Post-Transcriptional Regulation of Alpha One Antitrypsin by a Proteasome Inhibitor

 , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
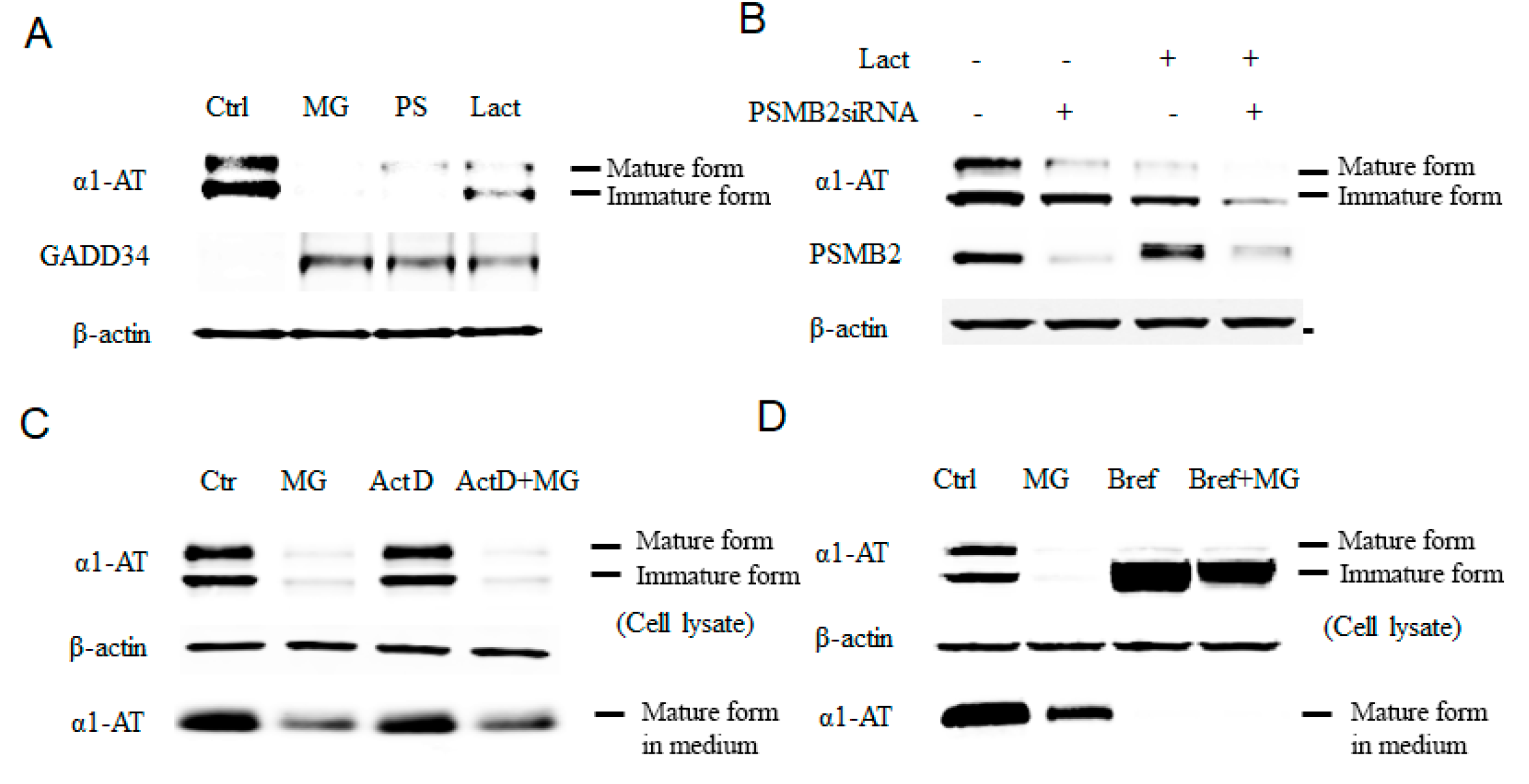
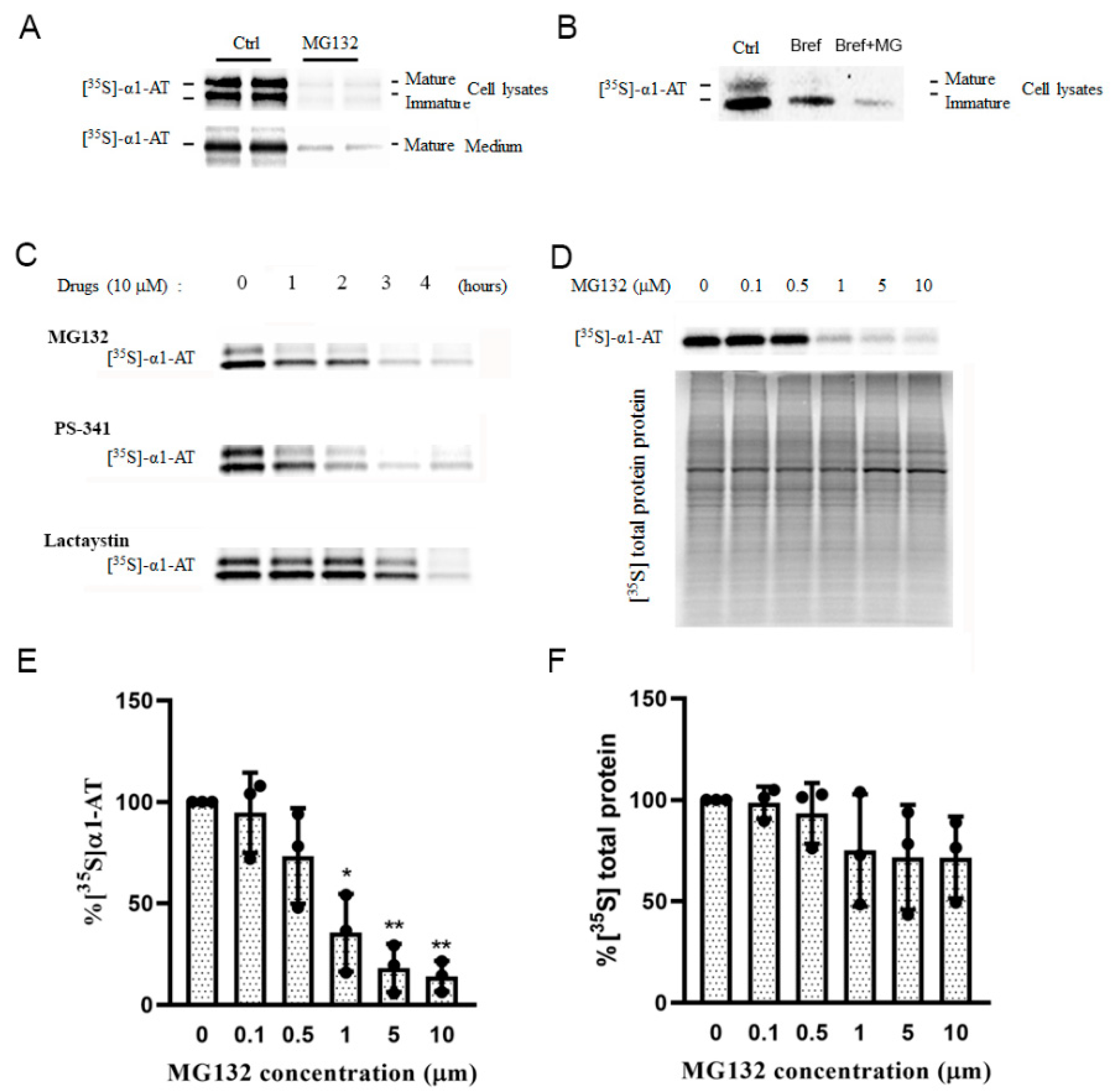
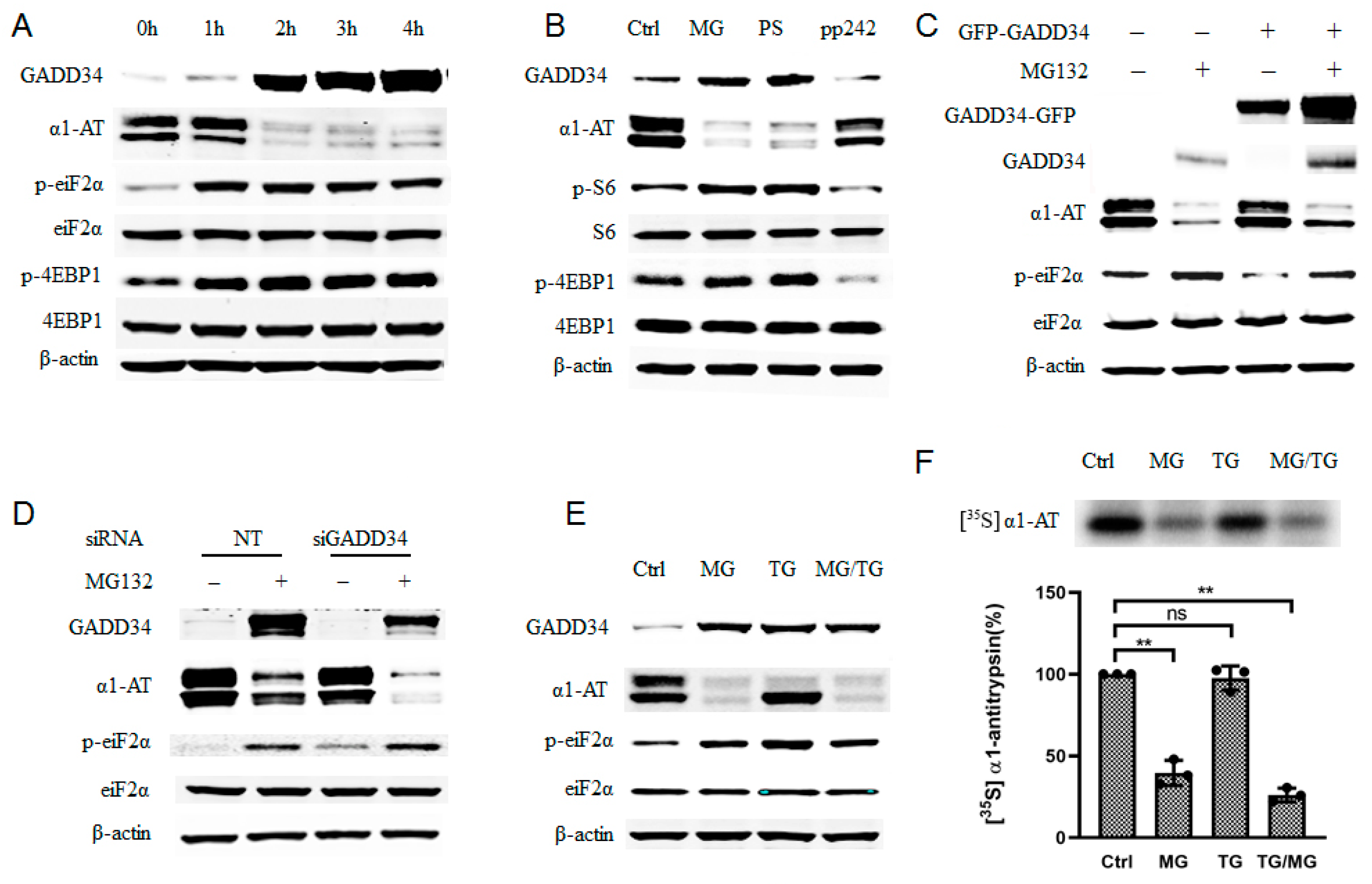
2.1. Alpha 1 Antitrypsin Was Down Regulated by Proteasome Inhibition
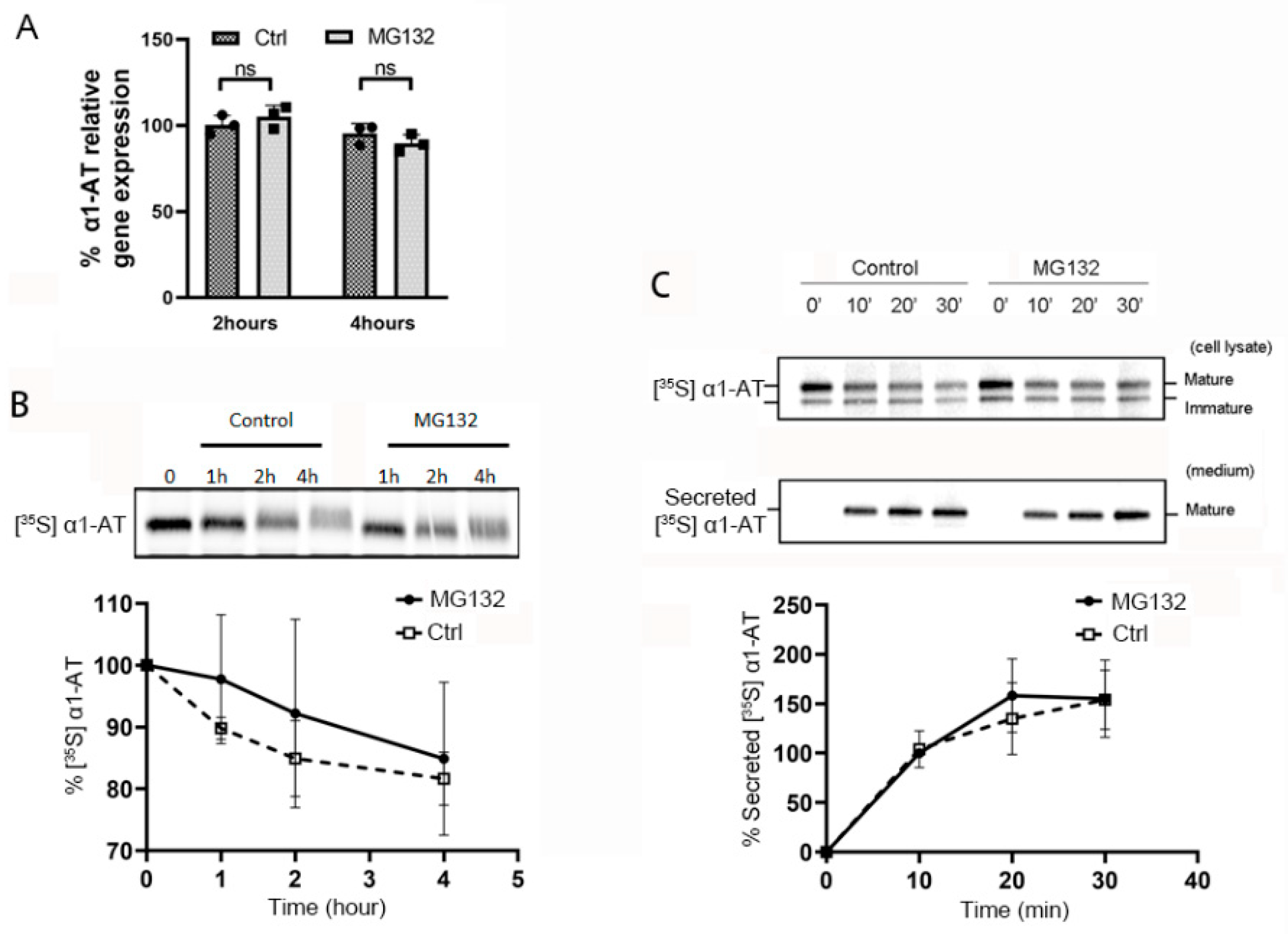
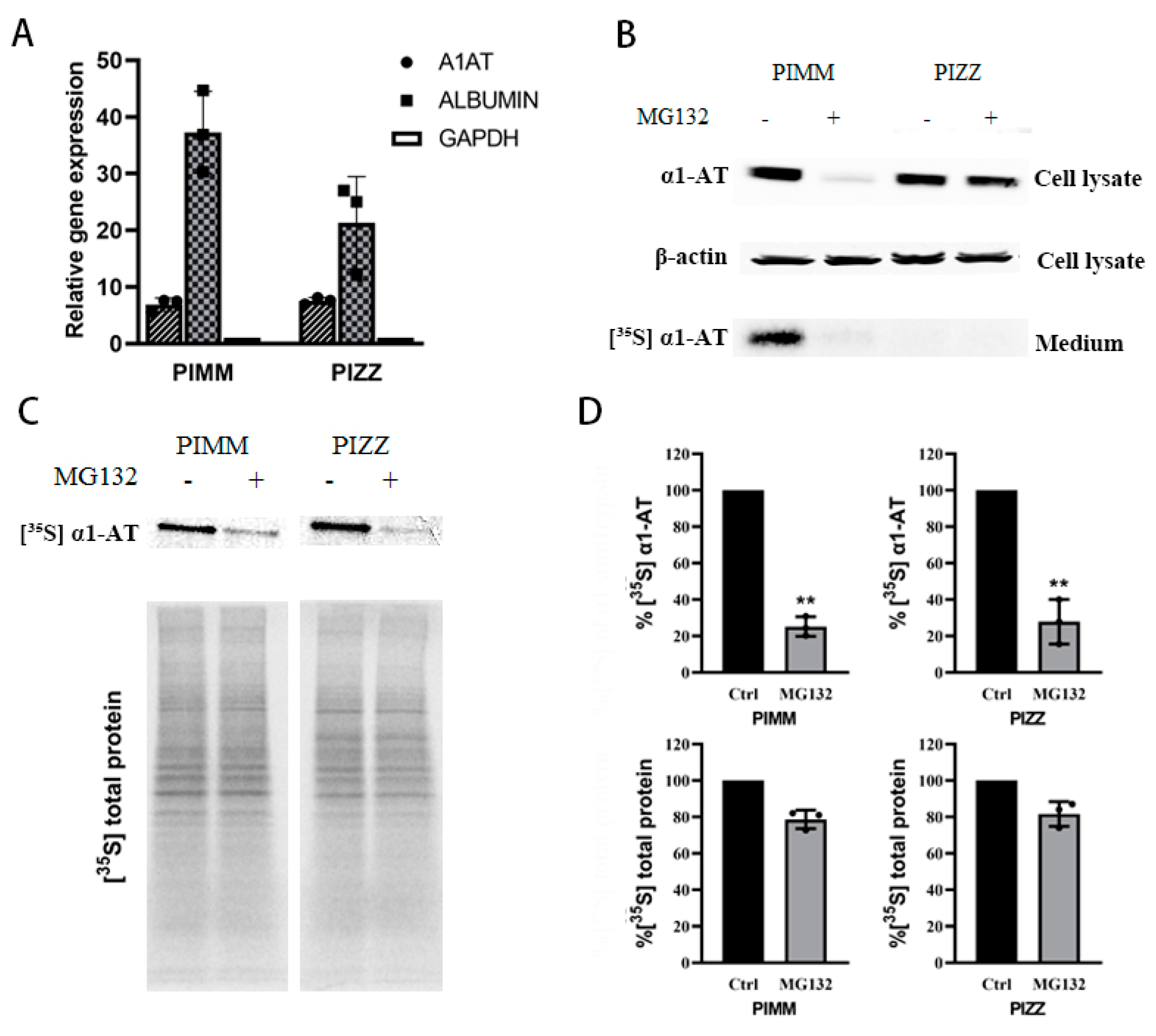
2.2. Proteasome Inhibitors Do Not Affect Transcription or Secretion of α1AT in Hepatocytes
2.3. Proteasome Inhibitors Selectively Inhibited α1AT Translation
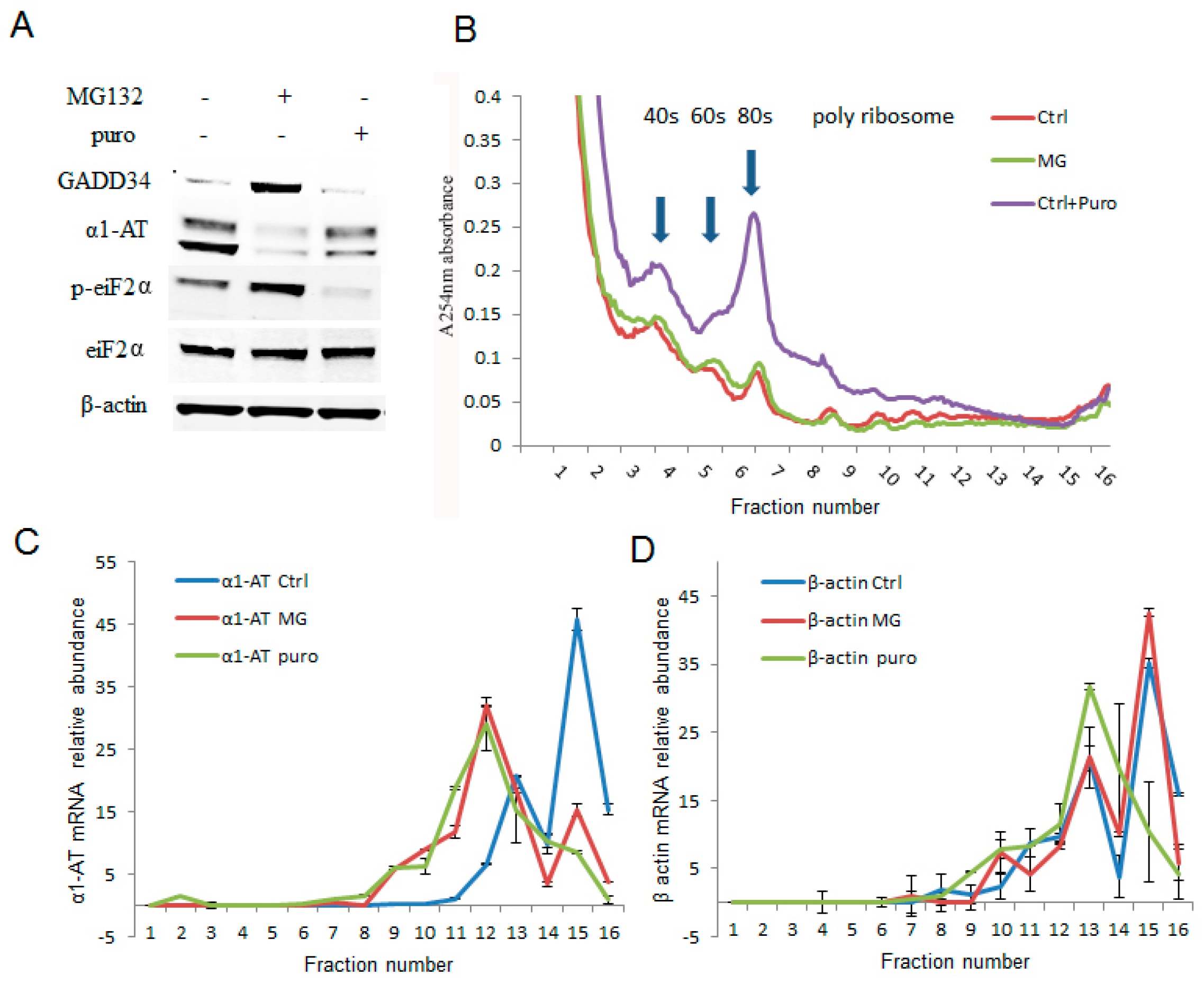
2.4. Phosphorylation of eIF2α Caused by Proteasome Inhibition Partially Contributing to α1AT mRNA Translation Inhibition
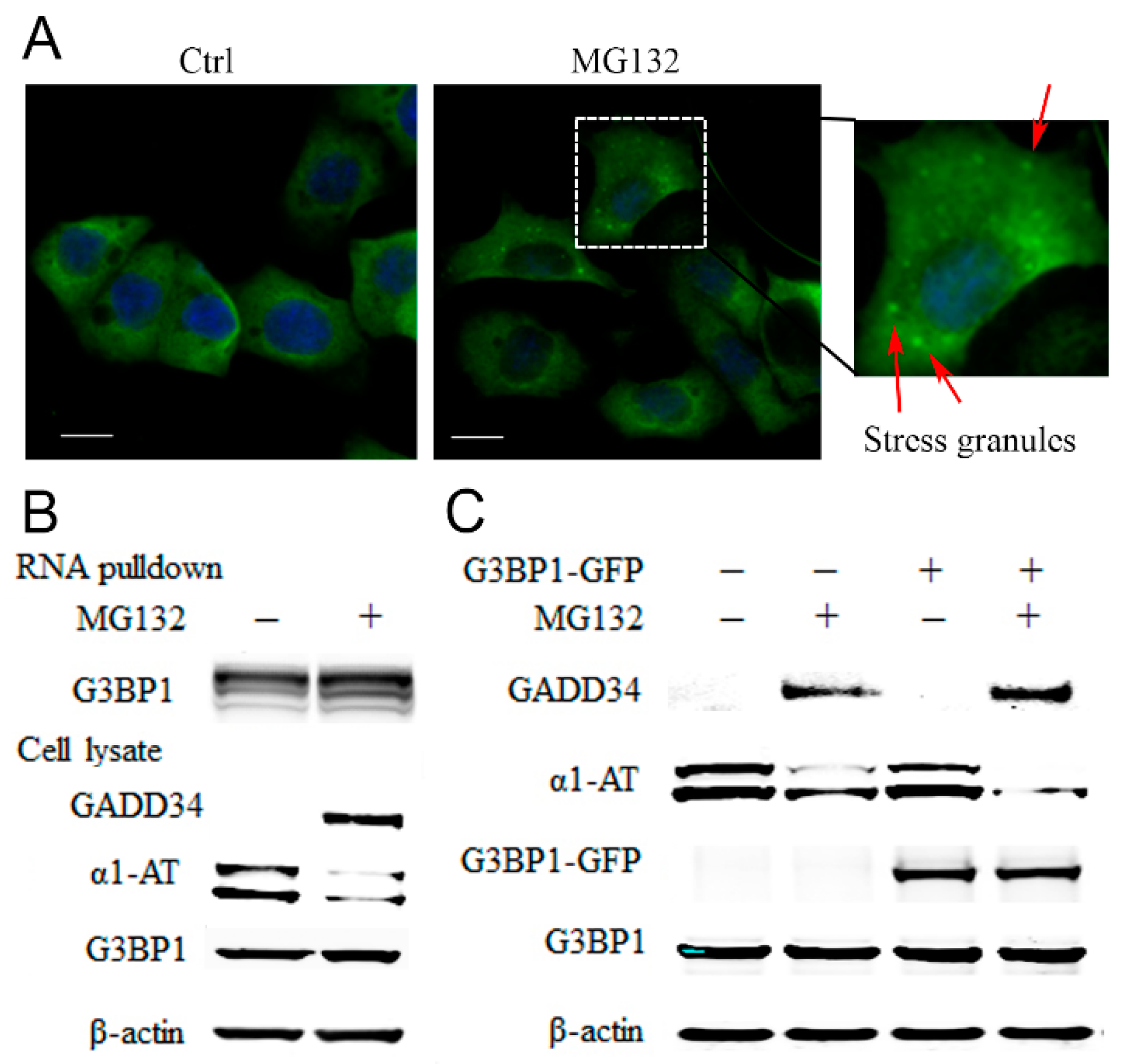
2.5. Proteasome Inhibitors Increased the Association of Stress Granule Proteins with α1AT mRNA and Inhibited Translation
2.6. Proteasome Inhibitors Inhibited the Translation of Mutant α1ATZ in α1ATD Disease-Specific Human iPSC Derived Hepatocyte
3. Discussion
4. Materials and Methods
4.1. Reagents Cells and Antibodys
4.2. qPCR, Northern Blot Analysis and Western Blot Analysis
4.3. Polysome Profiling by Sucrose Gradient Fractionation
4.4. RNA Interference-Mediated Gene Knockdown and Plasmids Transfection
4.5. Radiolabeling Study
4.6. Plasmids Construction
4.7. Biotinylated RNA Pull-Down Assay
4.8. Immunofluorescence Staining
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Wood, A.M.; Stockley, R.A. Alpha one antitrypsin deficiency: From gene to treatment. Respiration 2007, 74, 481–492. [Google Scholar] [CrossRef] [PubMed]
