Development of a Radiofluorinated Adenosine A2B Receptor Antagonist as Potential Ligand for PET Imaging

 , , ,
, , ,  ,
,  , , and
, , and
Abstract
:1. Introduction
2. Results and Discussion
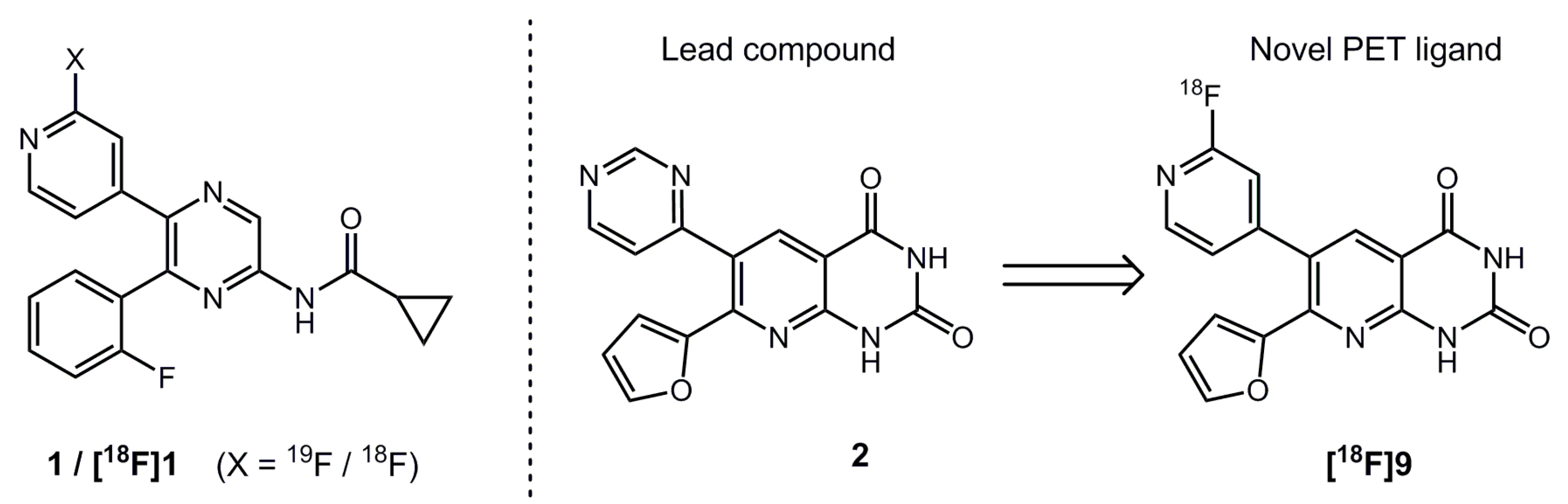
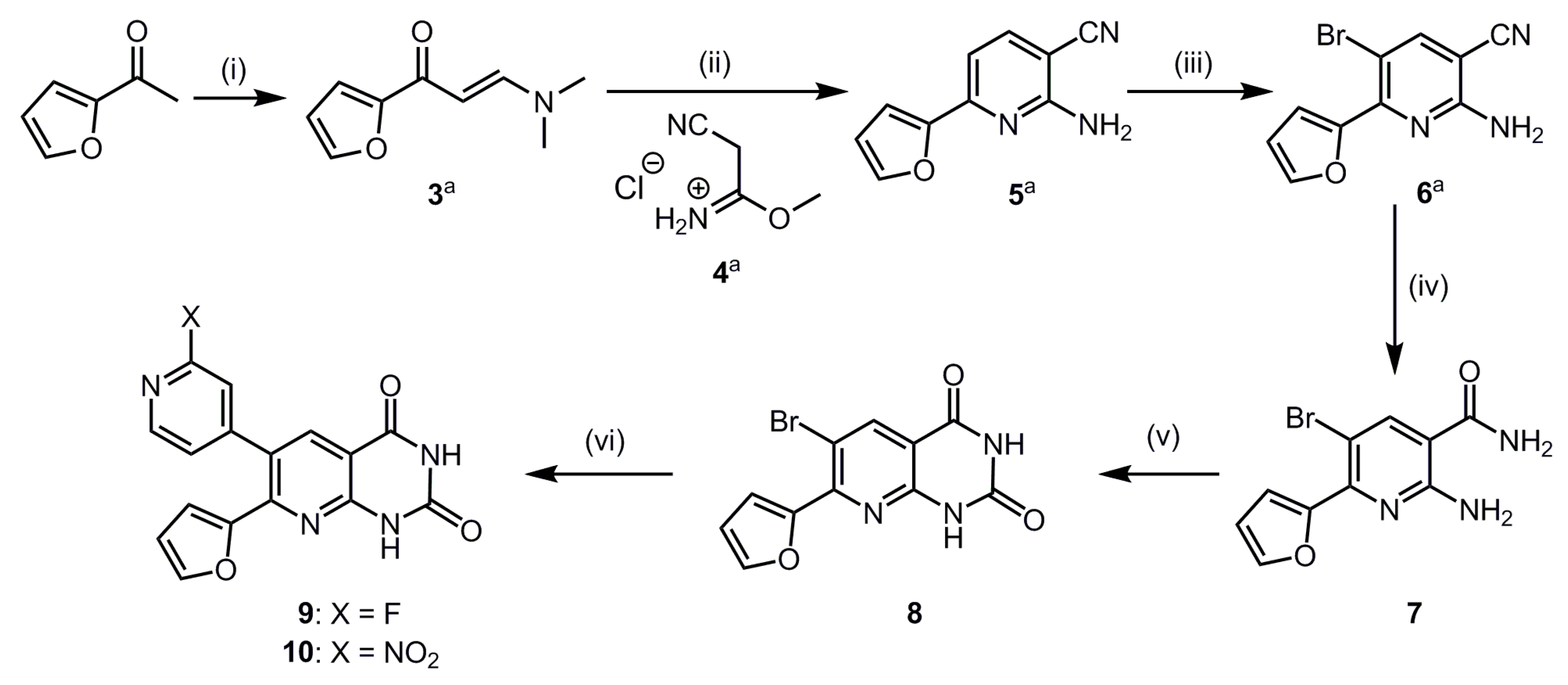
2.1. Organic Synthesis and In Vitro Binding Studies
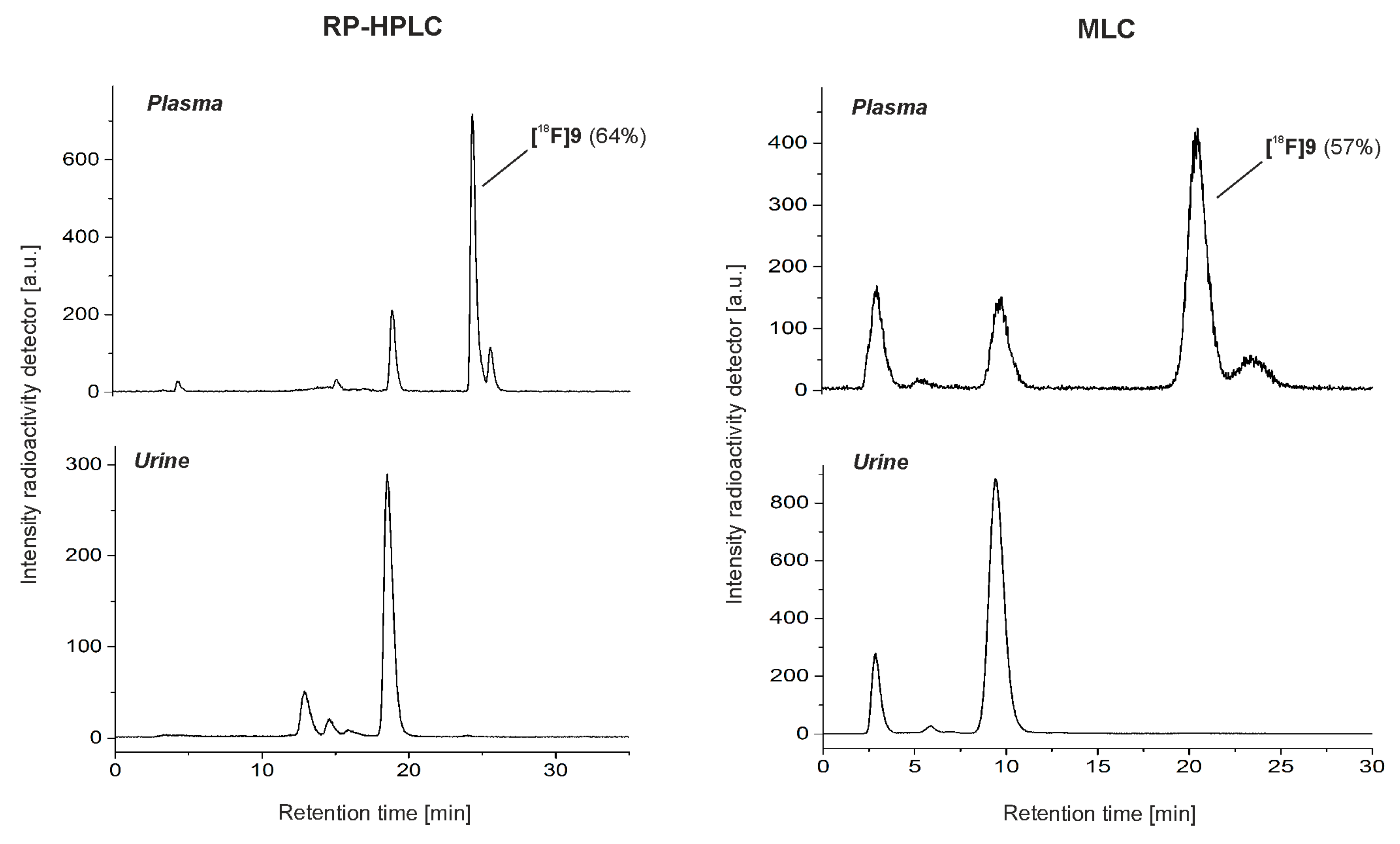
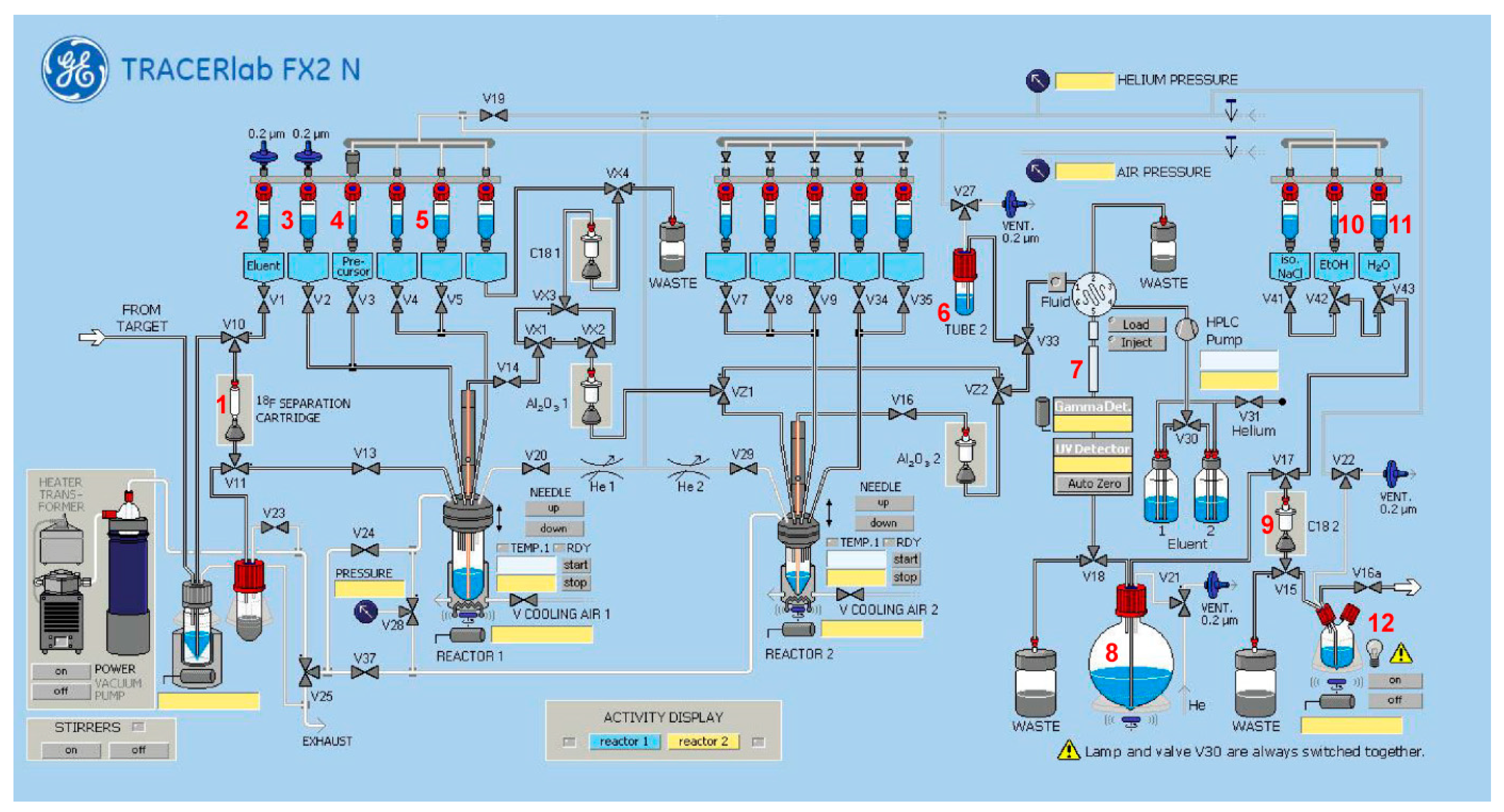
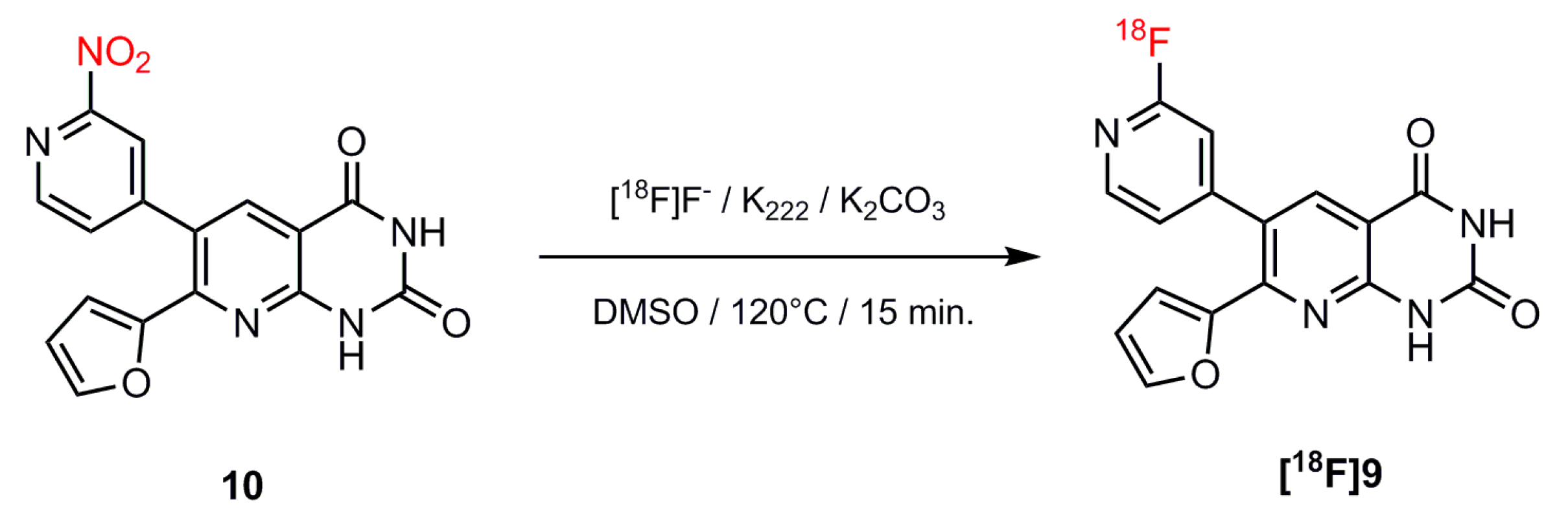
2.2. Automated Radiosynthesis and Characterization of [18F]9
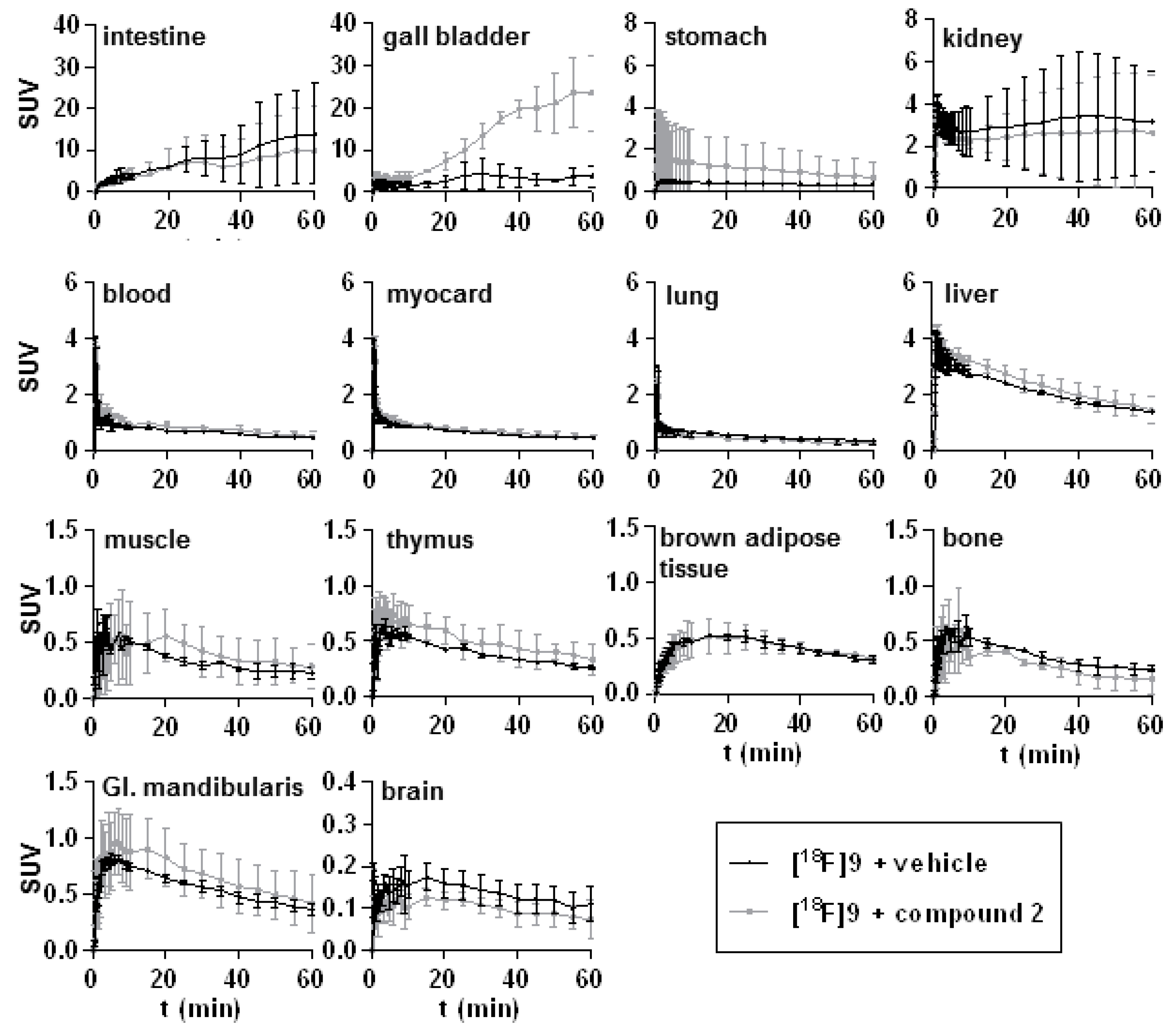
2.3. In Vivo Studies of [18F]9 in Mice
3. Materials and Methods
3.1. Organic Chemistry
3.1.1. General
3.1.2. 7-(Furan-2-yl)-6-(pyrimidin-4-yl)pyrido[2,3-d]pyrimidine-2,4-(1H,3H)-dione (2)
3.1.3. Compounds 3–6
3.1.4. 2-Amino-5-bromo-6-(furan-2-yl)nicotinamide (7)
3.1.5. 6-Bromo-7-(furan-2-yl)pyrido[2,3-d]pyrimidine-2,4(1H,3H)-dione (8)
3.1.6. 6-(2-Fluoropyridin-4-yl)-7-(furan-2-yl)pyrido[2,3-d]pyrimidine-2,4(1H,3H)-dione (9)
3.1.7. 7-(Furan-2-yl)-6-(2-nitropyridin-4-yl)pyrido[2,3-d]pyrimidine-2,4(1H,3H)-dione (10)
3.2. In Vitro Radioligand Binding Experiments
3.3. Radiochemistry
3.3.1. General
3.3.2. Radiosynthesis
3.4. In Vivo Studies in Mice
