Immune Response Dysfunction in Chronic Lymphocytic Leukemia: Dissecting Molecular Mechanisms and Microenvironmental Conditions

 ,
, 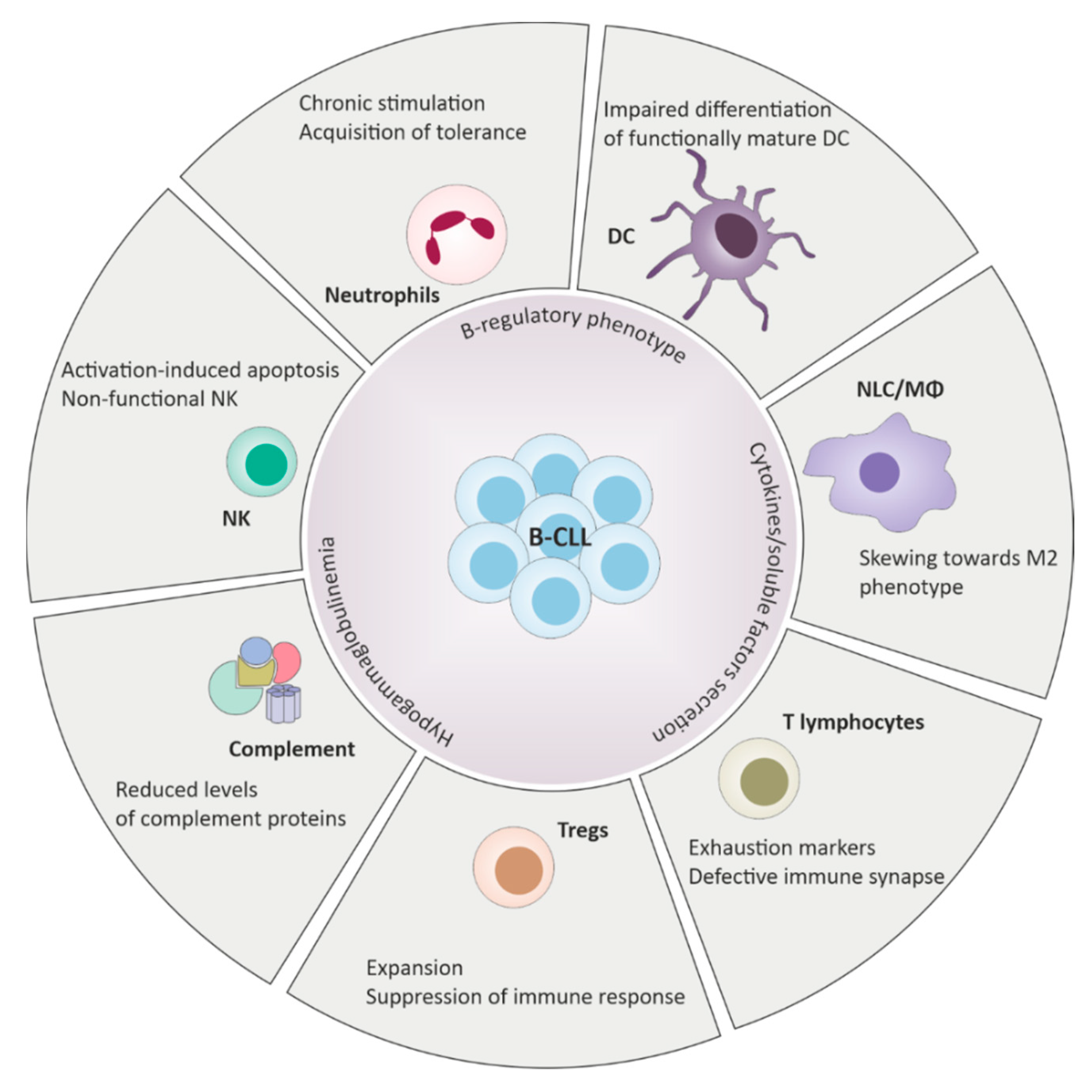{kind=link}
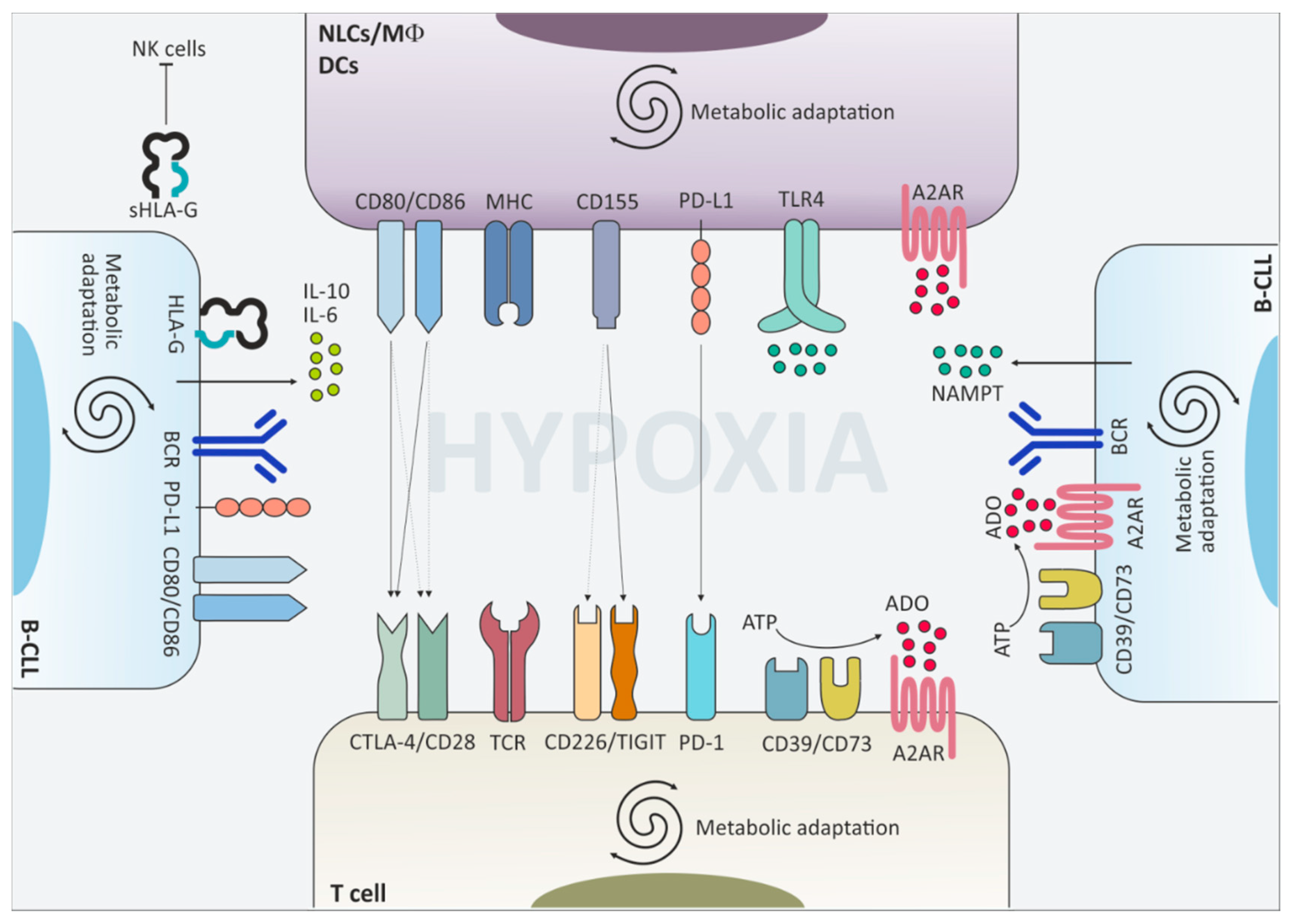{kind=link}
Abstract
:1. Introduction
2. Innate Immune Response Dysfunction in Chronic Lymphocytic Leukemia (CLL)
3. Adaptive Immune Response Dysfunction in CLL
4. Molecular Mechanisms Driving Immune Response Dysfunction in CLL
4.1. Cytotoxic T Lymphocyte Antigen-4 (CTLA-4)/B7
4.2. Programmed Cell Death 1 (PD-1)/Programmed Cell Death Ligand 1 (PD-L1)
4.3. T Cell Immunoreceptor with Immunoglobulin and Immunoreceptor Tyrosine-Based Inhibition Motif (ITIM) Domains (TIGIT)
4.4. HLA-G
5. Environmental Mechanisms Driving Immune Response Dysfunction in CLL
5.1. The B Cell Receptor
5.2. Hypoxia and Metabolic Adaptation
5.3. Adenosine Signaling
6. Concluding Remarks
Acknowledgments
Conflicts of Interest
Abbreviations
| IL-10 | interleukin-10 |
| SOCS5 | suppressor of cytokine signaling 5 |
| STAT6 | Signal transducer and activator of transcription 6 |
| HLA | Human leukocyte antigen |
| TNF | tumor necrosis factor |
| FOXP3 | forkhead box P3 |
| SLAM | signaling lymphocytic activation molecules |
| IFNg | interferon gamma |
| SHIP1 | Src homology 2 (SH2) domain containing inositol polyphosphate 5-phosphatase 1 |
| NF-kB | nuclear factor kB |
| PI3K | Phosphoinositide 3-kinases |
| MAPK | mitogen-activated protein kinase |
| VEGFA | vascular endothelial growth factor A |
| IRF4 | interferon regulatory factor |
References
- Chiorazzi, N.; Rai, K.R.; Ferrarini, M. Chronic lymphocytic leukemia. N. Engl. J. Med. 2005, 352, 804–815. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Puente, X.S.; Jares, P.; Campo, E. Chronic lymphocytic leukemia and mantle cell lymphoma: Crossroads of genetic and microenvironment interactions. Blood 2018, 131, 2283–2296. [Google Scholar] [CrossRef]
- Solomon, B.M.; Rabe, K.G.; Slager, S.L.; Brewer, J.D.; Cerhan, J.R.; Shanafelt, T.D. Overall and cancer-specific survival of patients with breast, colon, kidney, and lung cancers with and without chronic lymphocytic leukemia: A SEER population-based study. J. Clin. Oncol. 2013, 31, 930–937. [Google Scholar] [CrossRef]
- Teh, B.W.; Tam, C.S.; Handunnetti, S.; Worth, L.J.; Slavin, M.A. Infections in patients with chronic lymphocytic leukaemia: Mitigating risk in the era of targeted therapies. Blood Rev. 2018, 32, 499–507. [Google Scholar] [CrossRef] [PubMed]
