Mechanistic Study of Triazole Based Aminodiol Derivatives in Leukemic Cells—Crosstalk between Mitochondrial Stress-Involved Apoptosis and Autophagy

 , ,
, ,
Abstract
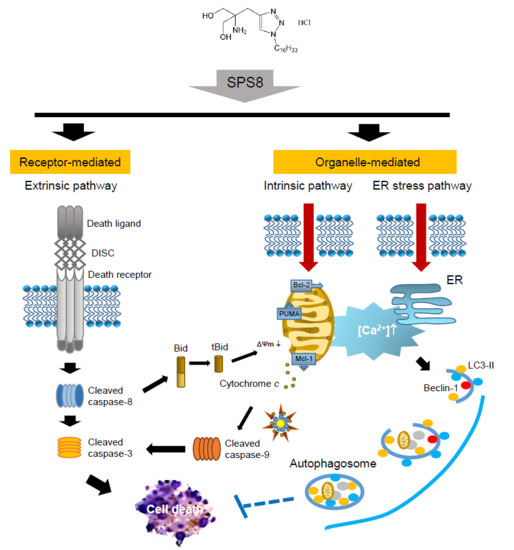
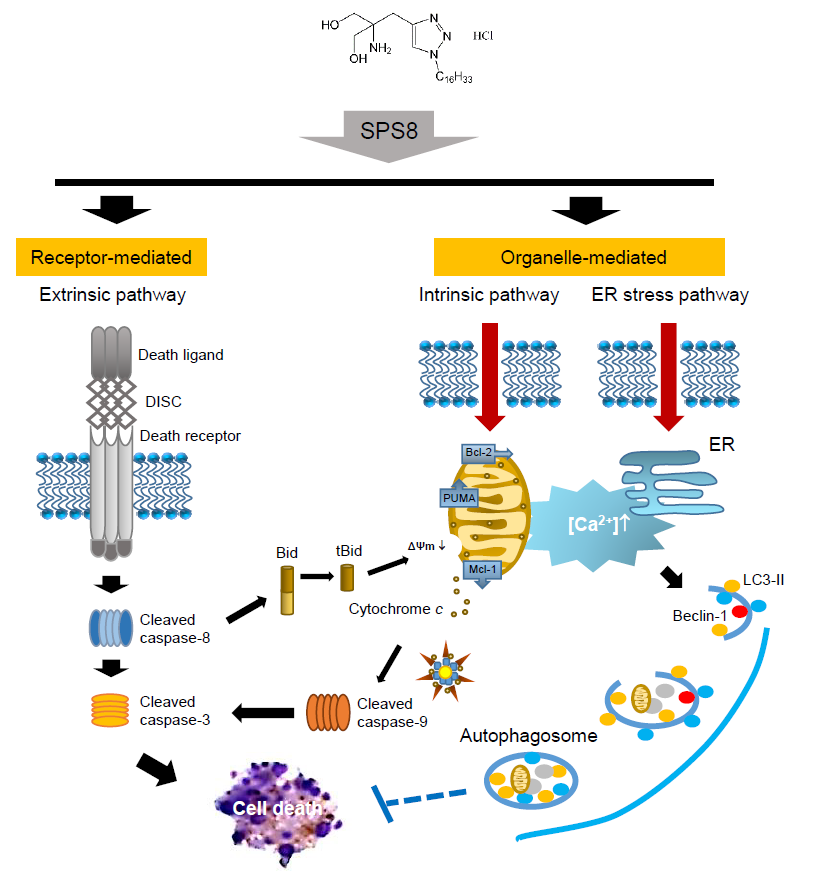
:
1. Introduction
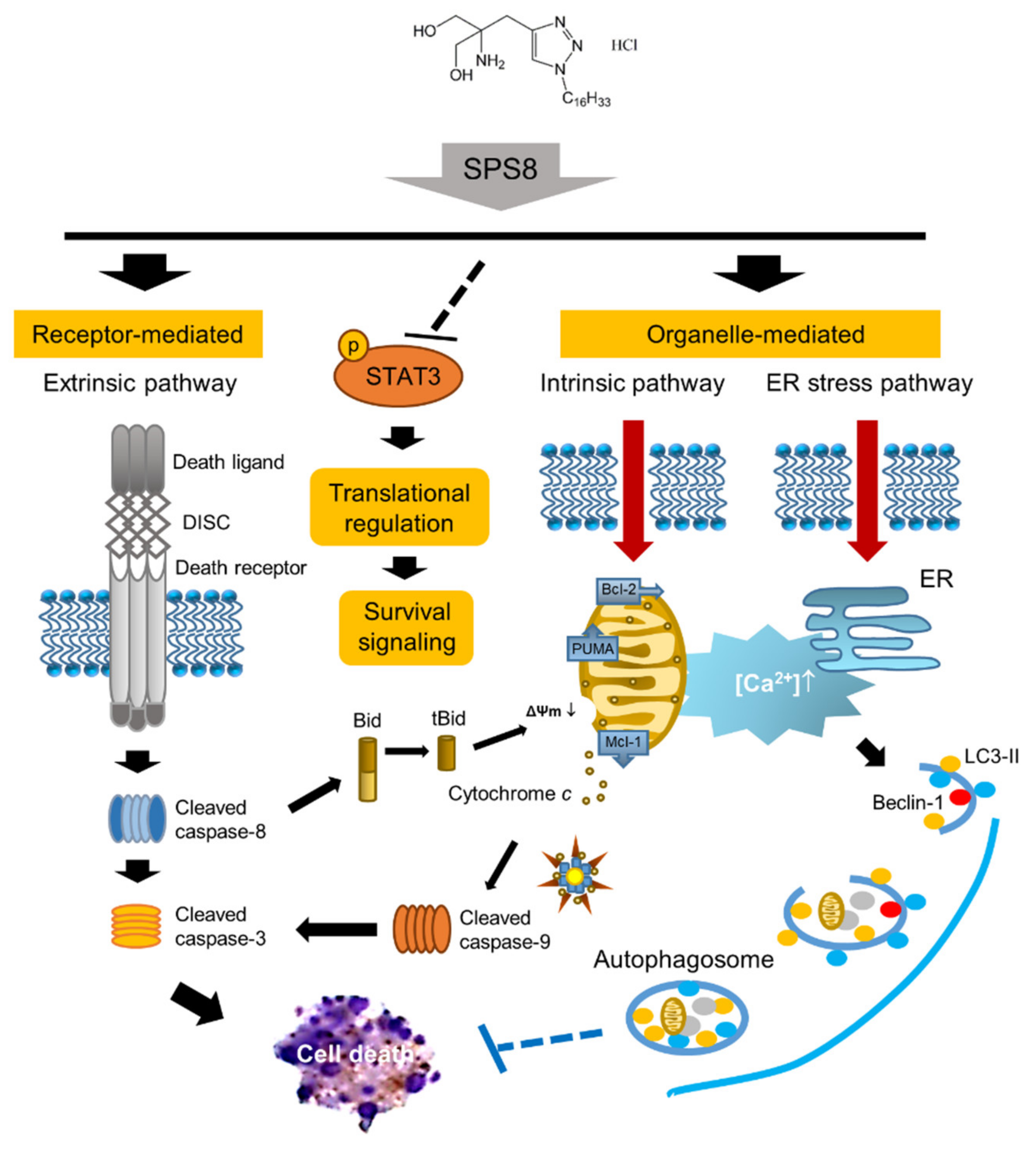
2. Results
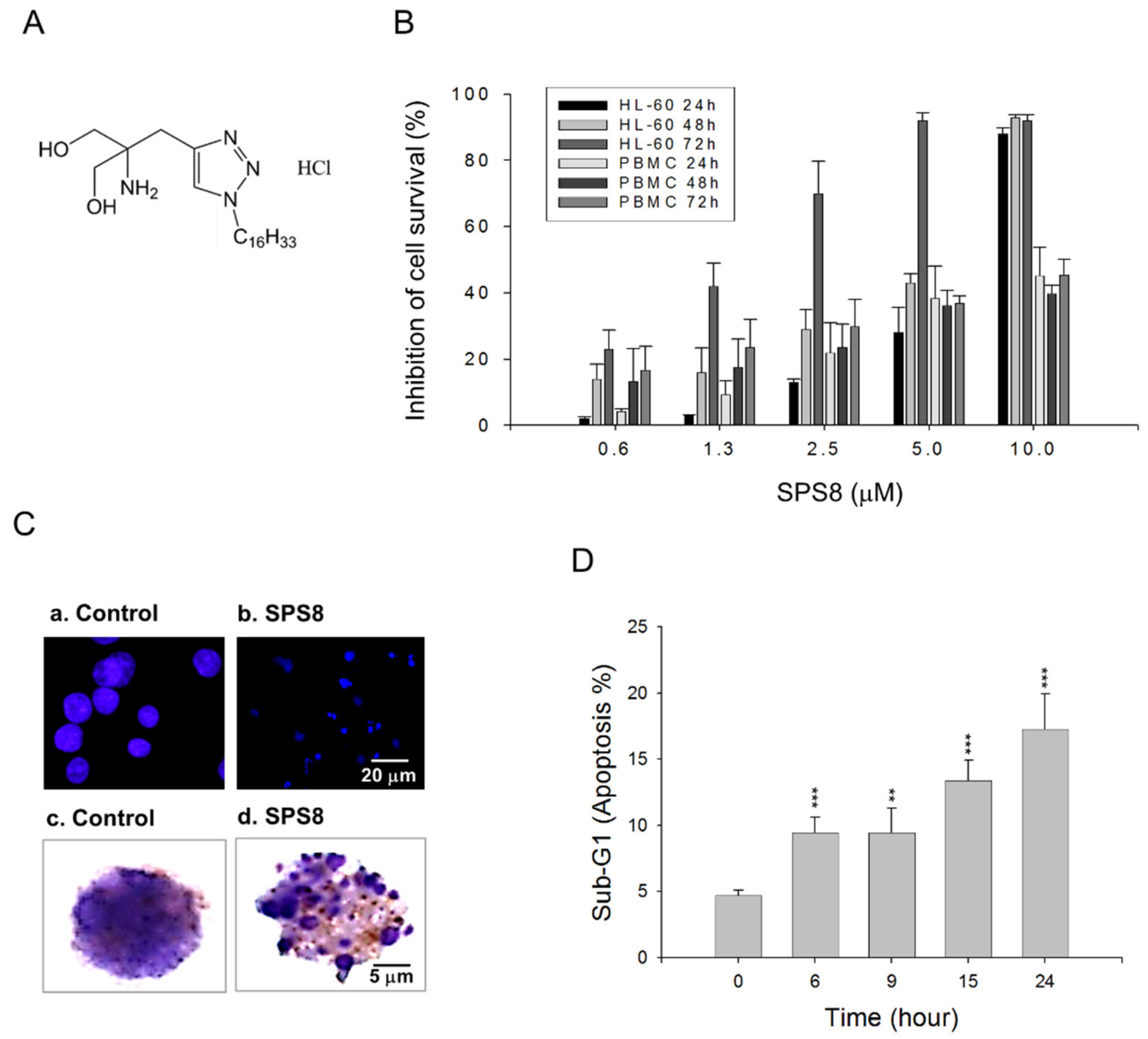
2.1. SPS8 Selectively Induces Cytotoxicity in APL HL-60 Cells
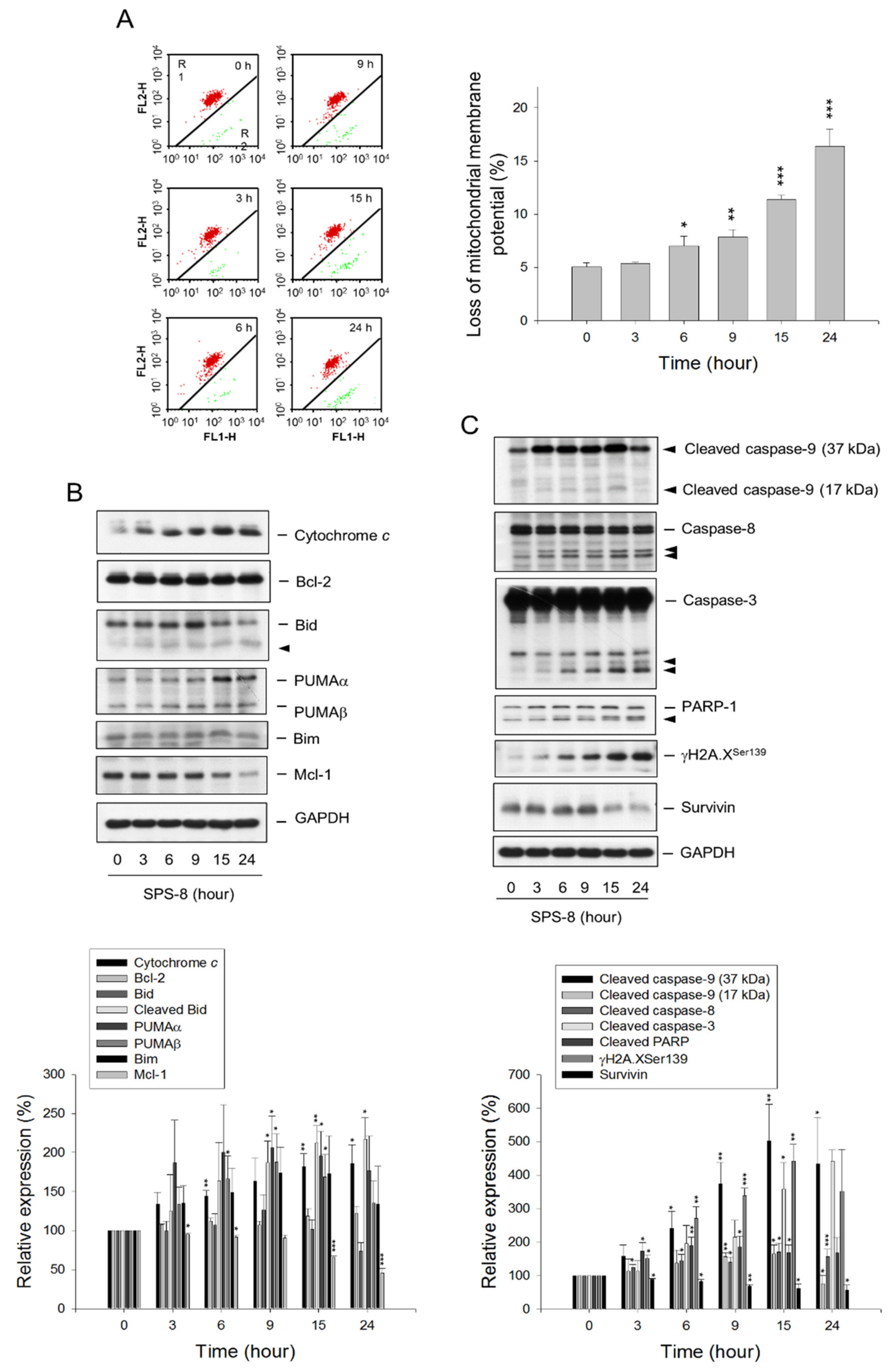
2.2. SPS8 Induces Mitochondrial Damage in Connecting Apoptotic Signaling Pathways
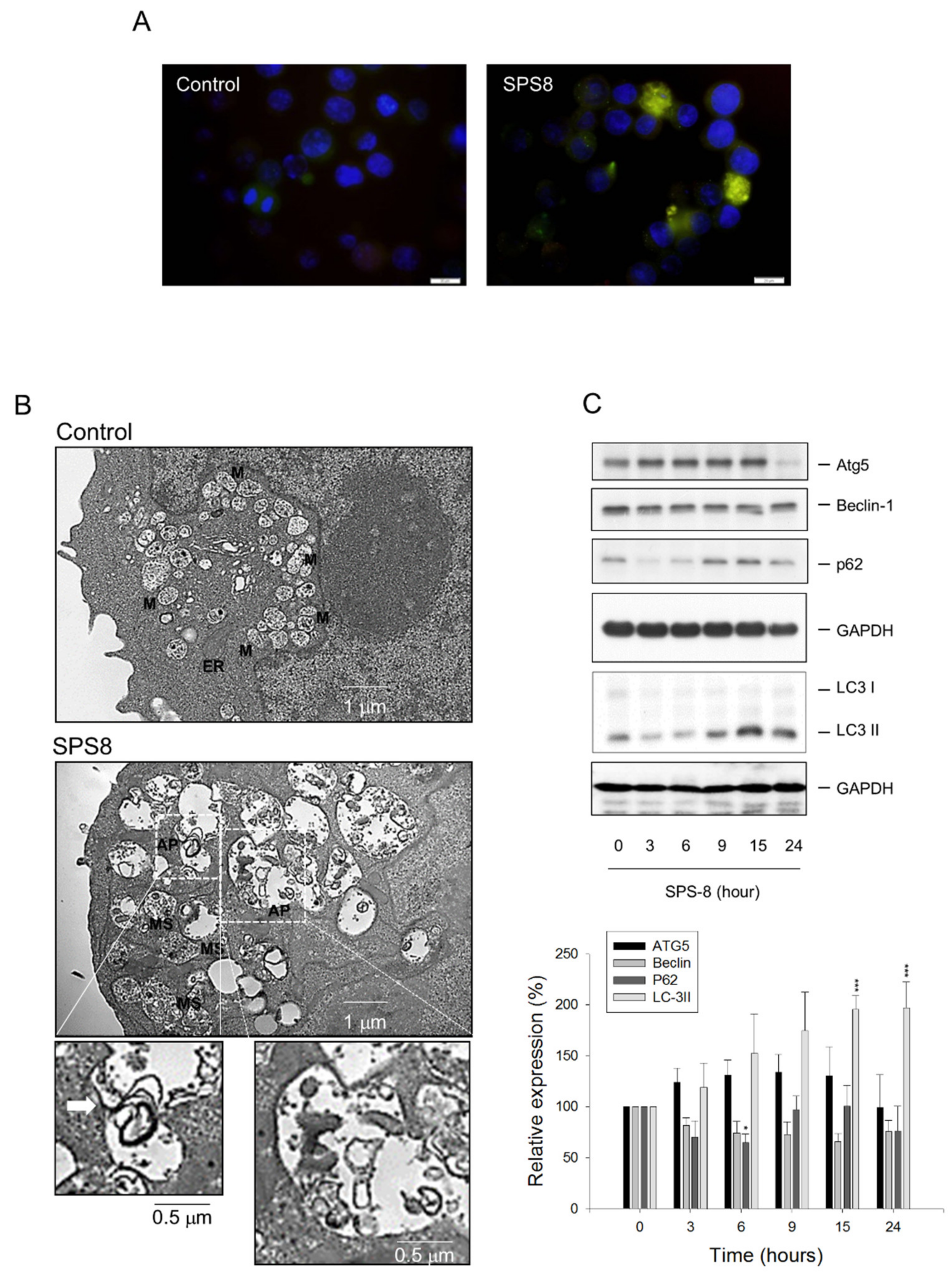
2.3. SPS8 Induces Autophagy in HL-60 Cells
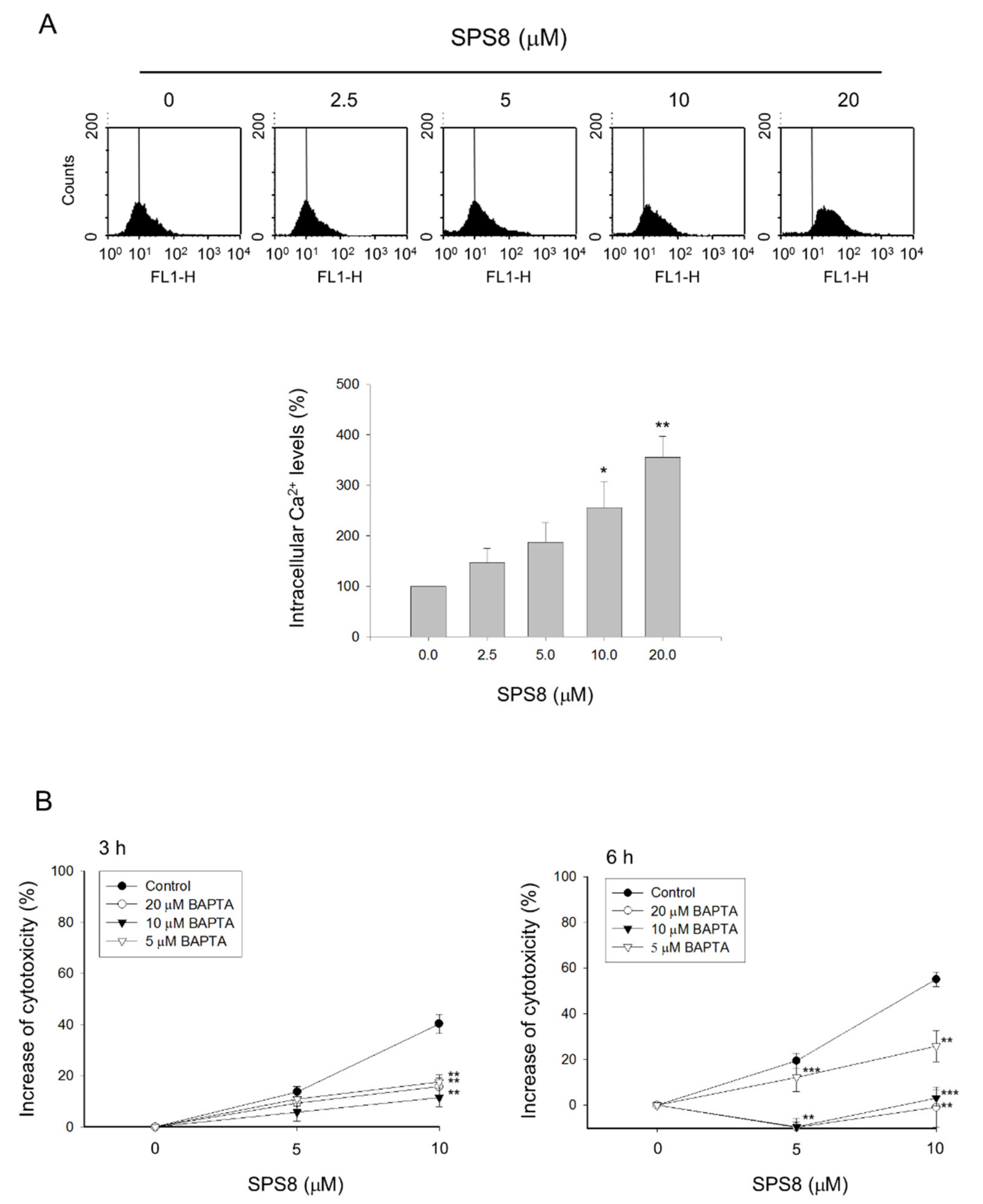
2.4. Calcium Plays a Role in the SPS8-Mediated Effect in HL-60 Cells
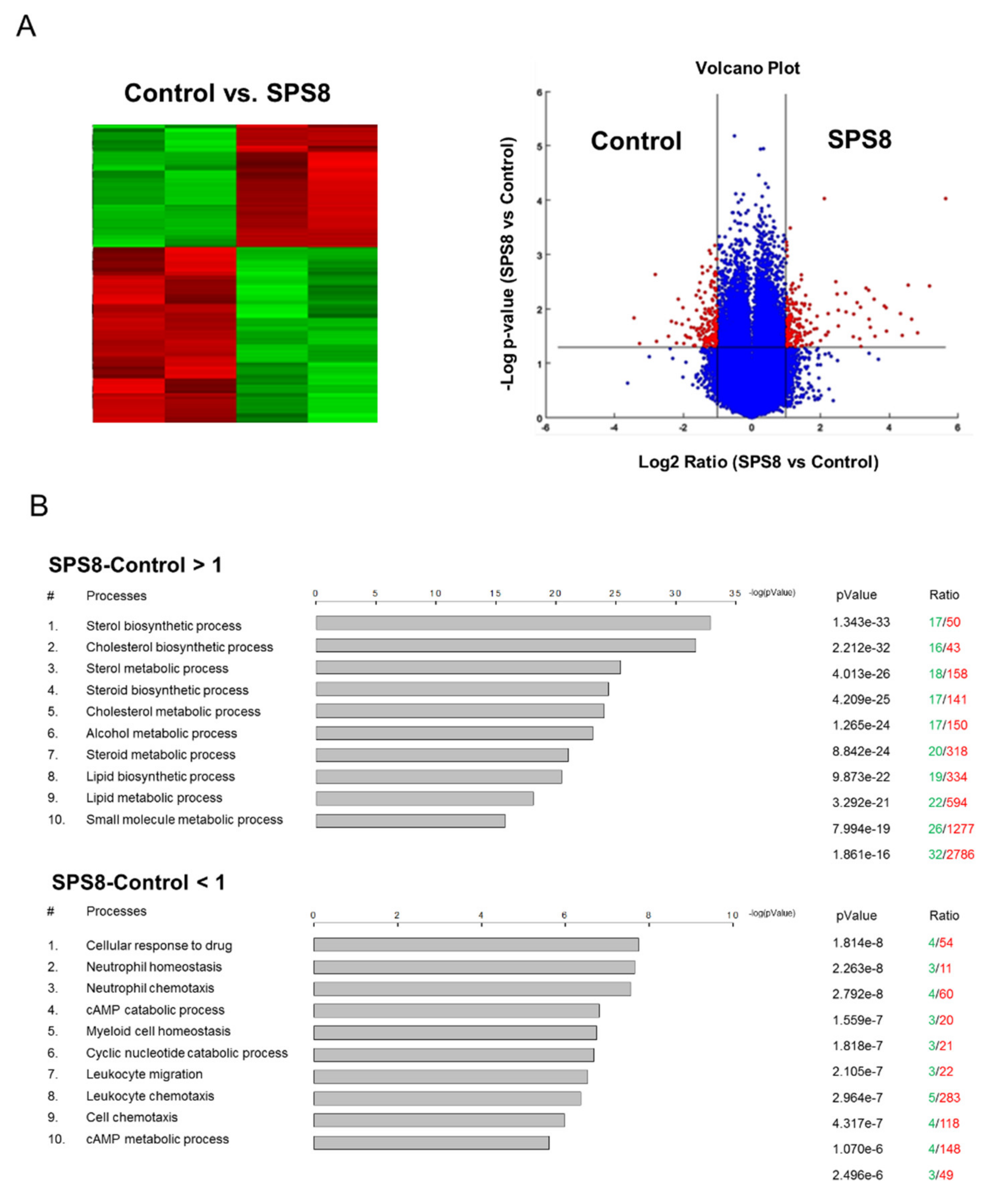
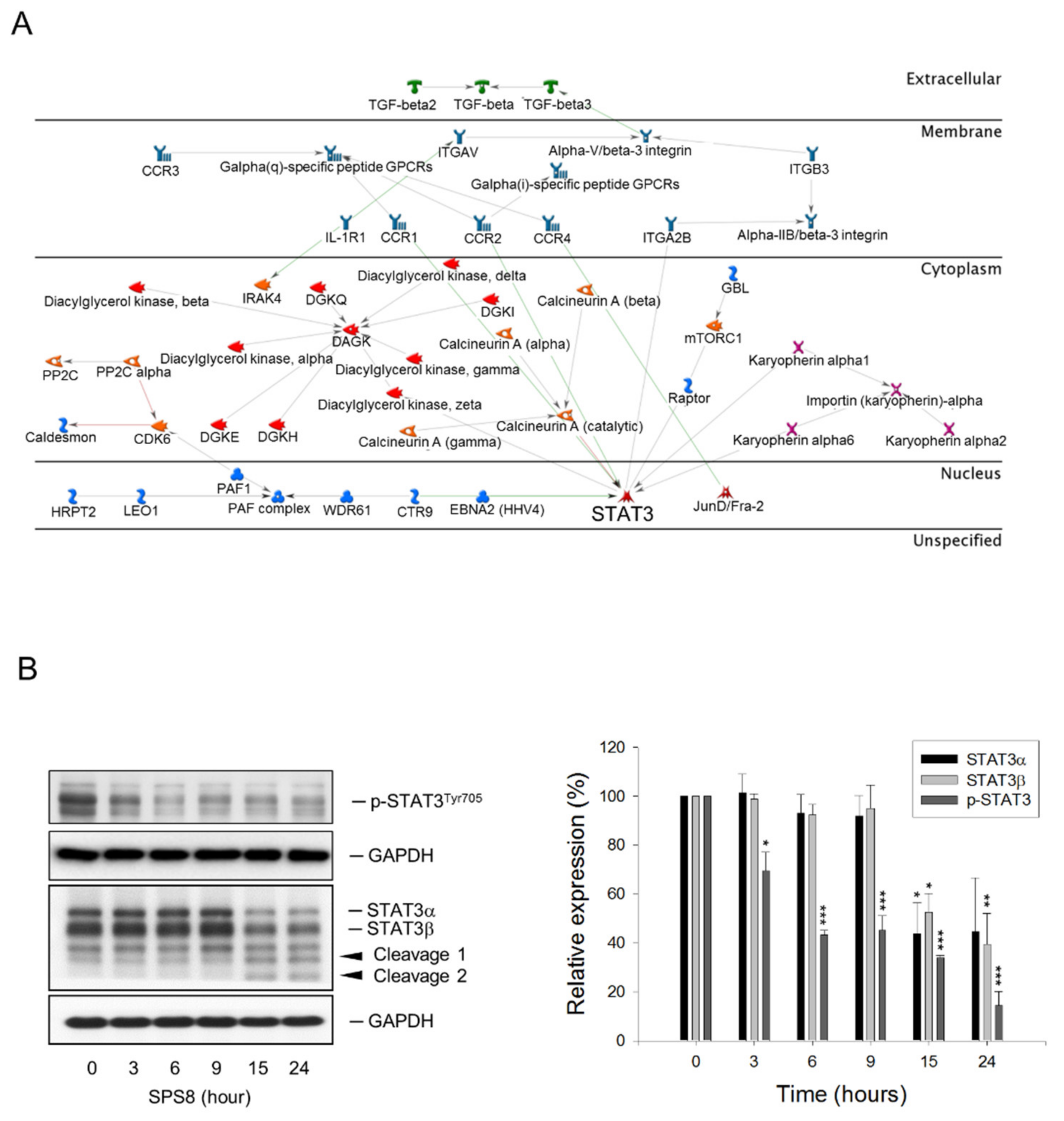
2.5. SPS8 Exhibits Transcriptional Regulation in HL-60 Cells
3. Discussion
4. Materials and Methods
4.1. Chemicals, Reagents, Antibodies and Cell Culture
4.2. Cell Viability Assay
4.3. Flow Cytometric Detection of Apoptosis
4.4. Measurement of ΔΨm
4.5. Confocal Microscopic Examination with DAPI Staining
4.6. Microscopic Observation of Cell Morphology
4.7. Transmission Electron Microscopy
4.8. Measurement of Intracellular Ca2+ Level
4.9. RNA Extraction, Microarray and Data Processing
4.10. Human Apoptosis Antibody Array
4.11. Western Blotting
4.12. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| AML | Acute myeloid leukemia |
| APL | Acute promyelocytic leukemia |
| ATRA | All-trans retinoic acid |
| CML | Chronic myeloid leukemia |
| ΔΨm | Mitochondrial membrane potential |
| DMSO | Dimethyl sulfoxide |
| ER | Endoplasmic reticulum |
| IAP | Inhibitor of apoptosis |
| JAK | Janus kinases |
| MTT | Methylthiazolyldiphenyl-tetrazolium bromide |
| PBMCs | Peripheral blood mononuclear cells |
| PI | Propidium iodine |
| RAR | Retinoic acid receptor |
| SREBP | Sterol-regulatory element–binding protein |
| STAT3 | Signal Transducer and Activator of Transcription 3 |
| tBid | Truncated Bid |
| TEM | Transmission electron microscopy |
References
- Redaelli, A.; Stephens, J.M.; Laskin, B.L.; Pashos, C.L.; Botteman, M.F. The burden and outcomes associated with four leukemias: AML, ALL, CLL and CML. Expert Rev. Anticancer Ther. 2003, 3, 311–329. [Google Scholar] [CrossRef] [PubMed]
- Cartledge Wolf, D.M.; Langhans, S.A. Moving myeloid leukemia drug discovery into the third dimension. Front. Pediatr. 2019, 7, 314. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grosso, D.A.; Hess, R.C.; Weiss, M.A. Immunotherapy in acute myeloid leukemia. Cancer 2015, 121, 2689–2704. [Google Scholar] [CrossRef] [PubMed]
