Transcriptional Activity and Stability of CD39+CD103+CD8+ T Cells in Human High-Grade Endometrial Cancer

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
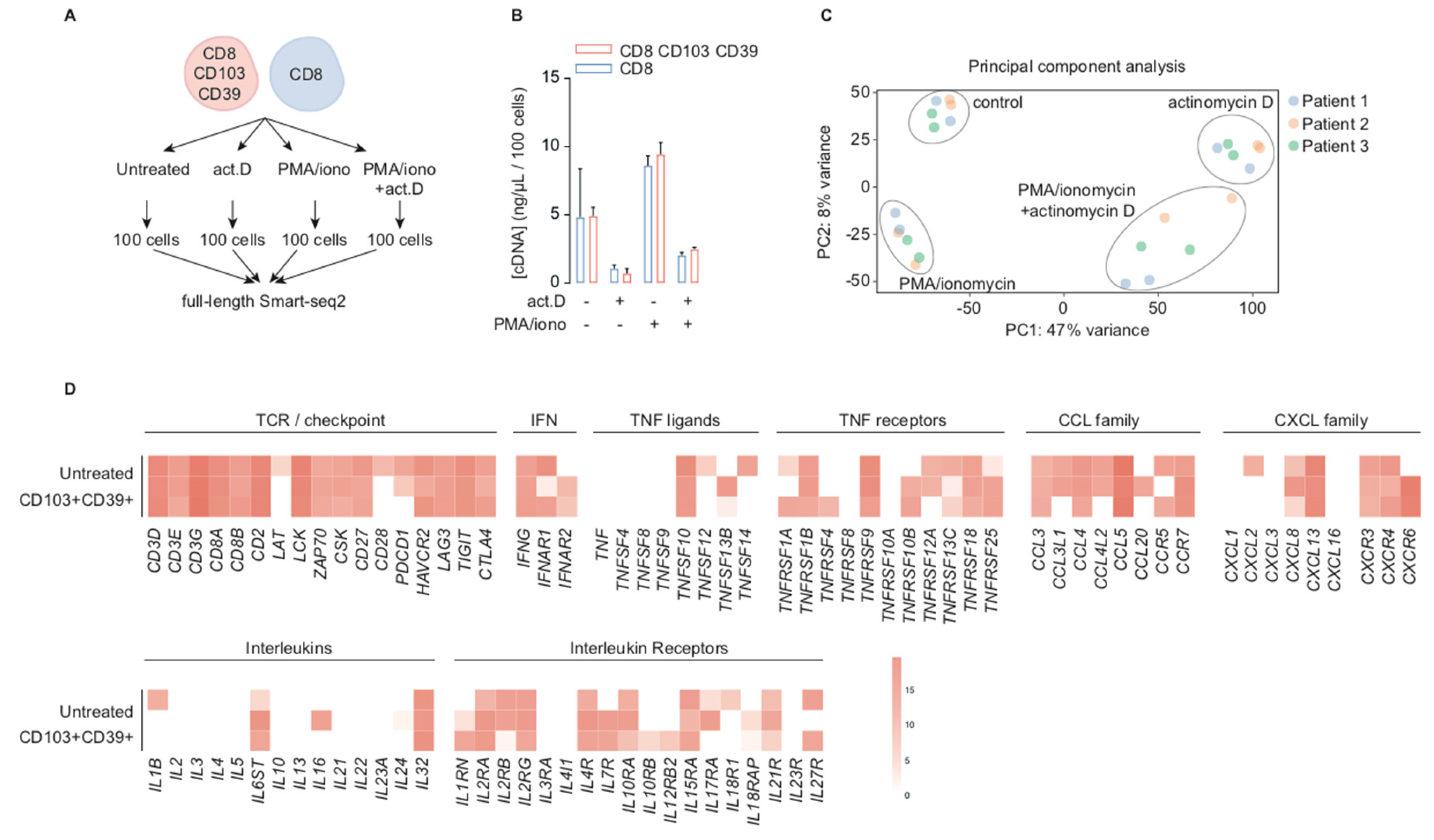
2.1. CD39+CD103+ TRM and CD8+ TIL are Transcriptionally Active in Situ
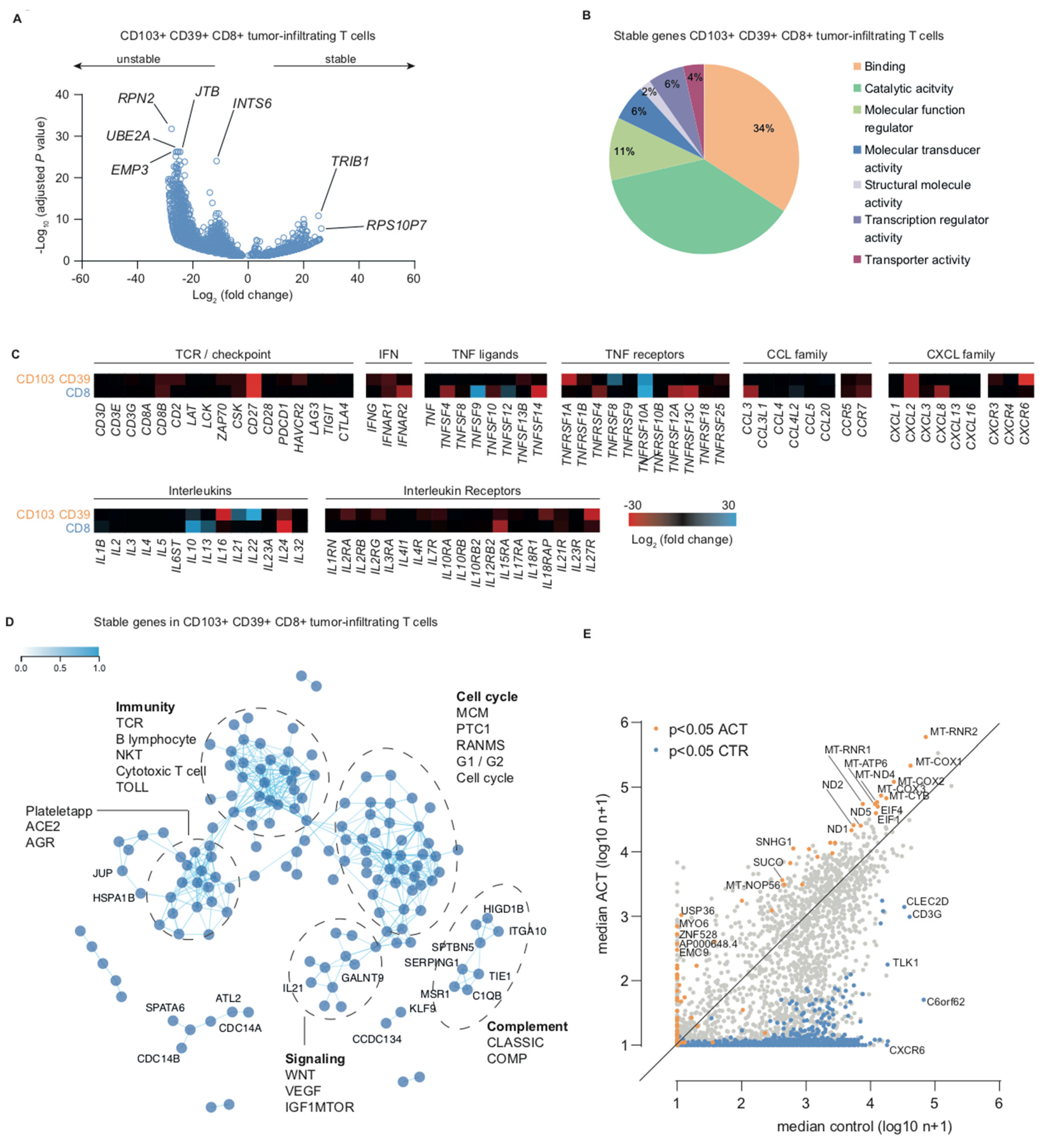
2.2. Transcription Inhibition by Actinomycin D Reveals Distinct Differences in Transcript Stability
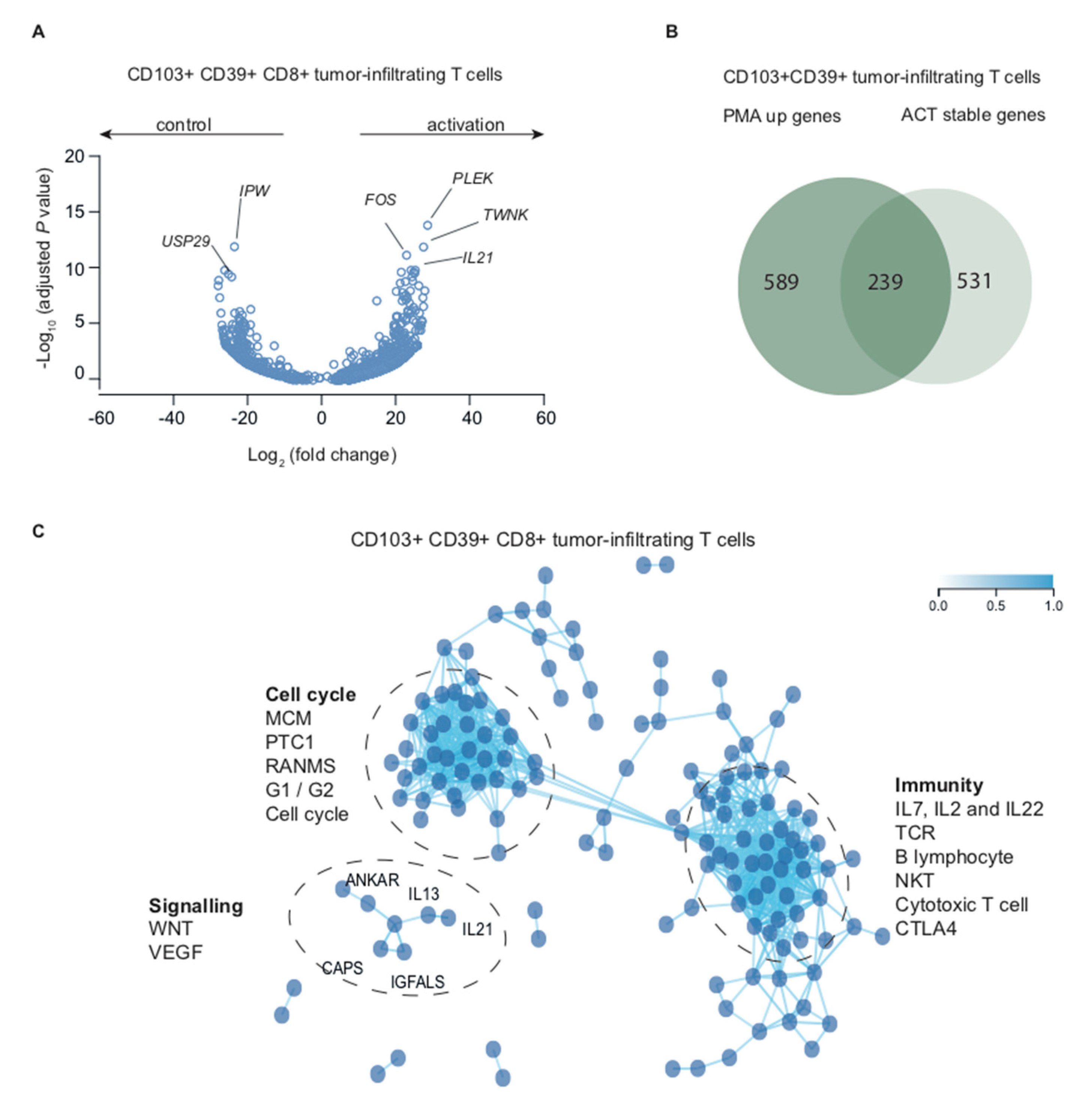
2.3. Stimulation with PMA/Ionomycin Induces an Activation-Responsive Genotype in Tumor-Infiltrating Lymphocytes
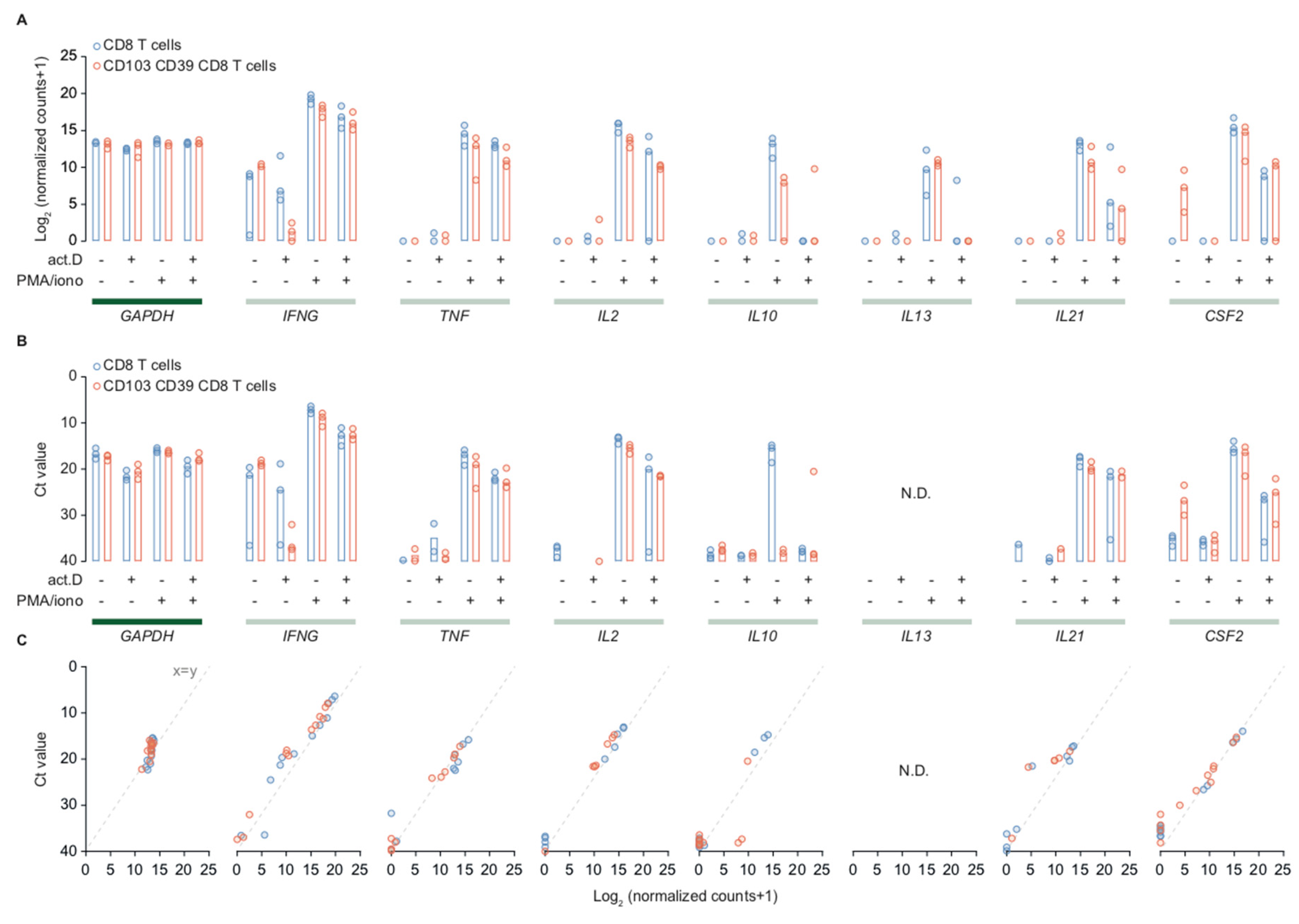
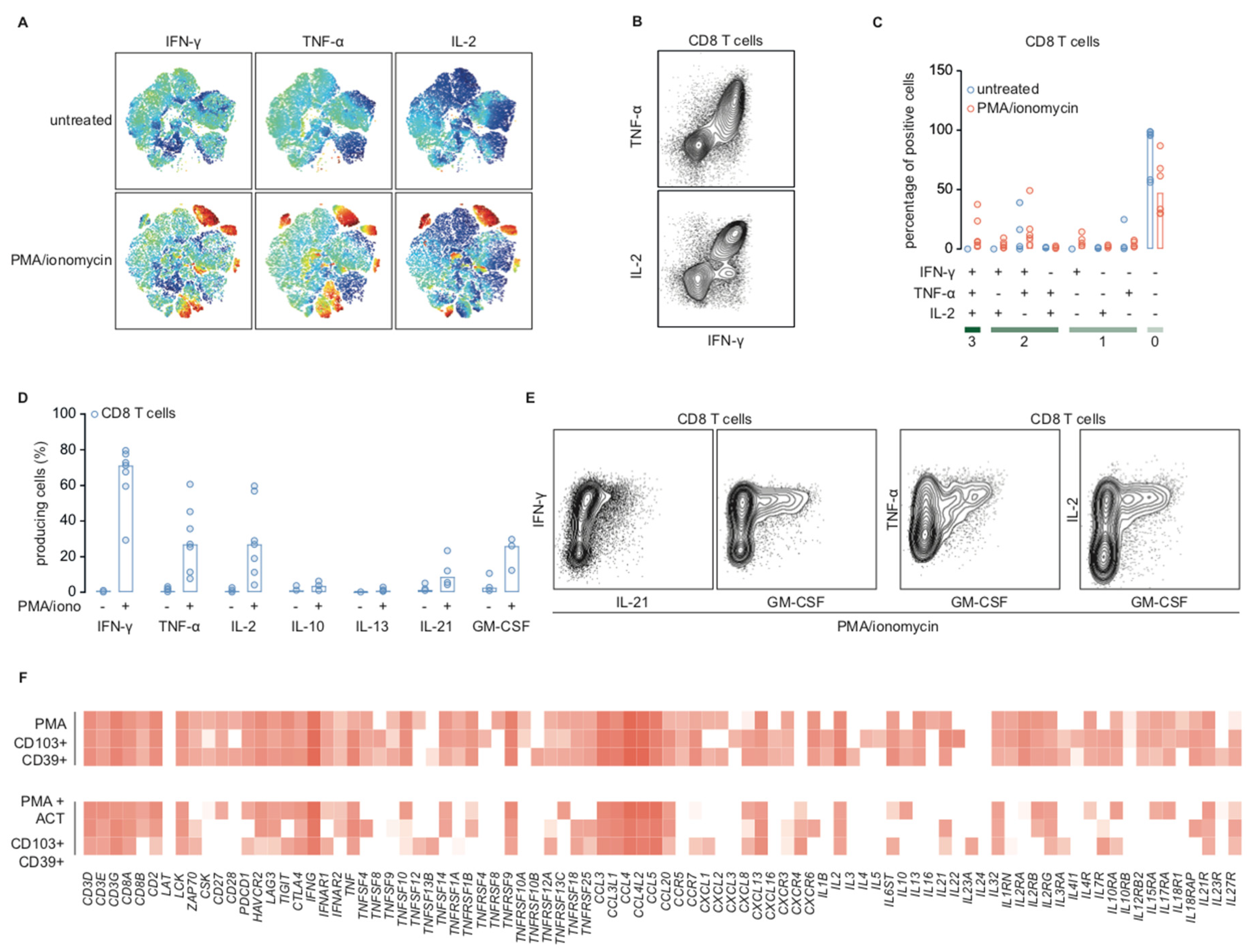
2.4. Cytotoxic T Cells in Endometrial Cancer are Polyfunctional T Cells that can Produce IL-21 and GM-CSF
2.5. TRM-Genes and PMA-Responsive Genes are Associated with Response to Immune Checkpoint Blockade in Publically Available Sequencing Data
3. Discussion
4. Materials and Methods
4.1. Tumor Material Preparation
4.2. Isolation of PBMCs from Healthy Volunteers
4.3. CD39+CD103+CD8+ and CD8+ T Cell Sorting
4.4. mRNA Sequencing CD39+CD103+CD8+ and CD8+ Tumor-Infiltrating Lymphocytes
4.5. qPCR of mRNA Sequencing Samples and Peripheral Blood CD8+ T Cells
4.6. Flow Cytometry
4.7. Publically Available Sequencing Data
4.8. Statistics
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| ACT | Actinomycin D |
| CD | Cluster of Differentiation |
| EC | Endometrial cancer |
| FCS | Fetal calf serum |
| FIGO | International Federation of Gynecology and Obstetrics |
| GM-CSF | Granulocyte Macrophage – Colony Stimulating Factor |
| ICB | Immune checkpoint blockade |
| IL | Interleukin |
| MMR | Mismatch repair |
| - dMMR | Mismatch repair deficient |
| - pMMR | Mismatch repair proficient |
| MSI | Microsatellite instability |
| MSS | Microsatellite stability |
| ND | Not determined |
| PBMC | Peripheral blood mononuclear cells |
| PD | Progressive disease |
| PRCR | Partial response/complete response |
| PMA | Phorbol myristate acetate |
| POLE | Polymerase epsilon |
| SD | Stable disease |
| TIL | Tumor-infiltrating lymphocytes |
| TRM | Tissue-resident memory cell |
| tSNE | t-Stochastic neighbor embedding |
| UNK | Unkown |
| UTR | Untranslated region |
References
- Barnes, T.A.; Amir, E. HYPE or HOPE: The prognostic value of infiltrating immune cells in cancer. Br. J. Cancer 2017. [Google Scholar] [CrossRef] [Green Version]
