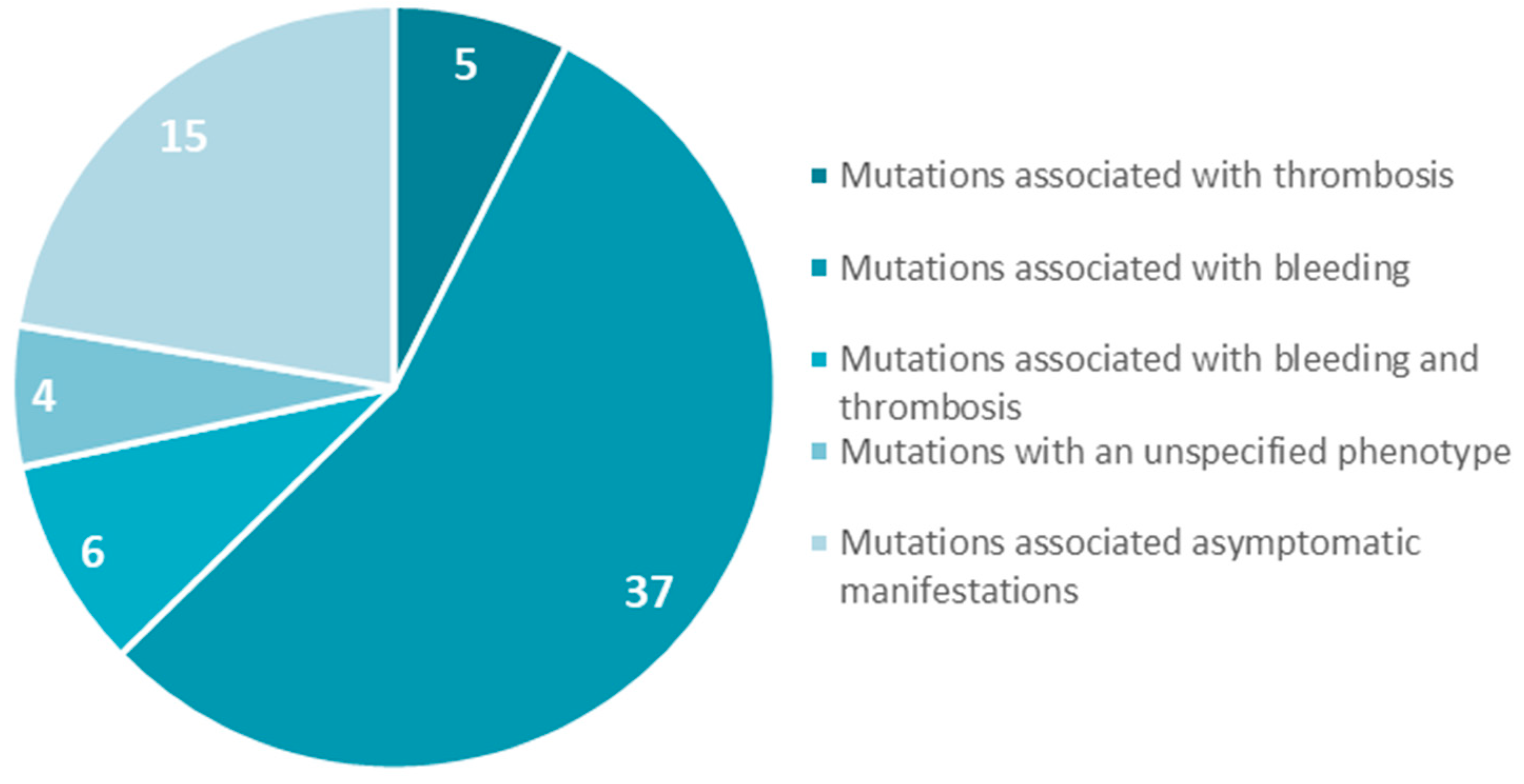
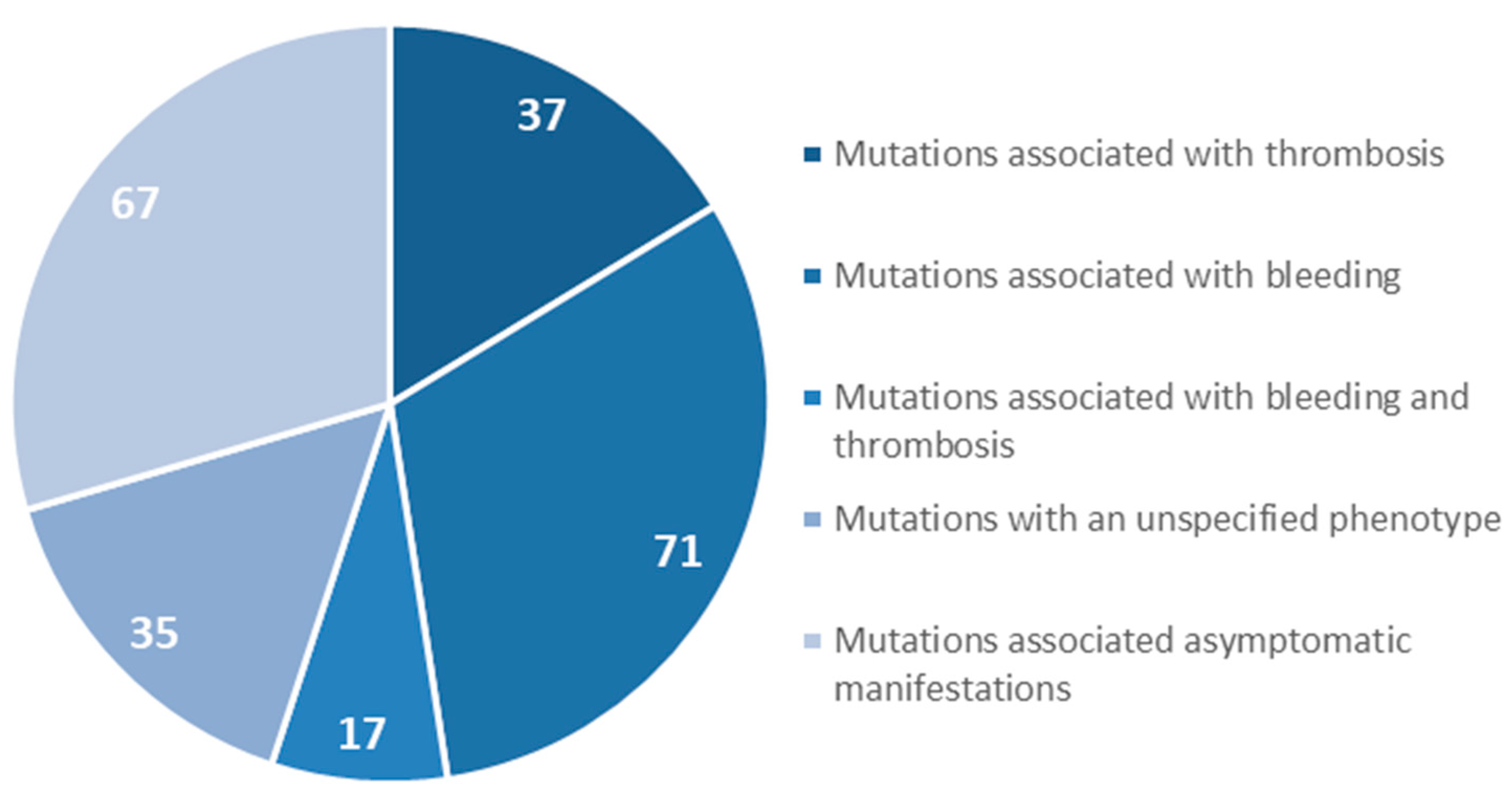
- Casini, A.; Blondon, M.; Tintillier, V.; Goodyer, M.; Sezgin, M.E.; Gunes, A.M.; Hanss, M.; De Moerloose, P.; Neerman-Arbez, M. Mutational Epidemiology of Congenital Fibrinogen Disorders. Thromb. Haemost. 2018, 118, 1867–1874. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tziomalos, K. Treatment of congenital fibrinogen deficiency: overview and recent findings. Vasc. Heal. Risk Manag. 2009, 5, 843. [Google Scholar] [CrossRef] [Green Version]
- Acharya, S.S.; DiMichele, D.M. Rare inherited disorders of fibrinogen. J. Thromb. Haemost. 2008, 14, 1151–1158. [Google Scholar] [CrossRef]
- Bornikova, L.; Peyvandi, F.; Allen, G.; Bernstein, J.; Manco-Johnson, M.J. Fibrinogen replacement therapy for congenital fibrinogen deficiency. J. Thromb. Haemost. 2011, 9, 1687–1704. [Google Scholar] [CrossRef]
- Mumford, A.D.; Ackroyd, S.; Alikhan, R.; Bowles, L.; Chowdary, P.; Grainger, J.; Mainwaring, J.; Mathias, M.; O’Connell, N. The BCSH Committee Guideline for the diagnosis and management of the rare coagulation disorders. Br. J. Haematol. 2014, 167, 304–326. [Google Scholar] [CrossRef]
- Kalina, U.; Stöhr, H.-A.; Bickhard, H.; Knaub, S.; Siboni, S.M.; Mannucci, P.M.; Peyvandi, F. Rotational thromboelastography for monitoring of fibrinogen concentrate therapy in fibrinogen deficiency. Blood Coagul. Fibrinolysis 2008, 19, 777–783. [Google Scholar] [CrossRef] [PubMed]
- Peng, H.T.; Nascimento, B.; Beckett, A. Thromboelastography and Thromboelastometry in Assessment of Fibrinogen Deficiency and Prediction for Transfusion Requirement: A Descriptive Review. BioMed Res. Int. 2018, 2018, 1–24. [Google Scholar] [CrossRef] [PubMed]
- Franchini, M.; Lippi, G. Fibrinogen replacement therapy: a critical review of the literature. High Speed Blood Transfus. Equip. 2011, 10, 23–27. [Google Scholar]