- Bertacchini, J.; Heidari, N.; Mediani, L.; Capitani, S.; Shahjahani, M.; Ahmadzadeh, A.; Saki, N. Targeting PI3K/AKT/mTOR network for treatment of leukemia. Cell. Mol. Life Sci. 2015, 72, 2337–2347. [Google Scholar] [CrossRef]
- Vaidya, A. Can systems biology approach help in finding more effective treatment for acute myeloid leukemia? Syst. Synth. Biol. 2014, 8, 165–167. [Google Scholar] [CrossRef] [Green Version]
- Lo-Coco, F.; Avvisati, G.; Vignetti, M.; Thiede, C.; Orlando, S.M.; Iacobelli, S.; Ferrara, F.; Fazi, P.; Cicconi, L.; Di Bona, E.; et al. Retinoic acid and arsenic trioxide for acute promyelocytic leukemia. N. Engl. J. Med. 2013, 369, 111–121. [Google Scholar] [CrossRef] [Green Version]
- De Braekeleer, E.; Douet-Guilbert, N.; De Braekeleer, M. RARA fusion genes in acute promyelocytic leukemia: A review. Expert Rev. Hematol. 2014, 7, 347–357. [Google Scholar] [CrossRef]
- Orfali, N.; McKenna, S.L.; Cahill, M.R.; Gudas, L.J.; Mongan, N.P. Retinoid receptor signaling and autophagy in acute promyelocytic leukemia. Exp. Cell Res. 2014, 324, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Breccia, M.; Latagliata, R.; Carmosino, I.; Cannella, L.; Diverio, D.; Guarini, A.; De Propris, M.S.; Petti, M.C.; Avvisati, G.; Cimino, G.; et al. Clinical and biological features of acute promyelocytic leukemia patients developing retinoic acid syndrome during induction treatment with all-trans retinoic acid and idarubicin. Haematologica 2008, 93, 1918–1920. [Google Scholar] [CrossRef]
- Patatanian, E.; Thompson, D.F. Retinoic acid syndrome: A review. J. Clin. Pharm. Ther. 2008, 33, 331–338. [Google Scholar] [CrossRef]
- Avvisati, G.; Tallman, M.S. All-trans retinoic acid in acute promyelocytic leukaemia. Best Pract. Res. Clin. Haematol. 2003, 16, 419–432. [Google Scholar] [CrossRef]
- Fenaux, P.; De Botton, S. Retinoic acid syndrome. Recognition, prevention and management. Drug Saf. 1998, 18, 273–279. [Google Scholar] [CrossRef] [PubMed]
- Stahl, M.; Tallman, M.S. Acute promyelocytic leukemia (APL): Remaining challenges towards a cure for all. Leuk. Lymphoma 2019, 60, 3107–3115. [Google Scholar] [CrossRef] [PubMed]
- Halicka, H.D.; Smolewski, P.; Darzynkiewicz, Z.; Dai, W.; Traganos, F. Arsenic trioxide arrests cells early in mitosis leading to apoptosis. Cell Cycle. 2002, 1, 201–209. [Google Scholar] [CrossRef] [Green Version]
- Takenokuchi, M.; Miyamoto, K.; Saigo, K.; Taniguchi, T. Bortezomib causes ER stress-related death of acute promyelocytic leukemia cells through excessive accumulation of PML-RARA. Anticancer Res. 2015, 35, 3307–3316. [Google Scholar]