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Fredholm, B.B.; IJzerman, A.P.; Jacobson, K.A.; Linden, J.; Müller, C.E. International Union of Basic and Clinical Pharmacology. LXXXI. Nomenclature and Classification of Adenosine Receptors–An Update. Pharmacol. Rev. 2011, 63, 1–34. [Google Scholar] [CrossRef]
- Fredholm, B.B.; IJzerman, A.P.; Jacobson, K.A.; Klotz, K.N.; Linden, J. International Union of Pharmacology. XXV. Nomenclature and classification of adenosine receptors. Pharmacol. Rev. 2001, 53, 527–552. [Google Scholar]
- Feoktistov, I.; Biaggioni, I. Adenosine A2B receptors. Pharmacol. Rev. 1997, 49, 381–402. [Google Scholar]
- Fredholm, B.B.; Irenius, E.; Kull, B.; Schulte, G. Comparison of the potency of adenosine as an agonist at human adenosine receptors expressed in Chinese hamster ovary cells. Biochem. Pharmacol. 2001, 61, 443–448. [Google Scholar] [CrossRef]
- Daly, J.W.; Butts-Lamb, P.; Padgett, W. Subclasses of adenosine receptors in the central nervous system: Interaction with caffeine and related methylxanthines. Cell Mol. Neurobiol. 1983, 3, 69–80. [Google Scholar] [CrossRef]
- Müller, C.E.; Baqi, Y.; Hinz, S.; Namasivayam, V. Medicinal chemistry of A2B adenosine receptors. In The Adenosine Receptors, Borea, P.A.; Varani, K., Gessi, S., Merighi, S., Vincenzi, F., Eds.; Humana Press: Totowa, NJ, USA, 2018; pp. 137–168. [Google Scholar]