- Balducci, L.; Dolan, D. Chronic Lymphocytic Leukemia in the Elderly: Epidemiology and Proposed Patient-Related Approach. Cancer Control 2015, 22, 3–6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thursky, K.A.; Worth, L.J.; Seymour, J.F.; Miles Prince, H.; Slavin, M.A. Spectrum of infection, risk and recommendations for prophylaxis and screening among patients with lymphoproliferative disorders treated with alemtuzumab*. Br. J. Haematol. 2006, 132, 3–12. [Google Scholar] [CrossRef]
- Morrison, V.A. Infectious complications of chronic lymphocytic leukaemia: Pathogenesis, spectrum of infection, preventive approaches. Best Pract. Res. Clin. Haematol. 2010, 23, 145–153. [Google Scholar] [CrossRef]
- Forconi, F.; Moss, P. Perturbation of the normal immune system in patients with CLL. Blood 2015, 126, 573–581. [Google Scholar] [CrossRef] [Green Version]
- Fattizzo, B.; Barcellini, W. Autoimmune Cytopenias in Chronic Lymphocytic Leukemia: Focus on Molecular Aspects. Front. Oncol. 2020, 9. [Google Scholar] [CrossRef]
- Burger, J.A.; Tsukada, N.; Burger, M.; Zvaifler, N.J.; Dell’Aquila, M.; Kipps, T.J. Blood-derived nurse-like cells protect chronic lymphocytic leukemia B cells from spontaneous apoptosis through stromal cell-derived factor-1. Blood 2000, 96, 2655–2663. [Google Scholar] [CrossRef]
- Tsukada, N.; Burger, J.A.; Zvaifler, N.J.; Kipps, T.J. Distinctive features of “nurselike” cells that differentiate in the context of chronic lymphocytic leukemia. Blood 2002, 99, 1030–1037. [Google Scholar] [CrossRef] [PubMed]
- Maffei, R.; Bulgarelli, J.; Fiorcari, S.; Bertoncelli, L.; Martinelli, S.; Guarnotta, C.; Castelli, I.; Deaglio, S.; Debbia, G.; De Biasi, S.; et al. The monocytic population in chronic lymphocytic leukemia shows altered composition and deregulation of genes involved in phagocytosis and inflammation. Haematologica 2013, 98, 1115–1123. [Google Scholar] [CrossRef] [PubMed]
- Filip, A.A.; Cisel, B.; Koczkodaj, D.; Wasik-Szczepanek, E.; Piersiak, T.; Dmoszynska, A. Circulating microenvironment of CLL: Are nurse-like cells related to tumor-associated macrophages? Blood Cells Mol. Dis. 2013, 50, 263–270. [Google Scholar] [CrossRef] [PubMed]
- Jurado-Camino, T.; Cordoba, R.; Esteban-Burgos, L.; Hernandez-Jimenez, E.; Toledano, V.; Hernandez-Rivas, J.A.; Ruiz-Sainz, E.; Cobo, T.; Siliceo, M.; Perez de Diego, R.; et al. Chronic lymphocytic leukemia: A paradigm of innate immune cross-tolerance. J. Immunol. 2015, 194, 719–727. [Google Scholar] [CrossRef] [Green Version]
- Nishio, M.; Endo, T.; Tsukada, N.; Ohata, J.; Kitada, S.; Reed, J.C.; Zvaifler, N.J.; Kipps, T.J. Nurselike cells express BAFF and APRIL, which can promote survival of chronic lymphocytic leukemia cells via a paracrine pathway distinct from that of SDF-1alpha. Blood 2005, 106, 1012–1020. [Google Scholar] [CrossRef] [Green Version]