- Ogretmen, B. Sphingolipid metabolism in cancer signaling and therapy. Nat. Rev. Cancer 2018, 18, 33–50. [Google Scholar] [CrossRef]
- Mesicek, J.; Lee, H.; Feldman, T.; Jiang, X.; Skobeleva, A.; Berdyshev, E.V.; Haimovitz-Friedman, A.; Fuks, Z.; Kolesnick, R. Ceramide synthases 2, 5, and 6 confer distinct roles in radiation-induced apoptosis in HeLa cells. Cell Signal. 2010, 22, 1300–1307. [Google Scholar] [CrossRef] [Green Version]
- Ogretmen, B.; Hannun, Y.A. Biologically active sphingolipids in cancer pathogenesis and treatment. Nat. Rev. Cancer 2004, 4, 604–616. [Google Scholar] [CrossRef]
- Swain, S.P.; Hou, D.R. Substituted 1,2,3-trizoles as antitumor agents. Inventors. National Central University, Assignee. U.S. Patent 9,738,613 B2, 22 August 2017. [Google Scholar]
- Ding, M.; Weng, X.Q.; Sheng, Y.; Wu, J.; Liang, C.; Cai, X. Dasatinib synergizes with ATRA to trigger granulocytic differentiation in ATRA resistant acute promyelocytic leukemia cell lines via Lyn inhibition-mediated activation of RAF-1/MEK/ERK. Food Chem. Toxicol. 2018, 119, 464–478. [Google Scholar] [CrossRef]
- Jung, Y.S.; Cheong, H.J.; Kim, S.J.; Kim, K.H.; Lee, N.; Park, H.S.; Won, J.H. Src family kinase inhibitor PP2 enhances differentiation of acute promyelocytic leukemia cell line induced by combination of all-trans-retinoic acid and arsenic trioxide. Leuk. Res. 2014, 38, 977–982. [Google Scholar] [CrossRef]
- Kivioja, J.L.; Thanasopoulou, A.; Kumar, A.; Kontro, M.; Yadav, B.; Majumder, M.M.; Javarappa, K.K.; Eldfors, S.; Schwaller, J.; Porkka, K.; et al. Dasatinib and navitoclax act synergistically to target NUP98-NSD1+/FLT3-ITD+ acute myeloid leukemia. Leukemia 2019, 33, 1360–1372. [Google Scholar] [CrossRef] [PubMed]
- Anderson, A.J.; Jackson, T.D.; Stroud, D.A.; Stojanovski, D. Mitochondria-hubs for regulating cellular biochemistry: Emerging concepts and networks. Open Biol. 2019, 9, 190126. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rogakou, E.P.; Nieves-Neira, W.; Boon, C.; Pommier, Y.; Bonner, W.M. Initiation of DNA fragmentation during apoptosis induces phosphorylation of H2AX histone at serine 139. J. Biol. Chem. 2000, 275, 9390–9395. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parzych, K.R.; Klionsky, D.J. An overview of autophagy: Morphology, mechanism, and regulation. Antioxid. Redox Signal. 2014, 20, 460–473. [Google Scholar] [CrossRef] [Green Version]
- Moreau, K.; Luo, S.; Rubinsztein, D.C. Cytoprotective roles for autophagy. Curr. Opin. Cell Biol. 2010, 22, 206–211. [Google Scholar] [CrossRef] [Green Version]