- Franciszkiewicz, K.; Le Floc’H, A.; Boutet, M.; Vergnon, I.; Schmitt, A.; Mami-Chouaib, F. CD103 or LFA-1 engagement at the immune synapse between cytotoxic T cells and tumor cells promotes maturation and regulates T-cell effector functions. Cancer Res. 2013, 73, 617–628. [Google Scholar] [CrossRef] [Green Version]
- Le Floc’h, A.; Jalil, A.; Franciszkiewicz, K.; Validire, P.; Vergnon, I.; Mami-Chouaib, F. Minimal engagement of CD103 on cytotoxic T lymphocytes with an E-cadherin-Fc molecule triggers lytic granule polarization via a phospholipase Cγ-dependent pathway. Cancer Res. 2011, 71, 328–338. [Google Scholar] [CrossRef] [Green Version]
- Komdeur, F.L.; Prins, T.M.; van de Wall, S.; Plat, A.; Wisman, G.B.A.; Hollema, H.; Daemen, T.; Church, D.N.; de Bruyn, M.; Nijman, H.W. CD103+ tumor-infiltrating lymphocytes are tumor-reactive intraepithelial CD8+ T cells associated with prognostic benefit and therapy response in cervical cancer. Oncoimmunology 2017. [Google Scholar] [CrossRef]
- Mokrani, M.; Klibi, J.; Bluteau, D.; Bismuth, G.; Mami-Chouaib, F. Smad and NFAT Pathways Cooperate To Induce CD103 Expression in Human CD8 T Lymphocytes. J. Immunol. 2014, 192, 2471–2479. [Google Scholar] [CrossRef] [Green Version]
- Komdeur, F.L.; Wouters, M.C.A.; Workel, H.H.; Tijans, A.M.; Terwindt, A.L.J.; Brunekreeft, K.L.; Plat, A.; Klip, H.G.; Eggink, F.A.; Leffers, N.; et al. CD103+ intraepithelial T cells in high-grade serous ovarian cancer are phenotypically diverse TCRαβ+ CD8αβ+ T cells that can be targeted for cancer immunotherapy. Oncotarget 2016. [Google Scholar] [CrossRef] [Green Version]
- Franciszkiewicz, K.; Le Floc’h, A.; Jalil, A.; Vigant, F.; Robert, T.; Vergnon, I.; Mackiewicz, A.; Benihoud, K.; Validire, P.; Chouaib, S.; et al. Intratumoral induction of CD103 triggers tumor-specific CTL function and CCR5-dependent T-cell retention. Cancer Res. 2009, 69, 6249–6255. [Google Scholar] [CrossRef] [Green Version]
- Abd Hamid, M.; Colin-York, H.; Khalid-Alham, N.; Browne, M.; Cerundolo, L.; Chen, J.L.; Yao, X.; Rosendo-Machado, S.; Waugh, C.; Maldonado-Perez, D.; et al. Self-Maintaining CD103+ Cancer-Specific T Cells Are Highly Energetic with Rapid Cytotoxic and Effector Responses. Cancer Immunol. Res. 2020. [Google Scholar] [CrossRef] [Green Version]
- Ganesan, A.-P.; Clarke, J.; Wood, O.; Garrido-Martin, E.M.; Chee, S.J.; Mellows, T.; Samaniego-Castruita, D.; Singh, D.; Seumois, G.; Alzetani, A.; et al. Tissue-resident memory features are linked to the magnitude of cytotoxic T cell responses in human lung cancer. Nat. Immunol. 2017, 18, 940–950. [Google Scholar] [CrossRef]