- Smith, N.; Bornikova, L.; Noetzli, L.; Guglielmone, H.; Minoldo, S.; Backos, D.; Ms, L.J.; Thornburg, C.; Escobar, M.; White-Adams, T.C.; et al. Identification and characterization of novel mutations implicated in congenital fibrinogen disorders. Res. Pr. Thromb. Haemost. 2018, 2, 800–811. [Google Scholar] [CrossRef]
- Levrat, E.; Aboukhamis, I.; De Moerloose, P.; Farho, J.; Chamaa, S.; Reber, G.; Fort, A.; Neerman-Arbez, M. A novel frameshift mutation in FGA (c.1846 del A) leading to congenital afibrinogenemia in a consanguineous Syrian family. Blood Coagul. Fibrinolysis 2011, 22, 148–150. [Google Scholar] [CrossRef]
- Grandone, E.; Tiscia, G.; Cappucci, F.; Favuzzi, G.; Santacroce, R.; Pisanelli, D.; Soli, F.; Legnani, C.; Rizzo, M.A.; Palareti, G.; et al. Clinical histories and molecular characterization of two afibrinogenemic patients: Insights into clinical management. Haemophilia 2011, 18, e16–e18. [Google Scholar] [CrossRef]
- Vu, D.; Bolton-Maggs, P.H.B.; Parr, J.R.; Morris, M.A.; De Moerloose, P.; Neerman-Arbez, M. Congenital afibrinogenemia: Identification and expression of a missense mutation in FGB impairing fibrinogen secretion. Blood 2003, 102, 4413–4415. [Google Scholar] [CrossRef]
- Mukaddam, A.; Patil, R.; Jadli, A.; Chandrakala, S.; Ghosh, K.; Shetty, S. Pradoxical bleeding and thrombosis in a patient with afibrinogenemia and fibrinogen Mumbain mutation. Am. J. Clin. Pathol. 2015, 143, 755–757. [Google Scholar] [CrossRef] [Green Version]
- Davis, R.L.; Mosesson, M.W.; Kerlin, B.A.; Canner, J.A.; Ruymann, F.B.; Brennan, S.O. Fibrinogen Columbus: A novel gamma Gly200Val mutation causing hypofibrinogenemia in a family with associated thrombophilia. Haematologica 2007, 92, 1151–1152. [Google Scholar] [CrossRef] [Green Version]
- Sheen, C.R.; Low, J.; Joseph, J.; Kotlyar, E.; George, P.M.; Brennan, S.O. Fibrinogen Darlinghurst: Hypofibrinogenemia caused by a W253G mutation in the gamma chain in a patient with both bleeding and trhombotic complications. Thromb. Haemost. 2006, 96, 685–687. [Google Scholar]