- Pérez-Hernández, M.; Arias, A.; Martínez-García, D.; Pérez-Tomás, R.; Quesada, R.; Soto-Cerrato, V. Targeting autophagy for cancer treatment and tumor chemosensitization. Cancers 2019, 11, E1599. [Google Scholar] [CrossRef] [Green Version]
- Cha-Molstad, H.; Lee, S.H.; Kim, J.G.; Sung, K.W.; Hwang, J.; Shim, S.M.; Ganipisetti, S.; McGuire, T.; Mook-Jung, I.; Ciechanover, A.; et al. Regulation of autophagic proteolysis by the N-recognin SQSTM1/p62 of the N-end rule pathway. Autophagy 2018, 14, 359–361. [Google Scholar] [CrossRef]
- Zhang, Y.; Mun, S.R.; Linares, J.F.; Ahn, J.; Towers, C.G.; Ji, C.H.; Fitzwalter, B.E.; Holden, M.R.; Mi, W.; Shi, X.; et al. ZZ-dependent regulation of p62/SQSTM1 in autophagy. Nat. Commun. 2018, 9, 4373. [Google Scholar] [CrossRef] [Green Version]
- Fan, Y.J.; Zong, W.X. The cellular decision between apoptosis and autophagy. Chin. J. Cancer 2013, 32, 121–129. [Google Scholar]
- Mukhopadhyay, S.; Panda, P.K.; Sinha, N.; Das, D.N.; Bhutia, S.K. Autophagy and apoptosis: Where do they meet? Apoptosis 2014, 19, 555–566. [Google Scholar] [CrossRef]
- Schaaf, M.B.; Houbaert, D.; Meçe, O.; To, S.K.; Ganne, M.; Maes, H.; Agostinis, P. Lysosomal Pathways and Autophagy Distinctively Control Endothelial Cell Behavior to Affect Tumor Vasculature. Front. Oncol. 2019, 9, 171. [Google Scholar] [CrossRef]
- Harr, M.W.; Distelhorst, C.W. Apoptosis and autophagy: Decoding calcium signals that mediate life or death. Cold Spring Harb. Perspect. Biol. 2010, 2, a005579. [Google Scholar] [CrossRef] [PubMed]
- Alsamri, H.; El Hasasna, H.; Al Dhaheri, Y.; Eid, A.H.; Attoub, S.; Iratni, R. Carnosol, a natural polyphenol, inhibits migration, metastasis, and tumor growth of breast cancer via a ROS-dependent proteasome degradation of STAT3. Front. Oncol. 2019, 9, 743. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Darnowski, J.W.; Goulette, F.A.; Guan, Y.J.; Chatterjee, D.; Yang, Z.F.; Cousens, L.P.; Chin, Y.E. Stat3 cleavage by caspases: Impact on full-length Stat3 expression, fragment formation, and transcriptional activity. J. Biol. Chem. 2006, 281, 17707–17717. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lengfelder, E.; Saussele, S.; Weisser, A.; Büchner, T.; Hehlmann, R. Treatment concepts of acute promyelocytic leukemia. Crit. Rev. Oncol. Hematol. 2005, 56, 261–274. [Google Scholar] [CrossRef]
- Cassier, P.A.; Castets, M.; Belhabri, A.; Vey, N. Targeting apoptosis in acute myeloid leukaemia. Br. J. Cancer 2017, 117, 1089–1098. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.S.; Cho, I.A.; Kang, K.R.; Lim, H.; Kim, T.H.; Yu, S.K.; Kim, H.J.; Lee, S.A.; Moon, S.M.; Chun, H.S.; et al. Reversine induces caspase-dependent apoptosis of human osteosarcoma cells through extrinsic and intrinsic apoptotic signaling pathways. Genes Genom. 2019, 41, 657–665. [Google Scholar] [CrossRef]