- Wang, B.; Wu, S.; Zeng, H.; Liu, Z.; Dong, W.; He, W.; Chen, X.; Dong, X.; Zheng, L.; Lin, T.; et al. CD103+ Tumor Infiltrating Lymphocytes Predict a Favorable Prognosis in Urothelial Cell Carcinoma of the Bladder. J. Urol. 2015, 194, 556–562. [Google Scholar] [CrossRef]
- Djenidi, F.; Adam, J.; Goubar, A.; Durgeau, A.; Meurice, G.; de Montpreville, V.; Validire, P.; Besse, B.; Mami-Chouaib, F. CD8+CD103+ Tumor-Infiltrating Lymphocytes Are Tumor-Specific Tissue-Resident Memory T Cells and a Prognostic Factor for Survival in Lung Cancer Patients. J. Immunol. 2015, 194, 3475–3486. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Webb, J.R.; Milne, K.; Watson, P.; DeLeeuw, R.J.; Nelson, B.H. Tumor-infiltrating lymphocytes expressing the tissue resident memory marker cd103 are associated with increased survival in high-grade serous ovarian cancer. Clin. Cancer Res. 2014, 20, 434–444. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Workel, H.H.; Komdeur, F.L.; Wouters, M.C.A.; Plat, A.; Klip, H.G.; Eggink, F.A.; Wisman, G.B.A.; Arts, H.J.G.; Oonk, M.H.M.; Mourits, M.J.E.; et al. CD103 defines intraepithelial CD8+ PD1+ tumour-infiltrating lymphocytes of prognostic significance in endometrial adenocarcinoma. Eur. J. Cancer 2016, 60, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Soslow, R.A.; Tornos, C.; Park, K.J.; Malpica, A.; Matias-Guiu, X.; Oliva, E.; Parkash, V.; Carlson, J.; Glenn McCluggage, W.; Blake Gilks, C. Endometrial Carcinoma Diagnosis: Use of FIGO Grading and Genomic Subcategories in Clinical Practice: Recommendations of the International Society of Gynecological Pathologists. Int. J. Gynecol. Pathol. 2019. [Google Scholar] [CrossRef] [Green Version]
- McAlpine, J.; Leon-Castillo, A.; Bosse, T. The rise of a novel classification system for endometrial carcinoma; integration of molecular subclasses. J. Pathol. 2018. [Google Scholar] [CrossRef]
- Eggink, F.A.; Van Gool, I.C.; Leary, A.; Pollock, P.M.; Crosbie, E.J.; Mileshkin, L.; Jordanova, E.S.; Adam, J.; Freeman-Mills, L.; Church, D.N.; et al. Immunological profiling of molecularly classified high-risk endometrial cancers identifies POLE-mutant and microsatellite unstable carcinomas as candidates for checkpoint inhibition. Oncoimmunology 2016, 6, e1264565. [Google Scholar] [CrossRef] [Green Version]
- Ramchander, N.C.; Ryan, N.A.J.; Walker, T.D.J.; Harries, L.; Bolton, J.; Bosse, T.; Evans, D.G.; Crosbie, E.J. Distinct Immunological Landscapes Characterize Inherited and Sporadic Mismatch Repair Deficient Endometrial Cancer. Front. Immunol. 2020. [Google Scholar] [CrossRef] [Green Version]