- Stoller, J.K.; Aboussouan, L.S. A review of alpha1-antitrypsin deficiency. Am. J. Respir. Crit. Care. Med. 2012, 185, 246–259. [Google Scholar] [CrossRef] [PubMed]
- Jeppsson, J.O.; Laurell, C.B. The amino acid substitutions of human alpha 1-antitrypsin M3, X. and Z. FEBS Lett 1988, 231, 327–330. [Google Scholar] [CrossRef] [Green Version]
- Lomas, D.A.; Mahadeva, R. Alpha1-antitrypsin polymerization and the serpinopathies: Pathobiology and prospects for therapy. J. Clin. Investig. 2002, 110, 1585–1590. [Google Scholar] [CrossRef] [PubMed]
- Wewers, M.D.; Crystal, R.G. Alpha-1 antitrypsin augmentation therapy. COPD 2013, 10, 64–67. [Google Scholar] [CrossRef]
- Stockley, R.A.; Miravitlles, M.; Vogelmeier, C.; Alpha One International, R. Augmentation therapy for alpha-1 antitrypsin deficiency: Towards a personalised approach. Orphanet J. Rare. Dis. 2013, 8, 149. [Google Scholar] [CrossRef] [Green Version]
- Gruntman, A.M.; Flotte, T.R. Progress with Recombinant Adeno-Associated Virus Vectors for Gene Therapy of Alpha-1 Antitrypsin Deficiency. Hum. Gene. Ther. Methods 2015, 26, 77–81. [Google Scholar] [CrossRef] [Green Version]
- Mueller, C.; Tang, Q.; Gruntman, A.; Blomenkamp, K.; Teckman, J.; Song, L.; Zamore, P.D.; Flotte, T.R. Sustained miRNA-mediated knockdown of mutant AAT with simultaneous augmentation of wild-type AAT has minimal effect on global liver miRNA profiles. Mol. Ther. 2012, 20, 590–600. [Google Scholar] [CrossRef] [Green Version]
- Kaushal, S.; Annamali, M.; Blomenkamp, K.; Rudnick, D.; Halloran, D.; Brunt, E.M.; Teckman, J.H. Rapamycin reduces intrahepatic alpha-1-antitrypsin mutant Z protein polymers and liver injury in a mouse model. Exp. Biol. Med. (Maywood) 2010, 235, 700–709. [Google Scholar] [CrossRef] [Green Version]
