HSP70-Mediated NLRP3 Inflammasome Suppression Underlies Reversal of Acute Kidney Injury Following Extracellular Vesicle and Focused Ultrasound Combination Therapy



Abstract
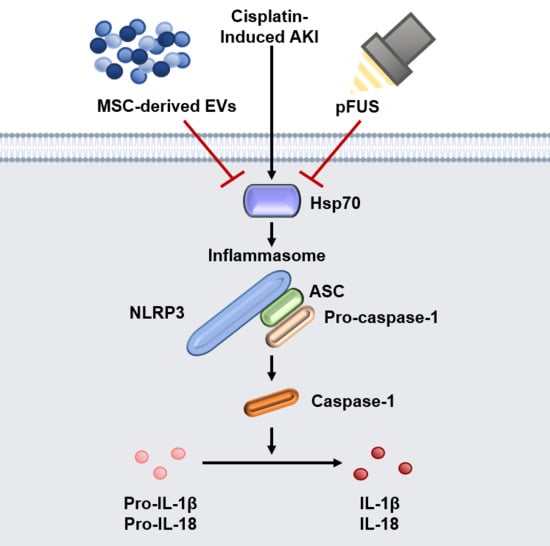
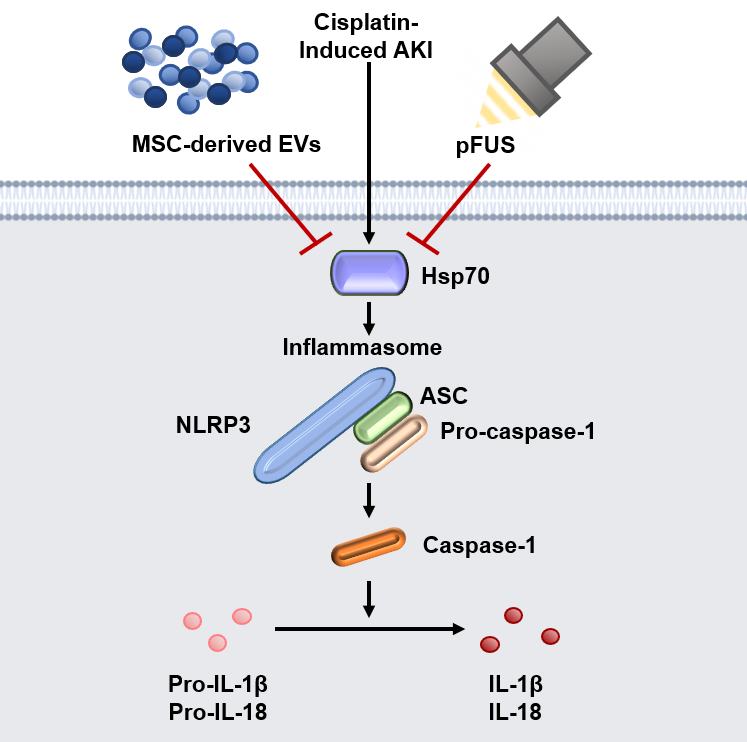
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
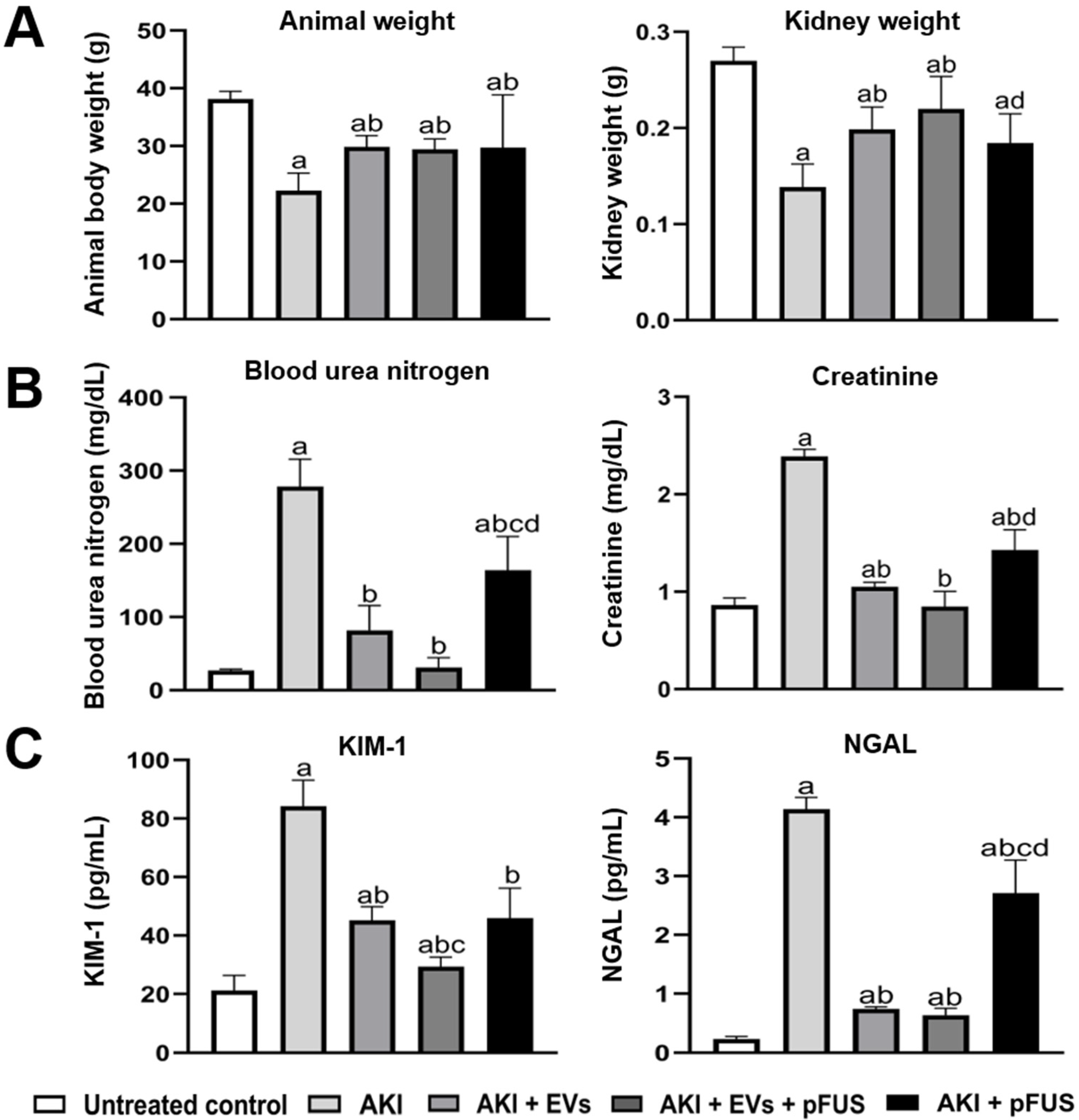
2.1. Reversal of AKI Using EVs and pFUS
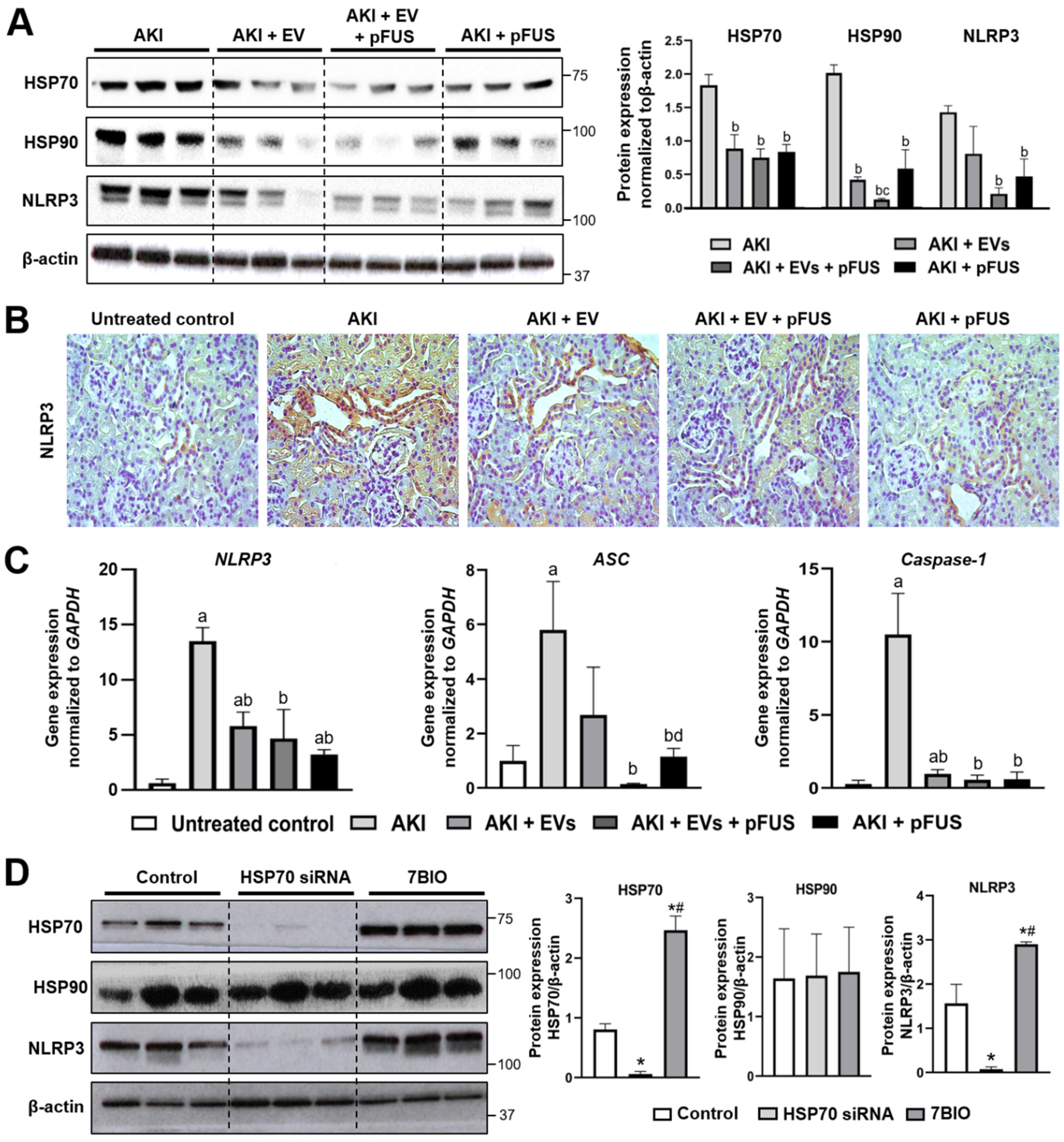
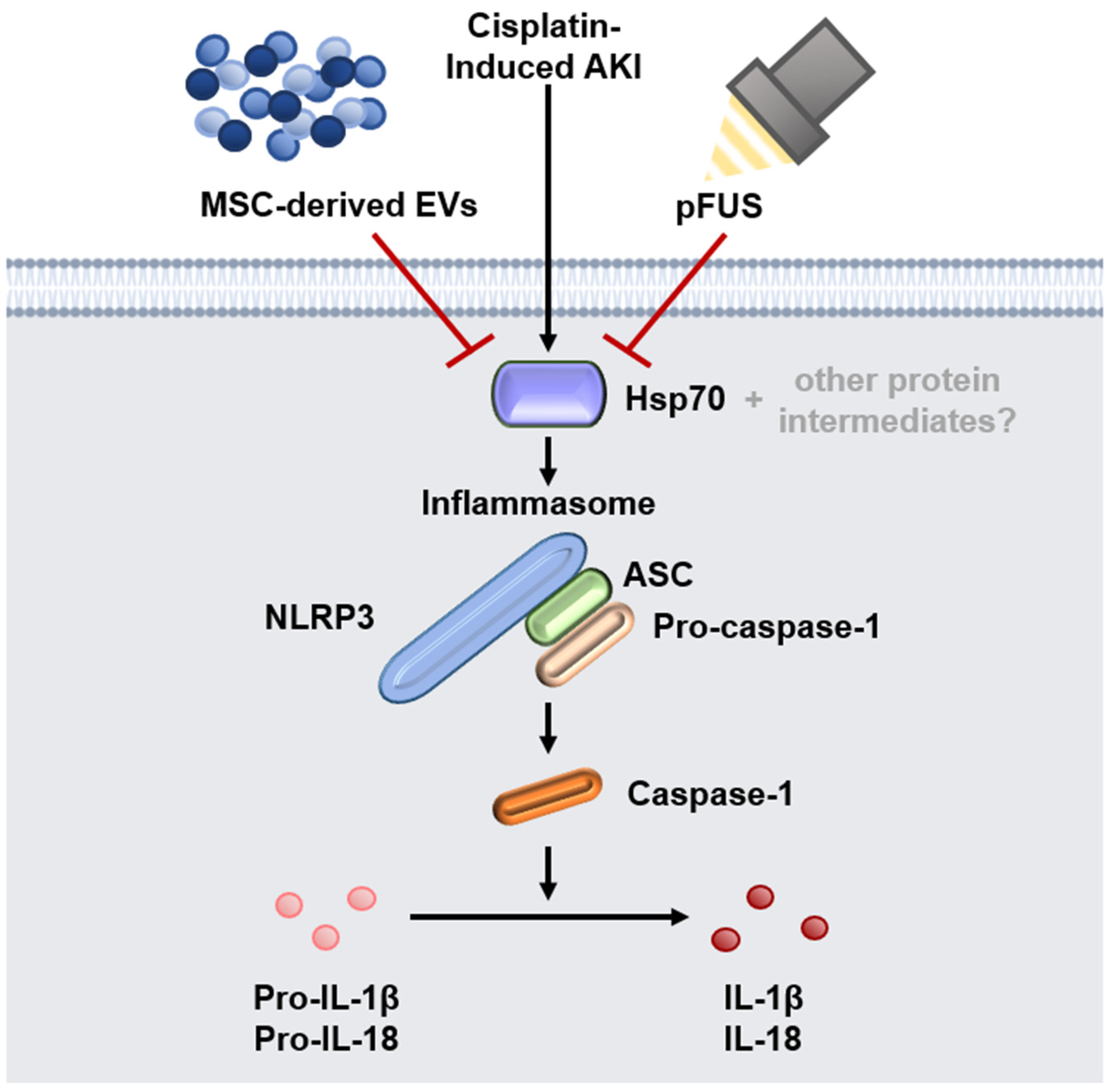
2.2. HSP70-Mediated Regulation of the NLRP3 Inflammasome
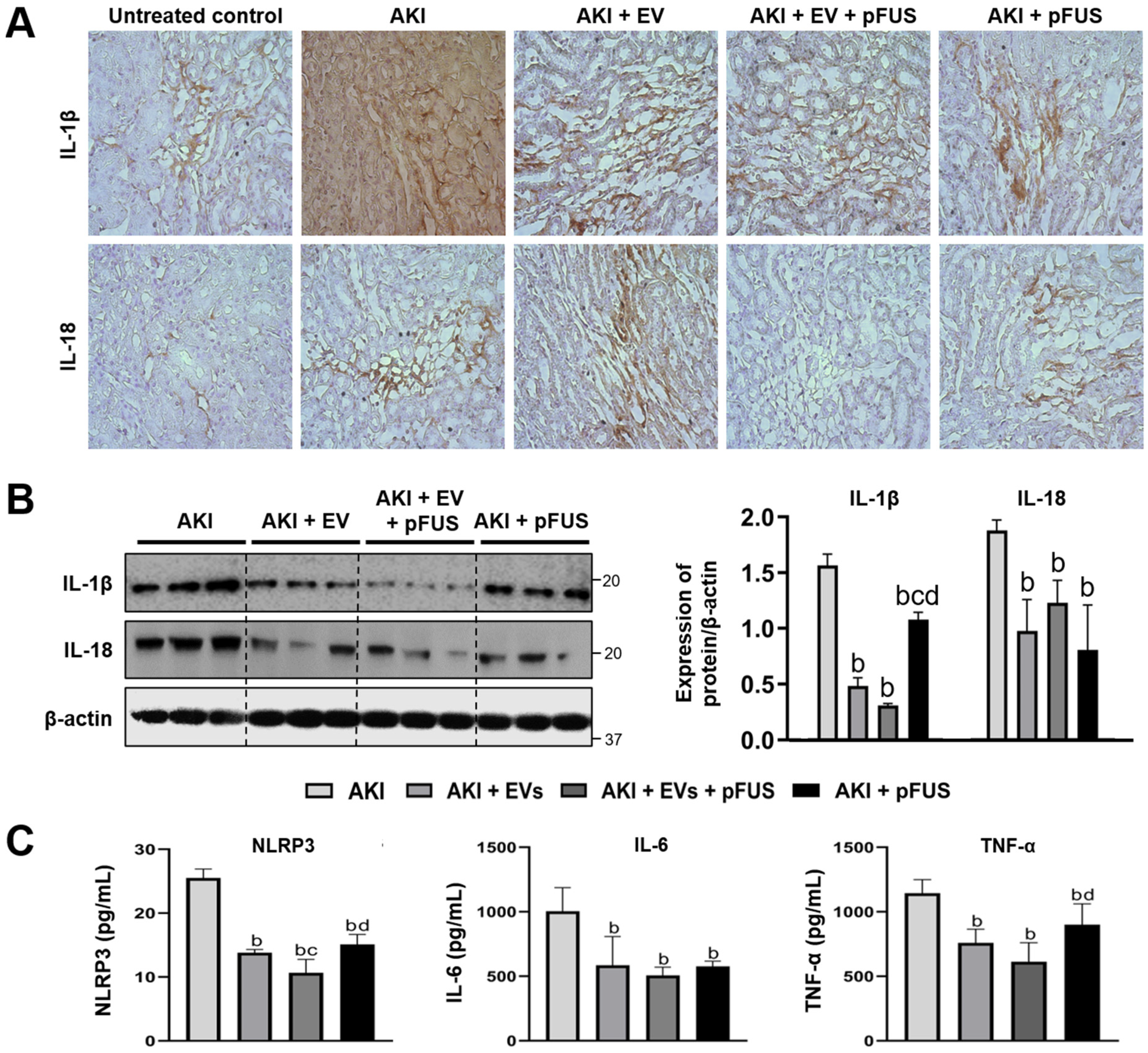
2.3. Suppression of Inflammation Following EV and pFUS Therapy
3. Discussion
4. Methods
4.1. Animal Experiments
4.2. Extracellular Vesicle Isolation and Purification
4.3. Pulsed Focused Ultrasound
4.4. Analysis of Kidney Function
4.5. Histology, Immunohistochemistry, and Immunofluorescence
4.6. NLRP3 Knockdown by siRNA and Overexpression by 7BIO
4.7. Western Blot Analysis
4.8. Quantitative Polymerase Chain Reaction
4.9. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Levey, A.S.; James, M.T. Acute Kidney Injury. Ann. Intern. Med. 2017, 167, ITC66–ITC80. [Google Scholar] [CrossRef] [PubMed]
- Susantitaphong, P.; Cruz, D.N.; Cerdá, J.; Abulfaraj, M.; Alqahtani, F.; Koulouridis, I.; Jaber, B.L. World incidence of AKI: A meta-analysis. Clin. J. Am. Soc. Nephrol. 2013, 8, 1482–1493. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rabb, H.; Griffin, M.D.; McKay, D.B.; Swaminathan, S.; Pickkers, P.; Rosner, M.H.; Kellum, J.A.; Ronco, C. Inflammation in AKI: Current Understanding, Key Questions, and Knowledge Gaps. J. Am. Soc. Nephrol. 2016, 27, 371–379. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Medzhitov, R. Inflammation 2010: New adventures of an old flame. Cell 2010, 140, 771–776. [Google Scholar] [CrossRef] [Green Version]