- Workel, H.H.; Lubbers, J.M.; Arnold, R.; Prins, T.M.; van der Vlies, P.; de Lange, K.; Bosse, T.; van Gool, I.C.; Eggink, F.A.; Wouters, M.C.A.; et al. A Transcriptionally Distinct CXCL13+CD103+CD8+ T-cell Population Is Associated with B-cell Recruitment and Neoantigen Load in Human Cancer. Cancer Immunol. Res. 2019. [Google Scholar] [CrossRef] [Green Version]
- Clarke, J.; Panwar, B.; Madrigal, A.; Singh, D.; Gujar, R.; Wood, O.; Chee, S.J.; Eschweiler, S.; King, E.V.; Awad, A.S.; et al. Single-cell transcriptomic analysis of tissue-resident memory T cells in human lung cancer. J. Exp. Med. 2019. [Google Scholar] [CrossRef]
- Duhen, T.; Duhen, R.; Montler, R.; Moses, J.; Moudgil, T.; De Miranda, N.F.; Goodall, C.P.; Blair, T.C.; Fox, B.A.; McDermott, J.E.; et al. Co-expression of CD39 and CD103 identifies tumor-reactive CD8 T cells in human solid tumors. Nat. Commun. 2018. [Google Scholar] [CrossRef] [Green Version]
- Simoni, Y.; Becht, E.; Fehlings, M.; Loh, C.Y.; Koo, S.L.; Teng, K.W.W.; Yeong, J.P.S.; Nahar, R.; Zhang, T.; Kared, H.; et al. Bystander CD8+ T cells are abundant and phenotypically distinct in human tumour infiltrates. Nature 2018. [Google Scholar] [CrossRef]
- Raczkowski, F.; Rissiek, A.; Ricklefs, I.; Heiss, K.; Schumacher, V.; Wundenberg, K.; Haag, F.; Koch-Nolte, F.; Tolosa, E.; Mittrücker, H.W. Cd39 is upregulated during activation of mouse and human t cells and attenuates the immune response to listeria monocytogenes. PLoS ONE 2018. [Google Scholar] [CrossRef]
- Canale, F.P.; Ramello, M.C.; Núñez, N.; Furlan, C.L.A.; Bossio, S.N.; Serrán, M.G.; Boari, J.T.; Del Castillo, A.; Ledesma, M.; Sedlik, C.; et al. CD39 expression defines cell exhaustion in tumor-infiltrating CD8+ T cells. Cancer Res. 2018. [Google Scholar] [CrossRef] [Green Version]
- Takenaka, M.C.; Robson, S.; Quintana, F.J. Regulation of the T Cell Response by CD39. Trends Immunol. 2016. [Google Scholar] [CrossRef] [Green Version]
- Allard, B.; Longhi, M.S.; Robson, S.C.; Stagg, J. The ectonucleotidases CD39 and CD73: Novel checkpoint inhibitor targets. Immunol. Rev. 2017. [Google Scholar] [CrossRef] [Green Version]
- Savas, P.; Virassamy, B.; Ye, C.; Salim, A.; Mintoff, C.P.; Caramia, F.; Salgado, R.; Byrne, D.J.; Teo, Z.L.; Dushyanthen, S.; et al. Single-cell profiling of breast cancer T cells reveals a tissue-resident memory subset associated with improved prognosis. Nat. Med. 2018. [Google Scholar] [CrossRef]