- Hidvegi, T.; Ewing, M.; Hale, P.; Dippold, C.; Beckett, C.; Kemp, C.; Maurice, N.; Mukherjee, A.; Goldbach, C.; Watkins, S.; et al. An autophagy-enhancing drug promotes degradation of mutant alpha1-antitrypsin Z and reduces hepatic fibrosis. Science 2010, 329, 229–232. [Google Scholar] [CrossRef]
- Mallya, M.; Phillips, R.L.; Saldanha, S.A.; Gooptu, B.; Brown, S.C.; Termine, D.J.; Shirvani, A.M.; Wu, Y.; Sifers, R.N.; Abagyan, R.; et al. Small molecules block the polymerization of Z alpha1-antitrypsin and increase the clearance of intracellular aggregates. J. Med. Chem. 2007, 50, 5357–5363. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berthelier, V.; Harris, J.B.; Estenson, K.N.; Baudry, J. Discovery of an inhibitor of Z-alpha1 antitrypsin polymerization. PLoS ONE 2015, 10, e0126256. [Google Scholar] [CrossRef] [PubMed]
- Kisselev, A.F.; Goldberg, A.L. Proteasome inhibitors: From research tools to drug candidates. Chem. Biol. 2001, 8, 739–758. [Google Scholar] [CrossRef] [Green Version]
- Adams, J. Proteasome inhibition: A novel approach to cancer therapy. Trends Mol. Med. 2002, 8, S49–S54. [Google Scholar] [CrossRef]
- Ciechanover, A.; DiGiuseppe, J.A.; Bercovich, B.; Orian, A.; Richter, J.D.; Schwartz, A.L.; Brodeur, G.M. Degradation of nuclear oncoproteins by the ubiquitin system in vitro. Proc. Natl. Acad. Sci. USA 1991, 88, 139–143. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pellom, S.T.; Shanker, A. Development of Proteasome Inhibitors as Therapeutic Drugs. J. Clin. Cell Immunol. 2012, S5, 5. [Google Scholar] [PubMed]
- Adams, J. Proteasome inhibitors as new anticancer drugs. Curr. Opin. Oncol. 2002, 14, 628–634. [Google Scholar] [CrossRef] [PubMed]