- Deaglio, S.; Vaisitti, T.; Bergui, L.; Bonello, L.; Horenstein, A.L.; Tamagnone, L.; Boumsell, L.; Malavasi, F. CD38 and CD100 lead a network of surface receptors relaying positive signals for B-CLL growth and survival. Blood 2005, 105, 3042–3050. [Google Scholar] [CrossRef]
- Ferretti, E.; Bertolotto, M.; Deaglio, S.; Tripodo, C.; Ribatti, D.; Audrito, V.; Blengio, F.; Matis, S.; Zupo, S.; Rossi, D.; et al. A novel role of the CX3CR1/CX3CL1 system in the cross-talk between chronic lymphocytic leukemia cells and tumor microenvironment. Leukemia 2011, 25, 1268–1277. [Google Scholar] [CrossRef] [Green Version]
- Orsini, E.; Guarini, A.; Chiaretti, S.; Mauro, F.R.; Foa, R. The circulating dendritic cell compartment in patients with chronic lymphocytic leukemia is severely defective and unable to stimulate an effective T-cell response. Cancer Res. 2003, 63, 4497–4506. [Google Scholar]
- Toniolo, P.A.; Liu, S.; Yeh, J.E.; Ye, D.Q.; Barbuto, J.A.; Frank, D.A. Deregulation of SOCS5 suppresses dendritic cell function in chronic lymphocytic leukemia. Oncotarget 2016, 7, 46301–46314. [Google Scholar] [CrossRef] [Green Version]
- Gustafson, M.P.; Abraham, R.S.; Lin, Y.; Wu, W.; Gastineau, D.A.; Zent, C.S.; Dietz, A.B. Association of an increased frequency of CD14+ HLA-DR lo/neg monocytes with decreased time to progression in chronic lymphocytic leukaemia (CLL). Br. J. Haematol. 2012, 156, 674–676. [Google Scholar] [CrossRef]
- Jitschin, R.; Braun, M.; Buttner, M.; Dettmer-Wilde, K.; Bricks, J.; Berger, J.; Eckart, M.J.; Krause, S.W.; Oefner, P.J.; Le Blanc, K.; et al. CLL-cells induce IDOhi CD14+HLA-DRlo myeloid-derived suppressor cells that inhibit T-cell responses and promote TRegs. Blood 2014, 124, 750–760. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huergo-Zapico, L.; Acebes-Huerta, A.; Gonzalez-Rodriguez, A.P.; Contesti, J.; Gonzalez-Garcia, E.; Payer, A.R.; Villa-Alvarez, M.; Fernandez-Guizan, A.; Lopez-Soto, A.; Gonzalez, S. Expansion of NK cells and reduction of NKG2D expression in chronic lymphocytic leukemia. Correlation with progressive disease. PLoS ONE 2014, 9, e108326. [Google Scholar] [CrossRef] [PubMed]
- MacFarlane, A.W.T.; Jillab, M.; Smith, M.R.; Alpaugh, R.K.; Cole, M.E.; Litwin, S.; Millenson, M.M.; Al-Saleem, T.; Cohen, A.D.; Campbell, K.S. NK cell dysfunction in chronic lymphocytic leukemia is associated with loss of the mature cells expressing inhibitory killer cell Ig-like receptors. Oncoimmunology 2017, 6, e1330235. [Google Scholar] [CrossRef] [PubMed]
