Fyn Tyrosine Kinase as Harmonizing Factor in Neuronal Functions and Dysfunctions


1
Division of Pharmacology, Department of Neuroscience, School of Medicine, University of Naples Federico II, 80131 Naples, Italy
2
Department of Biomedicine, Aarhus University, 8000 Aarhus C, Denmark
*
Author to whom correspondence should be addressed.
Int. J. Mol. Sci. 2020, 21(12), 4444; https://doi.org/10.3390/ijms21124444
Submission received: 18 May 2020
/
Revised: 19 June 2020
/
Accepted: 20 June 2020
/
Published: 22 June 2020
(This article belongs to the Special Issue Kinase Signal Transduction 2.0)
Abstract
:Fyn is a non-receptor or cytoplasmatic tyrosine kinase (TK) belonging to the Src family kinases (SFKs) involved in multiple transduction pathways in the central nervous system (CNS) including synaptic transmission, myelination, axon guidance, and oligodendrocyte formation. Almost one hundred years after the original description of Fyn, this protein continues to attract extreme interest because of its multiplicity of actions in the molecular signaling pathways underlying neurodevelopmental as well as neuropathologic events. This review highlights and summarizes the most relevant recent findings pertinent to the role that Fyn exerts in the brain, emphasizing aspects related to neurodevelopment and synaptic plasticity. Fyn is a common factor in healthy and diseased brains that targets different proteins and shapes different transduction signals according to the neurological conditions. We will primarily focus on Fyn-mediated signaling pathways involved in neuronal differentiation and plasticity that have been subjected to considerable attention lately, opening the fascinating scenario to target Fyn TK for the development of potential therapeutic interventions for the treatment of CNS injuries and certain neurodegenerative disorders like Alzheimer’s disease.
1. Introduction
Fyn is a non-receptor or cytoplasmatic tyrosine kinase belonging to the Src family kinases (SFKs) consisting of 11 members (Blk, Brk, Fgr, Frk, Hck, Lck, Lyn, c-Src, Srm, and c-Yes) in humans [1].
Src oncogene has been identified and characterized in the early 20th century when Peyton Rous discovered a particle smaller than a bacterium, which later became known as the Rous chicken sarcoma virus, that could be transmitted from bird to bird. Rous’ virus (RSV) was later found expressed in a truncated evolutionary conserved form called c-Src (Src) in uninfected vertebrate cells [2]. Indeed, Src is highly conserved among metazoans, and a Src ortholog has also been shown expressed in unicellular choanoflagellates [3,4].
In mammals, Src, Fyn, and Yes (SYF) are ubiquitously expressed, while the other family members display more restricted expression profile [5].
Fyn is primarily involved in several transduction pathways in the central nervous system (CNS) including myelination, axon guidance, and oligodendrocytes formation. Indeed, when disrupted these pathways, Fyn can contribute to the development of severe brain pathologies, such as Alzheimer’s disease (AD) and multiple sclerosis (MS) [6,7,8].
In the peripheral immune system, Fyn plays an important role in the regulation and functions of T-cell and B-cell receptor signaling and in the differentiation of natural killer cells [1,9,10]. A number of additional biological functions in which Fyn activity is involved has been extensively reported and includes growth factor and cytokine receptor signaling, ion channel function, platelet activation, fertilization, entry into mitosis [11,12].
On the other side, Fyn upregulation and genetic alterations have been associated with some malignancies as well as to several neuronal dysfunctions. In fact, when overexpressed, Fyn influences cell growth and proliferation and causes morphogenic transformations and alterations of mitogenic signals [13,14,15,16]. In addition, Fyn controls integrin adhesion and cell-cell interactions, all clinical features identified in cancer [15,17].
In this review, we will focus our interest on the role exerted by Fyn in the brain. The complexity of Fyn in the brain is mirrored in a myriad of neurological activities. However, we, by no means, expect to guide the reader through all of them, but rather highlight the role of Fyn either in synchronizing and optimizing functional neuronal networks or in exacerbating impaired or dysfunctional pathways in neuronal diseases that have no current cure. To this aim, several studies on the function that Fyn plays in developing neurons will be explored and emerging evidence about the role of Fyn in mature neurons as well as in the onset of brain disorders will be reviewed and discussed.
2. Fyn Structure and Activation Mechanisms
Fyn is 59kDa non-receptor protein tyrosine-kinase (TK) member of Src family [15] comprising 537 amino acids encoded by the Fyn gene, located on chromosome 6q21 [18]. Three isoforms of Fyn have been shown to arise via alternative splicing of exon 7 [15]: the isoform 1 (Fyn[B], “canonical sequence”) is the first identified; the isoform 2 (Fyn[T]) that tends to be expressed in T-cells and differs from the isoform 1 in the linker region between the SH2 and the SH1 domain [11]; the isoform 3 has been found in the blood cells and differs from the isoform type 1 as missing the sequence 233-287 [18]. FynT contains exon 7B (159 bp) and is expressed in thymocytes, splenocytes, and some hematolymphoid cell lines, while FynB (168 bp) includes exon 7A and it is more ubiquitous in its expression, although it accumulates principally in the brain [15].
Fyn, similarly, to the other Src members, is composed of several functional parts connected in a single protein chain (Figure 1). Beside the catalytic domain (SH1), Fyn contains two small, mutually unrelated, non-catalytic domains called SH2 and SH3 (Src-homology regions 2 and 3), of about 100 and 60 amino acids, respectively. SH2 and SH3 domains interact with other proteins, and these interactions regulate the TK activity.
Fyn exists in two conformations: active and inactive. Fyn activation (as for the other Src members) depends on the ligand binding to the SH2 and/or SH3 domains and on the phosphorylation/dephosphorylation of two critical tyrosine residues, Tyr528 in the isoform 2, FynT, (corresponding to Tyr531 in isoform 1, FynB) and Tyr417 (the activating Tyr residue) of the isoform 2, FynT, (corresponding to Tyr420 in isoform 1, FynB).
Multiple regulator mechanisms have been proposed leading to Fyn activation. Indeed, the dynamic control of Tyr420 and Tyr531 phosphorylation in the brain provides an important level of regulation of Fyn activity and its ability to interact with other proteins. When Tyr531 is not phosphorylated, Fyn adopts an “open” conformation that allows the interaction of the catalytic domain and the SH2 domain with their specific substrates (Figure 1). Conversely, when Tyr531 is phosphorylated, Fyn folds such that the tail and the SH2 domain interact to each other, preventing the two domains to bind their substrates and making Fyn inactive [9,19,20]. This suggests that increasing the local concentration of either an SH2 or an SH3 ligand would be expected to shift the equilibrium toward the “on” state. Particularly, potent activators would have binding sites for both the SH2 and SH3 domains, and consequently, decreased levels of such ligands would allow Fyn to revert to “the off state” [8]. This destabilization of SH2-SH3 interaction has been suggested as one of the most important mechanisms of Fyn activity regulation [11].
It is not worth that, the replacement of Tyr531 with phenylalanine is sufficient to convert Fyn protooncogene to oncogene [35,36].
Csk is a 50 kDa cytosolic TK differing from the other Src members because of the lack in both, the myristylation and the unique sequences [37,38,39]. Csk phosphorylates Fyn at Tyr531 [40] and promotes the intermolecular binding between SH2 domain and the C-tail, ultimately forcing the kinase into the inactive state [41]. Differently, the striatal enriched phosphatase (STEP), dephosphorylates Fyn at Tyr420 residue in the postsynaptic densities (PSDs) of striatal neurons and thus inactivates Fyn [42].
On the other side, Platelet-Derived Growth factor (PDGF) has been reported to stimulate the intrinsic kinase activity of Fyn by promoting its Tyr420 phosphorylation at N-terminal tail thereby activating Fyn intrinsic kinase activity and allowing it to interact with other proteins [16,43]. In addition, PDGF promotes Fyn-dependent migration of oligodendrocyte (OL) progenitors either during the brain development or lesion repair, when OLs migrate long distances before reaching the site of myelin formation [44].
Of note, an intriguing role of PDGF in initiating Fyn DNA synthesis has been also reported [45].
In addition, PDGF-mediated Fyn phosphorylation activates the serine/threonine Cdk5, which phosphorylates microtubule-associated proteins (MAP) such as MAP1B and promotes both microtubule assembly and stabilization during the early phases of neurons and OL differentiation and migration [43,46].
Interestingly, the receptor-like protein tyrosine phosphatase α (PTPα), highly expressed in the brain, regulates Fyn activity by triggering the dephosphorylation of Tyr531 residue on the Fyn C-tail [47,48,49]. PTPα has a very short extracellular domain with no adhesion motifs, which is the reason why it differs from most of the other receptor-like PTPs [48,50,51,52]. Overexpression of PTPα results in the acquisition of oncogenic Fyn phenotype [47] and PTPα deficient mice show a reduction in Fyn kinase activity [53]. PTPα mutant mice, in which the cysteine residues 414S (C414S) and 704S (C704S) are replaced with serine in the catalytic domains active site, fails to dephosphorylate Fyn [49,53].
Notably, Wang and colleagues recently described a novel critical upstream regulator of Fyn signaling, Receptor-type protein tyrosine phosphatase alpha (RPTPα), that appears to be required for oligodendrocyte progenitor differentiation and myelination [54].
3. Fyn in the Brain: Distribution and Function
Fyn is one of the highest expressed Src TK in the brain with a distribution in all the limbic regions as well as in cerebellum and striatum [55,56].
During embryonic development, a large amount of Fyn has been detected in the cerebral cortex, in the cerebellum, telencephalon, and brain stem [55,56,57]. Such extensive Fyn distribution is evident during brain development and it persists in adult brain (Figure 2) [55].
The broad distribution of Fyn in the brain reflects its critical role. Indeed, Fyn tempers excitatory and inhibitory synaptic transmission stimuli and regulates mechanisms related to learning and memory processes [55,58,59] (Figure 3).
Consistently, Fyn-deficient mice, show an aberrant distribution of neocortical neurons at E16 specifically in layer II-III [60] with a thin cerebral cortex, axonal degeneration, and a decreased number of oligodendrocytes [61]. Fyn mutant mice carrying a mutation on the regulatory residue Tyr531 (Y531F) display early lethality, reduced weight, hyperactivity, persistent tremor, lack of coordination and altered locomotion behavior [56]. In addition, mice carrying a point mutation in the SH2 domain (FynR176A) show an impaired neural migration in the cerebral cortex [62].
3.1. Fyn Promotes Myelination in the CNS
Myelin consists of multiple concentric layers of glial cell membrane, highly enriched in lipid (about 70% of the dry weight of myelin) and containing approximately 30% of proteins [67].
Myelin basic protein (MBP) [68] and the proteolipid protein (PLP/DM20) [69], are the two major myelin proteins in the CNS. MBP represents the 30% of total protein content and it is localized at opposing cytoplasmic faces of the myelin lamellae thus playing a role in myelin compaction and in the adhesion of the internal leaflets of the specialized oligodendroglia plasma membrane [70,71,72,73,74].
Umemori et al. (1999) [75] and White et al., (2008) [64] described the involvement of Fyn in the transactivation of the MBP gene in the initial stages of myelination. Shortly, MBP mRNA translation is essential for myelination and this process is regulated by the binding of the trans-acting factor heterogeneous nuclear ribonucleoprotein (hnRNP) A2 to the cis-acting A2 response element (A2RE). Hence, Fyn phosphorylates the hnRNP-A2, thus stimulating the MBP translation and initiating myelination processes [64].
In support to the critical role exerted by Fyn in myelination, Fyn deficient mice show a severe hypomyelination in the forebrain and a significant reduction in the amount of MBP compared to wild type [76]. Conversely, Fyn overexpression triggers myelinization in oligodendrocytes [76].
Interestingly, Lyn and Src knockout (KO) mice show no significant deficit in myelin formation, featuring the possibility that Fyn is the only Src family member that plays a role in myelination [66].
In addition, Fyn kinase-dead mutant mice, with a point mutation in the Fyn ATP binding site (FynK296R), show a severe myelin deficit in the forebrain with a reduced number of oligodendrocytes [66], suggesting that Fyn is required for the oligodendrocyte maturation. In agreement, Osterhout et al., (1999) revealed that Fyn activation is one of the earliest events as oligodendrocyte progenitor cells differentiate [77].
Fyn activation may occur by several pathways. Particularly, mechanisms involving neuronal adhesion molecule L1 [64], integrins [78,79] or gamma chain of immunoglobulin receptor (γFcR) [80] have been previously reported. In particular, the evidence that γFcR activates Fyn signaling cascade and consequently promotes OL differentiation and myelin repair has opened an unexplored scenario for the use of γFcR in the treatment of demyelinating diseases [80].
Of interest, a regulatory mechanism of MBP expression via Fyn mRNA transcriptional inhibition has been described. Accordingly, MiR-125a-3p, that it is particularly abundant in the CNS, directly binds the 3′UTR region of Fyn mRNA and inhibits Fyn expression [81]. This, in turn, controls MBP transactivation, resulting in a delay in the process of oligodendroglia maturation [81].
Another myelin constituent is MAG (myelin-associated protein), an adhesion member of the immunoglobulin superfamily expressed exclusively in myelinating oligodendrocytes and Schwann cells [82]. MAG binds Fyn at SH2 and SH3 domains and triggers Fyn activation [63]. Interestingly, an impressive colocalization of Fyn and MAG in fiber tract and oligodendrocytes during the early stage of myelination has been reported [63]. MAG appears to coordinate OL survival and differentiation through Fyn signals during the initial phase of myelination. Consistently, the genetic abrogation of MAG and Fyn causes a diffuse hypomyelination and exacerbates the phenotype observed in Fyn KO mice [64].
Whether or not Fyn contributes to the severity of diseases in which myelin growth or structure is damaged, such as multiple sclerosis (MS) or ischemic and traumatic brain injury [83], or neuropsychiatric diseases such as schizophrenia [84] or neurodegenerative diseases, such as Alzheimer’s disease [85,86] is largely controverted. Of interest, some single nucleotide polymorphisms (SNPs) in Fyn gene and/or in Fyn-related genes have been detected in schizophrenic patients pointing on Fyn genetic variants as a potential susceptibility factor in myelination disorders [87,88,89].
3.2. Fyn Mediates Oligodendrocytes Differentiation and Maturation
Fyn is involved in differentiation and/or maturation of oligodendrocytes (OL) [77]. Studies on primary cultures of differentiating oligodendrocytes demonstrate that Fyn is expressed in the cell body and throughout the processes, both in progenitor and mature oligodendrocytes. In particular, Fyn expression is two or three-fold more, and its kinase activity is 10-30 times higher in mature OL rather than in progenitors [77]. Accordingly, Fyn inhibitors, such as pyrazolopyrimidine derivates PP1 and PP2, prevent OL differentiation causing thick and irregular myelinization and the formation of clamps [77].
In addition, also the number of OL is regulated by Fyn as immunostaining experiments reveal that the absence of Fyn results in a reduction of 40–50% of the number of OL. However, whether this reduction is due to a reduced proliferation or to an increased neuronal death, or even to other mechanisms deserve further clarifications [61,66].
Relevantly, Klein et al. (2002) [90] reported that Tau binds the Fyn SH3 domain, and both are expressed in OL processes and soma. The deletion of -PPXX- Tau motif results in the disruption of Fyn-Tau interaction and causes the decrease in the number and length of OL processes thus emphasizing the significance of Fyn-Tau interaction also in OL process formation [90].