- De Raucourt, E.; de Mazancourt, P.; Maghzal, G.J.; Brennan, S.O.; Mosesson, M.W. Fibrinogen Saint—Germain II: Hypofibrinogenemia due to heterozygous γ N345S mutation. Thromb. Haemost. 2005, 94, 965–968. [Google Scholar]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera—A visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef] [Green Version]
- Horellou, M.H.; Chevreaud, C.; Mathieux, V.; Conard, J.; de Mazancourt, P. Fibrinogen Parix IX: A case of asymptomatic hypofibrinogenemia with Bβ Y236C and Bβ IVS7—1C→C mutations. J. Thromb. Haemost. 2006, 4, 1134–1136. [Google Scholar] [CrossRef] [PubMed]
- Okumura, N.; Terasawa, F.; Tanaka, H.; Hirota, M.; Ota, H.; Kitano, K.; Kiyosawa, K.; Lord, S.T. Analysis of Fibrinogen Gamma-Chain Truncations Shows the C-terminus, Particularly gammaIle387, Is Essential for Assembly and Secretion of This Multichain Protein. Blood 2002, 99, 3654–3660. [Google Scholar] [CrossRef] [PubMed]
- Asselta, R.; Robusto, M.; Braidotti, P.; Peyvandi, F.; Nastasio, S.; D’Antiga, L.; Perisic, V.N.; Maggiore, G.; Caccia, S.; Duga, S. Hepatic fibrinogen storage disease: Identification of two novel mutations (p.Asp316Asn, fibrinogen Pisa and p.Gly366Ser, fibrinogen Beograd) impacting on fibrinogen ?-module. J. Thromb. Haemost. 2015, 13, 1459–1467. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ariëns, R.A.S. Fibrin(ogen) and thrombotic disease. J. Thromb. Haemost. 2013, 11, 294–305. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Type and Subtypes | Descriptions |
|---|---|
| Afibrinogenemia | |
| Afibrinogenemia | Afibrinogenemia and bleeding phenotype or asymptomatic individuals |
| A. Afibrinogenemia with a thrombotic phenotype | Afibrinogenemia and thrombotic phenotype |
| Hypofibrinogenemia | |
| A. Severe hypofibrinogenemia | Functional fibrinogen level ˂ 0.5 g/L |
| B. Moderate hypofibrinogenemia | Functional fibrinogen level between 0.5–0.9 g/L |
| C. Mild hypofibrinogenemia | Functional fibrinogen level between 1 g/L and lower limit of normal value |
| D. Hypofibrinogenemia with fibrinogen storage disease | Familial hypofibrinogenemia with histologically proven accumulation of fibrin in hepatocytes |