- Manukyan, G.; Papajik, T.; Gajdos, P.; Mikulkova, Z.; Urbanova, R.; Gabcova, G.; Kudelka, M.; Turcsanyi, P.; Ryznerova, P.; Prochazka, V.; et al. Neutrophils in chronic lymphocytic leukemia are permanently activated and have functional defects. Oncotarget 2017, 8, 84889–84901. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fust, G.; Miszlay, Z.; Czink, E.; Varga, L.; Paloczi, K.; Szegedi, G.; Hollan, S.R. C1 and C4 abnormalities in chronic lymphocytic leukaemia and their significance. Immunol. Lett. 1987, 14, 255–259. [Google Scholar] [CrossRef]
- DiLillo, D.J.; Weinberg, J.B.; Yoshizaki, A.; Horikawa, M.; Bryant, J.M.; Iwata, Y.; Matsushita, T.; Matta, K.M.; Chen, Y.; Venturi, G.M.; et al. Chronic lymphocytic leukemia and regulatory B cells share IL-10 competence and immunosuppressive function. Leukemia 2013, 27, 170–182. [Google Scholar] [CrossRef] [Green Version]
- Rossi, D.; De Paoli, L.; Rossi, F.M.; Cerri, M.; Deambrogi, C.; Rasi, S.; Zucchetto, A.; Capello, D.; Gattei, V.; Gaidano, G. Early stage chronic lymphocytic leukaemia carrying unmutated IGHV genes is at risk of recurrent infections during watch and wait. Br. J. Haematol. 2008, 141, 734–736. [Google Scholar] [CrossRef]
- Vardi, A.; Vlachonikola, E.; Karypidou, M.; Stalika, E.; Bikos, V.; Gemenetzi, K.; Maramis, C.; Siorenta, A.; Anagnostopoulos, A.; Pospisilova, S.; et al. Restrictions in the T-cell repertoire of chronic lymphocytic leukemia: High-throughput immunoprofiling supports selection by shared antigenic elements. Leukemia 2017, 31, 1555–1561. [Google Scholar] [CrossRef]
- Riches, J.C.; Gribben, J.G. Immunomodulation and immune reconstitution in chronic lymphocytic leukemia. Semin. Hematol. 2014, 51, 228–234. [Google Scholar] [CrossRef]
- Riches, J.C.; Davies, J.K.; McClanahan, F.; Fatah, R.; Iqbal, S.; Agrawal, S.; Ramsay, A.G.; Gribben, J.G. T cells from CLL patients exhibit features of T-cell exhaustion but retain capacity for cytokine production. Blood 2013, 121, 1612–1621. [Google Scholar] [CrossRef]
- Kabanova, A.; Sanseviero, F.; Candi, V.; Gamberucci, A.; Gozzetti, A.; Campoccia, G.; Bocchia, M.; Baldari, C.T. Human Cytotoxic T Lymphocytes Form Dysfunctional Immune Synapses with B Cells Characterized by Non-Polarized Lytic Granule Release. Cell Rep. 2016, 15, 2313. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, L.; Flies, D.B. Molecular mechanisms of T cell co-stimulation and co-inhibition. Nat. Rev. Immunol. 2013, 13, 227–242. [Google Scholar] [CrossRef] [PubMed]