3.3. Fyn and Semaphorins in Neurodevelopment and Neurodevelopment-Associated Disorders
Fyn has been largely associated with neurodevelopment and in particular alterations in its expression levels of activity have been linked to the onset of deficits in the behavioral and emotional spectrum. Indeed, Fyn deficient mice have shown abnormal hippocampal development [94] and mice in which the Fyn gene is replaced by LacZ, show an increased sensitivity to stress and emotional abnormalities, also justified by the presence of lesions in the limbic system [56,57,60,95]. Consistently, increased expression of Fyn in the prefrontal cortex seems to contribute to the pathogenesis of schizophrenia [96] and SNPs along the Fyn gene sequence have been related to several development-related disorders [97]. In this context, rs6916861, rs3730353, and rs706895 Fyn polymorphisms were found risk factors for schizophrenia in the Chinese-Han population [98] and rs6916861 and rs3730353 for bipolar disorder [88].
Fyn controls also the BDNF/TrkB and NGF/TrkA pathways which are both involved in neurodevelopment as well as neurodegenerative processes [99,100,101,102,103]. Of note, BDNF gene polymorphisms have been hypothesized to play a possible role in the pathogenesis of autism spectrum disorder (ASD) via Fyn activity dysregulation [104,105,106,107,108,109,110].
Interestingly, Fyn promotes semaphorin receptor (plexin) phosphorylation and facilitates semaphorin and plexin interaction [111,112,113] that is crucial in mediating events related to neuronal polarization and migration [114,115], synapse formation [116,117], axonal pruning [118,119] and dendritic arborization [120,121,122] (Figure 4).
By contrast, in pathological conditions, following mechanisms still not well characterized, Fyn mediates Plexin-A2 hyperphosphorylation thus activating downstream signaling that have been associated with several neurodegenerative diseases such as Alzheimer’s and Parkinson’s disease, multiple sclerosis and Amyotrophic lateral sclerosis [123,124,125,126]. Adapted from Sasaki et al. (2002) [111].
Of relevance, Schafer et al., identified genetic variants of Fyn, as well as semaphorin and plexin, in patients with neurodevelopment-associated disorders and autism [97,113,127,128] (Table 1).
Fyn after interacting with plexin tyrosine sites, activates cyclin-dependent kinase 5 (Cdk5) [111]. Cdk5 is a member of proline-directed serine/threonine kinase family, expressed in proliferating cells that, unlike other family members, has a restricted role in the nervous system [129]. In turn, Cdk5 phosphorylates Tau protein and activates a cascade of events finally resulting in growth cone collapse. Of note, Fyn or Cdk5 pharmacological inhibitors prevent all these events [111] (Figure 4).
On the other hand, Cdk5 also phosphorylates other proteins, such as collapsin response mediator protein-2 (CRMP-2) [130] that appears to be important in mediating Sema3A signaling and in the control of axon formation and microtubule polymerization through its binding to tubulin heterodimers [131,132] (Figure 4). A mutation in Ser522 prevents Cdk5 phosphorylation and protects from Sema3A-induced growth cone collapse [133].
Emerging evidence suggests a role of Sema3A in promoting dendritic branching and neurites formation in cortical neurons via Fyn activation both these events are not observed in neurons exposed to PP2 (Fyn selective inhibitor) or in Fyn‒/‒ mice [120]. Of note, studies on Fyn‒/‒ or Sema3A‒/‒ mice, show that the lack of these two proteins decreases dendritic spine density in layer V of pyramidal neurons, whereas layer III seems not to be affected, highlighting the importance of Sema3A-Fyn pathway in regulating spine morphogenesis and density in cerebral cortex [120].
Although there are, as yet, no examples of neuronal diseases in which alterations in Fyn signal affects semaphorin-plexin interactions and downstream pathways, these are likely to exist.
Dysregulation of semaphorins has been detected in damaged CNS axons and particularly their expression has been found strongly upregulated in MS oligodendrocytes located near the injury site [134,135] (Figure 4).
Changes in the endothelial tight junction structure on the blood–brain barrier (BBB) have been detected in MS brains and associated with Sema3A and Sema4D overactivation [134,135]. Of note these changes have been related to the aberrant activation of plexin-Fyn-AKT signaling pathway and TK inhibitors have been proposed as potential strategy to ameliorate these deficits [7] (Figure 4).
An abnormal neurohistological pattern of semaphorins has been detected in adult brain tissues and in the affected cortex and hippocampus of AD brains [136]. In particular, Sema3A was found hyperphosphorylated in AD brains along with plexins, CRMP-2, and microtubule-associated protein 1B (MAP-1B), that are all molecular targets of Fyn [137]. Of note, semaphorin (as well as their receptor) gene variants have been associated with AD [138], opening the relatively underexplored possibility that alterations in semaphorin pathways may become potential susceptibility factors of AD.
In addition, Venkova et al. hypothesized a role of Sema3A in promoting distal axonopathy and muscle denervation observed in the SOD1G93A mouse model of ALS [139]. This was described to occur through the activation of Fyn/Cdk5 and Glycogen synthase kinase 3 beta (GSK3β) pathways as well as CRMP-2 phosphorylation ultimately leading to microtubule instability and actin cytoskeletal rearrangements [42] (Figure 4).
3.4. Fyn Controls Neuronal Migration
Fyn plays an important role in neuronal migration and cortical lamination [62].
A point mutation in the Fyn SH2 domain (FynR176A) impaired neuronal migration and neuronal morphogenesis by inducing neuronal aggregation and branching [62]. In addition, the double KO mouse of Fyn, as well as of Src, shows a reeler-like phenotype [140].
Reelin is an extracellular glycoprotein with a prominent activity in the control of neuronal migration and cellular layer formation in the developing brain [141,142,143]. Reeler mutant mice lacking reelin expression [144] exhibit a neurological phenotype characterized by ataxia and a typical “reeling” gate consisting in widespread defects in neuronal lamination in the developing forebrain, and in cerebellar hypoplasia, due to the failure of radially-migrating neurons to reach their destination and to the failure of Purkinje cells to form a cellular layer, respectively [145,146]. Similar phenotypes have been described in patients carrying reelin homozygous mutations, characterized by lissencephaly with cerebellar hypoplasia [147].
In addition, reelin activates RasGRF1/CaMKII pathway [148] and promotes dendrites maturation, synaptogenesis, synaptic transmission, and plasticity, thus controlling the formation and function of synaptic circuits during the development and in adult brain [149,150,151,152]. Consistently, heterozygous reelin mutations have been associated with lateral temporal epilepsy [153], and have been pointed as a risk factor in autism [154]. The molecular composition of the dendritic spines is also affected by reelin via an unidentified mechanism involving NMDA receptor [155,156].
A crosstalk between Fyn and reelin has been well characterized and elucidated and it is largely dependent on Disabled-1 (Dab1), whose phosphorylation is required for neuronal migration. Phospho-mutant Dab1 mice [157,158], double Fyn/Src KO mice [140], as well as spontaneous or genetically engineered Dab1 KO mice, all show similar reeler-like phenotypes [159,160,161,162,163,164].
Dab-1 binds the cytoplasmic tail of lipoprotein receptors, including ApoER2 and VLDLR [144] and upon reelin binding, becomes phosphorylated on tyrosine residues by Fyn and Src [157].
The binding of Reelin to ApoER2 triggers ApoER2, Dab1, and NMDA receptors clustering and the activation of Fyn, finally resulting in the NMDAR phosphorylation, in an increased Ca2+ influx and in neurotoxicity [165,166].
Other molecules have been implicated in reelin-dependent dendrite outgrowth such as the amyloid precursor protein (APP) [167], which binds Dab1 via its cytoplasmic tail [157,168]. Reelin signaling also exerts a protective effect against β-amyloid at the synapse, underscoring the potential relevance of this “developmental” factor for neurodegenerative disorders [149,169,170,171].
3.5. Fyn in Synaptic Regulation and Dysregulation
Strategically located at the postsynaptic density of glutamatergic synapses, Fyn is conceived to target local synaptic substrates and to regulate the strength and efficacy of synaptic transmission [172]. Fyn phosphorylates ionotropic glutamate receptors and other synaptic proteins, thereby modulating their expression and synaptic signaling [8,173]. Consequently, deregulations or dysfunctions in Fyn signaling have been related to synaptic deficits or neurodegenerative processes [6,174,175,176,177].
There are three classes of iGluRs: α-amino-3-hydroxy-5-methylisoxazole-4-propionic acid receptors (AMPAR), N-methyl-d-aspartate receptors (NMDAR), and kainate receptors [178].
NMDARs form functional channels gathering GluN1 (formerly known as NR1) with GluN2 subunits, mainly GluN2A (NR2A) and GluN2B (NR2B). AMPARs are assembled by four subunits (GluA1–4, previously named GluR1–4) [178,179].
All NMDAR and AMPAR subunits display an intracellular domain that has been found to accommodate dynamic protein–protein interactions and tyrosine phosphorylation [173,180,181,182].
In particular, Fyn phosphorylates the NR2A and NR2B subunits and consequently controls the subcellular or subsynaptic distributions of NMDA receptor and potentiate NMDAR currents [183]. Indeed, among 25 tyrosine residues in the C-terminal cytoplasmic region of NR2B, 7 of these are phosphorylated by Fyn in vitro [184].
NMDA phosphorylation is promoted by the presence of two PSD proteins: PSD-93 and PSD-95 [185,186]. PSD-95 forms a complex with Fyn and NMDA that appears to enhance NMDA activity [187]. Of note, a genetic deletion of PSD-93, results in a lower expression of Fyn and in the reduction in NR2A and NR2B phosphorylation [185].
After ischemic insults in the adult brain, Fyn promotes assembly and remodeling of the PSD complexes [19]. Fyn interacts directly with the α1c subunit of L-type voltage-gated calcium channel (L-VGCC) and increases Ca2+ influx and cytotoxicity [188]. Fyn also interacts with PSD-95 associated GTPase, SynGAP that has been reported increased during ischemic events, further triggering neurotoxic processes [189]. In addition, PSD-93 and Fyn/NR2B association and NR2B tyrosine phosphorylation is increased in adult brain after ischemia [190].
As a consequence, tyrosine-phosphorylated PSD-93 binds to Csk, a negative regulator of Fyn activity, and thereby inactivates Fyn [191].
Fyn phosphorylation at Tyr1472 (Y1472) of NR2B subunit, is also important to stabilize synaptic localization of NMDA receptor, by preventing the interaction with clathrin adaptor protein (AP2) and the consequent internalization [192,193,194,195].
Consistently, NR2B phosphorylation at Tyr1472 and Tyr1336 (Y1336) sites selectively enriched NR2B/NMDAR abundance in synaptic versus extrasynaptic compartments, respectively [195].
Of relevance, the aberrant Fyn-mediated phosphorylation of NR2A at Tyr1252 (Y1252) and NR2B at Tyr1472, as well as increased calpain activity, are associated with brain injury and mortality in response to neonatal hypoxic ischemia (HI) [19,196].
In particular, phosphorylation of Tyr1472 contributes to excitotoxic cell death by increasing NMDAR responses to glutamate [196]. In addition, phosphorylation of NR2B at Tyr1472 also activates reactive oxygen species (ROS) in a calcium-independent manner further supporting a detrimental role of Fyn in neuronal plasticity under neurotoxic stimuli [19,196].
Fyn phosphorylates AMPAR at Tyr876 (Y876) of GluR2 thus disrupting the association of GluR2 with glutamate receptor interacting proteins 1 and 2 (GRIP1/2), triggering endocytosis of GluR2 and ultimately leading to reduction of the surface expressed AMPARs [197].
Modulation of NMDA receptor controls cognitive function in prefrontal cortex and depends on dopamine D1 receptor [198]. The activation of dopamine D1 receptor triggers a rapid redistribution of NMDA receptors and increase NMDA expression at postsynaptic level [199].
Fyn phosphorylates the metabotropic glutamate receptor (mGluR) too [200,201]. Specifically, Fyn phosphorylates mGluR1a at Tyr937 (Y937) promoting its surface expression and controlling mGluR1a-dependent signaling transduction [200,201].
Fyn phosphorylation induces LTP in CA1 hippocampal areas [94,202]. Consistently, genetic ablation of Fyn results in LTP deficits [203] and gross structural changes in the dentate gyrus [163]. Interestingly, the role of Fyn appears to be restricted to specific developmental stages as it is not detectable in animals at less than 14 weeks [204].
Finally, Fyn activates protein tyrosine kinase 2 beta (Pyk2), encoded by the AD risk gene PTK2B, [205,206] and regulating synaptic plasticity. As the disease progresses, Aβ has been proposed to activate the Fyn phosphatase, striatal-enriched protein tyrosine phosphatase (STEP), eventually inactivating Fyn, which leads to the loss of synapses and dendritic spine collapse [207,208,209].
In mouse models, Fyn has been implicated as a downstream target of Amyloid β (Aβ) [209] (Figure 5). Accordingly, Aβ oligomers from the brains of AD patients activate Fyn after interacting with PrP(C) [177,210]. Aβ-Fyn interaction results in NR2B subunit phosphorylation, endocytosis of NMDARs and finally in synaptic deficiencies [182,183].
Notably, Fyn upregulation has been associated with increased APP Tyr682 residue phosphorylation in human AD neurons. Such hyperphosphorylation has been described to precede APP amyloidogenic processing, amyloid β accumulation, and neurodegenerative events [6,211] (Figure 5).
Fyn modulates cytoskeletal dynamics by phosphorylating and consequently inducing delocalization of proteins involved in cytoskeletal organization, such as Tau [212]. On the other side, Tau protein is hyperphosphorylated and abnormally folded in AD causing the lack in Tau ability to bind and stabilize microtubules in the axon. This loss of Tau function confers increasing aggregation properties triggering the formation of Tau tangles in AD [174,213] (Figure 5).
Although largely investigated, the role of Fyn in AD onset and/or progression is only partially understood. A large body of evidence has underlined the critical role of Fyn in balancing Tyr phosphorylation content of numerous neuronal proteins, including Tau and APP [6,214]. Consistently, the increased tyrosine phosphorylation of target proteins can be blocked by the addition of TK inhibitors suggesting that Fyn hyperactivity might be pharmacologically targeted to delay degenerative processes in AD [6,174,176,215,216].
Haass et al., recently described Fyn-APP/Aβ-Tau as a toxic triad. [217]. This toxicity appears to be related to the altered Tau redistribution in AD neurons, resulting in an enhanced Fyn-mediated phosphorylation of Tau either directly at the level of Tyr18 or indirectly at the level of Ser/Thre residues through GSK-3β activation [214,217,218,219].
Fyn expression in the brain is influenced by AD status and genetic content. An upregulation of Fyn and Tau proteins have been reported in a subset of neurons from AD tissues in the initial stages of neurodegenerative processes [214,218].
Fyn distribution and levels are altered in AD brains [220,221], and the genetic ablation of Fyn counteracts Aβ oligomers toxicity in hippocampal slices [222], suggesting that Aβ may derange synaptic functions through the aberrant activation of Fyn-related pathways [13,223]. Accordingly, an increased Fyn activity sensitizes neurons to Aβ-induced neuronal toxicity and exacerbates the Aβ-related increase in neuronal activity [13].