- Chen, D.; Frezza, M.; Schmitt, S.; Kanwar, J.; Dou, Q.P. Bortezomib as the first proteasome inhibitor anticancer drug: Current status and future perspectives. Curr. Cancer. Drug. Targets 2011, 11, 239–253. [Google Scholar] [CrossRef] [Green Version]
- Wunderlich, A.; Arndt, T.; Fischer, M.; Roth, S.; Ramaswamy, A.; Greene, B.H.; Brendel, C.; Hinterseher, U.; Bartsch, D.K.; Hoffmann, S. Targeting the proteasome as a promising therapeutic strategy in thyroid cancer. J. Surg. Oncol. 2012, 105, 357–364. [Google Scholar] [CrossRef]
- Ding, Q.; Dimayuga, E.; Markesbery, W.R.; Keller, J.N. Proteasome inhibition induces reversible impairments in protein synthesis. FASEB J. 2006, 20, 1055–1063. [Google Scholar] [CrossRef] [Green Version]
- Konstantinova, I.M.; Tsimokha, A.S.; Mittenberg, A.G. Role of proteasomes in cellular regulation. Int. Rev. Cell Mol. Biol. 2008, 267, 59–124. [Google Scholar] [CrossRef]
- Qu, D.; Teckman, J.H.; Omura, S.; Perlmutter, D.H. Degradation of a mutant secretory protein, alpha1-antitrypsin Z, in the endoplasmic reticulum requires proteasome activity. J. Biol. Chem. 1996, 271, 22791–22795. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Teckman, J.H.; Burrows, J.; Hidvegi, T.; Schmidt, B.; Hale, P.D.; Perlmutter, D.H. The proteasome participates in degradation of mutant alpha 1-antitrypsin Z in the endoplasmic reticulum of hepatoma-derived hepatocytes. J. Biol. Chem. 2001, 276, 44865–44872. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bathurst, I.C.; Stenflo, J.; Errington, D.M.; Carrell, R.W. Translation and processing of normal (PiMM) and abnormal (PiZZ) human alpha 1-antitrypsin. FEBS Lett. 1983, 153, 270–274. [Google Scholar] [CrossRef] [Green Version]