- Kurts, C.; Panzer, U.; Anders, H.J.; Rees, A.J. The immune system and kidney disease: Basic concepts and clinical implications. Nat. Rev. Immunol. 2013, 13, 738–753. [Google Scholar] [CrossRef]
- Vilaysane, A.; Chun, J.; Seamone, M.E.; Wang, W.; Chin, R.; Hirota, S.; Li, Y.; Clark, S.A.; Tschopp, J.; Trpkov, K.; et al. The NLRP3 inflammasome promotes renal inflammation and contributes to CKD. J. Am. Soc. Nephrol. 2010, 21, 1732–1744. [Google Scholar] [CrossRef] [Green Version]
- Valles, P.G.; Lorenzo, A.G.; Bocanegra, V.; Valles, R. Acute kidney injury: What part do toll-like receptors play? Int. J. Nephrol. Renov. Dis. 2014, 7, 241–251. [Google Scholar] [CrossRef] [Green Version]
- Bolisetty, S.; Agarwal, A. Neutrophils in acute kidney injury: Not neutral any more. Kidney Int. 2009, 75, 674–676. [Google Scholar] [CrossRef] [Green Version]
- Shigeoka, A.A.; Mueller, J.L.; Kambo, A.; Mathison, J.C.; King, A.J.; Hall, W.F.; Correia Jda, S.; Ulevitch, R.J.; Hoffman, H.M.; McKay, D.B. An inflammasome-independent role for epithelial-expressed Nlrp3 in renal ischemia-reperfusion injury. J. Immunol. 2010, 185, 6277–6285. [Google Scholar] [CrossRef] [Green Version]
- Shigeoka, A.A.; Kambo, A.; Mathison, J.C.; King, A.J.; Hall, W.F.; da Silva Correia, J.; Ulevitch, R.J.; McKay, D.B. Nod1 and nod2 are expressed in human and murine renal tubular epithelial cells and participate in renal ischemia reperfusion injury. J. Immunol. 2010, 184, 2297–2304. [Google Scholar] [CrossRef]
- Shigeoka, A.A.; Holscher, T.D.; King, A.J.; Hall, F.W.; Kiosses, W.B.; Tobias, P.S.; Mackman, N.; McKay, D.B. TLR2 is constitutively expressed within the kidney and participates in ischemic renal injury through both MyD88-dependent and -independent pathways. J. Immunol. 2007, 178, 6252–6258. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, M.G.; Koo, T.Y.; Yan, J.J.; Lee, E.; Han, K.H.; Jeong, J.C.; Ro, H.; Kim, B.S.; Jo, S.K.; Oh, K.H.; et al. IL-2/anti-IL-2 complex attenuates renal ischemia-reperfusion injury through expansion of regulatory T cells. J. Am. Soc. Nephrol. 2013, 24, 1529–1536. [Google Scholar] [CrossRef] [Green Version]
- Wang, M.; Yuan, Q.; Xie, L. Mesenchymal Stem Cell-Based Immunomodulation: Properties and Clinical Application. Stem. Cells Int. 2018, 2018, 3057624. [Google Scholar] [CrossRef] [PubMed]
- Herrera, M.B.; Bussolati, B.; Bruno, S.; Fonsato, V.; Romanazzi, G.M.; Camussi, G. Mesenchymal stem cells contribute to the renal repair of acute tubular epithelial injury. Int. J. Mol. Med. 2004, 14, 1035–1041. [Google Scholar] [CrossRef]
- Morigi, M.; Imberti, B.; Zoja, C.; Corna, D.; Tomasoni, S.; Abbate, M.; Rottoli, D.; Angioletti, S.; Benigni, A.; Perico, N.; et al. Mesenchymal stem cells are renotropic, helping to repair the kidney and improve function in acute renal failure. J. Am. Soc. Nephrol. 2004, 15, 1794–1804. [Google Scholar] [CrossRef]
- Morigi, M.; De Coppi, P. Cell therapy for kidney injury: Different options and mechanisms—Mesenchymal and amniotic fluid stem cells. Nephron Exp. Nephrol. 2014, 126, 59. [Google Scholar] [CrossRef]
- Lai, R.C.; Yeo, R.W.; Lim, S.K. Mesenchymal stem cell exosomes. Semin Cell Dev. Biol. 2015, 40, 82–88. [Google Scholar] [CrossRef]
- Ullah, M.; Liu, D.D.; Thakor, A.S. Mesenchymal Stromal Cell Homing: Mechanisms and Strategies for Improvement. iScience 2019, 15, 421–438. [Google Scholar] [CrossRef] [Green Version]
- Schubert, R.; Sann, J.; Frueh, J.T.; Ullrich, E.; Geiger, H.; Baer, P.C. Tracking of Adipose-Derived Mesenchymal Stromal/Stem Cells in a Model of Cisplatin-Induced Acute Kidney Injury: Comparison of Bioluminescence Imaging versus qRT-PCR. Int. J. Mol. Sci. 2018, 19, 2564. [Google Scholar] [CrossRef] [Green Version]