Interestingly, compensatory mechanisms that limit Fyn hyperactivity have been also reported in mice model of AD, overexpressing hAPP, consisting in a significant increase in phosphatase STEP levels, which dephosphorylates and inactivates Fyn [13] and consequently, reduces NMDA receptor phosphorylation and internalization and prevent synaptic dysfunctions [13,182,221].
miR-106b whose expression is reduced in AD tissue, directly binds to the 3′UTR of Fyn mRNA, and consequently deregulates Fyn mRNA expression resulting in decreased Fyn mRNA levels mostly in the frontal cortex. This finally results in a decreased Tau phosphorylation at Tyr18 [224].
Two SNPs located in the FYN gene, rs7768046, and rs1621289 (the first tagging the long isoform promoter region and the second located within the 3′ UTR region of FYN) appear to be associated with increased total Tau (t-Tau) levels in AD cerebrospinal fluid (CSF) [225].
Fyn phosphorylates also α-synuclein, a presynaptic protein that has been found hyperphosphorylated in neurons of Parkinson’s disease (PD) and AD patients [226]. Consistently, Fyn-mediated α-synuclein phosphorylation at Tyr125 was inhibited by the TyrKI inhibitor, PP2 [226]. In addition, studies performed in Fyn KO mice have prospected the possibility to use Fyn TK inhibitors to prevent or recover the altered dopamine-dependent trafficking of striatal NMDAR observed in PD neurons [227].
4. Conclusions
In summary, there is mounting and compelling evidence implicating Fyn as a key factor in modulating physiologic neuronal pathways and development. These observations raise the questions of whether and which alterations in these molecular pathways lead to neuronal deficiencies and dysfunctions and whether targeting Fyn can have disease-modifying effects in these conditions. These questions seem to find an answer in prior work from several groups including some of our studies where it has been suggested that the pharmacologic targeting of Fyn may be therapeutically efficacious in dysfunctional neurons in which Fyn activity is impaired.
However, despite the encouraging evidence that targeting known and emerging elements of Fyn signaling cascades may become a promising therapeutic strategy in neuronal dysfunctions, Fyn still remains a challenging target, with broad expression throughout the body and significant homology with other members of the Src family kinases, likely leading to unintended off-target effects.
Overall, this review pinpoints the importance of Fyn in a multitude of neuronal function and dysfunction opening up the necessity of the development of innovative and selective therapeutic strategies to control Fyn activity and prevent brain diseases.
Author Contributions
All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by MIUR/PRIN, grant number 2017T9JNLT.
Conflicts of Interest
The authors declare no conflict of interest.
Abbreviations
| AMPAR | α-amino-3-hydroxy-5-methylisoxazole-4-propionic acid receptor |
| AD | Alzheimer’s Disease |
| APP | Amyloid Precursor Protein |
| ApoER2 | Apolipoprotein E receptor 2 |
| AP2 | Clathrin adaptor protein |
| ASD | Autism Spectrum Disorder |
| A2RE | A2 response element |
| BBB | blood–brain barrier |
| Cdk5 | CiclinA2-dependent kinase 5 |
| CRMP-2 | Collapsin response mediator protein-2 |
| CDT | Carboxy-terminal domains |
| CNS | Central Nervous System |
| DAB1 | Intracellular adaptor protein |
| GAP-domain | GTPase activating domain |
| GRIP1/2 | Glutamate receptor interacting proteins 1 and 2 |
| GSK3β | Glycogen synthase kinase 3 beta |
| HI | Hypoxic–ischemia |
| HnRNP A2 | Heterogeneous nuclear ribonucleoprotein A2 |
| IPT | Immunoglobulin-plexin-transcription domain |
| LTP | Long-term potential |
| L-VGCC | L-type voltage-gated calcium channel |
| MAG | Myelin-associated glycoprotein |
| MAP | Microtubule-associated proteins |
| MBP | Myelin basic protein |
| MGLUR | Metabotropic glutamate receptor |
| MS | Multiple sclerosis |
| NGF | Nerve Growth Factor |
| NMDA | N-methyl-d-aspartate |
| NMDAR | N-methyl-d-aspartate receptor |
| NRTK | Non-Receptor Tyrosine Kinase |
| OL | Oligodendrocytes |
| OTK | Off-track kinases |
| PD | Parkinson Disease |
| PDGF | Platelet-Derived Growth factor |
| PLP | Proteolipid protein |
| PP1,PP2 | Pyrazolopyrimidine |
| PrPC | Prion protein |
| PSD | Postsynaptic density |
| PSI | Plexin-semaphorin-integrin domain |
| PYK2 | Protein tyrosine kinase 2 beta |
| ROS | Reactive oxygen species |
| RPTPα | Receptor-like Protein Tyrosine Phosphatase α |
| RSV | Rous’ virus |
| SFK | Src Family Kinases |
| SNPs | Single nucleotide polymorphisms |
| STEP | Striatal Enriched Phosphatase |
| TK | Tyrosine Kinase |
| VLDL-R | Very-low density lipoprotein receptor |
| Γ-FCR | Γ-chain of immunoglobulin receptor |
References
- Rudd, C.E.; Janssen, O.; Prasad, K.V.; Raab, M.; da Silva, A.; Telfer, J.C.; Yamamoto, M. src-related protein tyrosine kinases and their surface receptors. Biochim. Biophys. Acta 1993, 1155, 239–266. [Google Scholar] [CrossRef]
- Cooper, J.A.; Howell, B. The when and how of Src regulation. Cell 1993, 73, 1051–1054. [Google Scholar] [CrossRef]
- Li, W.; Young, S.L.; King, N.; Miller, W.T. Signaling properties of a non-metazoan Src kinase and the evolutionary history of Src negative regulation. J. Biol. Chem. 2008, 283, 15491–15501. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Segawa, Y.; Suga, H.; Iwabe, N.; Oneyama, C.; Akagi, T.; Miyata, T.; Okada, M. Functional development of Src tyrosine kinases during evolution from a unicellular ancestor to multicellular animals. Proc. Natl. Acad. Sci. USA 2006, 103, 12021–12026. [Google Scholar] [CrossRef] [Green Version]
- Reinecke, J.; Caplan, S. Endocytosis and the Src family of non-receptor tyrosine kinases. Biomol. Concepts 2014, 5, 143–155. [Google Scholar] [CrossRef] [Green Version]
- Matrone, C.; Iannuzzi, F.; Annunziato, L. The, Y682ENPTY687 motif of APP: Progress and insights toward a targeted therapy for Alzheimer’s disease patients. Ageing. Res. Rev. 2019, 52, 120–128. [Google Scholar] [CrossRef]
- Franklin, R.J.; Ffrench-Constant, C. Remyelination in the CNS: From biology to therapy. Nat. Rev. Neurosci. 2008, 9, 839–855. [Google Scholar] [CrossRef]
- Schenone, S.; Brullo, C.; Musumeci, F.; Biava, M.; Falchi, F.; Botta, M. Fyn kinase in brain diseases and cancer: The search for inhibitors. Curr. Med. Chem. 2011, 18, 2921–2942. [Google Scholar] [CrossRef] [PubMed]
- Resh, M.D. Fyn, a Src family tyrosine kinase. Int. J. Biochem. Cell Biol. 1998, 30, 1159–1162. [Google Scholar] [CrossRef]
- Saito, Y.D.; Jensen, A.R.; Salgia, R.; Posadas, E.M. Fyn: A novel molecular target in cancer. Cancer 2010, 116, 1629–1637. [Google Scholar] [CrossRef]
- Thomas, S.M.; Brugge, J.S. Cellular functions regulated by Src family kinases. Annu. Rev. Cell. Dev. Biol. 1997, 13, 513–609. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sicheri, F.; Kuriyan, J. Structures of Src-family tyrosine kinases. Curr. Opin. Struct. Biol. 1997, 7, 777–785. [Google Scholar] [CrossRef]
- Chin, J.; Palop, J.J.; Puoliväli, J.; Massaro, C.; Bien-Ly, N.; Gerstein, H.; Scearce-Levie, K.; Masliah, E.; Mucke, L. Fyn kinase induces synaptic and cognitive impairments in a transgenic mouse model of Alzheimer’s disease. J. Neurosci. 2005, 25, 9694–9703. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chun, J.T.; Crispino, M.; Tocco, G. The dual response of protein kinase Fyn to neural trauma: Early induction in neurons and delayed induction in reactive astrocytes. Exp. Neurol. 2004, 185, 109–119. [Google Scholar] [CrossRef]
- Cooke, M.P.; Perlmutter, R.M. Expression of a novel form of the fyn proto-oncogene in hematopoietic cells. New. Biol. 1989, 1, 66–74. [Google Scholar]
- Ding, Q.; Stewart, J.; Olman, M.A.; Klobe, M.R.; Gladson, C.L. The pattern of enhancement of Src kinase activity on platelet-derived growth factor stimulation of glioblastoma cells is affected by the integrin engaged. J. Biol. Chem. 2003, 278, 39882–39891. [Google Scholar] [CrossRef] [Green Version]
- Abram, C.L.; Courtneidge, S.A. Src family tyrosine kinases and growth factor signaling. Exp. Cell Res. 2000, 254, 1–13. [Google Scholar] [CrossRef]
- Goldsmith, J.F.; Hall, C.G.; Atkinson, T.P. Identification of an alternatively spliced isoform of the fyn tyrosine kinase. Biochem. Biophys. Res. Commun. 2002, 298, 501–504. [Google Scholar] [CrossRef]
- Knox, R.; Jiang, X. Fyn in Neurodevelopment and Ischemic Brain Injury. Dev. Neurosci. 2015, 37, 311–320. [Google Scholar] [CrossRef] [Green Version]
- Gonfloni, S.; Weijland, A.; Kretzschmar, J.; Superti-Furga, G. Crosstalk between the catalytic and regulatory domains allows bidirectional regulation of Src. Nat. Struct. Biol. 2000, 7, 281–286. [Google Scholar] [CrossRef]
- Kaplan, J.M.; Mardon, G.; Bishop, J.M.; Varmus, H.E. The first seven amino acids encoded by the v-src oncogene act as a myristylation signal: Lysine 7 is a critical determinant. Mol. Cell. Biol. 1988, 8, 2435–2441. [Google Scholar] [CrossRef] [Green Version]
- Resh, M.D. Interaction of tyrosine kinase oncoproteins with cellular membranes. Biochim. Biophys. Acta 1993, 1155, 307–322. [Google Scholar] [CrossRef]
- Rawat, A.; Nagaraj, R. Determinants of membrane association in the SH4 domain of Fyn: Roles of N-terminus myristoylation and side-chain thioacylation. Biochim. Biophys. Acta 2010, 1798, 1854–1863. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alland, L.; Peseckis, S.M.; Atherton, R.E.; Berthiaume, L.; Resh, M.D. Dual myristylation and palmitylation of Src family member p59fyn affects subcellular localization. J. Biol. Chem. 1994, 269, 16701–16705. [Google Scholar]
- Resh, M.D. Myristylation and palmitylation of Src family members: The fats of the matter. Cell 1994, 76, 411–413. [Google Scholar] [CrossRef]
- Patwardhan, P.; Resh, M.D. Myristoylation and membrane binding regulate c-Src stability and kinase activity. Mol. Cell. Biol. 2010, 30, 4094–4107. [Google Scholar] [CrossRef] [Green Version]
- Koch, C.A.; Anderson, D.; Moran, M.F.; Ellis, C.; Pawson, T. SH2 and SH3 domains: Elements that control interactions of cytoplasmic signaling proteins. Science 1991, 252, 668–674. [Google Scholar] [CrossRef] [PubMed]
- Mayer, B.J.; Baltimore, D. Signalling through SH2 and SH3 domains. Trends Cell Biol. 1993, 3, 8–13. [Google Scholar] [CrossRef]
- Cohen, G.B.; Ren, R.; Baltimore, D. Modular binding domains in signal transduction proteins. Cell 1995, 80, 237–248. [Google Scholar] [CrossRef] [Green Version]
- Songyang, Z.; Shoelson, S.E.; Chaudhuri, M.; Gish, G.; Pawson, T.; Haser, W.G.; King, F.; Roberts, T.; Ratnofsky, S.; Lechleider, R.J.; et al. SH2 domains recognize specific phosphopeptide sequences. Cell 1993, 72, 767–778. [Google Scholar] [CrossRef]
- Pawson, T. Protein modules and signalling networks. Nature 1995, 373, 573–580. [Google Scholar] [CrossRef] [PubMed]
- Brown, M.T.; Cooper, J.A. Regulation, substrates and functions of src. Biochim. Biophys. Acta 1996, 1287, 121–149. [Google Scholar] [CrossRef]
- Pucheta-Martínez, E.; Saladino, G.; Morando, M.A.; Martinez-Torrecuadrada, J.; Lelli, M.; Sutto, L.; D’Amelio, N.; Gervasio, F.L. An Allosteric Cross-Talk Between the Activation Loop and the ATP Binding Site Regulates the Activation of Src Kinase. Sci. Rep. 2016, 6, 24235. [Google Scholar] [CrossRef] [PubMed]
- Krämer-Albers, E.M.; White, R. From axon-glial signalling to myelination: The integrating role of oligodendroglial Fyn kinase. Cell. Mol. Life Sci. 2011, 68, 2003–2012. [Google Scholar] [CrossRef] [PubMed]
- Cartwright, C.A.; Simantov, R.; Kaplan, P.L.; Hunter, T.; Eckhart, W. Alterations in pp60c-src accompany differentiation of neurons from rat embryo striatum. Mol. Cell. Biol. 1987, 7, 1830–1840. [Google Scholar] [CrossRef] [Green Version]
- Reynolds, A.B.; Vila, J.; Lansing, T.J.; Potts, W.M.; Weber, M.J.; Parsons, J.T. Activation of the oncogenic potential of the avian cellular src protein by specific structural alteration of the carboxy terminus. EMBO J. 1987, 6, 2359–2364. [Google Scholar] [CrossRef]
- Okada, M.; Nakagawa, H. A protein tyrosine kinase involved in regulation of pp60c-src function. J. Biol. Chem. 1989, 264, 20886–20893. [Google Scholar]
- Cooper, J.A.; Gould, K.L.; Cartwright, C.A.; Hunter, T. Tyr527 is phosphorylated in pp60c-src: Implications for regulation. Science 1986, 231, 1431–1434. [Google Scholar] [CrossRef]
- Nada, S.; Okada, M.; MacAuley, A.; Cooper, J.A.; Nakagawa, H. Cloning of a complementary DNA for a protein-tyrosine kinase that specifically phosphorylates a negative regulatory site of p60c-src. Nature 1991, 351, 69–72. [Google Scholar] [CrossRef]
