TMT-Based Quantitative Proteomic Analysis Reveals the Crucial Biological Pathways Involved in Self-Incompatibility Responses in Camellia oleifera

Abstract
:1. Introduction
2. Results
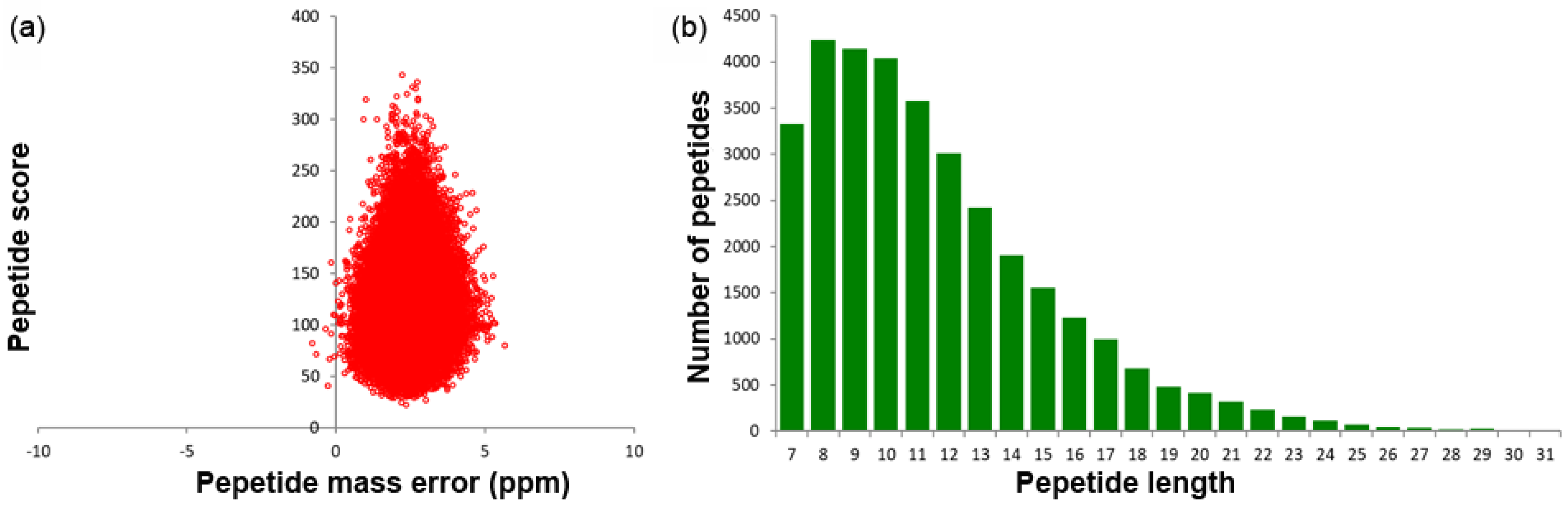
2.1. Identified Protein Overview
2.2. Protein Annotation
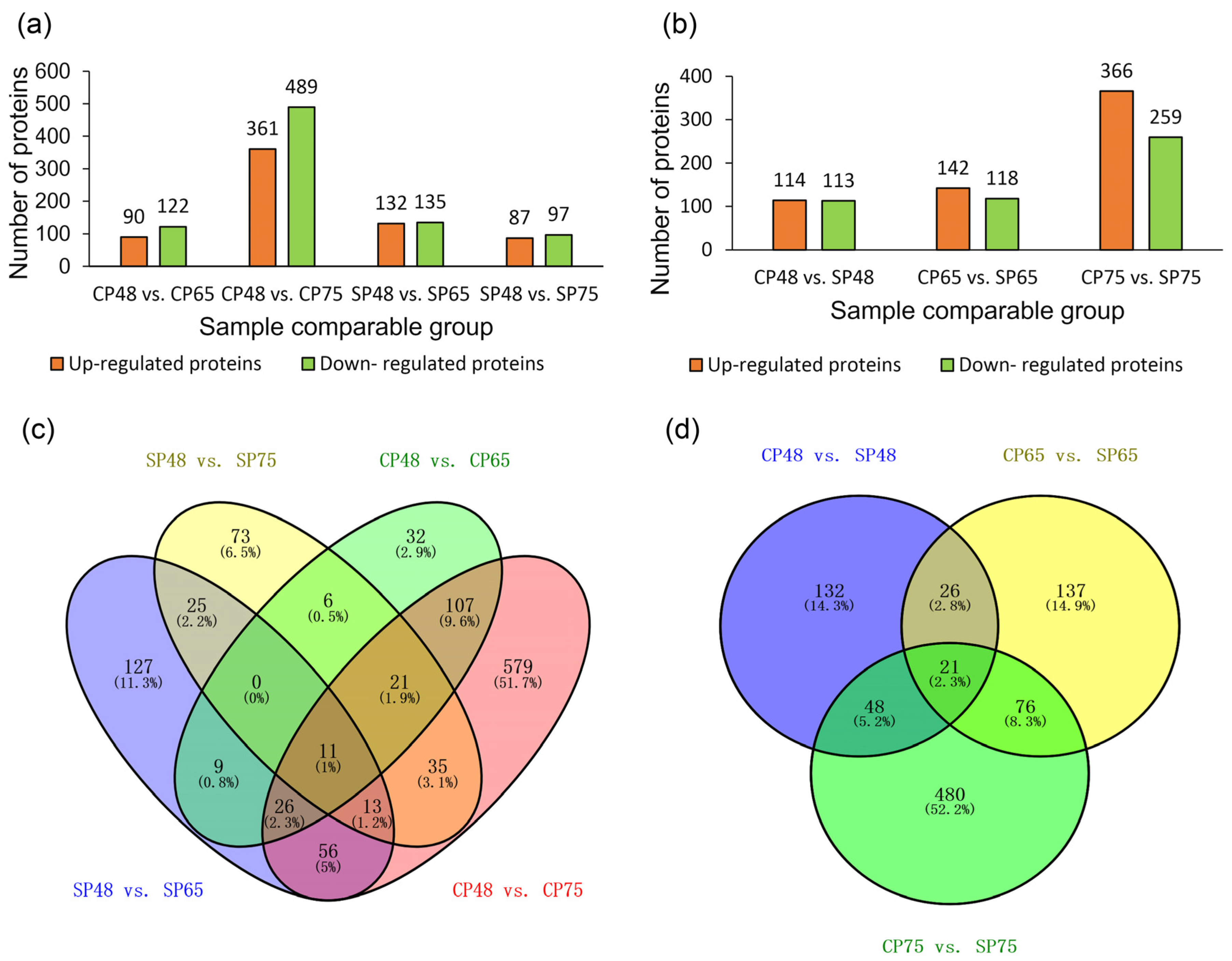
2.3. Identification and Analysis of Differentially Abundant Proteins
2.4. GO Enrichment Analysis of Pistils between Self- and Cross-Pollinated in C. oleifera
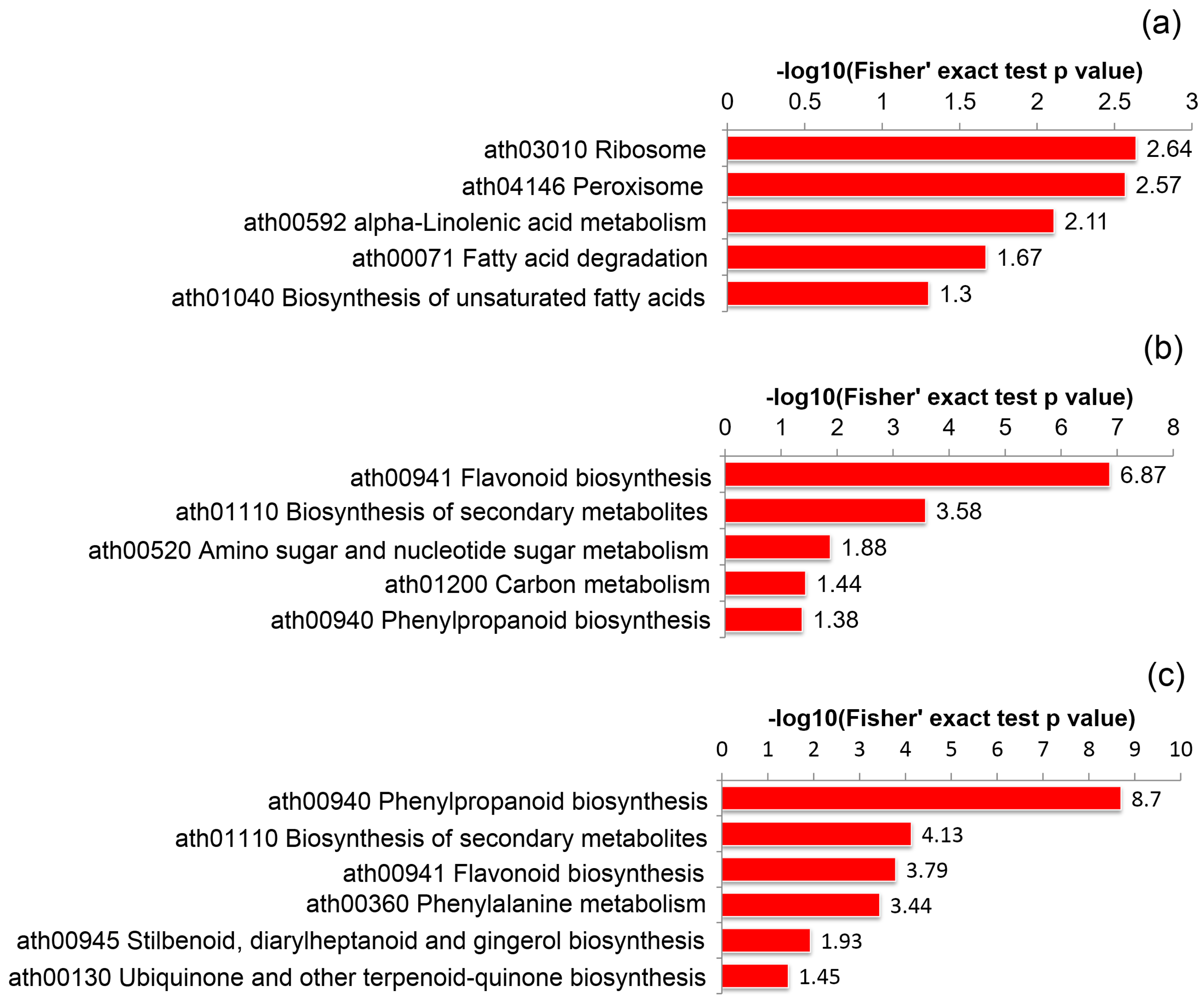
2.5. KEGG Pathway Enrichment Analysis of Self- and Cross Pollinated Pistils in C. Oleifera
2.6. Protein Domain Enrichment Analysis of Pistils between Self- and Cross-Pollinated in C. oleifera
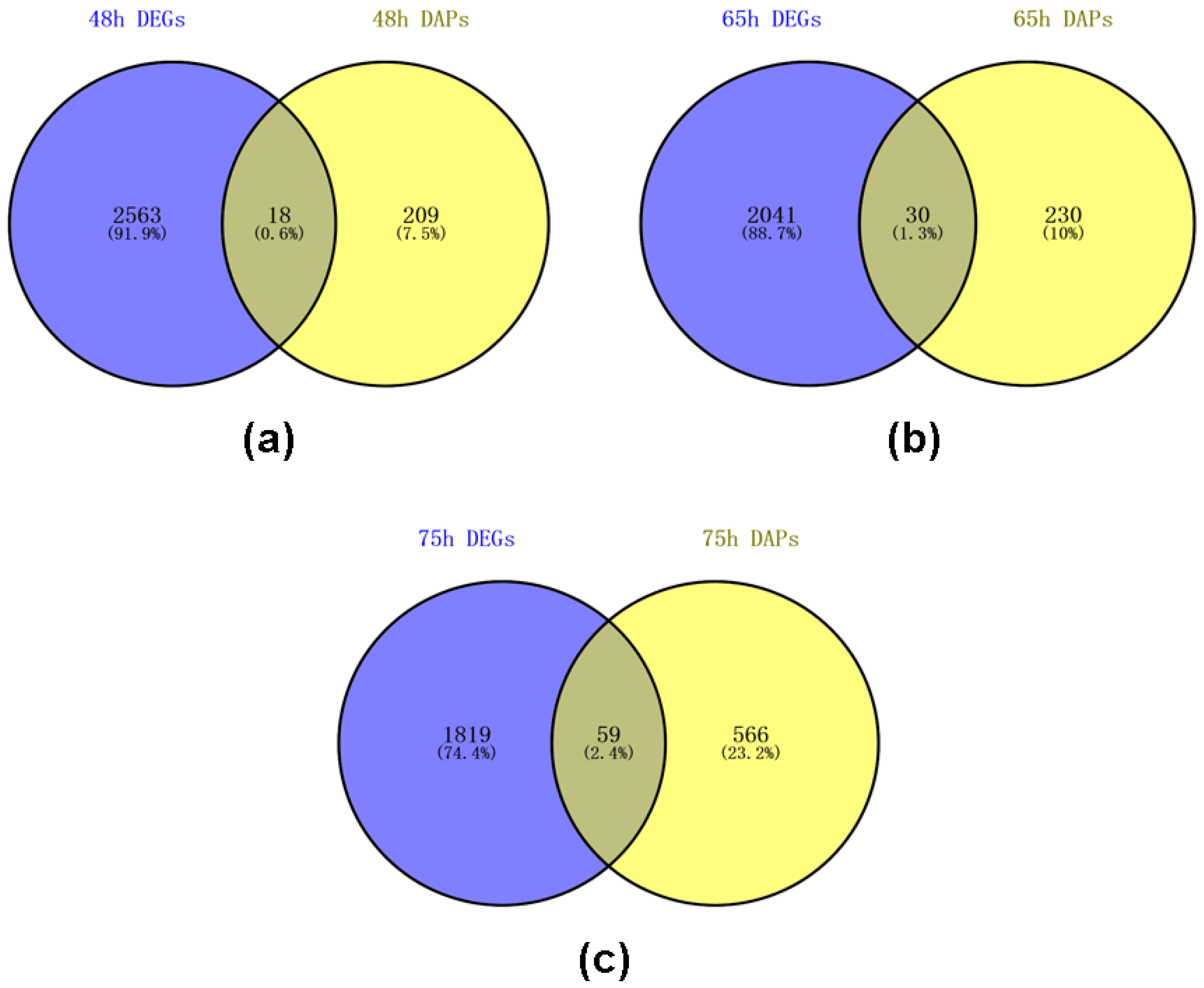
2.7. Venn Analysis of DEGs and DAPs of Self- and Cross Pollinated Pistils in C. oleifera Based on Transcriptome and Proteome Data
3. Discussion
3.1. Proteins Participated in Pollen Tube Growth in C. oleifera
3.2. Peroxisome and Lipid Metabolism Might Act A Key Role in SI Response of C. oleifera
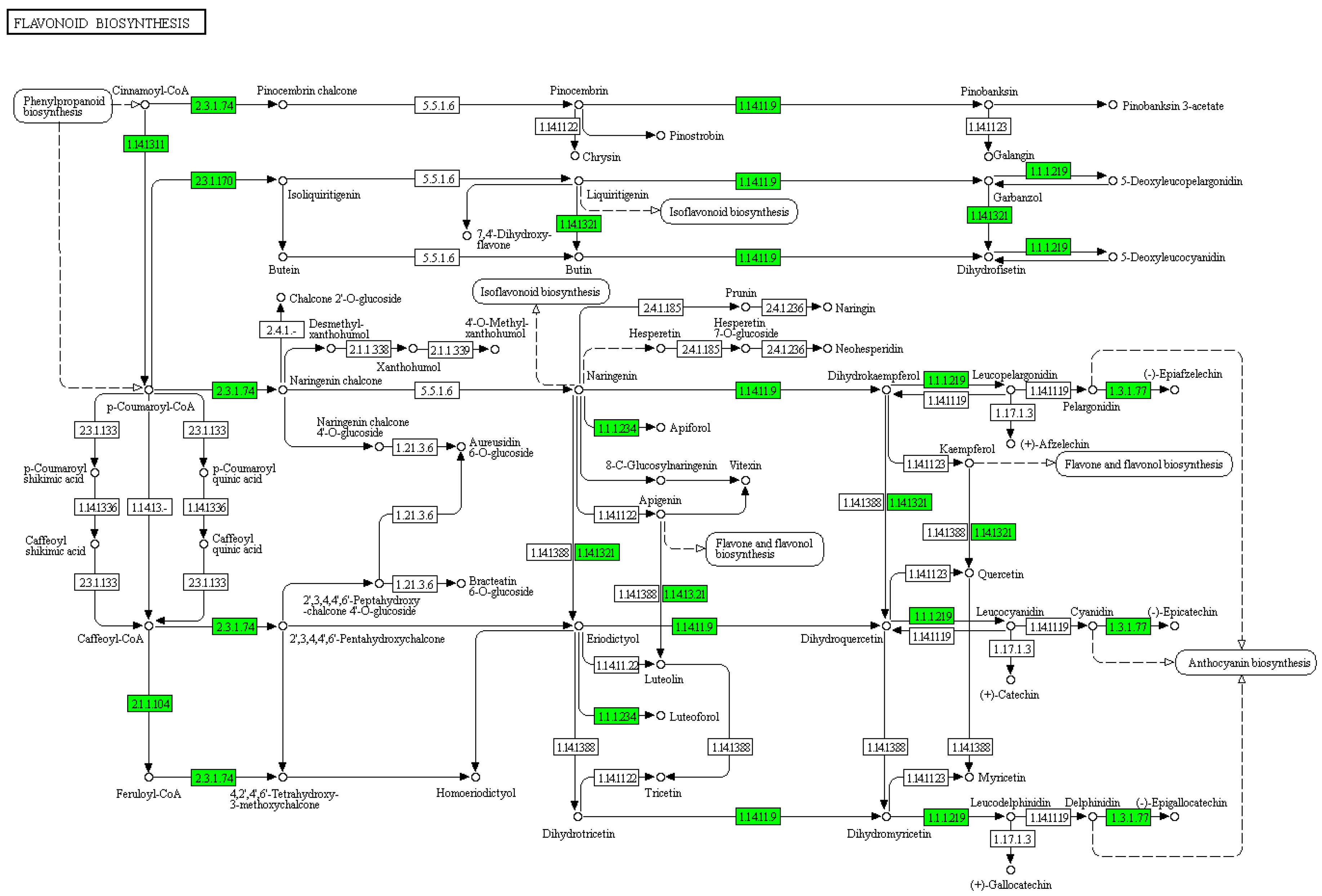
3.3. Flavonoids May Negatively Regulate SI Response in C. oleifera
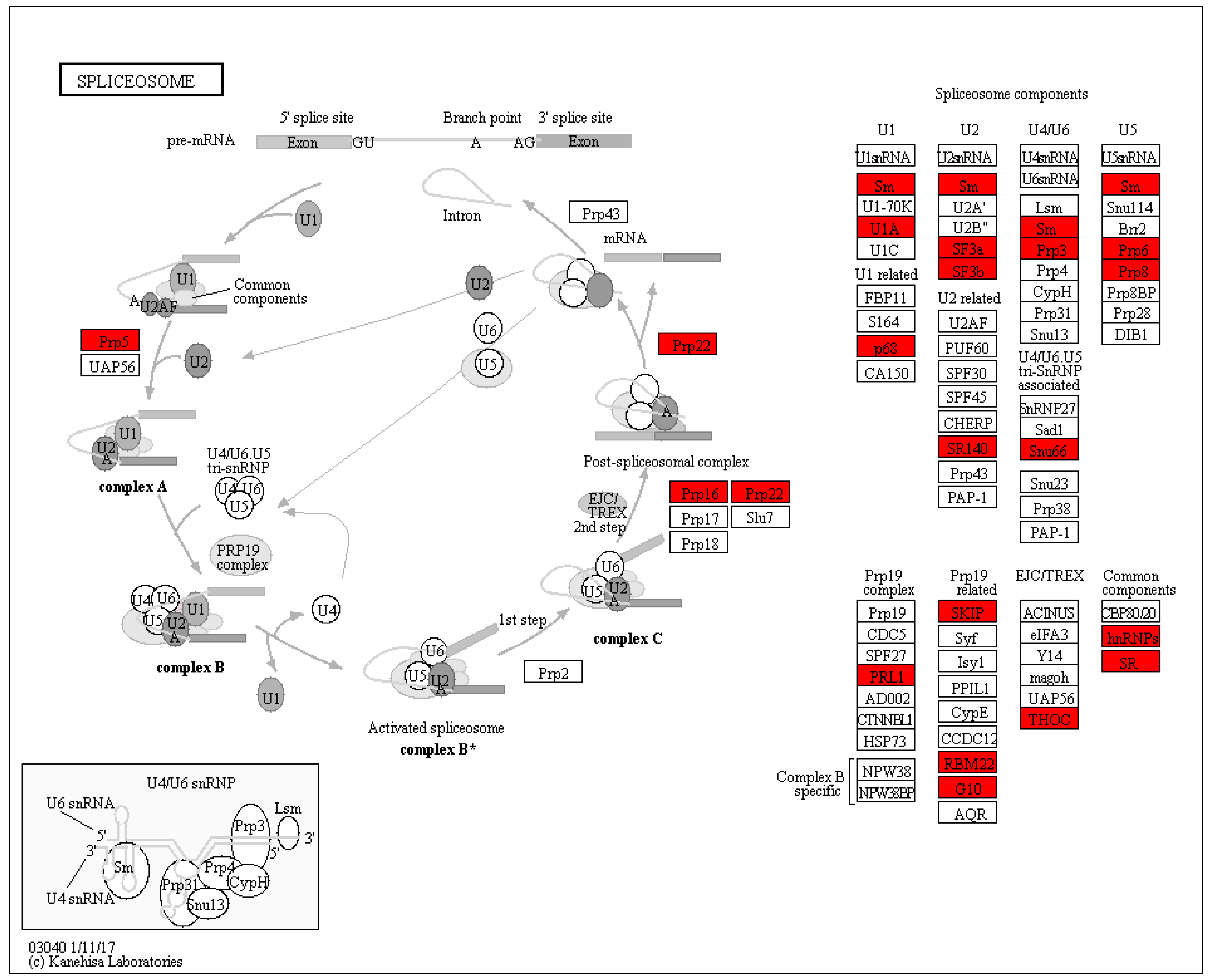
3.4. Splicesome Increased in SI Response in C. oleifera
3.5. Hypothetical Model Occurring in SI Response in C. oleifera
4. Materials and Methods
4.1. Plant Materials
4.2. Pollination Treatment
4.3. Protein Extraction
4.4. Trypsin Digestion
4.5. TMT Labeling
4.6. HPLC Fractionation
4.7. LC-MS/MS Analysis
4.8. Database Search
4.9. Data Bioinformatics Analyses
4.9.1. GO Annotation
4.9.2. Domain Annotation
4.9.3. KEGG Pathway Annotation
4.9.4. Subcellular Localization
4.9.5. Enrichment of Gene Ontology Analysis
4.9.6. Enrichment of Pathway Analysis
4.9.7. Enrichment of Protein Domain Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| ABA | Abscisic acid |
| AGC | Automatic gain control |
| ANR | Anthocyanidin reductase |
| APEM9 | Peroxisome morphology9 |
| ATO | Atropos |
| ATP | Phosphoenolpyruvate carboxykinase |
| CHS | Chalcone synthase |
| CP48 | 48 h after cross-pollination |
| CP65 | 65 h after cross-pollination |
| CP75 | 75 h after cross-pollination |
| DAPs | Differentially abundant proteins |
| DEGs | Differentially expressed genes |
| DFR | Dihydroflavonol4-reductase |
| FDR | false discovery rate |
| FLS | Flavanol synthase |
| GO | Gene Ontology |
| GSI | Gametophytic SI |