- Derakhshan, A.; Chen, Z.; Van Waes, C. Therapeutic small molecules target inhibitor of apoptosis proteins in cancers with deregulation of extrinsic and intrinsic cell death pathways. Clin. Cancer Res. 2017, 23, 1379–1387. [Google Scholar] [CrossRef] [Green Version]
- Liu, X.; Kim, C.N.; Yang, J.; Jemmerson, R.; Wang, X. Induction of apoptotic program in cell-free extracts: Requirement for dATP and cytochrome c. Cell 1996, 86, 147–157. [Google Scholar] [CrossRef] [Green Version]
- Wei, M.C.; Lindsten, T.; Mootha, V.K.; Weiler, S.; Gross, A.; Ashiya, M.; Thompson, C.B.; Korsmeyer, S.J. tBID, a membrane-targeted death ligand, oligomerizes BAK to release cytochrome C. Genes Dev. 2000, 14, 2060–2071. [Google Scholar]
- Youle, R.J.; Strasser, A. The BCL-2 protein family: Opposing activities that mediate cell death. Nat. Rev. Mol. Cell Biol. 2008, 9, 47–59. [Google Scholar] [CrossRef] [PubMed]
- Erlacher, M.; Labi, V.; Manzl, C.; Böck, G.; Tzankov, A.; Häcker, G.; Michalak, E.; Strasser, A.; Villunger, A. Puma cooperates with Bim, the rate-limiting BH3-only protein in cell death during lymphocyte development, in apoptosis induction. J. Exp. Med. 2006, 203, 2939–2951. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bose, P.; Grant, S. Mcl-1 as a therapeutic target in Acute Myelogenous Leukemia (AML). Leuk. Res. Rep. 2013, 2, 12–14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eisenberg-Lerner, A.; Bialik, S.; Simon, H.U.; Kimchi, A. Life and death partners: Apoptosis, autophagy and the cross-talk between them. Cell Death Differ. 2009, 16, 966–975. [Google Scholar] [CrossRef]
- Folkerts, H.; Hilgendorf, S.; Wierenga, A.T.J.; Jaques, J.; Mulder, A.B.; Coffer, P.J.; Schuringa, J.J.; Vellenga, E. Inhibition of autophagy as a treatment strategy for p53 wild-type acute myeloid leukemia. Cell Death Dis. 2017, 8, e2927. [Google Scholar] [CrossRef] [Green Version]
- Yousefi, S.; Perozzo, R.; Schmid, I.; Ziemiecki, A.; Schaffner, T.; Scapozza, L.; Brunner, T.; Simon, H.U. Calpain-mediated cleavage of Atg5 switches autophagy to apoptosis. Nat. Cell Biol. 2006, 8, 1124–1132. [Google Scholar] [CrossRef]