- Okada, M. Regulation of the SRC family kinases by Csk. Int. J. Biol. Sci. 2012, 8, 1385–1397. [Google Scholar] [CrossRef] [Green Version]
- Nada, S.; Yagi, T.; Takeda, H.; Tokunaga, T.; Nakagawa, H.; Ikawa, Y.; Okada, M.; Aizawa, S. Constitutive activation of Src family kinases in mouse embryos that lack Csk. Cell 1993, 73, 1125–1135. [Google Scholar] [CrossRef]
- Nguyen, T.H.; Liu, J.; Lombroso, P.J. Striatal enriched phosphatase 61 dephosphorylates Fyn at phosphotyrosine 420. J. Biol. Chem. 2002, 277, 24274–24279. [Google Scholar] [CrossRef] [Green Version]
- Hansen, K.; Alonso, G.; Courtneidge, S.A.; Rönnstrand, L.; Heldin, C.H. PDGF-induced phosphorylation of Tyr28 in the N-terminus of Fyn affects Fyn activation. Biochem. Biophys. Res. Commun. 1997, 241, 355–362. [Google Scholar] [CrossRef] [PubMed]
- Gartlehner, G.; Hansen, R.A.; Carson, S.S.; Lohr, K.N. Efficacy and safety of inhaled corticosteroids in patients with COPD: A systematic review and meta-analysis of health outcomes. Ann. Fam. Med. 2006, 4, 253–262. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Twamley-Stein, G.M.; Pepperkok, R.; Ansorge, W.; Courtneidge, S.A. The Src family tyrosine kinases are required for platelet-derived growth factor-mediated signal transduction in NIH 3T3 cells. Proc. Natl. Acad. Sci. USA 1993, 90, 7696–7700. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hansen, K.; Rönnstrand, L.; Claesson-Welsh, L.; Heldin, C.H. Phosphorylation of a 72-kDa protein in PDGF-stimulated cells which forms complex with c-Crk, c-Fyn and Eps15. FEBS Lett. 1997, 409, 195–200. [Google Scholar] [CrossRef] [Green Version]
- Zheng, X.M.; Wang, Y.; Pallen, C.J. Cell transformation and activation of pp60c-src by overexpression of a protein tyrosine phosphatase. Nature 1992, 359, 336–339. [Google Scholar] [CrossRef]
- Sap, J.; D’Eustachio, P.; Givol, D.; Schlessinger, J. Cloning and expression of a widely expressed receptor tyrosine phosphatase. Proc. Natl. Acad. Sci. USA 1990, 87, 6112–6116. [Google Scholar] [CrossRef] [Green Version]
- Bhandari, V.; Lim, K.L.; Pallen, C.J. Physical and functional interactions between receptor-like protein-tyrosine phosphatase alpha and p59fyn. J. Biol. Chem. 1998, 273, 8691–8698. [Google Scholar] [CrossRef] [Green Version]
- Matthews, R.J.; Cahir, E.D.; Thomas, M.L. Identification of an additional member of the protein-tyrosine-phosphatase family: Evidence for alternative splicing in the tyrosine phosphatase domain. Proc. Natl. Acad. Sci. USA 1990, 87, 4444–4448. [Google Scholar] [CrossRef] [Green Version]
- Kaplan, R.; Morse, B.; Huebner, K.; Croce, C.; Howk, R.; Ravera, M.; Ricca, G.; Jaye, M.; Schlessinger, J. Cloning of three human tyrosine phosphatases reveals a multigene family of receptor-linked protein-tyrosine-phosphatases expressed in brain. Proc. Natl. Acad. Sci. USA 1990, 87, 7000–7004. [Google Scholar] [CrossRef] [Green Version]
- Krueger, N.X.; Streuli, M.; Saito, H. Structural diversity and evolution of human receptor-like protein tyrosine phosphatases. EMBO J. 1990, 9, 3241–3252. [Google Scholar] [CrossRef] [PubMed]
- Ponniah, S.; Wang, D.Z.; Lim, K.L.; Pallen, C.J. Targeted disruption of the tyrosine phosphatase PTPalpha leads to constitutive downregulation of the kinases Src and Fyn. Curr. Biol. 1999, 9, 535–538. [Google Scholar] [CrossRef] [Green Version]
- Wang, P.S.; Wang, J.; Xiao, Z.C.; Pallen, C.J. Protein-tyrosine phosphatase alpha acts as an upstream regulator of Fyn signaling to promote oligodendrocyte differentiation and myelination. J. Biol. Chem. 2009, 284, 33692–33702. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Umemori, H.; Wanaka, A.; Kato, H.; Takeuchi, M.; Tohyama, M.; Yamamoto, T. Specific expressions of Fyn and Lyn, lymphocyte antigen receptor-associated tyrosine kinases, in the central nervous system. Brain Res. Mol. Brain. Res. 1992, 16, 303–310. [Google Scholar] [CrossRef]
- Yagi, T.; Shigetani, Y.; Okado, N.; Tokunaga, T.; Ikawa, Y.; Aizawa, S. Regional localization of Fyn in adult brain; studies with mice in which fyn gene was replaced by lacZ. Oncogene 1993, 8, 3343–3351. [Google Scholar]
- Yagi, T.; Shigetani, Y.; Furuta, Y.; Nada, S.; Okado, N.; Ikawa, Y.; Aizawa, S. Fyn expression during early neurogenesis in mouse embryos. Oncogene 1994, 9, 2433–2440. [Google Scholar] [PubMed]
- Bixby, J.L.; Jhabvala, P. Tyrosine phosphorylation in early embryonic growth cones. J. Neurosci. 1993, 13, 3421–3432. [Google Scholar] [CrossRef] [Green Version]
- Sudol, M.; Hanafusa, H. Cellular proteins homologous to the viral yes gene product. Mol. Cell. Biol. 1986, 6, 2839–2846. [Google Scholar] [CrossRef] [Green Version]
- Yuasa, S.; Hattori, K.; Yagi, T. Defective neocortical development in Fyn-tyrosine-kinase-deficient mice. Neuroreport 2004, 15, 819–822. [Google Scholar] [CrossRef]
- Goto, J.; Tezuka, T.; Nakazawa, T.; Sagara, H.; Yamamoto, T. Loss of Fyn tyrosine kinase on the C57BL/6 genetic background causes hydrocephalus with defects in oligodendrocyte development. Mol. Cell. Neurosci. 2008, 38, 203–212. [Google Scholar] [CrossRef] [PubMed]
- Lu, X.; Hu, X.; Song, L.; An, L.; Duan, M.; Chen, S.; Zhao, S. The SH2 domain is crucial for function of Fyn in neuronal migration and cortical lamination. BMB Rep. 2015, 48, 97–102. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Umemori, H.; Sato, S.; Yagi, T.; Aizawa, S.; Yamamoto, T. Initial events of myelination involve Fyn tyrosine kinase signalling. Nature 1994, 367, 572–576. [Google Scholar] [CrossRef] [PubMed]
- White, R.; Gonsior, C.; Krämer-Albers, E.M.; Stöhr, N.; Hüttelmaier, S.; Trotter, J. Activation of oligodendroglial Fyn kinase enhances translation of mRNAs transported in hnRNP A2-dependent RNA granules. J. Cell Biol. 2008, 181, 579–586. [Google Scholar] [CrossRef] [PubMed]
- Biffiger, K.; Bartsch, S.; Montag, D.; Aguzzi, A.; Schachner, M.; Bartsch, U. Severe hypomyelination of the murine CNS in the absence of myelin-associated glycoprotein and fyn tyrosine kinase. J. Neurosci. 2000, 20, 7430–7437. [Google Scholar] [CrossRef] [Green Version]
- Sperber, B.R.; Boyle-Walsh, E.A.; Engleka, M.J.; Gadue, P.; Peterson, A.C.; Stein, P.L.; Scherer, S.S.; McMorris, F.A. A unique role for Fyn in CNS myelination. J. Neurosci. 2001, 21, 2039–2047. [Google Scholar] [CrossRef]
- Baumann, N.; Pham-Dinh, D. Biology of oligodendrocyte and myelin in the mammalian central nervous system. Physiol. Rev. 2001, 81, 871–927. [Google Scholar] [CrossRef]
- Steinman, L. Multiple sclerosis: A coordinated immunological attack against myelin in the central nervous system. Cell 1996, 85, 299–302. [Google Scholar] [CrossRef] [Green Version]
- Greer, J.M.; Lees, M.B. Myelin proteolipid protein--the first 50 years. Int. J. Biochem. Cell Biol. 2002, 34, 211–215. [Google Scholar] [CrossRef]
- Stadelmann, C.; Timmler, S.; Barrantes-Freer, A.; Simons, M. Myelin in the Central Nervous System: Structure, Function, and Pathology. Physiol. Rev. 2019, 99, 1381–1431. [Google Scholar] [CrossRef]
- Roach, A.; Boylan, K.; Horvath, S.; Prusiner, S.B.; Hood, L.E. Characterization of cloned cDNA representing rat myelin basic protein: Absence of expression in brain of shiverer mutant mice. Cell 1983, 34, 799–806. [Google Scholar] [CrossRef]
- Roach, A.; Takahashi, N.; Pravtcheva, D.; Ruddle, F.; Hood, L. Chromosomal mapping of mouse myelin basic protein gene and structure and transcription of the partially deleted gene in shiverer mutant mice. Cell 1985, 42, 149–155. [Google Scholar] [CrossRef]
- Readhead, C.; Popko, B.; Takahashi, N.; Shine, H.D.; Saavedra, R.A.; Sidman, R.L.; Hood, L. Expression of a myelin basic protein gene in transgenic shiverer mice: Correction of the dysmyelinating phenotype. Cell 1987, 48, 703–712. [Google Scholar] [CrossRef]
- Lemke, G. Unwrapping the genes of myelin. Neuron 1988, 1, 535–543. [Google Scholar] [CrossRef]
- Umemori, H.; Kadowaki, Y.; Hirosawa, K.; Yoshida, Y.; Hironaka, K.; Okano, H.; Yamamoto, T. Stimulation of Myelin Basic Protein Gene Transcription by Fyn Tyrosine Kinase for Myelination. J. Neurosci. 1999, 19, 1393–1397. [Google Scholar] [CrossRef] [Green Version]
- Krämer, E.M.; Klein, C.; Koch, T.; Boytinck, M.; Trotter, J. Compartmentation of Fyn kinase with glycosylphosphatidylinositol-anchored molecules in oligodendrocytes facilitates kinase activation during myelination. J. Biol. Chem. 1999, 274, 29042–29049. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Osterhout, D.J.; Wolven, A.; Wolf, R.M.; Resh, M.D.; Chao, M.V. Morphological differentiation of oligodendrocytes requires activation of Fyn tyrosine kinase. J. Cell Biol. 1999, 145, 1209–1218. [Google Scholar] [CrossRef]
- Colognato, H.; Ramachandrappa, S.; Olsen, I.M.; ffrench-Constant, C. Integrins direct Src family kinases to regulate distinct phases of oligodendrocyte development. J. Cell Biol. 2004, 167, 365–375. [Google Scholar] [CrossRef] [Green Version]
- Liang, X.; Draghi, N.A.; Resh, M.D. Signaling from integrins to Fyn to Rho family GTPases regulates morphologic differentiation of oligodendrocytes. J. Neurosci. 2004, 24, 7140–7149. [Google Scholar] [CrossRef]
- Nakahara, J.; Tan-Takeuchi, K.; Seiwa, C.; Gotoh, M.; Kaifu, T.; Ujike, A.; Inui, M.; Yagi, T.; Ogawa, M.; Aiso, S.; et al. Signaling via immunoglobulin Fc receptors induces oligodendrocyte precursor cell differentiation. Dev. Cell 2003, 4, 841–852. [Google Scholar] [CrossRef] [Green Version]
- Ninio-Many, L.; Grossman, H.; Shomron, N.; Chuderland, D.; Shalgi, R. microRNA-125a-3p reduces cell proliferation and migration by targeting Fyn. J. Cell Sci. 2013, 126, 2867–2876. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Filbin, M.T. The muddle with MAG. Mol. Cell. Neurosci. 1996, 8, 84–92. [Google Scholar] [CrossRef] [PubMed]
- Rosenzweig, S.; Carmichael, S.T. The axon-glia unit in white matter stroke: Mechanisms of damage and recovery. Brain Res. 2015, 1623, 123–134. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haroutunian, V.; Katsel, P.; Roussos, P.; Davis, K.L.; Altshuler, L.L.; Bartzokis, G. Myelination, oligodendrocytes, and serious mental illness. Glia 2014, 62, 1856–1877. [Google Scholar] [CrossRef]
- Araque Caballero, M.; Suárez-Calvet, M.; Duering, M.; Franzmeier, N.; Benzinger, T.; Fagan, A.M.; Bateman, R.J.; Jack, C.R.; Levin, J.; Dichgans, M.; et al. White matter diffusion alterations precede symptom onset in autosomal dominant Alzheimer’s disease. Brain 2018, 141, 3065–3080. [Google Scholar] [CrossRef]
- Allen, M.; Wang, X.; Burgess, J.D.; Watzlawik, J.; Serie, D.J.; Younkin, C.S.; Nguyen, T.; Malphrus, K.G.; Lincoln, S.; Carrasquillo, M.M.; et al. Conserved brain myelination networks are altered in Alzheimer’s and other neurodegenerative diseases. Alzheimers Dement. 2018, 14, 352–366. [Google Scholar] [CrossRef]
- Hattori, K.; Fukuzako, H.; Hashiguchi, T.; Hamada, S.; Murata, Y.; Isosaka, T.; Yuasa, S.; Yagi, T. Decreased expression of Fyn protein and disbalanced alternative splicing patterns in platelets from patients with schizophrenia. Psychiatry Res. 2009, 168, 119–128. [Google Scholar] [CrossRef]
- Szczepankiewicz, A.; Rybakowski, J.K.; Skibinska, M.; Dmitrzak-Weglarz, M.; Leszczynska-Rodziewicz, A.; Wilkosc, M.; Hauser, J. FYN kinase gene: Another glutamatergic gene associated with bipolar disorder? Neuropsychobiology 2009, 59, 178–183. [Google Scholar] [CrossRef]
- Szczepankiewicz, A.; Skibinska, M.; Suwalska, A.; Hauser, J.; Rybakowski, J.K. The association study of three FYN polymorphisms with prophylactic lithium response in bipolar patients. Hum. Psychopharmacol. 2009, 24, 287–291. [Google Scholar] [CrossRef]
- Klein, C.; Kramer, E.M.; Cardine, A.M.; Schraven, B.; Brandt, R.; Trotter, J. Process outgrowth of oligodendrocytes is promoted by interaction of fyn kinase with the cytoskeletal protein tau. J. Neurosci. 2002, 22, 698–707. [Google Scholar] [CrossRef] [Green Version]
- Belkadi, A.; LoPresti, P. Truncated Tau with the Fyn-binding domain and without the microtubule-binding domain hinders the myelinating capacity of an oligodendrocyte cell line. J. Neurochem. 2008, 107, 351–360. [Google Scholar] [CrossRef] [PubMed]
- Baranzini, S.E.; Galwey, N.W.; Wang, J.; Khankhanian, P.; Lindberg, R.; Pelletier, D.; Wu, W.; Uitdehaag, B.M.; Kappos, L.; Polman, C.H.; et al. Pathway and network-based analysis of genome-wide association studies in multiple sclerosis. Hum. Mol. Genet. 2009, 18, 2078–2090. [Google Scholar] [CrossRef] [PubMed]