| KEGG | Kyoto Encyclopedia of Genes and Genomes |
| LSI | Late self-incompatibility |
| LRR | Leucine-rich repeat |
| NCED | 9-cis-epoxycarotenoid dioxygenase |
| PCD | Programmed cell death |
| PSM | peptide-spectrum match |
| RLKs | Receptor-like kinase |
| SFB | S haplotype-specific F-box protein gene |
| SI | Self-incompatibility |
| SLF | S-locus F-box protein |
| SSI | Sporophytic SI |
| SWA1 | Slow walker1 |
| SP48 | 48 h after self-pollination |
| SP65 | 65 h after self-pollination |
| SP75 | 75 h after self-pollination |
| TMT | Tandem mass tag |
| UTP15 | U3 small nucleolar ribonucleoprotein15 |
References
- Wheeler, M.J.; De Graaf, B.H.J.; Hadjiosif, N.; Perry, R.M.; Poulter, N.S.; Osman, K.; Vatovec, S.; Harper, A.; Franklin, F.C.H.; Franklin-Tong, V.E. Identification of the pollen self-incompatibility determinant in Papaver rhoeas. Nature 2009, 459, 992–995. [Google Scholar] [CrossRef]
- De Nettancourt, D. Incompatibility in angiosperms. Sex. Plant Reprod. 1997, 10, 185–199. [Google Scholar] [CrossRef]
- Franklin-Tong, V.E.; Ride, J.P.; Franklin, F.C.H. Recombinant stigmatic self-incompatibility (S-) protein elicits a Ca2+ transient in pollen of Papaver rhoeas. Plant J. 1995, 8, 299–307. [Google Scholar] [CrossRef]
- Zhou, Q.; Jia, J.; Huang, X.; Yan, X.; Cheng, L.; Chen, S.; Li, X.; Peng, X.; Liu, G. The large-scale investigation of gene expression in Leymus chinensis stigmas provides a valuable resource for understanding the mechanisms of poaceae self-incompatibility. BMC Genom. 2014, 15, 399. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, S.; Ding, F.; He, X.; Luo, C.; Huang, G.; Hu, Y. Characterization of the ‘Xiangshui’ lemon transcriptome by de novo assembly to discover genes associated with self-incompatibility. Mol. Genet. Genom. 2014, 290, 365–375. [Google Scholar] [CrossRef] [PubMed]
- Liao, T.; Yuan, D.-Y.; Zou, F.; Gao, C.; Yang, Y.; Zhang, L.; Tan, X.-F. Self-Sterility in Camellia oleifera May Be Due to the Prezygotic Late-Acting Self-Incompatibility. PLoS ONE 2014, 9, e99639. [Google Scholar] [CrossRef]
- Takayama, S.; Isogai, A. SELF-INCOMPATIBILITY IN PLANTS. Annu. Rev. Plant Boil. 2005, 56, 467–489. [Google Scholar] [CrossRef] [Green Version]
- Gibbs, P.E. Late-acting self-incompatibility-the pariah breeding system in flowering plants. New Phytol. 2014, 203, 717–734. [Google Scholar] [CrossRef]