- Wirawan, E.; Vande Walle, L.; Kersse, K.; Cornelis, S.; Claerhout, S.; Vanoverberghe, I.; Roelandt, R.; De Rycke, R.; Verspurten, J.; Declercq, W.; et al. Caspase-mediated cleavage of Beclin-1 inactivates Beclin-1-induced autophagy and enhances apoptosis by promoting the release of proapoptotic factors from mitochondria. Cell Death Dis. 2010, 1, e18. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Yan, J.; Wang, L.; Xiao, F.; Yang, Y.; Guo, X.; Wang, H. Beclin1 inhibition promotes autophagy and decreases gemcitabine-induced apoptosis in Miapaca2 pancreatic cancer cells. Cancer Cell Int. 2013, 13, 26. [Google Scholar] [CrossRef] [Green Version]
- Yokoyama, C.; Wang, X.; Briggs, M.R.; Admon, A.; Wu, J.; Hua, X.; Goldstein, J.L.; Brown, M.S. SREBP-1, a basic-helix-loop-helix-leucine zipper protein that controls transcription of the low density lipoprotein receptor gene. Cell 1993, 75, 187–197. [Google Scholar] [CrossRef]
- Hua, X.; Yokoyama, C.; Wu, J.; Briggs, M.R.; Brown, M.S.; Goldstein, J.L.; Wang, X. SREBP-2, a second basic-helix-loop-helix-leucine zipper protein that stimulates transcription by binding to a sterol regulatory element. Proc. Natl. Acad. Sci. USA 1993, 90, 11603–11607. [Google Scholar] [CrossRef] [Green Version]
- Horton, J.D.; Goldstein, J.L.; Brown, M.S. SREBPs: Activators of the complete program of cholesterol and fatty acid synthesis in the liver. J. Clin. Invest. 2002, 109, 1125–1131. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.A.; Liu, W.X.; Durnaoglu, S.; Lee, S.K.; Lian, J.; Lehner, R.; Ahnn, J.; Agellon, L.B.; Michalak, M. Loss of calreticulin uncovers a critical role for calcium in regulating cellular lipid homeostasis. Sci. Rep. 2017, 7, 5941. [Google Scholar] [CrossRef] [PubMed]
- Colgan, S.M.; Hashimi, A.A.; Austin, R.C. Endoplasmic reticulum stress and lipid dysregulation. Expert Rev. Mol. Med. 2011, 13, e4. [Google Scholar] [CrossRef] [PubMed]
- Caldenhoven, E.; van Dijk, T.B.; Solari, R.; Armstrong, J.; Raaijmakers, J.A.; Lammers, J.W.; Koenderman, L.; de Groot, R.P. STAT3beta, a splice variant of transcription factor STAT3, is a dominant negative regulator of transcription. J. Biol. Chem. 1996, 271, 13221–13227. [Google Scholar] [CrossRef] [Green Version]
- Ng, I.H.; Ng, D.C.; Jans, D.A.; Bogoyevitch, M.A. Selective STAT3-α or -β expression reveals spliceform-specific phosphorylation kinetics, nuclear retention and distinct gene expression outcomes. Biochem. J. 2012, 447, 125–136. [Google Scholar] [CrossRef]
- Nguyen-Jackson, H.; Panopoulos, A.D.; Zhang, H.; Li, H.S.; Watowich, S.S. STAT3 controls the neutrophil migratory response to CXCR2 ligands by direct activation of G-CSF-induced CXCR2 expression and via modulation of CXCR2 signal transduction. Blood 2010, 115, 3354–3363. [Google Scholar] [CrossRef] [Green Version]