- Borea, P.A.; Gessi, S.; Merighi, S.; Vincenzi, F.; Varani, K. Pharmacology of Adenosine Receptors: The State of the Art. Physiol. Rev. 2018, 98, 1591–1625. [Google Scholar] [CrossRef] [PubMed]
- Antonioli, L.; Blandizzi, C.; Pacher, P.; Hasko, G. Immunity, inflammation and cancer: A leading role for adenosine. Nat. Rev. Cancer 2013, 13, 842–857. [Google Scholar] [CrossRef] [PubMed]
- Gao, Z.G.; Jacobson, K.A. A2B Adenosine Receptor and Cancer. Int. J. Mol. Sci. 2019, 18, 5139. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sepulveda, C.; Palomo, I.; Fuentes, E. Role of adenosine A2B receptor overexpression in tumor progression. Life Sci. 2016, 166, 92–99. [Google Scholar] [CrossRef]
- Sorrentino, C.; Morello, S. Role of adenosine in tumor progression: Focus on A2B receptor as potential therapeutic target. J. Cancer Metastasis Treat. 2017, 3, 127–138. [Google Scholar] [CrossRef] [Green Version]
- Hinz, S.; Navarro, G.; Borroto-Escuela, D.; Seibt, B.F.; Ammon, Y.C.; de Filippo, E.; Danish, A.; Lacher, S.K.; Cervinkova, B.; Rafehi, M.; et al. Adenosine A2A receptor ligand recognition and signaling is blocked by A2B receptors. Oncotarget 2018, 9, 13593–13611. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Petroni, D.; Giacomelli, C.; Taliani, S.; Barresi, E.; Robello, M.; Daniele, S.; Bartoli, A.; Burchielli, S.; Pardini, S.; Salvadori, P.A.; et al. Toward PET imaging of A2B adenosine receptors: A carbon-11 labeled triazinobenzimidazole tracer: Synthesis and imaging of a new A2B PET tracer. Nucl. Med. Biol. 2016, 43, 309–317. [Google Scholar] [CrossRef] [PubMed]
- Lindemann, M.; Hinz, S.; Deuther-Conrad, W.; Namasivayam, V.; Dukic-Stefanovic, S.; Teodoro, R.; Toussaint, M.; Kranz, M.; Juhl, C.; Steinbach, J.; et al. Radiosynthesis and in vivo evaluation of a fluorine-18 labeled pyrazine based radioligand for PET imaging of the adenosine A2B receptor. Bioorg. Med. Chem. 2018, 26, 4650–4663. [Google Scholar] [CrossRef] [PubMed]
- Eastwood, P.; Esteve, C.; Gonzalez, J.; Fonquerna, S.; Aiguade, J.; Carranco, I.; Domenech, T.; Aparici, M.; Miralpeix, M.; Alberti, J.; et al. Discovery of LAS101057: A Potent, Selective, and Orally Efficacious A2B Adenosine Receptor Antagonist. ACS Med. Chem. Lett. 2011, 2, 213–218. [Google Scholar] [CrossRef] [Green Version]
- Eastwood, P.; Gonzalez, J.; Paredes, S.; Fonquerna, S.; Cardus, A.; Alonso, J.A.; Nueda, A.; Domenech, T.; Reinoso, R.F.; Vidal, B. Discovery of potent and selective bicyclic A2B adenosine receptor antagonists via bioisosteric amide replacement. Bioorg. Med. Chem. Lett. 2010, 20, 1634–1637. [Google Scholar] [CrossRef]