- Park, S.L.; Buzzai, A.; Rautela, J.; Hor, J.L.; Hochheiser, K.; Effern, M.; McBain, N.; Wagner, T.; Edwards, J.; McConville, R.; et al. Tissue-resident memory CD8+ T cells promote melanoma–immune equilibrium in skin. Nature 2019. [Google Scholar] [CrossRef]
- Edwards, J.; Wilmott, J.S.; Madore, J.; Gide, T.N.; Quek, C.; Tasker, A.; Ferguson, A.; Chen, J.; Hewavisenti, R.; Hersey, P.; et al. CD103+ tumor-resident CD8+ T cells are associated with improved survival in immunotherapy-naïve melanoma patients and expand significantly during anti-PD-1 treatment. Clin. Cancer Res. 2018. [Google Scholar] [CrossRef] [Green Version]
- Bhattacharya, A.; Bense, R.D.; Urzúa-Traslaviña, C.G.; de Vries, E.G.E.; van Vugt, M.A.T.M.; Fehrmann, R.S.N. Transcriptional effects of copy number alterations in a large set of human cancers. Nat. Commun. 2020. [Google Scholar] [CrossRef]
- Riaz, N.; Havel, J.J.; Makarov, V.; Desrichard, A.; Urba, W.J.; Sims, J.S.; Hodi, F.S.; Martín-Algarra, S.; Mandal, R.; Sharfman, W.H.; et al. Tumor and Microenvironment Evolution during Immunotherapy with Nivolumab. Cell 2017. [Google Scholar] [CrossRef] [Green Version]
- Willinger, T.; Freeman, T.; Hasegawa, H.; McMichael, A.J.; Callan, M.F.C. Molecular Signatures Distinguish Human Central Memory from Effector Memory CD8 T Cell Subsets. J. Immunol. 2005. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, X.; Zhang, Y.; Zheng, L.; Zheng, C.; Song, J.; Zhang, Q.; Kang, B.; Liu, Z.; Jin, L.; Xing, R.; et al. Global characterization of T cells in non-small-cell lung cancer by single-cell sequencing. Nat. Med. 2018. [Google Scholar] [CrossRef]
- Kim, Y.; Shin, Y.; Kang, G.H. Prognostic significance of CD103+ immune cells in solid tumor: A systemic review and meta-analysis. Sci. Rep. 2019. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hugo, W.; Zaretsky, J.M.; Sun, L.; Song, C.; Moreno, B.H.; Hu-Lieskovan, S.; Berent-Maoz, B.; Pang, J.; Chmielowski, B.; Cherry, G.; et al. Genomic and Transcriptomic Features of Response to Anti-PD-1 Therapy in Metastatic Melanoma. Cell 2016. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shields, B.D.; Koss, B.; Taylor, E.M.; Storey, A.J.; West, K.L.; Byrum, S.D.; Mackintosh, S.G.; Edmondson, R.; Mahmoud, F.; Shalin, S.C.; et al. Loss of E-cadherin inhibits CD103 antitumor activity and reduces checkpoint blockade responsiveness in melanoma. Cancer Res. 2019. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Szabo, P.A.; Levitin, H.M.; Miron, M.; Snyder, M.E.; Senda, T.; Yuan, J.; Cheng, Y.L.; Bush, E.C.; Dogra, P.; Thapa, P.; et al. Single-cell transcriptomics of human T cells reveals tissue and activation signatures in health and disease. Nat. Commun. 2019. [Google Scholar] [CrossRef]