| Dysfibrinogenemia | |
| A. Dysfibrinogenemia | Dysfibrinogenemia and bleeding phenotype, thrombotic phenotype or asymptomatic individuals |
| B. Thrombotic-related dysfibrinogenemia | Dysfibrinogenemia patients carriers of a thrombotic fibrinogen mutation * or suffering from thrombotic events with a first-degree familial thrombotic history (relatives with the same genotype) without any other thrombophilia |
| Hypodysfibrinogenema | |
| A. Severehypodysfibrinogenemia | Antigenic fibrinogen level ˂ 0.5 g/L |
| B. Moderate hypodysfibrinogenemia | Antigenic fibrinogen level between 0.5–0.9 g/L |
| C. Mild hypodysfibrinogenemia | Antigenic fibrinogen level between 1 g/L and lower limit of normal value |
| Name/Origin | Plasma Protein | Native Protein | Gene | Gene Status | Type | Haemorraghes | Numbers of Studied Family Member/Positive Numbers of Mutation | Numbers of Thrombotic Complications | Other Thrombophilic States | References |
|---|---|---|---|---|---|---|---|---|---|---|
| Fibrinogen Bβ Chain Mutations Associated with Thrombosis | ||||||||||
| PARIS IX | Bβ(236) Tyr>Cys | p.Tyr266Cys | 5909A>G IVS7+1G>C | Compound | Hypofib. | Yes | 2/1 | 2 | Not listed | [6] |
| ALGERIAN | Bβ(269) Tyr>His | p.Tyr299His | c.895T>C | Homozyg. | Afib. | Yes | 1/1 | 1 | Heterozygous Factor V Leiden mutation | [48] |
| NORTHERN ITALY | Bβ(277) Ala>Ser | p.Ala307Ser | c.919G>T | Homozyg. | Afib. | No | 3/1 | 3 | No other thrombophilic state | [67] |
| MARTIN II | Bβ(338) Tyr>His | p.Tyr368His | c.1102T>C | Homozyg. | Hypofib. | No | 4/2 | 5 | No other thrombophilic state | [26] |
| GENEVA | Bβ(17) Arg>Stop Bβ(414) Gly>Ser | p.Arg47Stop p.Gly444Ser | c.139C>T c.1330G>C | Compound | Afib. | Yes | 4/2 | 2 | No other thrombophilic state | [68] |
| MUMBAI | Bβ(434) Gly>Asp | p.Gly464Asp | c.G1391A | Homozyg. | Afib. | Yes | 1/1 | 1 | Heterozygous PAI 4G/5G polymorphism | [69] |
| PORTUGUESE | Bβ(442) Gly>Val | p.Gly472Val | c.1415G>T | Homozyg. | Hypofib. | No | 1/1 | 5 | No other thrombophilic state | [48] |