- Eastwood, P.; Gonzalez, J.; Paredes, S.; Nueda, A.; Domenech, T.; Alberti, J.; Vidal, B. Discovery of N-(5,6-diarylpyridin-2-yl)amide derivatives as potent and selective A2B adenosine receptor antagonists. Bioorg. Med. Chem. Lett. 2010, 20, 1697–1700. [Google Scholar] [CrossRef]
- Harada, H.; Asano, O.; Miyazawa, S.; Ueda, M.; Yasuda, M.; Yasuda, N. Aminopyridine Compounds and Use Thereof as Drugs. U.S. Patent 2004/006082, 1 August 2004. [Google Scholar]
- Troschütz, R.; Dennstedt, T. Synthesis of Substituted 2-Aminonicotinonitriles. Arch. Pharm. 1994, 327, 33–40. [Google Scholar] [CrossRef]
- Pham, S.M.; Chen, J.F.; Ansari, A.; Jadhavar, P.S.; Patil, V.S.; Khan, F.; Ramachandran, S.A.; Argaval, A.K.; Chakravarty, S. 1,8-Naphthyridinone Compounds and Uses Thereof. WO Patent 2019018583, 24 January 2019. [Google Scholar]
- Miyaura, N.; Suzuki, A. Stereoselective Synthesis of Arylated (E)-Alkenes by the Reaction of Alk-1-Enylboranes with Aryl Halides in the Presence of Palladium Catalyst. J. Chem. Soc. Chem. Comm. 1979, 866–867. [Google Scholar] [CrossRef]
- Miyaura, N.; Yamada, K.; Suzuki, A. New Stereospecific Cross-Coupling by the Palladium-Catalyzed Reaction of 1-Alkenylboranes with 1-Alkenyl or 1-Alkynyl Halides. Tetrahedron Lett. 1979, 20, 3437–3440. [Google Scholar] [CrossRef] [Green Version]