- Shevach, E.M. Mechanisms of foxp3+ T regulatory cell-mediated suppression. Immunity 2009, 30, 636–645. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- D’Arena, G.; Rossi, G.; Vannata, B.; Deaglio, S.; Mansueto, G.; D’Auria, F.; Statuto, T.; Simeon, V.; De Martino, L.; Marandino, A.; et al. Regulatory T-cells in chronic lymphocytic leukemia and autoimmune diseases. Mediterr. J. Hematol. Infect. Dis. 2012, 4, e2012053. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sakaguchi, S.; Yamaguchi, T.; Nomura, T.; Ono, M. Regulatory T cells and immune tolerance. Cell 2008, 133, 775–787. [Google Scholar] [CrossRef] [Green Version]
- Jak, M.; Mous, R.; Remmerswaal, E.B.; Spijker, R.; Jaspers, A.; Yague, A.; Eldering, E.; Van Lier, R.A.; Van Oers, M.H. Enhanced formation and survival of CD4+ CD25hi Foxp3+ T-cells in chronic lymphocytic leukemia. Leuk. Lymphoma 2009, 50, 788–801. [Google Scholar] [CrossRef]
- Mpakou, V.E.; Ioannidou, H.D.; Konsta, E.; Vikentiou, M.; Spathis, A.; Kontsioti, F.; Kontos, C.K.; Velentzas, A.D.; Papageorgiou, S.; Vasilatou, D.; et al. Quantitative and qualitative analysis of regulatory T cells in B cell chronic lymphocytic leukemia. Leuk. Res. 2017, 60, 74–81. [Google Scholar] [CrossRef]
- Bagnara, D.; Kaufman, M.S.; Calissano, C.; Marsilio, S.; Patten, P.E.; Simone, R.; Chum, P.; Yan, X.J.; Allen, S.L.; Kolitz, J.E.; et al. A novel adoptive transfer model of chronic lymphocytic leukemia suggests a key role for T lymphocytes in the disease. Blood 2011, 117, 5463–5472. [Google Scholar] [CrossRef]
- Rudd, C.E.; Taylor, A.; Schneider, H. CD28 and CTLA-4 coreceptor expression and signal transduction. Immunol. Rev. 2009, 229, 12–26. [Google Scholar] [CrossRef] [Green Version]
- Ramsay, A.G. Immune checkpoint blockade immunotherapy to activate anti-tumour T-cell immunity. Br. J. Haematol. 2013, 162, 313–325. [Google Scholar] [CrossRef]
- Qureshi, O.S.; Zheng, Y.; Nakamura, K.; Attridge, K.; Manzotti, C.; Schmidt, E.M.; Baker, J.; Jeffery, L.E.; Kaur, S.; Briggs, Z.; et al. Trans-endocytosis of CD80 and CD86: A molecular basis for the cell-extrinsic function of CTLA-4. Science 2011, 332, 600–603. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Greenwald, R.J.; Boussiotis, V.A.; Lorsbach, R.B.; Abbas, A.K.; Sharpe, A.H. CTLA-4 regulates induction of anergy in vivo. Immunity 2001, 14, 145–155. [Google Scholar] [CrossRef]
- Peggs, K.S.; Quezada, S.A.; Chambers, C.A.; Korman, A.J.; Allison, J.P. Blockade of CTLA-4 on both effector and regulatory T cell compartments contributes to the antitumor activity of anti-CTLA-4 antibodies. J. Exp. Med. 2009, 206, 1717–1725. [Google Scholar] [CrossRef] [Green Version]
- Frydecka, I.; Kosmaczewska, A.; Bocko, D.; Ciszak, L.; Wolowiec, D.; Kuliczkowski, K.; Kochanowska, I. Alterations of the expression of T-cell-related costimulatory CD28 and downregulatory CD152 (CTLA-4) molecules in patients with B-cell chronic lymphocytic leukaemia. Br. J. Cancer 2004, 90, 2042–2048. [Google Scholar] [CrossRef] [Green Version]