| Fibrinogen γ Chain Mutations Associated with Thrombosis | ||||||||||
| COLUMBUS | γ(200) Gly>Val | p.Gly226Val | c.677G>T | Heterozyg. | Hypofib. | Yes | 8/2 | 2 | Heterozygous Factor V Leiden, MTHFR C677T mutations | [70] |
| MARTIN III | γ(249) Glu>Stop | p.Glu275Stop | c.823G>T | Heterozyg. | Hypofib. | No | 1/1 | 3 | No other thrombophilic state | [5] |
| DARLINGHURST | γ(253) Trp>Gly | p.Trp279Gly | c.835T>G | Homozyg. | Hypofib. | Yes | 2/1 | 2 | No other thrombophilic state | [71] |
| SAINT GERMAIN II | γ(345) Asn>Ser | p.Asn371Ser | 7687A>G | Heterozyg. | Hypofib. | No | 3/1 | 2 | Heterozygous Factor V Leiden mutation, prothrombin G20210 mutation | [72] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Simurda, T.; Brunclikova, M.; Asselta, R.; Caccia, S.; Zolkova, J.; Kolkova, Z.; Loderer, D.; Skornova, I.; Hudecek, J.; Lasabova, Z.; et al. Genetic Variants in the FGB and FGG Genes Mapping in the Beta and Gamma Nodules of the Fibrinogen Molecule in Congenital Quantitative Fibrinogen Disorders Associated with a Thrombotic Phenotype. Int. J. Mol. Sci. 2020, 21, 4616. https://doi.org/10.3390/ijms21134616
Simurda T, Brunclikova M, Asselta R, Caccia S, Zolkova J, Kolkova Z, Loderer D, Skornova I, Hudecek J, Lasabova Z, et al. Genetic Variants in the FGB and FGG Genes Mapping in the Beta and Gamma Nodules of the Fibrinogen Molecule in Congenital Quantitative Fibrinogen Disorders Associated with a Thrombotic Phenotype. International Journal of Molecular Sciences. 2020; 21(13):4616. https://doi.org/10.3390/ijms21134616
Chicago/Turabian StyleSimurda, Tomas, Monika Brunclikova, Rosanna Asselta, Sonia Caccia, Jana Zolkova, Zuzana Kolkova, Dusan Loderer, Ingrid Skornova, Jan Hudecek, Zora Lasabova, and et al. 2020. "Genetic Variants in the FGB and FGG Genes Mapping in the Beta and Gamma Nodules of the Fibrinogen Molecule in Congenital Quantitative Fibrinogen Disorders Associated with a Thrombotic Phenotype" International Journal of Molecular Sciences 21, no. 13: 4616. https://doi.org/10.3390/ijms21134616