- Lv, L.L.; Wu, W.J.; Feng, Y.; Li, Z.L.; Tang, T.T.; Liu, B.C. Therapeutic application of extracellular vesicles in kidney disease: Promises and challenges. J. Cell Mol. Med. 2018, 22, 728–737. [Google Scholar] [CrossRef]
- Phinney, D.G.; Pittenger, M.F. Concise Review: MSC-Derived Exosomes for Cell-Free Therapy. Stem Cells 2017, 35, 851–858. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ullah, M.; Liu, D.D.; Rai, S.; Razavi, M.; Choi, J.; Wang, J.; Concepcion, W.; Thakor, A.S. A Novel Approach to Deliver Therapeutic Extracellular Vesicles Directly into the Mouse Kidney via Its Arterial Blood Supply. Cells 2020, 9, 937. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schrepfer, S.; Deuse, T.; Reichenspurner, H.; Fischbein, M.P.; Robbins, R.C.; Pelletier, M.P. Stem cell transplantation: The lung barrier. Transplant. Proc. 2007, 39, 573–576. [Google Scholar] [CrossRef] [PubMed]
- Santeramo, I.; Herrera Perez, Z.; Illera, A.; Taylor, A.; Kenny, S.; Murray, P.; Wilm, B.; Gretz, N. Human kidney-derived cells ameliorate acute kidney injury without engrafting into renal tissue. Stem Cells Transl. Med. 2017, 6, 1373–1384. [Google Scholar] [CrossRef]
- Leibacher, J.; Henschler, R. Biodistribution, migration and homing of systemically applied mesenchymal stem/stromal cells. Stem Cell Res. Ther. 2016, 7, 7. [Google Scholar] [CrossRef] [Green Version]
- Eggenhofer, E.; Benseler, V.; Kroemer, A.; Popp, F.C.; Geissler, E.K.; Schlitt, H.J.; Baan, C.C.; Dahlke, M.H.; Hoogduijn, M.J. Mesenchymal stem cells are short-lived and do not migrate beyond the lungs after intravenous infusion. Front. Immunol. 2012, 3, 297. [Google Scholar] [CrossRef] [Green Version]