- Allen, A.M.; Hiscock, S.J.; Franklin-Tong, V.E. Evolution and Phylogeny of Self-Incompatibility Systems in Angiosperms. In Self-Incompatibility in Flowering Plants; Springer: Berlin/Heidelberg, Germany, 2008; pp. 73–101. [Google Scholar]
- Zhou, Q.; Zheng, Y. Comparative De Novo Transcriptome Analysis of Fertilized Ovules in Xanthoceras sorbifolium Uncovered a Pool of Genes Expressed Specifically or Preferentially in the Selfed Ovule That Are Potentially Involved in Late-Acting Self-Incompatibility. PLoS ONE 2015, 10, e0140507. [Google Scholar] [CrossRef] [Green Version]
- Stein, J.C.; Howlett, B.; Boyes, D.C.; Nasrallah, M.E.; Nasrallah, J.B. Molecular cloning of a putative receptor protein kinase gene encoded at the self-incompatibility locus of Brassica oleracea. Proc. Natl. Acad. Sci. USA 1991, 88, 8816–8820. [Google Scholar] [CrossRef] [Green Version]
- Watanabe, M.; Suzuki, G.; Takayama, S.; Franklin-Tong, V.E. Milestones Identifying Self-Incompatibility Genes in Brassica Species: From Old Stories to New Findings. In Self-Incompatibility in Flowering Plants; Springer Science and Business Media LLC: Berlin, Germany, 2008; pp. 151–172. [Google Scholar]
- Schopfer, C.R. The Male Determinant of Self-Incompatibility in Brassica. Science 1999, 286, 1697–1700. [Google Scholar] [CrossRef] [PubMed]
- Takasaki, T.; Hatakeyama, K.; Suzuki, G.; Watanabe, M.; Isogai, A.; Hinata, K. The S receptor kinase determines self-incompatibility in Brassica stigma. Nature 2000, 403, 913–916. [Google Scholar] [CrossRef] [PubMed]
- Murase, K.; Shiba, H.; Iwano, M.; Che, F.-S.; Watanabe, M.; Isogai, A.; Takayama, S. A Membrane-Anchored Protein Kinase Involved in Brassica Self-Incompatibility Signaling. Science 2004, 303, 1516–1519. [Google Scholar] [CrossRef] [PubMed]
- Kakita, M.; Murase, K.; Iwano, M.; Matsumoto, T.; Watanabe, M.; Shiba, H.; Isogai, A.; Takayama, S. Two Distinct Forms of M-Locus Protein Kinase Localize to the Plasma Membrane and Interact Directly with S-Locus Receptor Kinase to Transduce Self-Incompatibility Signaling in Brassica rapa. Plant Cell 2007, 19, 3961–3973. [Google Scholar] [CrossRef] [Green Version]
- Murfett, J.; Atherton, T.L.; Mou, B.; Gassert, C.S.; McClure, B. S-RNase expressed in transgenic Nicotiana causes S-allele-specific pollen rejection. Nature 1994, 367, 563–566. [Google Scholar] [CrossRef]
- Sijacic, P.; Wang, X.; Skirpan, A.L.; Wang, Y.; Dowd, P.E.; McCubbin, A.G.; Huang, S.; Kao, T.-H. Identification of the pollen determinant of S-RNase-mediated self-incompatibility. Nature 2004, 429, 302–305. [Google Scholar] [CrossRef]