- Motta, M.; Rassenti, L.; Shelvin, B.J.; Lerner, S.; Kipps, T.J.; Keating, M.J.; Wierda, W.G. Increased expression of CD152 (CTLA-4) by normal T lymphocytes in untreated patients with B-cell chronic lymphocytic leukemia. Leukemia 2005, 19, 1788–1793. [Google Scholar] [CrossRef] [Green Version]
- Hodi, F.S.; O’Day, S.J.; McDermott, D.F.; Weber, R.W.; Sosman, J.A.; Haanen, J.B.; Gonzalez, R.; Robert, C.; Schadendorf, D.; Hassel, J.C.; et al. Improved survival with ipilimumab in patients with metastatic melanoma. N. Engl. J. Med. 2010, 363, 711–723. [Google Scholar] [CrossRef] [PubMed]
- Ciszak, L.; Frydecka, I.; Wolowiec, D.; Szteblich, A.; Kosmaczewska, A. Patients with chronic lymphocytic leukaemia (CLL) differ in the pattern of CTLA-4 expression on CLL cells: The possible implications for immunotherapy with CTLA-4 blocking antibody. Tumour. Biol. 2016, 37, 4143–4157. [Google Scholar] [CrossRef] [Green Version]
- Kosmaczewska, A.; Ciszak, L.; Suwalska, K.; Wolowiec, D.; Frydecka, I. CTLA-4 overexpression in CD19+/CD5+ cells correlates with the level of cell cycle regulators and disease progression in B-CLL patients. Leukemia 2005, 19, 301–304. [Google Scholar] [CrossRef] [Green Version]
- Joshi, A.D.; Hegde, G.V.; Dickinson, J.D.; Mittal, A.K.; Lynch, J.C.; Eudy, J.D.; Armitage, J.O.; Bierman, P.J.; Bociek, R.G.; Devetten, M.P.; et al. ATM, CTLA4, MNDA, and HEM1 in high versus low CD38 expressing B-cell chronic lymphocytic leukemia. Clin. Cancer Res. 2007, 13, 5295–5304. [Google Scholar] [CrossRef] [Green Version]
- Mittal, A.K.; Chaturvedi, N.K.; Rohlfsen, R.A.; Gupta, P.; Joshi, A.D.; Hegde, G.V.; Bociek, R.G.; Joshi, S.S. Role of CTLA4 in the proliferation and survival of chronic lymphocytic leukemia. PLoS ONE 2013, 8, e70352. [Google Scholar] [CrossRef] [Green Version]
- Greaves, P.; Gribben, J.G. The role of B7 family molecules in hematologic malignancy. Blood 2013, 121, 734–744. [Google Scholar] [CrossRef] [PubMed]
- Grzywnowicz, M.; Karabon, L.; Karczmarczyk, A.; Zajac, M.; Skorka, K.; Zaleska, J.; Wlasiuk, P.; Chocholska, S.; Tomczak, W.; Bojarska-Junak, A.; et al. The function of a novel immunophenotype candidate molecule PD-1 in chronic lymphocytic leukemia. Leuk. Lymphoma 2015, 56, 2908–2913. [Google Scholar] [CrossRef] [PubMed]