- Fischer, U.M.; Harting, M.T.; Jimenez, F.; Monzon-Posadas, W.O.; Xue, H.; Savitz, S.I.; Laine, G.A.; Cox, C.S., Jr. Pulmonary passage is a major obstacle for intravenous stem cell delivery: The pulmonary first-pass effect. Stem Cells Dev. 2009, 18, 683–692. [Google Scholar] [CrossRef]
- Lou, G.; Chen, Z.; Zheng, M.; Liu, Y. Mesenchymal stem cell-derived exosomes as a new therapeutic strategy for liver diseases. Exp. Mol. Med. 2017, 49, e346. [Google Scholar] [CrossRef]
- Liu, D.D.; Ullah, M.; Concepcion, W.; Dahl, J.J.; Thakor, A.S. The role of ultrasound in enhancing mesenchymal stromal cell-based therapies. Stem Cells Transl. Med. 2020. [Google Scholar] [CrossRef] [Green Version]
- Ziadloo, A.; Burks, S.R.; Gold, E.M.; Lewis, B.K.; Chaudhry, A.; Merino, M.J.; Frenkel, V.; Frank, J.A. Enhanced homing permeability and retention of bone marrow stromal cells by noninvasive pulsed focused ultrasound. Stem Cells 2012, 30, 1216–1227. [Google Scholar] [CrossRef] [Green Version]
- Burks, S.R.; Nguyen, B.A.; Tebebi, P.A.; Kim, S.J.; Bresler, M.N.; Ziadloo, A.; Street, J.M.; Yuen, P.S.; Star, R.A.; Frank, J.A. Pulsed focused ultrasound pretreatment improves mesenchymal stromal cell efficacy in preventing and rescuing established acute kidney injury in mice. Stem Cells 2015, 33, 1241–1253. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burks, S.R.; Nagle, M.E.; Bresler, M.N.; Kim, S.J.; Star, R.A.; Frank, J.A. Mesenchymal stromal cell potency to treat acute kidney injury increased by ultrasound-activated interferon-gamma/interleukin-10 axis. J. Cell Mol. Med. 2018, 22, 6015–6025. [Google Scholar] [CrossRef] [PubMed]
- Ullah, M.; Liu, D.D.; Rai, S.; Dadhania, A.; Jonnakuti, S.; Concepcion, W.; Thakor, A.S. Reversing Acute Kidney Injury Using Pulsed Focused Ultrasound and MSC Therapy: A Role for HSP-Mediated PI3K/AKT Signaling. Mol. Ther. Methods Clin. Dev. 2020, 17, 683–694. [Google Scholar] [CrossRef] [PubMed]