- Chen, Y.; Yu, M.; Zheng, Y.; Fu, G.; Xin, G.; Zhu, W.; Luo, L.; Burns, R.; Li, Q.Z.; Dent, A.L.; et al. CXCR5+PD-1+ follicular helper CD8 T cells control B cell tolerance. Nat. Commun. 2019. [Google Scholar] [CrossRef] [Green Version]
- Casey, K.A.; Mescher, M.F. IL-21 Promotes Differentiation of Naive CD8 T Cells to a Unique Effector Phenotype. J. Immunol. 2007. [Google Scholar] [CrossRef]
- Novy, P.; Huang, X.; Leonard, W.J.; Yang, Y. Intrinsic IL-21 Signaling Is Critical for CD8 T Cell Survival and Memory Formation in Response to Vaccinia Viral Infection. J. Immunol. 2011. [Google Scholar] [CrossRef] [Green Version]
- Hinrichs, C.S.; Spolski, R.; Paulos, C.M.; Gattinoni, L.; Kerstann, K.W.; Palmer, D.C.; Klebanoff, C.A.; Rosenberg, S.A.; Leonard, W.J.; Restifo, N.P. IL-2 and IL-21 confer opposing differentiation programs to CD8+ T cells for adoptive immunotherapy. Blood 2008. [Google Scholar] [CrossRef] [Green Version]
- Xiao, L.; Jia, L.; Bai, L.; He, L.; Yang, B.; Wu, C.; Li, H. Phenotypic and functional characteristics of IL-21-expressing CD8+ T cells in human nasal polyps. Sci. Rep. 2016. [Google Scholar] [CrossRef] [Green Version]
- Wei, X.X.; Chan, S.; Kwek, S.; Lewis, J.; Dao, V.; Zhang, L.; Cooperberg, M.R.; Ryan, C.J.; Lin, A.M.; Friedlander, T.W.; et al. Systemic GM-CSF recruits effector T cells into the tumor microenvironment in localized prostate cancer. Cancer Immunol. Res. 2016. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, J.; López-Moyado, I.F.; Seo, H.; Lio, C.W.J.; Hempleman, L.J.; Sekiya, T.; Yoshimura, A.; Scott-Browne, J.P.; Rao, A. NR4A transcription factors limit CAR T cell function in solid tumours. Nature 2019. [Google Scholar] [CrossRef] [PubMed]
- Shehata, H.M.; Khan, S.; Chen, E.; Fields, P.E.; Flavell, R.A.; Sanjabi, S. Lack of Sprouty 1 and 2 enhances survival of effector CD8+ T cells and yields more protective memory cells. Proc. Natl. Acad. Sci. USA 2018. [Google Scholar] [CrossRef] [Green Version]