- International Multiple Sclerosis Genetics Consortium. Network-based multiple sclerosis pathway analysis with GWAS data from 15,000 cases and 30,000 controls. Am. J. Hum. Genet. 2013, 92, 854–865. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grant, S.G.; O’Dell, T.J.; Karl, K.A.; Stein, P.L.; Soriano, P.; Kandel, E.R. Impaired long-term potentiation, spatial learning, and hippocampal development in fyn mutant mice. Science 1992, 258, 1903–1910. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yagi, T.; Aizawa, S.; Tokunaga, T.; Shigetani, Y.; Takeda, N.; Ikawa, Y. A role for Fyn tyrosine kinase in the suckling behaviour of neonatal mice. Nature 1993, 366, 742–745. [Google Scholar] [CrossRef]
- Ohnuma, T.; Kato, H.; Arai, H.; McKenna, P.J.; Emson, P.C. Expression of Fyn, a non-receptor tyrosine kinase in prefrontal cortex from patients with schizophrenia and its correlation with clinical onset. Brain Res. Mol. Brain. Res. 2003, 112, 90–94. [Google Scholar] [CrossRef]
- Schafer, S.T.; Paquola, A.C.M.; Stern, S.; Gosselin, D.; Ku, M.; Pena, M.; Kuret, T.J.M.; Liyanage, M.; Mansour, A.A.; Jaeger, B.N.; et al. Pathological priming causes developmental gene network heterochronicity in autistic subject-derived neurons. Nat. Neurosci. 2019, 22, 243–255. [Google Scholar] [CrossRef]
- Wu, L.; Huang, Y.; Li, J.; Zhao, H.; Du, H.; Jin, Q.; Zhao, X.; Ma, H.; Zhu, G. Association study of the Fyn gene with schizophrenia in the Chinese-Han population. Psychiatr. Genet. 2013, 23, 39–40. [Google Scholar] [CrossRef]
- Matrone, C.; Ciotti, M.T.; Mercanti, D.; Marolda, R.; Calissano, P. NGF and BDNF signaling control amyloidogenic route and Abeta production in hippocampal neurons. Proc. Natl. Acad. Sci. USA 2008, 105, 13139–13144. [Google Scholar] [CrossRef] [Green Version]
- Matrone, C.; Marolda, R.; Ciafre, S.; Ciotti, M.T.; Mercanti, D.; Calissano, P. Tyrosine kinase nerve growth factor receptor switches from prosurvival to proapoptotic activity via Abeta-mediated phosphorylation. Proc. Natl. Acad. Sci. USA 2009, 106, 11358–11363. [Google Scholar] [CrossRef] [Green Version]
- Houlton, J.; Abumaria, N.; Hinkley, S.F.R.; Clarkson, A.N. Therapeutic Potential of Neurotrophins for Repair After Brain Injury: A Helping Hand From Biomaterials. Front. Neurosci. 2019, 13, 790. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peckham, H.; Giuffrida, L.; Wood, R.; Gonsalvez, D.; Ferner, A.; Kilpatrick, T.J.; Murray, S.S.; Xiao, J. Fyn is an intermediate kinase that BDNF utilizes to promote oligodendrocyte myelination. Glia 2016, 64, 255–269. [Google Scholar] [CrossRef] [PubMed]
- Nagappan, G.; Lu, B. Activity-dependent modulation of the BDNF receptor TrkB: Mechanisms and implications. Trends Neurosci. 2005, 28, 464–471. [Google Scholar] [CrossRef] [PubMed]
- Yoo, H.J.; Yang, S.Y.; Cho, I.H.; Park, M.; Kim, S.A. Polymorphisms of BDNF gene and autism spectrum disorders: Family based association study with korean trios. Psychiatry Investig. 2014, 11, 319–324. [Google Scholar] [CrossRef] [Green Version]
- Yamada, K.; Nabeshima, T. Brain-derived neurotrophic factor/TrkB signaling in memory processes. J. Pharmacol. Sci. 2003, 91, 267–270. [Google Scholar] [CrossRef] [Green Version]
- Mizuno, M.; Yamada, K.; He, J.; Nakajima, A.; Nabeshima, T. Involvement of BDNF receptor TrkB in spatial memory formation. Learn. Mem. 2003, 10, 108–115. [Google Scholar] [CrossRef] [Green Version]
- Yamada, K.; Nabeshima, T. Interaction of BDNF/TrkB signaling with NMDA receptor in learning and memory. Drug News Perspect. 2004, 17, 435–438. [Google Scholar] [CrossRef]
- Chao, M.V. Trophic factors: An evolutionary cul-de-sac or door into higher neuronal function? J. Neurosci. Res. 2000, 59, 353–355. [Google Scholar] [CrossRef]
- Mizuno, M.; Yamada, K.; Olariu, A.; Nawa, H.; Nabeshima, T. Involvement of brain-derived neurotrophic factor in spatial memory formation and maintenance in a radial arm maze test in rats. J. Neurosci. 2000, 20, 7116–7121. [Google Scholar] [CrossRef] [Green Version]
- Iwasaki, Y.; Gay, B.; Wada, K.; Koizumi, S. Association of the Src family tyrosine kinase Fyn with TrkB. J. Neurochem. 1998, 71, 106–111. [Google Scholar] [CrossRef]
- Sasaki, Y.; Cheng, C.; Uchida, Y.; Nakajima, O.; Ohshima, T.; Yagi, T.; Taniguchi, M.; Nakayama, T.; Kishida, R.; Kudo, Y.; et al. Fyn and Cdk5 mediate semaphorin-3A signaling, which is involved in regulation of dendrite orientation in cerebral cortex. Neuron 2002, 35, 907–920. [Google Scholar] [CrossRef] [Green Version]
- Franco, M.; Tamagnone, L. Tyrosine phosphorylation in semaphorin signalling: Shifting into overdrive. EMBO Rep. 2008, 9, 865–871. [Google Scholar] [CrossRef] [PubMed]
- Suda, S.; Iwata, K.; Shimmura, C.; Kameno, Y.; Anitha, A.; Thanseem, I.; Nakamura, K.; Matsuzaki, H.; Tsuchiya, K.J.; Sugihara, G.; et al. Decreased expression of axon-guidance receptors in the anterior cingulate cortex in autism. Mol. Autism 2011, 2, 14. [Google Scholar] [CrossRef] [Green Version]
- Chen, G.; Sima, J.; Jin, M.; Wang, K.Y.; Xue, X.J.; Zheng, W.; Ding, Y.Q.; Yuan, X.B. Semaphorin-3A guides radial migration of cortical neurons during development. Nat. Neurosci. 2008, 11, 36–44. [Google Scholar] [CrossRef]
- Renaud, J.; Kerjan, G.; Sumita, I.; Zagar, Y.; Georget, V.; Kim, D.; Fouquet, C.; Suda, K.; Sanbo, M.; Suto, F.; et al. Plexin-A2 and its ligand, Sema6A, control nucleus-centrosome coupling in migrating granule cells. Nat. Neurosci. 2008, 11, 440–449. [Google Scholar] [CrossRef]
- Orr, B.O.; Fetter, R.D.; Davis, G.W. Retrograde semaphorin-plexin signalling drives homeostatic synaptic plasticity. Nature 2017, 550, 109–113. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tillo, M.; Ruhrberg, C.; Mackenzie, F. Emerging roles for semaphorins and VEGFs in synaptogenesis and synaptic plasticity. Cell Adh. Migr. 2012, 6, 541–546. [Google Scholar] [CrossRef]
- Bagri, A.; Cheng, H.J.; Yaron, A.; Pleasure, S.J.; Tessier-Lavigne, M. Stereotyped pruning of long hippocampal axon branches triggered by retraction inducers of the semaphorin family. Cell 2003, 113, 285–299. [Google Scholar] [CrossRef] [Green Version]
- Low, L.K.; Liu, X.B.; Faulkner, R.L.; Coble, J.; Cheng, H.J. Plexin signaling selectively regulates the stereotyped pruning of corticospinal axons from visual cortex. Proc. Natl. Acad. Sci. USA 2008, 105, 8136–8141. [Google Scholar] [CrossRef] [Green Version]
- Morita, A.; Yamashita, N.; Sasaki, Y.; Uchida, Y.; Nakajima, O.; Nakamura, F.; Yagi, T.; Taniguchi, M.; Usui, H.; Katoh-Semba, R.; et al. Regulation of dendritic branching and spine maturation by semaphorin3A-Fyn signaling. J. Neurosci. 2006, 26, 2971–2980. [Google Scholar] [CrossRef]
- Makihara, H.; Nakai, S.; Ohkubo, W.; Yamashita, N.; Nakamura, F.; Kiyonari, H.; Shioi, G.; Jitsuki-Takahashi, A.; Nakamura, H.; Tanaka, F.; et al. CRMP1 and CRMP2 have synergistic but distinct roles in dendritic development. Genes. Cells 2016, 21, 994–1005. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ziak, J.; Weissova, R.; Jeřábková, K.; Janikova, M.; Maimon, R.; Petrasek, T.; Pukajova, B.; Kleisnerova, M.; Wang, M.; Brill, M.S.; et al. CRMP2 mediates Sema3F-dependent axon pruning and dendritic spine remodeling. EMBO Rep. 2020, 21, e48512. [Google Scholar] [CrossRef]
- Eixarch, H.; Gutiérrez-Franco, A.; Montalban, X.; Espejo, C. Semaphorins 3A and 7A: Potential immune and neuroregenerative targets in multiple sclerosis. Trends Mol. Med. 2013, 19, 157–164. [Google Scholar] [CrossRef] [PubMed]
- Gil, V.; Del Río, J.A. Functions of Plexins/Neuropilins and Their Ligands during Hippocampal Development and Neurodegeneration. Cells 2019, 8, 206. [Google Scholar] [CrossRef] [Green Version]
- Lee, W.S.; Lee, W.H.; Bae, Y.C.; Suk, K. Axon Guidance Molecules Guiding Neuroinflammation. Exp. Neurobiol. 2019, 28, 311–319. [Google Scholar] [CrossRef] [PubMed]
- Pasterkamp, R.J. Getting neural circuits into shape with semaphorins. Nat. Rev. Neurosci. 2012, 13, 605–618. [Google Scholar] [CrossRef] [PubMed]
- Kenney, J.W.; Frankland, P.W. Making connections. Elife 2014, 3. [Google Scholar] [CrossRef]
- Mah, S.; Nelson, M.R.; Delisi, L.E.; Reneland, R.H.; Markward, N.; James, M.R.; Nyholt, D.R.; Hayward, N.; Handoko, H.; Mowry, B.; et al. Identification of the semaphorin receptor PLXNA2 as a candidate for susceptibility to schizophrenia. Mol. Psychiatry 2006, 11, 471–478. [Google Scholar] [CrossRef] [Green Version]
- Su, S.C.; Tsai, L.H. Cyclin-dependent kinases in brain development and disease. Annu. Rev. Cell Dev. Biol. 2011, 27, 465–491. [Google Scholar] [CrossRef]
- Uchida, Y.; Ohshima, T.; Sasaki, Y.; Suzuki, H.; Yanai, S.; Yamashita, N.; Nakamura, F.; Takei, K.; Ihara, Y.; Mikoshiba, K.; et al. Semaphorin3A signalling is mediated via sequential Cdk5 and GSK3beta phosphorylation of CRMP2: Implication of common phosphorylating mechanism underlying axon guidance and Alzheimer’s disease. Genes Cells 2005, 10, 165–179. [Google Scholar] [CrossRef]
- Fukata, Y.; Itoh, T.J.; Kimura, T.; Ménager, C.; Nishimura, T.; Shiromizu, T.; Watanabe, H.; Inagaki, N.; Iwamatsu, A.; Hotani, H.; et al. CRMP-2 binds to tubulin heterodimers to promote microtubule assembly. Nat. Cell Biol. 2002, 4, 583–591. [Google Scholar] [CrossRef] [PubMed]
- Inagaki, N.; Chihara, K.; Arimura, N.; Ménager, C.; Kawano, Y.; Matsuo, N.; Nishimura, T.; Amano, M.; Kaibuchi, K. CRMP-2 induces axons in cultured hippocampal neurons. Nat. Neurosci. 2001, 4, 781–782. [Google Scholar] [CrossRef] [PubMed]
- Brown, M.; Jacobs, T.; Eickholt, B.; Ferrari, G.; Teo, M.; Monfries, C.; Qi, R.Z.; Leung, T.; Lim, L.; Hall, C. Alpha2-chimaerin, cyclin-dependent Kinase 5/p35, and its target collapsin response mediator protein-2 are essential components in semaphorin 3A-induced growth-cone collapse. J. Neurosci. 2004, 24, 8994–9004. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mecollari, V.; Nieuwenhuis, B.; Verhaagen, J. A perspective on the role of class III semaphorin signaling in central nervous system trauma. Front. Cell. Neurosci. 2014, 8, 328. [Google Scholar] [CrossRef] [Green Version]
- Van Battum, E.Y.; Brignani, S.; Pasterkamp, R.J. Axon guidance proteins in neurological disorders. Lancet Neurol. 2015, 14, 532–546. [Google Scholar] [CrossRef]
- Hirsch, E.; Hu, L.J.; Prigent, A.; Constantin, B.; Agid, Y.; Drabkin, H.; Roche, J. Distribution of semaphorin IV in adult human brain. Brain Res. 1999, 823, 67–79. [Google Scholar] [CrossRef]
- Good, P.F.; Alapat, D.; Hsu, A.; Chu, C.; Perl, D.; Wen, X.; Burstein, D.E.; Kohtz, D.S. A role for semaphorin 3A signaling in the degeneration of hippocampal neurons during Alzheimer’s disease. J. Neurochem. 2004, 91, 716–736. [Google Scholar] [CrossRef]
- Villa, C.; Venturelli, E.; Fenoglio, C.; De Riz, M.; Scalabrini, D.; Cortini, F.; Serpente, M.; Cantoni, C.; Bresolin, N.; Scarpini, E.; et al. Candidate gene analysis of semaphorins in patients with Alzheimer’s disease. Neurol. Sci. 2010, 31, 169–173. [Google Scholar] [CrossRef]
- Venkova, K.; Christov, A.; Kamaluddin, Z.; Kobalka, P.; Siddiqui, S.; Hensley, K. Semaphorin 3A signaling through neuropilin-1 is an early trigger for distal axonopathy in the SOD1G93A mouse model of amyotrophic lateral sclerosis. J. Neuropathol. Exp. Neurol. 2014, 73, 702–713. [Google Scholar] [CrossRef] [Green Version]
- Kuo, G.; Arnaud, L.; Kronstad-O’Brien, P.; Cooper, J.A. Absence of Fyn and Src causes a reeler-like phenotype. J. Neurosci. 2005, 25, 8578–8586. [Google Scholar] [CrossRef]
- D’Arcangelo, G.; Miao, G.G.; Chen, S.C.; Soares, H.D.; Morgan, J.I.; Curran, T. A protein related to extracellular matrix proteins deleted in the mouse mutant reeler. Nature 1995, 374, 719–723. [Google Scholar] [CrossRef] [PubMed]
- D’Arcangelo, G.; Curran, T. Reeler: New tales on an old mutant mouse. Bioessays 1998, 20, 235–244. [Google Scholar] [CrossRef]
- D’Arcangelo, G.; Homayouni, R.; Keshvara, L.; Rice, D.S.; Sheldon, M.; Curran, T. Reelin is a ligand for lipoprotein receptors. Neuron 1999, 24, 471–479. [Google Scholar] [CrossRef] [Green Version]
- Trommsdorff, M.; Gotthardt, M.; Hiesberger, T.; Shelton, J.; Stockinger, W.; Nimpf, J.; Hammer, R.E.; Richardson, J.A.; Herz, J. Reeler/Disabled-like disruption of neuronal migration in knockout mice lacking the VLDL receptor and ApoE receptor 2. Cell 1999, 97, 689–701. [Google Scholar] [CrossRef] [Green Version]
- Miyata, T.; Nakajima, K.; Mikoshiba, K.; Ogawa, M. Regulation of Purkinje cell alignment by reelin as revealed with CR-50 antibody. J. Neurosci. 1997, 17, 3599–3609. [Google Scholar] [CrossRef] [PubMed]
- Goffinet, A.M. The embryonic development of the cerebellum in normal and reeler mutant mice. Anat. Embryol. 1983, 168, 73–86. [Google Scholar] [CrossRef]
- Hong, S.E.; Shugart, Y.Y.; Huang, D.T.; Shahwan, S.A.; Grant, P.E.; Hourihane, J.O.; Martin, N.D.; Walsh, C.A. Autosomal recessive lissencephaly with cerebellar hypoplasia is associated with human RELN mutations. Nat. Genet. 2000, 26, 93–96. [Google Scholar] [CrossRef]