- Ishiyama, T.; Murata, M.; Miyaura, N. Palladium(O)-Catalyzed Cross-Coupling Reaction of Alkoxydiboron with Haloarenes—A Direct Procedure for Arylboronic Esters. J. Org. Chem. 1995, 60, 7508–7510. [Google Scholar] [CrossRef]
- Khaledi, M.G. Micelles as separation media in high-performance liquid chromatography and high-performance capillary electrophoresis: Overview and perspective. J. Chromatogr. A 1997, 780, 3–40. [Google Scholar] [CrossRef]
- Sun, Y.; Huang, P. Adenosine A2B Receptor: From Cell Biology to Human Diseases. Front. Chem. 2016, 4, 37. [Google Scholar] [CrossRef] [PubMed]
- Borrmann, T.; Hinz, S.; Bertarelli, D.C.; Li, W.; Florin, N.C.; Scheiff, A.B.; Müller, C.E. 1-Alkyl-8-(piperazine-1-sulfonyl)phenylxanthines: Development and characterization of adenosine A2B receptor antagonists and a new radioligand with subnanomolar affinity and subtype specificity. J. Med. Chem. 2009, 52, 3994–4006. [Google Scholar] [CrossRef]
- Alnouri, M.W.; Jepards, S.; Casari, A.; Schiedel, A.C.; Hinz, S.; Müller, C.E. Selectivity is species-dependent: Characterization of standard agonists and antagonists at human, rat, and mouse adenosine receptors. Purinergic Signal. 2015, 11, 389–407. [Google Scholar] [CrossRef] [Green Version]