- Yokosuka, T.; Takamatsu, M.; Kobayashi-Imanishi, W.; Hashimoto-Tane, A.; Azuma, M.; Saito, T. Programmed cell death 1 forms negative costimulatory microclusters that directly inhibit T cell receptor signaling by recruiting phosphatase SHP2. J. Exp. Med. 2012, 209, 1201–1217. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Francisco, L.M.; Salinas, V.H.; Brown, K.E.; Vanguri, V.K.; Freeman, G.J.; Kuchroo, V.K.; Sharpe, A.H. PD-L1 regulates the development, maintenance, and function of induced regulatory T cells. J. Exp. Med. 2009, 206, 3015–3029. [Google Scholar] [CrossRef]
- Liu, Y.; Yu, Y.; Yang, S.; Zeng, B.; Zhang, Z.; Jiao, G.; Zhang, Y.; Cai, L.; Yang, R. Regulation of arginase I activity and expression by both PD-1 and CTLA-4 on the myeloid-derived suppressor cells. Cancer Immunol. Immunother. 2009, 58, 687–697. [Google Scholar] [CrossRef]
- Nunes, C.; Wong, R.; Mason, M.; Fegan, C.; Man, S.; Pepper, C. Expansion of a CD8(+)PD-1(+) replicative senescence phenotype in early stage CLL patients is associated with inverted CD4:CD8 ratios and disease progression. Clin. Cancer Res. 2012, 18, 678–687. [Google Scholar] [CrossRef] [Green Version]
- Korona-Glowniak, I.; Grywalska, E.; Grzegorczyk, A.; Rolinski, J.; Glowniak, A.; Malm, A. Bacterial Colonization in Patients with Chronic Lymphocytic Leukemia and Factors Associated with Infections and Colonization. J. Clin. Med. 2019, 8. [Google Scholar] [CrossRef] [Green Version]
- Brusa, D.; Serra, S.; Coscia, M.; Rossi, D.; D’Arena, G.; Laurenti, L.; Jaksic, O.; Fedele, G.; Inghirami, G.; Gaidano, G.; et al. The PD-1/PD-L1 axis contributes to T-cell dysfunction in chronic lymphocytic leukemia. Haematologica 2013, 98, 953–963. [Google Scholar] [CrossRef]
- Xu-Monette, Z.Y.; Zhou, J.; Young, K.H. PD-1 expression and clinical PD-1 blockade in B-cell lymphomas. Blood 2018, 131, 68–83. [Google Scholar] [CrossRef] [Green Version]
- Grzywnowicz, M.; Karczmarczyk, A.; Skorka, K.; Zajac, M.; Zaleska, J.; Chocholska, S.; Tomczak, W.; Giannopoulos, K. Expression of Programmed Death 1 Ligand in Different Compartments of Chronic Lymphocytic Leukemia. Acta Haematol. 2015, 134, 255–262. [Google Scholar] [CrossRef]
- Haderk, F.; Schulz, R.; Iskar, M.; Cid, L.L.; Worst, T.; Willmund, K.V.; Schulz, A.; Warnken, U.; Seiler, J.; Benner, A.; et al. Tumor-derived exosomes modulate PD-L1 expression in monocytes. Sci. Immunol. 2017, 2. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, J.; Xu, X.; Lee, E.J.; Shull, A.Y.; Pei, L.; Awan, F.; Wang, X.; Choi, J.H.; Deng, L.; Xin, H.B.; et al. Phenotypic alteration of CD8+ T cells in chronic lymphocytic leukemia is associated with epigenetic reprogramming. Oncotarget 2016, 7, 40558–40570. [Google Scholar] [CrossRef] [PubMed]