- McClure, B. New views of S-RNase-based self-incompatibility. Curr. Opin. Plant Boil. 2006, 9, 639–646. [Google Scholar] [CrossRef]
- Goldraij, A.; Kondo, K.; Lee, C.B.; Hancock, C.N.; Sivaguru, M.; Vazquez-Santana, S.; Kim, S.; Phillips, T.E.; Cruz-Garcia, F.; McClure, B. Compartmentalization of S-RNase and HT-B degradation in self-incompatible Nicotiana. Nature 2006, 439, 805–810. [Google Scholar] [CrossRef]
- Thomas, S.; Franklin-Tong, V.E. Self-incompatibility triggers programmed cell death in Papaver pollen. Nature 2004, 429, 305–309. [Google Scholar] [CrossRef]
- Ladoux, T.; Friar, E.A. Late-Acting Self-Incompatibility inIpomopsis tenuifolia(Gray) V. Grant (Polemoniaceae). Int. J. Plant Sci. 2006, 167, 463–471. [Google Scholar] [CrossRef]
- Cope, F.W. The mechanism of pollen incompatibility in Theobroma cacao L. Heredity 1962, 17, 157–182. [Google Scholar] [CrossRef] [Green Version]
- Seavey, S.R.; Bawa, K. Late-acting self-incompatibility in angiosperms. Bot. Rev. 1986, 52, 195–219. [Google Scholar] [CrossRef]
- Lipow, S.R.; Wyatt, R. Single gene control of postzygotic self-incompatibility in poke milkweed, Asclepias exaltata L. Genetics 2000, 154, 893–907. [Google Scholar] [PubMed]
- Sage, T.L.; Bertin, R.I.; Williams, E.G. Ovarian and Other Late-Acting Self-Incompatibility Systems; Springer Science and Business Media LLC: Berlin, Germany, 1994; Volume 2, pp. 116–140. [Google Scholar]
- Sage, T.; Williams, E. Structure, ultrastructure, and histochemistry of the pollen tube pathway in the milkweed Asclepias exaltata L. Sex. Plant Reprod. 1995, 8, 257–265. [Google Scholar] [CrossRef]
- Yu, Y.; Ren, S.; Tan, K. Study on climatic regionalization and layer and belt distribution of oiltea camellia quality in China. J. Natl. Res. 1999, 14, 123–127. [Google Scholar]
- Lee, C.-P.; Yen, G.-C. Antioxidant Activity and Bioactive Compounds of Tea Seed (Camellia oleiferaAbel.) Oil. J. Agric. Food Chem. 2006, 54, 779–784. [Google Scholar] [CrossRef]
- He, Y.; Song, Q.; Chen, S.; Wu, Y.; Zheng, G.; Feng, J.; Yang, Z.; Lin, W.; Li, Y.; Chen, H. Transcriptome analysis of self- and cross-pollinated pistils revealing candidate unigenes of self-incompatibility in Camellia oleifera. J. Hortic. Sci. Biotechnol. 2019, 95, 19–31. [Google Scholar] [CrossRef]