- Klotz, K.N.; Lohse, M.J.; Schwabe, U.; Cristalli, G.; Vittori, S.; Grifantini, M. 2-Chloro-N6-[3H]cyclopentyladenosine ([3H]CCPA)-a high affinity agonist radioligand for A1 adenosine receptors. Naunyn Schmiedebergs Arch. Pharmacol. 1989, 340, 679–683. [Google Scholar] [CrossRef]
- Müller, C.E.; Maurinsh, J.; Sauer, R. Binding of [3H]MSX-2 (3-(3-hydroxypropyl)-7-methyl-8-(m-methoxystyryl)-1-propargylxanthine) to rat striatal membranes-a new, selective antagonist radioligand for A2A adenosine receptors. Eur. J. Pharm. Sci. 2000, 10, 259–265. [Google Scholar] [CrossRef]
- Müller, C.E.; Diekmann, M.; Thorand, M.; Ozola, V. [3H]8-Ethyl-4-methyl-2-phenyl-(8R)-4,5,7,8-tetrahydro-1H-imidazo[2,1-i]-purin-5-one ([3H]PSB-11), a novel high-affinity antagonist radioligand for human A3 adenosine receptors. Bioorg. Med. Chem. Lett. 2002, 12, 501–503. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | Ki in nM a | Selectivity Ratio Ki(Ax)/Ki(A2B) | ||||||
|---|---|---|---|---|---|---|---|---|
| A2B | A2A | A1 | A3 | A2A/A2B | A1/A2B | A3/A2B | ||
| 1 | 4.24 ± 0.04 b | 55.0 ± 6.1 b | 19.0 ± 5.2 b | 796 ± 26 b | 13 | 4.5 | 188 | |
| 2 | 2.51 ± 1.1 | 98.8 ± 28.3 | > 1000 | > 1000 | 39 | > 400 | > 400 | |