- Martine, P.; Rebe, C. Heat Shock Proteins and Inflammasomes. Int. J. Mol. Sci. 2019, 20, 4508. [Google Scholar] [CrossRef] [Green Version]
- Mayor, A.; Martinon, F.; De Smedt, T.; Petrilli, V.; Tschopp, J. A crucial function of SGT1 and HSP90 in inflammasome activity links mammalian and plant innate immune responses. Nat. Immunol. 2007, 8, 497–503. [Google Scholar] [CrossRef]
- Piippo, N.; Korhonen, E.; Hytti, M.; Skottman, H.; Kinnunen, K.; Josifovska, N.; Petrovski, G.; Kaarniranta, K.; Kauppinen, A. Hsp90 inhibition as a means to inhibit activation of the NLRP3 inflammasome. Sci. Rep. 2018, 8, 6720. [Google Scholar] [CrossRef] [Green Version]
- Zuo, Y.; Wang, J.; Liao, F.; Yan, X.; Li, J.; Huang, L.; Liu, F. Inhibition of Heat Shock Protein 90 by 17-AAG Reduces Inflammation via P2X7 Receptor/NLRP3 Inflammasome Pathway and Increases Neurogenesis After Subarachnoid Hemorrhage in Mice. Front. Mol. Neurosci. 2018, 11, 401. [Google Scholar] [CrossRef] [Green Version]
- Asea, A.; Kraeft, S.K.; Kurt-Jones, E.A.; Stevenson, M.A.; Chen, L.B.; Finberg, R.W.; Koo, G.C.; Calderwood, S.K. HSP70 stimulates cytokine production through a CD14-dependant pathway, demonstrating its dual role as a chaperone and cytokine. Nat. Med. 2000, 6, 435–442. [Google Scholar] [CrossRef]
- Yombo, D.J.K.; Mentink-Kane, M.M.; Wilson, M.S.; Wynn, T.A.; Madala, S.K. Heat shock protein 70 is a positive regulator of airway inflammation and goblet cell hyperplasia in a mouse model of allergic airway inflammation. J. Biol. Chem. 2019, 294, 15082–15094. [Google Scholar] [CrossRef]