- DiBattista, A.M.; Dumanis, S.B.; Song, J.M.; Bu, G.; Weeber, E.; Rebeck, G.W.; Hoe, H.S. Very low density lipoprotein receptor regulates dendritic spine formation in a RasGRF1/CaMKII dependent manner. Biochim. Biophys. Acta 2015, 1853, 904–917. [Google Scholar] [CrossRef] [Green Version]
- Pujadas, L.; Gruart, A.; Bosch, C.; Delgado, L.; Teixeira, C.M.; Rossi, D.; de Lecea, L.; Martínez, A.; Delgado-García, J.M.; Soriano, E. Reelin regulates postnatal neurogenesis and enhances spine hypertrophy and long-term potentiation. J. Neurosci. 2010, 30, 4636–4649. [Google Scholar] [CrossRef] [Green Version]
- Niu, S.; Yabut, O.; D’Arcangelo, G. The Reelin signaling pathway promotes dendritic spine development in hippocampal neurons. J. Neurosci. 2008, 28, 10339–10348. [Google Scholar] [CrossRef]
- Iafrati, J.; Orejarena, M.J.; Lassalle, O.; Bouamrane, L.; Gonzalez-Campo, C.; Chavis, P. Reelin, an extracellular matrix protein linked to early onset psychiatric diseases, drives postnatal development of the prefrontal cortex via GluN2B-NMDARs and the mTOR pathway. Mol. Psychiatry 2014, 19, 417–426. [Google Scholar] [CrossRef] [Green Version]
- Kim, M.; Jeong, Y.; Chang, Y.C. Extracellular matrix protein reelin regulate dendritic spine density through CaMKIIβ. Neurosci. Lett. 2015, 599, 97–101. [Google Scholar] [CrossRef]
- Dazzo, E.; Fanciulli, M.; Serioli, E.; Minervini, G.; Pulitano, P.; Binelli, S.; Di Bonaventura, C.; Luisi, C.; Pasini, E.; Striano, S.; et al. Heterozygous reelin mutations cause autosomal-dominant lateral temporal epilepsy. Am. J. Hum. Genet. 2015, 96, 992–1000. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Rubeis, S.; He, X.; Goldberg, A.P.; Poultney, C.S.; Samocha, K.; Cicek, A.E.; Kou, Y.; Liu, L.; Fromer, M.; Walker, S.; et al. Synaptic, transcriptional and chromatin genes disrupted in autism. Nature 2014, 515, 209–215. [Google Scholar] [CrossRef]
- Groc, L.; Choquet, D.; Stephenson, F.A.; Verrier, D.; Manzoni, O.J.; Chavis, P. NMDA receptor surface trafficking and synaptic subunit composition are developmentally regulated by the extracellular matrix protein Reelin. J. Neurosci. 2007, 27, 10165–10175. [Google Scholar] [CrossRef]
- Ventruti, A.; Kazdoba, T.M.; Niu, S.; D’Arcangelo, G. Reelin deficiency causes specific defects in the molecular composition of the synapses in the adult brain. Neuroscience 2011, 189, 32–42. [Google Scholar] [CrossRef] [PubMed]
- Howell, B.W.; Lanier, L.M.; Frank, R.; Gertler, F.B.; Cooper, J.A. The disabled 1 phosphotyrosine-binding domain binds to the internalization signals of transmembrane glycoproteins and to phospholipids. Mol. Cell. Biol. 1999, 19, 5179–5188. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Howell, B.W.; Herrick, T.M.; Hildebrand, J.D.; Zhang, Y.; Cooper, J.A. Dab1 tyrosine phosphorylation sites relay positional signals during mouse brain development. Curr. Biol. 2000, 10, 877–885. [Google Scholar] [CrossRef] [Green Version]
- Howell, B.W.; Gertler, F.B.; Cooper, J.A. Mouse disabled (mDab1): A Src binding protein implicated in neuronal development. EMBO J. 1997, 16, 121–132. [Google Scholar] [CrossRef] [Green Version]
- Sheldon, M.; Rice, D.S.; D’Arcangelo, G.; Yoneshima, H.; Nakajima, K.; Mikoshiba, K.; Howell, B.W.; Cooper, J.A.; Goldowitz, D.; Curran, T. Scrambler and yotari disrupt the disabled gene and produce a reeler-like phenotype in mice. Nature 1997, 389, 730–733. [Google Scholar] [CrossRef] [PubMed]
- Yoneshima, H.; Nagata, E.; Matsumoto, M.; Yamada, M.; Nakajima, K.; Miyata, T.; Ogawa, M.; Mikoshiba, K. A novel neurological mutant mouse, yotari, which exhibits reeler-like phenotype but expresses CR-50 antigen/reelin. Neurosci. Res. 1997, 29, 217–223. [Google Scholar] [CrossRef]
- Ware, M.L.; Fox, J.W.; González, J.L.; Davis, N.M.; Lambert de Rouvroit, C.; Russo, C.J.; Chua, S.C., Jr.; Goffinet, A.M.; Walsh, C.A. Aberrant splicing of a mouse disabled homolog, mdab1, in the scrambler mouse. Neuron 1997, 19, 239–249. [Google Scholar] [CrossRef] [Green Version]
- Kojima, N.; Wang, J.; Mansuy, I.M.; Grant, S.G.; Mayford, M.; Kandel, E.R. Rescuing impairment of long-term potentiation in fyn-deficient mice by introducing Fyn transgene. Proc. Natl. Acad. Sci. USA 1997, 94, 4761–4765. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kojima, T.; Nakajima, K.; Mikoshiba, K. The disabled 1 gene is disrupted by a replacement with L1 fragment in yotari mice. Brain. Res. Mol. Brain Res. 2000, 75, 121–127. [Google Scholar] [CrossRef]
- Beffert, U.; Weeber, E.J.; Durudas, A.; Qiu, S.; Masiulis, I.; Sweatt, J.D.; Li, W.P.; Adelmann, G.; Frotscher, M.; Hammer, R.E.; et al. Modulation of synaptic plasticity and memory by Reelin involves differential splicing of the lipoprotein receptor Apoer2. Neuron 2005, 47, 567–579. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Y.; Beffert, U.; Ertunc, M.; Tang, T.S.; Kavalali, E.T.; Bezprozvanny, I.; Herz, J. Reelin modulates NMDA receptor activity in cortical neurons. J. Neurosci. 2005, 25, 8209–8216. [Google Scholar] [CrossRef]
- Hoe, H.S.; Lee, K.J.; Carney, R.S.; Lee, J.; Markova, A.; Lee, J.Y.; Howell, B.W.; Hyman, B.T.; Pak, D.T.; Bu, G.; et al. Interaction of reelin with amyloid precursor protein promotes neurite outgrowth. J. Neurosci. 2009, 29, 7459–7473. [Google Scholar] [CrossRef]
- Homayouni, R.; Rice, D.S.; Sheldon, M.; Curran, T. Disabled-1 binds to the cytoplasmic domain of amyloid precursor-like protein 1. J. Neurosci. 1999, 19, 7507–7515. [Google Scholar] [CrossRef]
- Durakoglugil, M.S.; Chen, Y.; White, C.L.; Kavalali, E.T.; Herz, J. Reelin signaling antagonizes beta-amyloid at the synapse. Proc. Natl. Acad. Sci. USA 2009, 106, 15938–15943. [Google Scholar] [CrossRef] [Green Version]
- Krstic, D.; Rodriguez, M.; Knuesel, I. Regulated proteolytic processing of Reelin through interplay of tissue plasminogen activator (tPA), ADAMTS-4, ADAMTS-5, and their modulators. PLoS ONE 2012, 7, e47793. [Google Scholar] [CrossRef]
- Pujadas, L.; Rossi, D.; Andrés, R.; Teixeira, C.M.; Serra-Vidal, B.; Parcerisas, A.; Maldonado, R.; Giralt, E.; Carulla, N.; Soriano, E. Reelin delays amyloid-beta fibril formation and rescues cognitive deficits in a model of Alzheimer’s disease. Nat. Commun. 2014, 5, 3443. [Google Scholar] [CrossRef] [PubMed]
- Kalia, L.V.; Gingrich, J.R.; Salter, M.W. Src in synaptic transmission and plasticity. Oncogene 2004, 23, 8007–8016. [Google Scholar] [CrossRef] [Green Version]
- Ohnishi, H.; Murata, Y.; Okazawa, H.; Matozaki, T. Src family kinases: Modulators of neurotransmitter receptor function and behavior. Trends Neurosci. 2011, 34, 629–637. [Google Scholar] [CrossRef] [PubMed]
- Nygaard, H.B. Targeting Fyn Kinase in Alzheimer’s Disease. Biol. Psychiatry 2018, 83, 369–376. [Google Scholar] [CrossRef] [PubMed]
- Kaufman, A.C.; Salazar, S.V.; Haas, L.T.; Yang, J.; Kostylev, M.A.; Jeng, A.T.; Robinson, S.A.; Gunther, E.C.; van Dyck, C.H.; Nygaard, H.B.; et al. Fyn inhibition rescues established memory and synapse loss in Alzheimer mice. Ann. Neurol. 2015, 77, 953–971. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nygaard, H.B.; van Dyck, C.H.; Strittmatter, S.M. Fyn kinase inhibition as a novel therapy for Alzheimer’s disease. Alzheimers Res. Ther. 2014, 6, 8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Um, J.W.; Nygaard, H.B.; Heiss, J.K.; Kostylev, M.A.; Stagi, M.; Vortmeyer, A.; Wisniewski, T.; Gunther, E.C.; Strittmatter, S.M. Alzheimer amyloid-β oligomer bound to postsynaptic prion protein activates Fyn to impair neurons. Nat. Neurosci. 2012, 15, 1227–1235. [Google Scholar] [CrossRef] [Green Version]
- Traynelis, S.F.; Wollmuth, L.P.; McBain, C.J.; Menniti, F.S.; Vance, K.M.; Ogden, K.K.; Hansen, K.B.; Yuan, H.; Myers, S.J.; Dingledine, R. Glutamate receptor ion channels: Structure, regulation, and function. Pharmacol. Rev. 2010, 62, 405–496. [Google Scholar] [CrossRef] [Green Version]
- Dingledine, R.; Borges, K.; Bowie, D.; Traynelis, S.F. The glutamate receptor ion channels. Pharmacol. Rev. 1999, 51, 7–61. [Google Scholar]
- Mao, L.M.; Guo, M.L.; Jin, D.Z.; Fibuch, E.E.; Choe, E.S.; Wang, J.Q. Post-translational modification biology of glutamate receptors and drug addiction. Front. Neuroanat. 2011, 5, 19. [Google Scholar] [CrossRef] [Green Version]
- Suzuki, T.; Okumura-Noji, K. NMDA receptor subunits epsilon 1 (NR2A) and epsilon 2 (NR2B) are substrates for Fyn in the postsynaptic density fraction isolated from the rat brain. Biochem. Biophys. Res. Commun. 1995, 216, 582–588. [Google Scholar] [CrossRef] [PubMed]
- Trepanier, C.H.; Jackson, M.F.; MacDonald, J.F. Regulation of NMDA receptors by the tyrosine kinase Fyn. FEBS J. 2012, 279, 12–19. [Google Scholar] [CrossRef] [PubMed]
- Yang, K.; Trepanier, C.; Sidhu, B.; Xie, Y.F.; Li, H.; Lei, G.; Salter, M.W.; Orser, B.A.; Nakazawa, T.; Yamamoto, T.; et al. Metaplasticity gated through differential regulation of GluN2A versus GluN2B receptors by Src family kinases. EMBO J. 2012, 31, 805–816. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakazawa, T.; Komai, S.; Tezuka, T.; Hisatsune, C.; Umemori, H.; Semba, K.; Mishina, M.; Manabe, T.; Yamamoto, T. Characterization of Fyn-mediated tyrosine phosphorylation sites on GluR epsilon 2 (NR2B) subunit of the N-methyl-D-aspartate receptor. J. Biol. Chem. 2001, 276, 693–699. [Google Scholar] [CrossRef] [Green Version]
- Sato, Y.; Tao, Y.X.; Su, Q.; Johns, R.A. Post-synaptic density-93 mediates tyrosine-phosphorylation of the N-methyl-D-aspartate receptors. Neuroscience 2008, 153, 700–708. [Google Scholar] [CrossRef] [Green Version]
- Tezuka, T.; Umemori, H.; Akiyama, T.; Nakanishi, S.; Yamamoto, T. PSD-95 promotes Fyn-mediated tyrosine phosphorylation of the N-methyl-D-aspartate receptor subunit NR2A. Proc. Natl. Acad. Sci. USA 1999, 96, 435–440. [Google Scholar] [CrossRef] [Green Version]
- Nakazawa, T.; Tezuka, T.; Yamamoto, T. Regulation of NMDA receptor function by Fyn-mediated tyrosine phosphorylation. Nihon Shinkei Seishin Yakurigaku Zasshi 2002, 22, 165–167. [Google Scholar]
- Hou, X.Y.; Zhang, G.Y.; Yan, J.Z.; Liu, Y. Increased tyrosine phosphorylation of alpha(1C) subunits of L-type voltage-gated calcium channels and interactions among Src/Fyn, PSD-95 and alpha(1C) in rat hippocampus after transient brain ischemia. Brain Res. 2003, 979, 43–50. [Google Scholar] [CrossRef]
- Pei, L.; Teves, R.L.; Wallace, M.C.; Gurd, J.W. Transient cerebral ischemia increases tyrosine phosphorylation of the synaptic RAS-GTPase activating protein, SynGAP. J. Cereb. Blood Flow Metab. 2001, 21, 955–963. [Google Scholar] [CrossRef] [Green Version]
- Zhang, M.; Li, Q.; Chen, L.; Li, J.; Zhang, X.; Chen, X.; Zhang, Q.; Shao, Y.; Xu, Y. PSD-93 deletion inhibits Fyn-mediated phosphorylation of NR2B and protects against focal cerebral ischemia. Neurobiol. Dis. 2014, 68, 104–111. [Google Scholar] [CrossRef]
- Nada, S.; Shima, T.; Yanai, H.; Husi, H.; Grant, S.G.; Okada, M.; Akiyama, T. Identification of PSD-93 as a substrate for the Src family tyrosine kinase Fyn. J. Biol. Chem. 2003, 278, 47610–47621. [Google Scholar] [CrossRef] [Green Version]
- Prybylowski, K.; Chang, K.; Sans, N.; Kan, L.; Vicini, S.; Wenthold, R.J. The synaptic localization of NR2B-containing NMDA receptors is controlled by interactions with PDZ proteins and AP-2. Neuron 2005, 47, 845–857. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Owen, D.J.; Evans, P.R. A structural explanation for the recognition of tyrosine-based endocytotic signals. Science 1998, 282, 1327–1332. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lavezzari, G.; McCallum, J.; Lee, R.; Roche, K.W. Differential binding of the AP-2 adaptor complex and PSD-95 to the C-terminus of the NMDA receptor subunit NR2B regulates surface expression. Neuropharmacology 2003, 45, 729–737. [Google Scholar] [CrossRef]
- Goebel-Goody, S.M.; Davies, K.D.; Alvestad Linger, R.M.; Freund, R.K.; Browning, M.D. Phospho-regulation of synaptic and extrasynaptic N-methyl-d-aspartate receptors in adult hippocampal slices. Neuroscience 2009, 158, 1446–1459. [Google Scholar] [CrossRef] [PubMed]
- Knox, R.; Zhao, C.; Miguel-Perez, D.; Wang, S.; Yuan, J.; Ferriero, D.; Jiang, X. Enhanced NMDA receptor tyrosine phosphorylation and increased brain injury following neonatal hypoxia-ischemia in mice with neuronal Fyn overexpression. Neurobiol. Dis. 2013, 51, 113–119. [Google Scholar] [CrossRef] [Green Version]
- Hayashi, T.; Huganir, R.L. Tyrosine phosphorylation and regulation of the AMPA receptor by SRC family tyrosine kinases. J. Neurosci. 2004, 24, 6152–6160. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seamans, J.K.; Yang, C.R. The principal features and mechanisms of dopamine modulation in the prefrontal cortex. Prog. Neurobiol. 2004, 74, 1–58. [Google Scholar] [CrossRef] [PubMed]