- Collins, S.; Waickman, A.; Basson, A.; Kupfer, A.; Licht, J.D.; Horton, M.R.; Powell, J.D. Regulation of CD4+ and CD8+ Effector Responses by Sprouty-1. PLoS ONE 2012. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Orlandini von Niessen, A.G.; Poleganov, M.A.; Rechner, C.; Plaschke, A.; Kranz, L.M.; Fesser, S.; Diken, M.; Löwer, M.; Vallazza, B.; Beissert, T.; et al. Improving mRNA-Based Therapeutic Gene Delivery by Expression-Augmenting 3′ UTRs Identified by Cellular Library Screening. Mol. Ther. 2019. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ahmed, A.; Thompson, J.; Emiliusen, L.; Murphy, S.; Beauchamp, R.D.; Suzuki, K.; Alemany, R.; Harrington, K.; Vile, R.G. A conditionally replicating adenovirus targeted to tumor cells through activated RAS/P-MAPK-selective mRNA stabilization. Nat. Biotechnol. 2003. [Google Scholar] [CrossRef]
- McNamara, M.A.; Nair, S.K.; Holl, E.K. RNA-Based Vaccines in Cancer Immunotherapy. J. Immunol. Res. 2015. [Google Scholar] [CrossRef] [Green Version]
- Van Hoecke, L.; Roose, K. How mRNA therapeutics are entering the monoclonal antibody field. J. Transl. Med. 2019. [Google Scholar] [CrossRef] [Green Version]
- Sahin, U.; Türeci, Ö. Personalized vaccines for cancer immunotherapy. Science 2018. [Google Scholar] [CrossRef] [Green Version]
- Lo, Y.S.; Tseng, W.H.; Chuang, C.Y.; Hou, M.H. The structural basis of actinomycin D-binding induces nucleotide flipping out, a sharp bend and a left-handed twist in CGG triplet repeats. Nucleic Acids Res. 2013. [Google Scholar] [CrossRef]
- Bensaude, O. Inhibiting eukaryotic transcription. Transcription 2011. [Google Scholar] [CrossRef] [Green Version]
- Perry, R.P.; Kelley, D.E. Inhibition of RNA synthesis by actinomycin D: Characteristic dose-response of different RNA species. J. Cell. Physiol. 1970. [Google Scholar] [CrossRef]
- Yan, F.; Liu, J.J.; Ip, V.; Jamieson, S.M.F.; McKeage, M.J. Role of platinum DNA damage-induced transcriptional inhibition in chemotherapy-induced neuronal atrophy and peripheral neurotoxicity. J. Neurochem. 2015. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Todd, R.C.; Lippard, S.J. Inhibition of transcription by platinum antitumor compounds. Metallomics 2009. [Google Scholar] [CrossRef] [Green Version]
- Hato, S.V.; Khong, A.; De Vries, I.J.M.; Lesterhuis, W.J. Molecular pathways: The immunogenic effects of platinum-based chemotherapeutics. Clin. Cancer Res. 2014. [Google Scholar] [CrossRef] [Green Version]
- Brunekreeft, K.L.; Paijens, S.T.; Wouters, M.C.A.; Komdeur, F.L.; Eggink, F.A.; Lubbers, J.M.; Workel, H.H.; Van Der Slikke, E.C.; Pröpper, N.E.J.; Leffers, N.; et al. Deep immune profiling of ovarian tumors identifies minimal MHC-I expression after neoadjuvant chemotherapy as negatively associated with T-cell-dependent outcome. Oncoimmunology 2020, 9, 1760705. [Google Scholar] [CrossRef]