| 1 ± 0 c | 181 ± 25 c | 1727 ± 617 c | 6267 ± 2322 c | |||||
| 9 | 2.51 ± 0.58 | 107 ± 15 | 149 ± 26 | 286 ± 10 | 43 | 59 | 114 | |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lindemann, M.; Moldovan, R.-P.; Hinz, S.; Deuther-Conrad, W.; Gündel, D.; Dukic-Stefanovic, S.; Toussaint, M.; Teodoro, R.; Juhl, C.; Steinbach, J.; et al. Development of a Radiofluorinated Adenosine A2B Receptor Antagonist as Potential Ligand for PET Imaging. Int. J. Mol. Sci. 2020, 21, 3197. https://doi.org/10.3390/ijms21093197
Lindemann M, Moldovan R-P, Hinz S, Deuther-Conrad W, Gündel D, Dukic-Stefanovic S, Toussaint M, Teodoro R, Juhl C, Steinbach J, et al. Development of a Radiofluorinated Adenosine A2B Receptor Antagonist as Potential Ligand for PET Imaging. International Journal of Molecular Sciences. 2020; 21(9):3197. https://doi.org/10.3390/ijms21093197
Chicago/Turabian StyleLindemann, Marcel, Rareş-Petru Moldovan, Sonja Hinz, Winnie Deuther-Conrad, Daniel Gündel, Sladjana Dukic-Stefanovic, Magali Toussaint, Rodrigo Teodoro, Cathleen Juhl, Jörg Steinbach, and et al. 2020. "Development of a Radiofluorinated Adenosine A2B Receptor Antagonist as Potential Ligand for PET Imaging" International Journal of Molecular Sciences 21, no. 9: 3197. https://doi.org/10.3390/ijms21093197