- Gao, C.; Wolf, M.E. Dopamine receptors regulate NMDA receptor surface expression in prefrontal cortex neurons. J. Neurochem. 2008, 106, 2489–2501. [Google Scholar] [CrossRef] [Green Version]
- Jin, D.Z.; Guo, M.L.; Xue, B.; Fibuch, E.E.; Choe, E.S.; Mao, L.M.; Wang, J.Q. Phosphorylation and feedback regulation of metabotropic glutamate receptor 1 by calcium/calmodulin-dependent protein kinase II. J. Neurosci. 2013, 33, 3402–3412. [Google Scholar] [CrossRef] [Green Version]
- Jin, D.Z.; Mao, L.M.; Wang, J.Q. An Essential Role of Fyn in the Modulation of Metabotropic Glutamate Receptor 1 in Neurons. Eneuro 2017, 4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, Y.F.; Kojima, N.; Tomizawa, K.; Moriwaki, A.; Matsushita, M.; Obata, K.; Matsui, H. Enhanced synaptic transmission and reduced threshold for LTP induction in fyn-transgenic mice. Eur. J. Neurosci. 1999, 11, 75–82. [Google Scholar] [CrossRef] [PubMed]
- O’Dell, T.J.; Hawkins, R.D.; Kandel, E.R.; Arancio, O. Tests of the roles of two diffusible substances in long-term potentiation: Evidence for nitric oxide as a possible early retrograde messenger. Proc. Natl. Acad. Sci. USA 1991, 88, 11285–11289. [Google Scholar] [CrossRef] [Green Version]
- Rosenblum, K.; Dudai, Y.; Richter-Levin, G. Long-term potentiation increases tyrosine phosphorylation of the N-methyl-D-aspartate receptor subunit 2B in rat dentate gyrus in vivo. Proc. Natl. Acad. Sci. USA 1996, 93, 10457–10460. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, Y.; Lu, W.; Ali, D.W.; Pelkey, K.A.; Pitcher, G.M.; Lu, Y.M.; Aoto, H.; Roder, J.C.; Sasaki, T.; Salter, M.W.; et al. CAKbeta/Pyk2 kinase is a signaling link for induction of long-term potentiation in CA1 hippocampus. Neuron 2001, 29, 485–496. [Google Scholar] [CrossRef] [Green Version]
- Qian, D.; Lev, S.; van Oers, N.S.; Dikic, I.; Schlessinger, J.; Weiss, A. Tyrosine phosphorylation of Pyk2 is selectively regulated by Fyn during TCR signaling. J. Exp. Med. 1997, 185, 1253–1259. [Google Scholar] [CrossRef] [Green Version]
- Yang, K.; Belrose, J.; Trepanier, C.H.; Lei, G.; Jackson, M.F.; MacDonald, J.F. Fyn, a potential target for Alzheimer’s disease. J. Alzheimers Dis. 2011, 27, 243–252. [Google Scholar] [CrossRef]
- Boehm, S.L.; Peden, L.; Chang, R.; Harris, R.A.; Blednov, Y.A. Deletion of the fyn-kinase gene alters behavioral sensitivity to ethanol. Alcohol. Clin. Exp. Res. 2003, 27, 1033–1040. [Google Scholar] [CrossRef]
- Boehm, J. A ‘danse macabre’: Tau and Fyn in STEP with amyloid beta to facilitate induction of synaptic depression and excitotoxicity. Eur. J. Neurosci. 2013, 37, 1925–1930. [Google Scholar] [CrossRef]
- Larson, M.; Sherman, M.A.; Amar, F.; Nuvolone, M.; Schneider, J.A.; Bennett, D.A.; Aguzzi, A.; Lesné, S.E. The complex PrP(c)-Fyn couples human oligomeric Aβ with pathological tau changes in Alzheimer’s disease. J. Neurosci. 2012, 32, 16857–16871. [Google Scholar] [CrossRef] [Green Version]
- Poulsen, E.T.; Iannuzzi, F.; Rasmussen, H.F.; Maier, T.J.; Enghild, J.J.; Jørgensen, A.L.; Matrone, C. An Aberrant Phosphorylation of Amyloid Precursor Protein Tyrosine Regulates Its Trafficking and the Binding to the Clathrin Endocytic Complex in Neural Stem Cells of Alzheimer’s Disease Patients. Front. Mol. Neurosci. 2017, 10, 59. [Google Scholar] [CrossRef] [Green Version]
- Ittner, L.M.; Ke, Y.D.; Delerue, F.; Bi, M.; Gladbach, A.; van Eersel, J.; Wölfing, H.; Chieng, B.C.; Christie, M.J.; Napier, I.A.; et al. Dendritic function of tau mediates amyloid-beta toxicity in Alzheimer’s disease mouse models. Cell 2010, 142, 387–397. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iqbal, K.; Liu, F.; Gong, C.X. Recent developments with tau-based drug discovery. Expert Opin. Drug Discov. 2018, 13, 399–410. [Google Scholar] [CrossRef] [PubMed]
- Lee, G.; Thangavel, R.; Sharma, V.M.; Litersky, J.M.; Bhaskar, K.; Fang, S.M.; Do, L.H.; Andreadis, A.; Van Hoesen, G.; Ksiezak-Reding, H. Phosphorylation of tau by fyn: Implications for Alzheimer’s disease. J. Neurosci. 2004, 24, 2304–2312. [Google Scholar] [CrossRef] [PubMed]
- Folch, J.; Petrov, D.; Ettcheto, M.; Pedrós, I.; Abad, S.; Beas-Zarate, C.; Lazarowski, A.; Marin, M.; Olloquequi, J.; Auladell, C.; et al. Masitinib for the treatment of mild to moderate Alzheimer’s disease. Expert Rev. Neurother. 2015, 15, 587–596. [Google Scholar] [CrossRef]
- Piette, F.; Belmin, J.; Vincent, H.; Schmidt, N.; Pariel, S.; Verny, M.; Marquis, C.; Mely, J.; Hugonot-Diener, L.; Kinet, J.P.; et al. Masitinib as an adjunct therapy for mild-to-moderate Alzheimer’s disease: A randomised, placebo-controlled phase 2 trial. Alzheimers Res. Ther. 2011, 3, 16. [Google Scholar] [CrossRef]
- Haass, C.; Mandelkow, E. Fyn-tau-amyloid: A toxic triad. Cell 2010, 142, 356–358. [Google Scholar] [CrossRef] [Green Version]
- Lee, G.; Newman, S.T.; Gard, D.L.; Band, H.; Panchamoorthy, G. Tau interacts with src-family non-receptor tyrosine kinases. J. Cell Sci. 1998, 111 Pt 21, 3167–3177. [Google Scholar]
- Hernandez, P.; Lee, G.; Sjoberg, M.; Maccioni, R.B. Tau phosphorylation by cdk5 and Fyn in response to amyloid peptide Abeta (25-35): Involvement of lipid rafts. J. Alzheimers Dis. 2009, 16, 149–156. [Google Scholar] [CrossRef] [Green Version]
- Shirazi, S.K.; Wood, J.G. The protein tyrosine kinase, fyn, in Alzheimer’s disease pathology. Neuroreport 1993, 4, 435–437. [Google Scholar] [CrossRef]
- Ho, O.H.; Delgado, J.Y.; O’Dell, T.J. Phosphorylation of proteins involved in activity-dependent forms of synaptic plasticity is altered in hippocampal slices maintained in vitro. J. Neurochem. 2004, 91, 1344–1357. [Google Scholar] [CrossRef] [PubMed]
- Lambert de Rouvroit, C.; Goffinet, A.M. The reeler mouse as a model of brain development. Adv. Anat. Embryol. Cell Biol. 1998, 150, 1–106. [Google Scholar] [PubMed]
- Chin, J.; Palop, J.J.; Yu, G.Q.; Kojima, N.; Masliah, E.; Mucke, L. Fyn kinase modulates synaptotoxicity, but not aberrant sprouting, in human amyloid precursor protein transgenic mice. J. Neurosci. 2004, 24, 4692–4697. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.; Zhao, J.; Lu, G. miR-106b inhibits tau phosphorylation at Tyr18 by targeting Fyn in a model of Alzheimer’s disease. Biochem. Biophys. Res. Commun. 2016, 478, 852–857. [Google Scholar] [CrossRef] [PubMed]
- Bekris, L.M.; Millard, S.; Lutz, F.; Li, G.; Galasko, D.R.; Farlow, M.R.; Quinn, J.F.; Kaye, J.A.; Leverenz, J.B.; Tsuang, D.W.; et al. Tau phosphorylation pathway genes and cerebrospinal fluid tau levels in Alzheimer’s disease. Am. J. Med. Genet. B Neuropsychiatr. Genet. 2012, 159, 874–883. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ellis, C.E.; Schwartzberg, P.L.; Grider, T.L.; Fink, D.W.; Nussbaum, R.L. alpha-synuclein is phosphorylated by members of the Src family of protein-tyrosine kinases. J. Biol. Chem. 2001, 276, 3879–3884. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sanz-Blasco, S.; Bordone, M.P.; Damianich, A.; Gomez, G.; Bernardi, M.A.; Isaja, L.; Taravini, I.R.; Hanger, D.P.; Avale, M.E.; Gershanik, O.S.; et al. The Kinase Fyn As a Novel Intermediate in L-DOPA-Induced Dyskinesia in Parkinson’s Disease. Mol. Neurobiol. 2018, 55, 5125–5136. [Google Scholar] [CrossRef] [Green Version]
- Dunah, A.W.; Sirianni, A.C.; Fienberg, A.A.; Bastia, E.; Schwarzschild, M.A.; Standaert, D.G. Dopamine D1-dependent trafficking of striatal N-methyl-D-aspartate glutamate receptors requires Fyn protein tyrosine kinase but not DARPP-32. Mol. Pharmacol. 2004, 65, 121–129. [Google Scholar] [CrossRef] [Green Version]
Figure 1.
Fyn structure. The SH4 domain, is a membrane-targeting domain containing signals for the appropriate subcellular localization and membrane attachment of Fyn [21]. This domain consists of an extreme N-terminal ‘Met-Gly-Cys’ motif followed by a polar region [9,22]. The N-terminal Gly2 seems to be an absolute requirement for myristylation. Cys3 and Cys6, are both palmitoylated via a thioester linkage [23], thus, allowing Fyn anchors to the plasma membrane [24,25,26]. Between the SH4 and the SH3 motifs, there is a short region called “the unique region” that it is likely to be required for the subcellular localization of Fyn [27]. The SH3 motif constitutes a small domain of 50 amino acids containing a consensus sequence XPXXPPPXXP [28] that allows the binding to amino acid sequences rich in proline residues [29]. The SH2 domain consists of about 100 amino acids and appears to facilitate the binding to phosphotyrosine residues and to hydrophobic sequences within the cytoplasmic tails of growth factor receptors (i.e., PDGF-R, CSF-1 R) [30,31]. The catalytic domain SH1 is responsible of TK specific activity. This domain is highly conserved among Src family members [32]. The kinase domain, that catalyses the transfer of the terminal ATP phosphate group to a tyrosine residue of a target protein, shows a typical bilobed structure consisting in a small N-terminal lobe, involved in the binding with ATP, and a larger C-terminal lobe, where an activation loop (A-loop) is present. The conserved Tyr420, crucial for Src activity, is included in this domain [33]. However, the major site of phosphorylation and regulation of Fyn TK activity is the short negative regulatory tail where Tyr531 is located [32]. Adapted from Krämer-Albers et al. (2011) [34].
Figure 1.
Fyn structure. The SH4 domain, is a membrane-targeting domain containing signals for the appropriate subcellular localization and membrane attachment of Fyn [21]. This domain consists of an extreme N-terminal ‘Met-Gly-Cys’ motif followed by a polar region [9,22]. The N-terminal Gly2 seems to be an absolute requirement for myristylation. Cys3 and Cys6, are both palmitoylated via a thioester linkage [23], thus, allowing Fyn anchors to the plasma membrane [24,25,26]. Between the SH4 and the SH3 motifs, there is a short region called “the unique region” that it is likely to be required for the subcellular localization of Fyn [27]. The SH3 motif constitutes a small domain of 50 amino acids containing a consensus sequence XPXXPPPXXP [28] that allows the binding to amino acid sequences rich in proline residues [29]. The SH2 domain consists of about 100 amino acids and appears to facilitate the binding to phosphotyrosine residues and to hydrophobic sequences within the cytoplasmic tails of growth factor receptors (i.e., PDGF-R, CSF-1 R) [30,31]. The catalytic domain SH1 is responsible of TK specific activity. This domain is highly conserved among Src family members [32]. The kinase domain, that catalyses the transfer of the terminal ATP phosphate group to a tyrosine residue of a target protein, shows a typical bilobed structure consisting in a small N-terminal lobe, involved in the binding with ATP, and a larger C-terminal lobe, where an activation loop (A-loop) is present. The conserved Tyr420, crucial for Src activity, is included in this domain [33]. However, the major site of phosphorylation and regulation of Fyn TK activity is the short negative regulatory tail where Tyr531 is located [32]. Adapted from Krämer-Albers et al. (2011) [34].

Figure 2.
Fyn expression in human brain. (A) Fyn mRNA expression during development (6–20 refers to embryonic days; P7 = 7 days post-hatch). Adapted from Bixby et al. (1993) [58]. (B) Fyn human expression in the different areas of adult brain (data extracted from ATLAS).
Figure 2.
Fyn expression in human brain. (A) Fyn mRNA expression during development (6–20 refers to embryonic days; P7 = 7 days post-hatch). Adapted from Bixby et al. (1993) [58]. (B) Fyn human expression in the different areas of adult brain (data extracted from ATLAS).

Figure 3.
Fyn functions in the brain. Adapted from Schenone et al. (2011) [8].
Figure 3.
Fyn functions in the brain. Adapted from Schenone et al. (2011) [8].

Figure 4.
A proposed mechanism by which Fyn and Cdk5 control Sema3A and PlexinA2 downstream signaling cascade either in physiologic (left) or in pathologic (right) conditions. Fyn phosphorylates PlexinA2 and promotes PlexinA2 interaction to Sema3A. Fyn also phosphorylates cyclin-dependent kinase-5 (Cdk5) and in turn collapse response mediator protein (CRMP). The activation of Cdk5 facilitates the suppression of Rac-PAK signaling leading to the actin dynamic regulation. On the other hand, when phosphorylated, Cdk5 activates Tau resulting in destabilization of microtubules. Both these events culminate in growth cone collapse, remodeling of synaptic network and regulation of dendrites and axonal orientation (Adapted from Sasaki et al. 2002) [111].