- Shahali, Y.; Sutra, J.-P.; Fasoli, E.; D’Amato, A.; Righetti, P.G.; Futamura, N.; Boschetti, E.; Sénéchal, H.; Poncet, P. Allergomic study of cypress pollen via combinatorial peptide ligand libraries. J. Proteom. 2012, 77, 101–110. [Google Scholar] [CrossRef]
- Qin, X.; Zeevaart, J.A.D. Overexpression of a 9-cis-Epoxycarotenoid Dioxygenase Gene in Nicotiana plumbaginifolia Increases Abscisic Acid and Phaseic Acid Levels and Enhances Drought Tolerance1. Plant Physiol. 2002, 128, 544–551. [Google Scholar] [CrossRef]
- Martínez-Andújar, C.; Ordiz, M.I.; Huang, Z.; Nonogaki, M.; Beachy, R.N.; Nonogaki, H. Induction of 9-cis-epoxycarotenoid dioxygenase in Arabidopsis thaliana seeds enhances seed dormancy. Proc. Natl. Acad. Sci. USA 2011, 108, 17225–17229. [Google Scholar] [CrossRef] [Green Version]
- Yoshida, K.; Kaothien-Nakayama, P.; Matsui, T.; Kawaoka, A.; Shinmyo, A. Molecular biology and application of plant peroxidase genes. Appl. Microbiol. Biotechnol. 2003, 60, 665–670. [Google Scholar] [CrossRef] [PubMed]
- McInnis, S.M.; Costa, L.M.; Gutierrez-Marcos, J.F.; Henderson, C.A.; Hiscock, S.J. Isolation and characterization of a polymorphic stigma-specific class III peroxidase gene from Senecio squalidus L. (Asteraceae). Plant Mol. Boil. 2005, 57, 659–677. [Google Scholar] [CrossRef] [PubMed]
- Taylor, L.P.; Hepler, P.K. POLLEN GERMINATION AND TUBE GROWTH. Annu. Rev. Plant Boil. 1997, 48, 461–491. [Google Scholar] [CrossRef] [PubMed]
- Jones, D. Phenylalanine ammonia-lyase: Regulation of its induction, and its role in plant development. Phytochemistry 1984, 23, 1349–1359. [Google Scholar] [CrossRef]
- Chai, S.; Ge, F.; Li, S.; Zhang, Y. The journey to glory: Receptor-like kinases in pollen tube growth. Sci. Bull. 2016, 61, 827–831. [Google Scholar] [CrossRef]
- Pan, R.; Liu, J.; Hu, J. Peroxisomes in plant reproduction and seed-related development. J. Integr. Plant Boil. 2019, 61, 784–802. [Google Scholar] [CrossRef] [Green Version]
- Kaur, N.; Reumann, S.; Hu, J. Peroxisome Biogenesis and Function. Arab. Book 2009, 7, e0123. [Google Scholar] [CrossRef]
- Kao, Y.-T.; Gonzalez, K.; Bartel, B. Peroxisome Function, Biogenesis, and Dynamics in Plants. Plant Physiol. 2017, 176, 162–177. [Google Scholar] [CrossRef] [Green Version]
- Pan, R.; Satkovich, J.; Chen, C.; Hu, J. The E3 ubiquitin ligase SP1-like 1 plays a positive role in peroxisome biogenesis in Arabidopsis. Plant J. 2018, 94, 836–846. [Google Scholar] [CrossRef] [Green Version]