- Pan, Y.R.; Chen, C.C.; Chan, Y.T.; Wang, H.J.; Chien, F.T.; Chen, Y.L.; Liu, J.L.; Yang, M.H. STAT3-coordinated migration facilitates the dissemination of diffuse large B-cell lymphomas. Nat. Commun. 2018, 9, 3696. [Google Scholar] [CrossRef]
- Mesbahi, Y.; Zekri, A.; Ghaffari, S.H.; Tabatabaie, P.S.; Ahmadian, S.; Ghavamzadeh, A. Blockade of JAK2/STAT3 intensifies the anti-tumor activity of arsenic trioxide in acute myeloid leukemia cells: Novel synergistic mechanism via the mediation of reactive oxygen species. Eur. J. Pharmacol. 2018, 834, 65–76. [Google Scholar] [CrossRef]
- Dong, S.; Chen, S.J.; Tweardy, D.J. Cross-talk between retinoic acid and STAT3 signaling pathways in acute promyelocytic leukemia. Leuk. Lymphoma 2003, 44, 2023–2029. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Up-Regulated Genes | ||
|---|---|---|
| Gene Title | Gene Symbol | log2ratio |
| 3-hydroxy-3-methylglutaryl-CoA synthase 1 (soluble) | HMGCS1 | 3.870 |
| insulin induced gene 1 | INSIG1 | 3.212 |
| methylsterol monooxygenase 1 | MSMO1 | 2.935 |
| 7-dehydrocholesterol reductase | DHCR7 | 2.759 |
| cyclin G2 | CCNG2 | 2.696 |
| 3-hydroxy-3-methylglutaryl-CoA reductase | HMGCR | 2.639 |
| TSC22 domain family, member 3 | TSC22D3 | 2.618 |
| low density lipoprotein receptor | LDLR | 2.503 |
| fatty acid desaturase 1 | FADS1 | 2.486 |
| StAR-related lipid transfer (START) domain containing 4 | STARD4 | 2.444 |
| squalene epoxidase | SQLE | 2.437 |
| lanosterol synthase (2,3-oxidosqualene-lanosterol cyclase) | LSS | 2.386 |
| solute carrier organic anion transporter family, member 4C1 | SLCO4C1 | 2.305 |
| tubulin, alpha 1a | TUBA1A | 2.275 |
| isopentenyl-diphosphate delta isomerase 1 | IDI1 | 2.257 |
| Farnesyl-diphosphate farnesyltransferase 1 | FDFT1 | 2.200 |
| RUSC1 antisense RNA 1 (non-protein coding) | RUSC1-AS1 | 2.074 |
| sterol-C5-desaturase (ERG3 delta-5-desaturase homolog, S. cerevisiae)-like | SC5DL | 1.802 |
| acyl-CoA synthetase short-chain family member 2 | ACSS2 | 1.797 |
| stearoyl-CoA desaturase (delta-9-desaturase) | SCD | 1.790 |
| acetyl-CoA acetyltransferase 2 | ACAT2 | 1.652 |
| chloride channel, voltage-sensitive 6 | CLCN6 | 1.641 |
| 24-dehydrocholesterol reductase | DHCR24 | 1.607 |
| methylenetetrahydrofolate reductase (NAD(P)H) | MTHFR | 1.603 |
| arrestin domain containing 3 | ARRDC3 | 1.582 |
| hydroxysteroid (17-beta) dehydrogenase 7 | HSD17B7 | 1.565 |
| syntaxin binding protein 1 | STXBP1 | 1.522 |
| chromosome 14 open reading frame 1 | C14orf1 | 1.512 |
| mevalonate kinase | MVK | 1.511 |
| Kruppel-like factor 6 | KLF6 | 1.481 |
| isocitrate dehydrogenase 1 (NADP+), soluble | IDH1 | 1.411 |