- Brush, M.H.; Shenolikar, S. Control of cellular GADD34 levels by the 26S proteasome. Mol. Cell Biol. 2008, 28, 6989–7000. [Google Scholar] [CrossRef] [Green Version]
- Isaacson, P.; Jones, D.B.; Millward-Sadler, G.H.; Judd, M.A.; Payne, S. Alpha-1-antitrypsin in human macrophages. J. Clin. Pathol. 1981, 34, 982–990. [Google Scholar] [CrossRef] [Green Version]
- Orci, L.; Tagaya, M.; Amherdt, M.; Perrelet, A.; Donaldson, J.G.; Lippincott-Schwartz, J.; Klausner, R.D.; Rothman, J.E. Brefeldin A, a drug that blocks secretion, prevents the assembly of non-clathrin-coated buds on Golgi cisternae. Cell 1991, 64, 1183–1195. [Google Scholar]
- Blobel, G.; Sabatini, D. Dissociation of Mammalian Polyribosomes into Subunits by Puromycin. Proc. Natl. Acad. Sci. USA 1971, 68, 390–394. [Google Scholar] [CrossRef] [Green Version]
- Sonenberg, N.; Hinnebusch, A.G. Regulation of translation initiation in eukaryotes: Mechanisms and biological targets. Cell 2009, 136, 731–745. [Google Scholar] [CrossRef] [Green Version]
- Hara, K.; Yonezawa, K.; Weng, Q.P.; Kozlowski, M.T.; Belham, C.; Avruch, J. Amino acid sufficiency and mTOR regulate p70 S6 kinase and eIF-4E BP1 through a common effector mechanism. J. Biol. Chem. 1998, 273, 14484–14494. [Google Scholar] [CrossRef] [Green Version]
- Feldman, M.E.; Apsel, B.; Uotila, A.; Loewith, R.; Knight, Z.A.; Ruggero, D.; Shokat, K.M. Active-site inhibitors of mTOR target rapamycin-resistant outputs of mTORC1 and mTORC2. PLoS Biol. 2009, 7, e38. [Google Scholar] [CrossRef]
- Thoreen, C.C.; Chantranupong, L.; Keys, H.R.; Wang, T.; Gray, N.S.; Sabatini, D.M. A unifying model for mTORC1-mediated regulation of mRNA translation. Nature 2012, 485, 109–113. [Google Scholar] [CrossRef] [PubMed]