- Martine, P.; Chevriaux, A.; Derangere, V.; Apetoh, L.; Garrido, C.; Ghiringhelli, F.; Rebe, C. HSP70 is a negative regulator of NLRP3 inflammasome activation. Cell Death Dis. 2019, 10, 256. [Google Scholar] [CrossRef] [Green Version]
- Tempany, C.M.; McDannold, N.J.; Hynynen, K.; Jolesz, F.A. Focused ultrasound surgery in oncology: Overview and principles. Radiology 2011, 259, 39–56. [Google Scholar] [CrossRef]
- Guo, H.; Callaway, J.B.; Ting, J.P. Inflammasomes: Mechanism of action, role in disease, and therapeutics. Nat. Med. 2015, 21, 677–687. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, W.; Wang, X.; Chun, J.; Vilaysane, A.; Clark, S.; French, G.; Bracey, N.A.; Trpkov, K.; Bonni, S.; Duff, H.J.; et al. Inflammasome-independent NLRP3 augments TGF-beta signaling in kidney epithelium. J. Immunol. 2013, 190, 1239–1249. [Google Scholar] [CrossRef] [Green Version]
- Oh, J.Y.; Ko, J.H.; Lee, H.J.; Yu, J.M.; Choi, H.; Kim, M.K.; Wee, W.R.; Prockop, D.J. Mesenchymal stem/stromal cells inhibit the NLRP3 inflammasome by decreasing mitochondrial reactive oxygen species. Stem Cells 2014, 32, 1553–1563. [Google Scholar] [CrossRef]
- Miteva, K.; Pappritz, K.; Sosnowski, M.; El-Shafeey, M.; Muller, I.; Dong, F.; Savvatis, K.; Ringe, J.; Tschope, C.; Van Linthout, S. Mesenchymal stromal cells inhibit NLRP3 inflammasome activation in a model of Coxsackievirus B3-induced inflammatory cardiomyopathy. Sci. Rep. 2018, 8, 2820. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, S.; Wu, H.; Han, D.; Ma, S.; Fan, W.; Wang, Y.; Zhang, R.; Fan, M.; Huang, Y.; Fu, X.; et al. A Novel Mechanism of Mesenchymal Stromal Cell-Mediated Protection against Sepsis: Restricting Inflammasome Activation in Macrophages by Increasing Mitophagy and Decreasing Mitochondrial ROS. Oxidative Med. Cell Longev. 2018, 2018, 3537609. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Lou, G.; Li, A.; Zhang, T.; Qi, J.; Ye, D.; Zheng, M.; Chen, Z. AMSC-derived exosomes alleviate lipopolysaccharide/d-galactosamine-induced acute liver failure by miR-17-mediated reduction of TXNIP/NLRP3 inflammasome activation in macrophages. EBioMedicine 2018, 36, 140–150. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, L.; Lu, F.B.; Chen, D.Z.; Wu, J.L.; Hu, E.D.; Xu, L.M.; Zheng, M.H.; Li, H.; Huang, Y.; Jin, X.Y.; et al. BMSCs-derived miR-223-containing exosomes contribute to liver protection in experimental autoimmune hepatitis. Mol. Immunol. 2018, 93, 38–46. [Google Scholar] [CrossRef]
- Chatterjee, S.; Burns, T.F. Targeting Heat Shock Proteins in Cancer: A Promising Therapeutic Approach. Int. J. Mol. Sci. 2017, 18, 1978. [Google Scholar] [CrossRef] [Green Version]




© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ullah, M.; Liu, D.D.; Rai, S.; Concepcion, W.; Thakor, A.S. HSP70-Mediated NLRP3 Inflammasome Suppression Underlies Reversal of Acute Kidney Injury Following Extracellular Vesicle and Focused Ultrasound Combination Therapy. Int. J. Mol. Sci. 2020, 21, 4085. https://doi.org/10.3390/ijms21114085
Ullah M, Liu DD, Rai S, Concepcion W, Thakor AS. HSP70-Mediated NLRP3 Inflammasome Suppression Underlies Reversal of Acute Kidney Injury Following Extracellular Vesicle and Focused Ultrasound Combination Therapy. International Journal of Molecular Sciences. 2020; 21(11):4085. https://doi.org/10.3390/ijms21114085
Chicago/Turabian StyleUllah, Mujib, Daniel D. Liu, Sravanthi Rai, Waldo Concepcion, and Avnesh S. Thakor. 2020. "HSP70-Mediated NLRP3 Inflammasome Suppression Underlies Reversal of Acute Kidney Injury Following Extracellular Vesicle and Focused Ultrasound Combination Therapy" International Journal of Molecular Sciences 21, no. 11: 4085. https://doi.org/10.3390/ijms21114085