| mevalonate (diphospho) decarboxylase | MVD | 1.379 |
| sialidase 1 (lysosomal sialidase) | NEU1 | 1.371 |
| IDI2 antisense RNA 1 (non-protein coding) | IDI2-AS1 | 1.325 |
| family with sequence similarity 117, member A | FAM117A | 1.322 |
| Kruppel-like factor 7 (ubiquitous) | KLF7 | 1.296 |
| lipin 1 | LPIN1 | 1.294 |
| methyltransferase like 7A | METTL7A | 1.224 |
| RAB33A, member RAS oncogene family | RAB33A | 1.163 |
| centromere protein I | CENPI | 1.159 |
| jun proto-oncogene | JUN | 1.148 |
| MCM3AP antisense RNA 1 (non-protein coding) | MCM3AP-AS1 | 1.129 |
| cytoplasmic FMR1 interacting protein 2 | CYFIP2 | 1.127 |
| transmembrane 7 superfamily member 2 | TM7SF2 | 1.119 |
| Kruppel-like factor 2 (lung) | KLF2 | 1.094 |
| kelch-like 24 (Drosophila) | KLHL24 | 1.092 |
| pantothenate kinase 3 | PANK3 | 1.070 |
| NAD(P) dependent steroid dehydrogenase-like | NSDHL | 1.059 |
| ELOVL fatty acid elongase 6 | ELOVL6 | 1.050 |
| transmembrane protein 97 | TMEM97 | 1.019 |
| Down-Regulated Genes | ||
| Gene Title | Gene Symbol | log2ratio |
| myosin regulatory light chain interacting protein | MYLIP | −1.036 |
| phosphodiesterase 4B, cAMP-specific | PDE4B | −1.052 |
| regulator of G-protein signaling 18 | RGS18 | −1.092 |
| T cell receptor delta variable 3 | TRDV3 | −1.116 |
| SLAM family member 8 | SLAMF8 | −1.167 |
| C-type lectin domain family 5, member A | CLEC5A | −1.177 |
| ankyrin repeat domain 22 | ANKRD22 | −1.356 |
| Fc fragment of IgA, receptor for | FCAR | −1.432 |
| chemokine (C-C motif) ligand 2 | CCL2 | −1.803 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chan, S.-H.; Leu, W.-J.; Swain, S.P.; Hsu, J.-L.; Hou, D.-R.; Guh, J.-H. Mechanistic Study of Triazole Based Aminodiol Derivatives in Leukemic Cells—Crosstalk between Mitochondrial Stress-Involved Apoptosis and Autophagy. Int. J. Mol. Sci. 2020, 21, 2470. https://doi.org/10.3390/ijms21072470
Chan S-H, Leu W-J, Swain SP, Hsu J-L, Hou D-R, Guh J-H. Mechanistic Study of Triazole Based Aminodiol Derivatives in Leukemic Cells—Crosstalk between Mitochondrial Stress-Involved Apoptosis and Autophagy. International Journal of Molecular Sciences. 2020; 21(7):2470. https://doi.org/10.3390/ijms21072470
Chicago/Turabian StyleChan, She-Hung, Wohn-Jenn Leu, Sharada Prasanna Swain, Jui-Ling Hsu, Duen-Ren Hou, and Jih-Hwa Guh. 2020. "Mechanistic Study of Triazole Based Aminodiol Derivatives in Leukemic Cells—Crosstalk between Mitochondrial Stress-Involved Apoptosis and Autophagy" International Journal of Molecular Sciences 21, no. 7: 2470. https://doi.org/10.3390/ijms21072470