- Werner, E.D.; Brodsky, J.L.; McCracken, A.A. Proteasome-dependent endoplasmic reticulum-associated protein degradation: An unconventional route to a familiar fate. Proc. Natl. Acad. Sci. USA 1996, 93, 13797–13801. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Novoa, I.; Zeng, H.; Harding, H.P.; Ron, D. Feedback inhibition of the unfolded protein response by GADD34-mediated dephosphorylation of eIF2alpha. J. Cell Biol. 2001, 153, 1011–1022. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsaytler, P.; Harding, H.P.; Ron, D.; Bertolotti, A. Selective inhibition of a regulatory subunit of protein phosphatase 1 restores proteostasis. Science 2011, 332, 91–94. [Google Scholar] [CrossRef]
- Proud, C.G. Signalling to translation: How signal transduction pathways control the protein synthetic machinery. Biochem. J. 2007, 403, 217–234. [Google Scholar] [CrossRef] [Green Version]
- Mazroui, R.; Di Marco, S.; Kaufman, R.J.; Gallouzi, I.E. Inhibition of the ubiquitin-proteasome system induces stress granule formation. Mol. Biol. Cell 2007, 18, 2603–2618. [Google Scholar] [CrossRef] [Green Version]
- Kedersha, N.; Anderson, P. Mammalian stress granules and processing bodies. Methods Enzymol. 2007, 431, 61–81. [Google Scholar] [CrossRef]
- Reineke, L.C.; Dougherty, J.D.; Pierre, P.; Lloyd, R.E. Large G3BP-induced granules trigger eIF2alpha phosphorylation. Mol. Biol. Cell 2012, 23, 3499–3510. [Google Scholar] [CrossRef]