- Li, X.; Li, H.-J.; Yuan, L.; Liu, M.; Shi, D.-Q.; Liu, J.; Yang, W.-C. Arabidopsis DAYU/ABERRANT PEROXISOME MORPHOLOGY9 Is a Key Regulator of Peroxisome Biogenesis and Plays Critical Roles during Pollen Maturation and Germination in Planta. Plant Cell 2014, 26, 619–635. [Google Scholar] [CrossRef] [Green Version]
- Wolters-Arts, M.; Lush, W.M.; Mariani, C. Lipids are required for directional pollen-tube growth. Nature 1998, 392, 818–821. [Google Scholar] [CrossRef] [PubMed]
- Footitt, S.; Dietrich, D.; Fait, A.; Fernie, A.R.; Holdsworth, M.J.; Baker, A.; Theodoulou, F. The COMATOSE ATP-Binding Cassette Transporter Is Required for Full Fertility in Arabidopsis1. Plant Physiol. 2007, 144, 1467–1480. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grotewold, E. THE GENETICS AND BIOCHEMISTRY OF FLORAL PIGMENTS. Annu. Rev. Plant Boil. 2006, 57, 761–780. [Google Scholar] [CrossRef] [PubMed]
- Mierziak, J.; Kostyn, K.; Kulma, A. Flavonoids as Important Molecules of Plant Interactions with the Environment. Molecules 2014, 19, 16240–16265. [Google Scholar] [CrossRef]
- Saito, K.; Yonekura-Sakakibara, K.; Nakabayashi, R.; Higashi, Y.; Yamazaki, M.; Tohge, T.; Fernie, A.R. The flavonoid biosynthetic pathway in Arabidopsis: Structural and genetic diversity. Plant Physiol. Biochem. 2013, 72, 21–34. [Google Scholar] [CrossRef] [Green Version]
- Martinez, V.; Mestre, T.C.; Rubio, F.; Girones-Vilaplana, A.; Moreno, D.A.; Mittler, R.; Rivero, R.M. Accumulation of Flavonols over Hydroxycinnamic Acids Favors Oxidative Damage Protection under Abiotic Stress. Front. Plant Sci. 2016, 7, 838. [Google Scholar] [CrossRef]
- Schenke, D.; Utami, H.P.; Zhou, Z.; Gallegos, M.-T.; Cai, D. Suppression of UV-B stress induced flavonoids by biotic stress: Is there reciprocal crosstalk? Plant Physiol. Biochem. 2019, 134, 53–63. [Google Scholar] [CrossRef]
- Winkel, B.S.J. Biosynthesis of flavonoids and effects of stress. Curr. Opin. Plant Boil. 2002, 5, 218–223. [Google Scholar] [CrossRef]
- Van Der Meer, I.M.; Stam, M.E.; Van Tunen, A.J.; Mol, J.N.; Stuitje, A.R. Antisense inhibition of flavonoid biosynthesis in petunia anthers results in male sterility. Plant Cell 1992, 4, 253–262. [Google Scholar]
- Mo, Y.; Nagel, C.; Taylor, L.P. Biochemical complementation of chalcone synthase mutants defines a role for flavonols in functional pollen. Proc. Natl. Acad. Sci. USA 1992, 89, 7213–7217. [Google Scholar] [CrossRef] [Green Version]