Figure 4.
A proposed mechanism by which Fyn and Cdk5 control Sema3A and PlexinA2 downstream signaling cascade either in physiologic (left) or in pathologic (right) conditions. Fyn phosphorylates PlexinA2 and promotes PlexinA2 interaction to Sema3A. Fyn also phosphorylates cyclin-dependent kinase-5 (Cdk5) and in turn collapse response mediator protein (CRMP). The activation of Cdk5 facilitates the suppression of Rac-PAK signaling leading to the actin dynamic regulation. On the other hand, when phosphorylated, Cdk5 activates Tau resulting in destabilization of microtubules. Both these events culminate in growth cone collapse, remodeling of synaptic network and regulation of dendrites and axonal orientation (Adapted from Sasaki et al. 2002) [111].

Figure 5.
Fyn target proteins in AD neurons. Left: When overactivated, Fyn promotes phosphorylation either of APP at Tyr682 or Tau at Tyr18. These target protein hyperphosphorylations activate downstream pathways finally resulting in AD-related neuronal dysfunction and death. Right: A proposed mechanism by which Fyn, after interacting with the Aβ oligomers-Prion like protein complex, phosphorylates NR2B receptor and triggers excitotoxicity.
Figure 5.
Fyn target proteins in AD neurons. Left: When overactivated, Fyn promotes phosphorylation either of APP at Tyr682 or Tau at Tyr18. These target protein hyperphosphorylations activate downstream pathways finally resulting in AD-related neuronal dysfunction and death. Right: A proposed mechanism by which Fyn, after interacting with the Aβ oligomers-Prion like protein complex, phosphorylates NR2B receptor and triggers excitotoxicity.


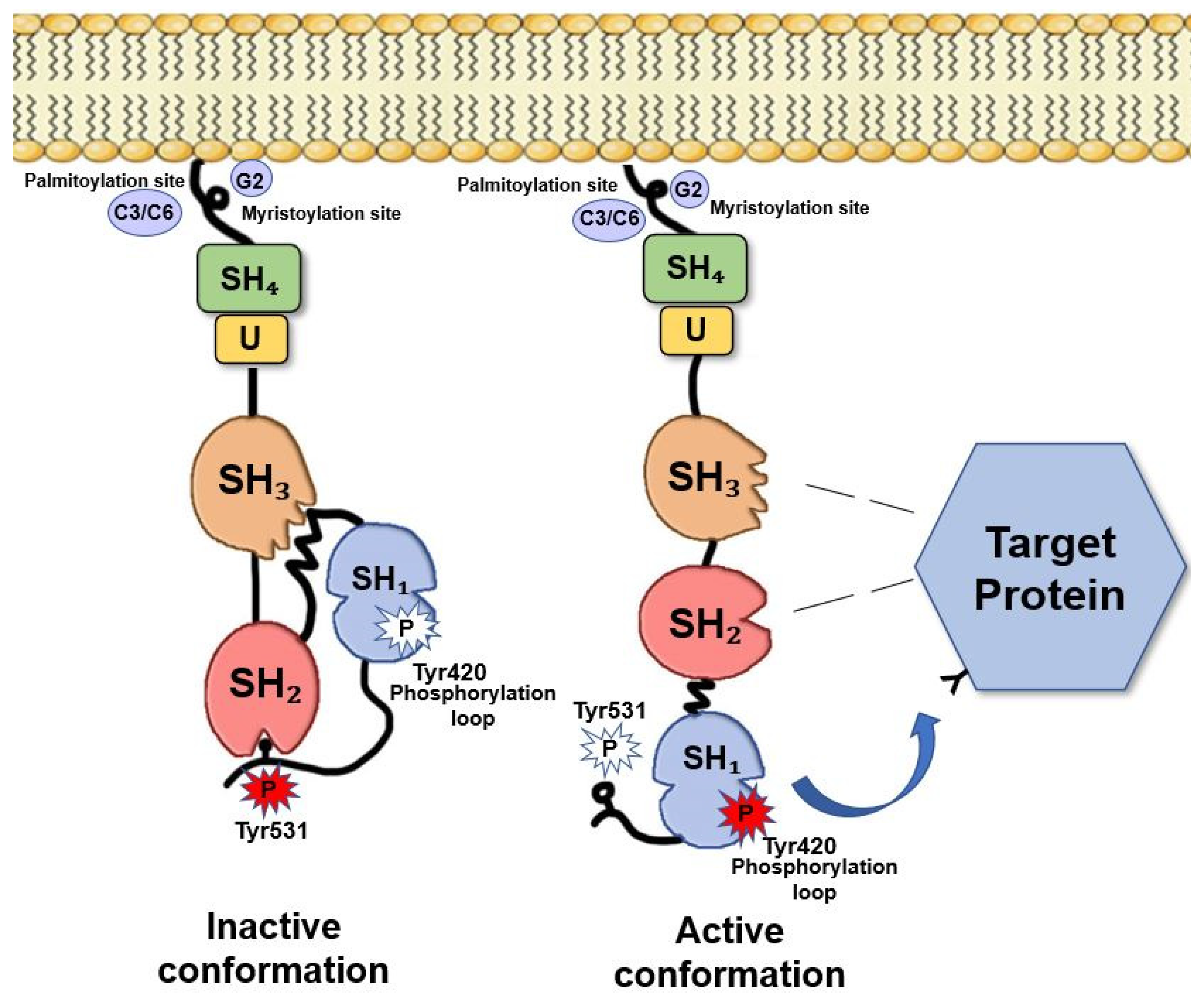{kind=link}
{kind=link}
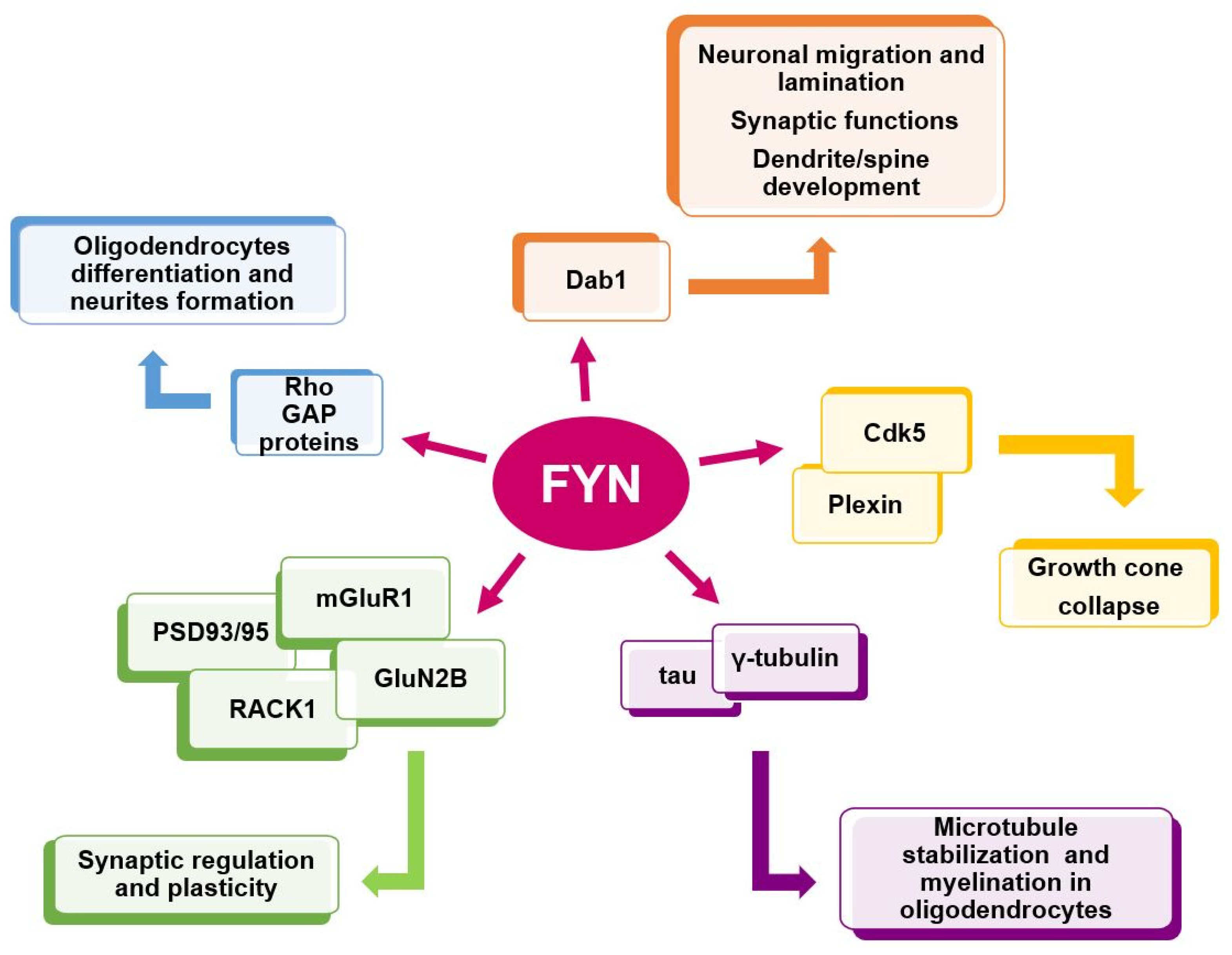{kind=link}
{kind=link}
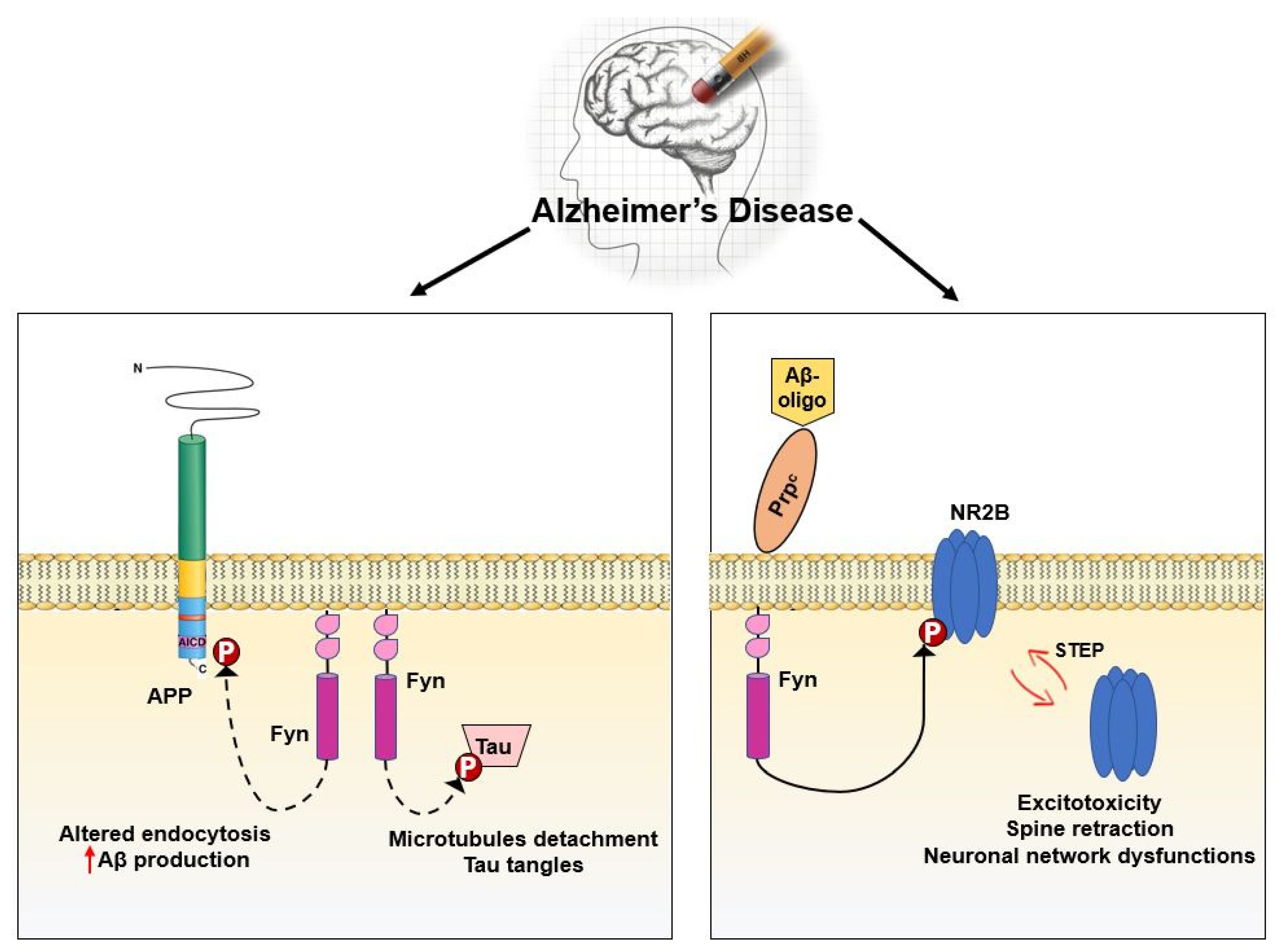{kind=link}
Table 1.
PlexinA4 (PLEXA4), semaphorin 5A (SEMA5A) and Fyn-related kinase (FRK) gene association analysis in ASD patients. Adapted from Schafer et al. (2019) [97].
Table 1.
PlexinA4 (PLEXA4), semaphorin 5A (SEMA5A) and Fyn-related kinase (FRK) gene association analysis in ASD patients. Adapted from Schafer et al. (2019) [97].
| Symbol and Name Gene | Support in Autism | N° of Studies Reporting the Evidence | Supporting Evidence |
|---|---|---|---|
| PLEXA4 (Plexin A4) | Functional | 3 | Copy number variations (CNVs) involving the PLXN-A4 gene were identified in two unrelated ASD case |
| SEMA5A (Semaphorin 5A) | Functional | 15 | Expression of the SEMA5A gene has been shown to be downregulated in some autistic individuals |
| FRK (Fyn-related kinase) | Genetic association | 3 | Genetic association has been found between the FRK gene and autism in two large cohorts (AGRE and ACC) of European ancestry and replicate in two other cohort (CAP and CART) |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Matrone, C.; Petrillo, F.; Nasso, R.; Ferretti, G. Fyn Tyrosine Kinase as Harmonizing Factor in Neuronal Functions and Dysfunctions. Int. J. Mol. Sci. 2020, 21, 4444. https://doi.org/10.3390/ijms21124444
AMA Style
Matrone C, Petrillo F, Nasso R, Ferretti G. Fyn Tyrosine Kinase as Harmonizing Factor in Neuronal Functions and Dysfunctions. International Journal of Molecular Sciences. 2020; 21(12):4444. https://doi.org/10.3390/ijms21124444
Chicago/Turabian StyleMatrone, Carmela, Federica Petrillo, Rosarita Nasso, and Gabriella Ferretti. 2020. "Fyn Tyrosine Kinase as Harmonizing Factor in Neuronal Functions and Dysfunctions" International Journal of Molecular Sciences 21, no. 12: 4444. https://doi.org/10.3390/ijms21124444
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.