- Rashid, S.T.; Corbineau, S.; Hannan, N.; Marciniak, S.J.; Miranda, E.; Alexander, G.; Huang-Doran, I.; Griffin, J.; Ahrlund-Richter, L.; Skepper, J.; et al. Modeling inherited metabolic disorders of the liver using human induced pluripotent stem cells. J. Clin. Investig. 2010, 120, 3127–3136. [Google Scholar] [CrossRef] [Green Version]
- Fairbanks, K.D.; Tavill, A.S. Liver disease in alpha 1-antitrypsin deficiency: A review. Am. J. Gastroenterol. 2008, 103, 2136–2141, quiz 2142. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.; Bai, L.; Chen, W.; Xu, S. The NF-kappaB activation pathways, emerging molecular targets for cancer prevention and therapy. Expert Opin. Ther. Targets 2010, 14, 45–55. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Traenckner, E.B.; Wilk, S.; Baeuerle, P.A. A proteasome inhibitor prevents activation of NF-kappa B and stabilizes a newly phosphorylated form of I kappa B-alpha that is still bound to NF-kappa B. EMBO J. 1994, 13, 5433–5441. [Google Scholar] [CrossRef] [PubMed]
- Jiang, H.Y.; Wek, R.C. Phosphorylation of the alpha-subunit of the eukaryotic initiation factor-2 (eIF2alpha) reduces protein synthesis and enhances apoptosis in response to proteasome inhibition. J. Biol. Chem. 2005, 280, 14189–14202. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alard, A.; Marboeuf, C.; Fabre, B.; Jean, C.; Martineau, Y.; Lopez, F.; Vende, P.; Poncet, D.; Schneider, R.J.; Bousquet, C.; et al. Differential Regulation of the Three Eukaryotic mRNA Translation Initiation Factor (eIF) 4Gs by the Proteasome. Front. Genet. 2019, 10, 254. [Google Scholar] [CrossRef] [PubMed]
- Wu, W.K.; Volta, V.; Cho, C.H.; Wu, Y.C.; Li, H.T.; Yu, L.; Li, Z.J.; Sung, J.J. Repression of protein translation and mTOR signaling by proteasome inhibitor in colon cancer cells. Biochem. Biophys. Res. Commun. 2009, 386, 598–601. [Google Scholar] [CrossRef]