- Ylstra, B.; Busscher, J.; Franken, J.; Hollman, P.C.; Mol, J.N.; Van Tunen, A.J. Flavonols and fertilization in Petunia hybrida: Localization and mode of action during pollen tube growth. Plant J. 1994, 6, 201–212. [Google Scholar] [CrossRef]
- Vogt, T.; Pollak, P.; Tarlyn, N.; Taylor, L.P. Pollination- or Wound-Induced Kaempferol Accumulation in Petunia Stigmas Enhances Seed Production. Plant Cell 1994, 6, 11–23. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, P.; Vogt, T.; Taylor, L.P. Uptake and metabolism of flavonols during in-vitro germination of Petunia hybrida (L.) pollen. Planta 1997, 202, 257–265. [Google Scholar] [CrossRef]
- Derksen, J.; Knuiman, B.; Ylstra, B.; Van Tunen, A.J. Pollen tubes of flavonol-deficient Petunia show striking alterations in wall structure leading to tube disruption. Planta 1999, 207, 575–581. [Google Scholar] [CrossRef]
- Miller, K.D.; Guyon, V.; Evans, J.N.S.; Shuttleworth, W.A.; Taylor, L.P. Purification, Cloning, and Heterologous Expression of a Catalytically Efficient Flavonol 3-O-Galactosyltransferase Expressed in the Male Gametophyte ofPetunia hybrida. J. Boil. Chem. 1999, 274, 34011–34019. [Google Scholar] [CrossRef] [Green Version]
- Guyon, V.N. Isolation and Characterization of cDNAs Expressed in the Early Stages of Flavonol-Induced Pollen Germination in Petunia. Plant Physiol. 2000, 123, 699–710. [Google Scholar] [CrossRef] [Green Version]
- Taylor, L.P.; Jorgensen, R. Conditional Male Fertility in Chalcone Synthase-Deficient Petunia. J. Hered. 1992, 83, 11–17. [Google Scholar] [CrossRef]
- Liu, Y.; Yan, Z.; Chen, N.; Di, X.; Huang, J.; Guo, G. Development and function of central cell in angiosperm female gametophyte. Genesis 2010, 48, 466–478. [Google Scholar] [CrossRef]
- Moll, C.; Von Lyncker, L.; Zimmermann, S.; Kägi, C.; Baumann, N.; Twell, D.; Grossniklaus, U.; Groß-Hardt, R. CLO/GFA1andATOare novel regulators of gametic cell fate in plants. Plant J. 2008, 56, 913–921. [Google Scholar] [CrossRef]
- Gross-Hardt, R.; Kägi, C.; Baumann, N.; Moore, J.M.; Baskar, R.; Gagliano, W.B.; Jürgens, G.; Grossniklaus, U.; Groß-Hardt, R. LACHESIS restricts gametic cell fate in the female gametophyte of Arabidopsis. PLoS Boil. 2007, 5, e47. [Google Scholar] [CrossRef]