- Böhm, S.; Montfort, A.; Pearce, O.M.T.; Topping, J.; Chakravarty, P.; Everitt, G.L.A.; Clear, A.; McDermott, J.R.; Ennis, D.; Dowe, T.; et al. Neoadjuvant chemotherapy modulates the immune microenvironment in metastases of tubo-ovarian high-grade serous carcinoma. Clin. Cancer Res. 2016. [Google Scholar] [CrossRef] [Green Version]
- Lo, C.S.; Sanii, S.; Kroeger, D.R.; Milne, K.; Talhouk, A.; Chiu, D.S.; Rahimi, K.; Shaw, P.A.; Clarke, B.A.; Nelson, B.H. Neoadjuvant chemotherapy of ovarian cancer results in three patterns of tumor-infiltrating lymphocyte response with distinct implications for immunotherapy. Clin. Cancer Res. 2017. [Google Scholar] [CrossRef] [Green Version]
- Pölcher, M.; Braun, M.; Friedrichs, N.; Rudlowski, C.; Bercht, E.; Fimmers, R.; Sauerwald, A.; Keyver-Paik, M.D.; Kübler, K.; Büttner, R.; et al. Foxp3+ cell infiltration and granzyme B+/Foxp3 + cell ratio are associated with outcome in neoadjuvant chemotherapy-treated ovarian carcinoma. Cancer Immunol. Immunother. 2010. [Google Scholar] [CrossRef]
- Khairallah, A.S.; Genestie, C.; Auguste, A.; Leary, A. Impact of neoadjuvant chemotherapy on the immune microenvironment in advanced epithelial ovarian cancer: Prognostic and therapeutic implications. Int. J. Cancer 2018. [Google Scholar] [CrossRef] [PubMed] [Green Version]





© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Workel, H.H.; van Rooij, N.; Plat, A.; Spierings, D.C.J.; Fehrmann, R.S.N.; Nijman, H.W.; de Bruyn, M. Transcriptional Activity and Stability of CD39+CD103+CD8+ T Cells in Human High-Grade Endometrial Cancer. Int. J. Mol. Sci. 2020, 21, 3770. https://doi.org/10.3390/ijms21113770
Workel HH, van Rooij N, Plat A, Spierings DCJ, Fehrmann RSN, Nijman HW, de Bruyn M. Transcriptional Activity and Stability of CD39+CD103+CD8+ T Cells in Human High-Grade Endometrial Cancer. International Journal of Molecular Sciences. 2020; 21(11):3770. https://doi.org/10.3390/ijms21113770
Chicago/Turabian StyleWorkel, Hagma H., Nienke van Rooij, Annechien Plat, Diana C.J. Spierings, Rudolf S. N. Fehrmann, Hans W. Nijman, and Marco de Bruyn. 2020. "Transcriptional Activity and Stability of CD39+CD103+CD8+ T Cells in Human High-Grade Endometrial Cancer" International Journal of Molecular Sciences 21, no. 11: 3770. https://doi.org/10.3390/ijms21113770