- Yerlikaya, A.; Kimball, S.R.; Stanley, B.A. Phosphorylation of eIF2alpha in response to 26S proteasome inhibition is mediated by the haem-regulated inhibitor (HRI) kinase. Biochem. J. 2008, 412, 579–588. [Google Scholar] [CrossRef] [Green Version]
- Obeng, E.A.; Carlson, L.M.; Gutman, D.M.; Harrington, W.J.; Lee, K.P.; Boise, L.H. Proteasome inhibitors induce a terminal unfolded protein response in multiple myeloma cells. Blood 2006, 107, 4907–4916. [Google Scholar] [CrossRef] [Green Version]
- Farkas, D.; Bhat, V.B.; Mandapati, S.; Wishnok, J.S.; Tannenbaum, S.R. Characterization of the secreted proteome of rat hepatocytes cultured in collagen sandwiches. Chem. Res. Toxicol. 2005, 18, 1132–1139. [Google Scholar] [CrossRef] [Green Version]







© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rao, L.; Xu, Y.; Reineke, L.C.; Bhattacharya, A.; Tyryshkin, A.; Shin, J.N.; Eissa, N.T. Post-Transcriptional Regulation of Alpha One Antitrypsin by a Proteasome Inhibitor. Int. J. Mol. Sci. 2020, 21, 4318. https://doi.org/10.3390/ijms21124318
Rao L, Xu Y, Reineke LC, Bhattacharya A, Tyryshkin A, Shin JN, Eissa NT. Post-Transcriptional Regulation of Alpha One Antitrypsin by a Proteasome Inhibitor. International Journal of Molecular Sciences. 2020; 21(12):4318. https://doi.org/10.3390/ijms21124318
Chicago/Turabian StyleRao, Lang, Yi Xu, Lucas Charles Reineke, Abhisek Bhattacharya, Alexey Tyryshkin, Jin Na Shin, and N. Tony Eissa. 2020. "Post-Transcriptional Regulation of Alpha One Antitrypsin by a Proteasome Inhibitor" International Journal of Molecular Sciences 21, no. 12: 4318. https://doi.org/10.3390/ijms21124318