- Ishida, T.; Maekawa, S.; Yanagisawa, S. The Pre-rRNA Processing Complex in Arabidopsis Includes Two WD40-Domain-Containing Proteins Encoded by Glucose-Inducible Genes and Plant-Specific Proteins. Mol. Plant 2016, 9, 312–315. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Total Spectrums | Matched Spetrums | Peptides | Unique Peptides | Identified Proteins | Quantifiable Proteins |
|---|---|---|---|---|---|
| 318,969 | 54,545 | 33,088 | 31,071 | 7666 | 6616 |
| Cellular Component | |
|---|---|
| intracellular non-membrane-bounded organelle | |
| non-membrane-bounded organelle | |
| ribonucleoprotein complex | |
| intracellular ribonucleoprotein complex | |
| ribosome | |
| Molecular Function | |
| structural molecule activity | |
| Biological Process | |
| oxidation-reduction process |
| Protein Accession | Protein Description | Subcellular Localization |
|---|---|---|
| c102785_g1 | Sigma factor sigb regulation protein rsbq | cytoplasm |
| c121964_g2 | 9-cis-epoxycarotenoid dioxygenase 3 protein family isoform 2 | mitochondria |
| c124653_g1 | Class III peroxidase | chloroplast |
| c130353_g1 | Alpha-copaene synthase | nucleus |
| c130529_g2 | Ubiquitin-40S ribosomal protein S27a isoform 1 | nucleus |
| c131407_g2 | Phenylalanine ammonia-lyase | chloroplast |
| c132935_g1 | Formamidase-like isoform 1 | nucleus |
| c134412_g1 | Terpene synthase | cytoplasm |
| c134849_g2 | (E)-beta-caryophyllene synthase | cytoskeleton |
| c135487_g1 | Receptor-like protein kinase HAIKU2-like | nucleus |
| c82718_g1 | 22.7 kDa class IV heat shock protein-like | chloroplast |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
He, Y.; Song, Q.; Wu, Y.; Ye, S.; Chen, S.; Chen, H. TMT-Based Quantitative Proteomic Analysis Reveals the Crucial Biological Pathways Involved in Self-Incompatibility Responses in Camellia oleifera. Int. J. Mol. Sci. 2020, 21, 1987. https://doi.org/10.3390/ijms21061987
He Y, Song Q, Wu Y, Ye S, Chen S, Chen H. TMT-Based Quantitative Proteomic Analysis Reveals the Crucial Biological Pathways Involved in Self-Incompatibility Responses in Camellia oleifera. International Journal of Molecular Sciences. 2020; 21(6):1987. https://doi.org/10.3390/ijms21061987
Chicago/Turabian StyleHe, Yifan, Qianqian Song, Yuefeng Wu, Shutao Ye, Shipin Chen, and Hui Chen. 2020. "TMT-Based Quantitative Proteomic Analysis Reveals the Crucial Biological Pathways Involved in Self-Incompatibility Responses in Camellia oleifera" International Journal of Molecular Sciences 21, no. 6: 1987. https://doi.org/10.3390/ijms21061987