- Lewinsky, H.; Barak, A.F.; Huber, V.; Kramer, M.P.; Radomir, L.; Sever, L.; Orr, I.; Mirkin, V.; Dezorella, N.; Shapiro, M.; et al. CD84 regulates PD-1/PD-L1 expression and function in chronic lymphocytic leukemia. J. Clin. Invest. 2018, 128, 5465–5478. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Long, M.; Beckwith, K.; Do, P.; Mundy, B.L.; Gordon, A.; Lehman, A.M.; Maddocks, K.J.; Cheney, C.; Jones, J.A.; Flynn, J.M.; et al. Ibrutinib treatment improves T cell number and function in CLL patients. J. Clin. Invest. 2017, 127, 3052–3064. [Google Scholar] [CrossRef]
- Kondo, K.; Shaim, H.; Thompson, P.A.; Burger, J.A.; Keating, M.; Estrov, Z.; Harris, D.; Kim, E.; Ferrajoli, A.; Daher, M.; et al. Ibrutinib modulates the immunosuppressive CLL microenvironment through STAT3-mediated suppression of regulatory B-cell function and inhibition of the PD-1/PD-L1 pathway. Leukemia 2018, 32, 960–970. [Google Scholar] [CrossRef]
- Niemann, C.U.; Herman, S.E.; Maric, I.; Gomez-Rodriguez, J.; Biancotto, A.; Chang, B.Y.; Martyr, S.; Stetler-Stevenson, M.; Yuan, C.M.; Calvo, K.R.; et al. Disruption of in vivo Chronic Lymphocytic Leukemia Tumor-Microenvironment Interactions by Ibrutinib--Findings from an Investigator-Initiated Phase II Study. Clin. Cancer Res. 2016, 22, 1572–1582. [Google Scholar] [CrossRef] [Green Version]
- Palma, M.; Krstic, A.; Pena Perez, L.; Berglof, A.; Meinke, S.; Wang, Q.; Blomberg, K.E.M.; Kamali-Moghaddam, M.; Shen, Q.; Jaremko, G.; et al. Ibrutinib induces rapid down-regulation of inflammatory markers and altered transcription of chronic lymphocytic leukaemia-related genes in blood and lymph nodes. Br. J. Haematol. 2018, 183, 212–224. [Google Scholar] [CrossRef] [Green Version]
- Dubovsky, J.A.; Chappell, D.L.; Harrington, B.K.; Agrawal, K.; Andritsos, L.A.; Flynn, J.M.; Jones, J.A.; Paulaitis, M.E.; Bolon, B.; Johnson, A.J.; et al. Lymphocyte cytosolic protein 1 is a chronic lymphocytic leukemia membrane-associated antigen critical to niche homing. Blood 2013, 122, 3308–3316. [Google Scholar] [CrossRef] [Green Version]
- Sagiv-Barfi, I.; Kohrt, H.E.; Czerwinski, D.K.; Ng, P.P.; Chang, B.Y.; Levy, R. Therapeutic antitumor immunity by checkpoint blockade is enhanced by ibrutinib, an inhibitor of both BTK and ITK. Proc. Natl. Acad. Sci. USA 2015, 112, E966–E972. [Google Scholar] [CrossRef] [Green Version]
- Ramsay, A.G.; Clear, A.J.; Fatah, R.; Gribben, J.G. Multiple inhibitory ligands induce impaired T-cell immunologic synapse function in chronic lymphocytic leukemia that can be blocked with lenalidomide: Establishing a reversible immune evasion mechanism in human cancer. Blood 2012, 120, 1412–1421. [Google Scholar] [CrossRef]
- McClanahan, F.; Riches, J.C.; Miller, S.; Day, W.P.; Kotsiou, E.; Neuberg, D.; Croce, C.M.; Capasso, M.; Gribben, J.G. Mechanisms of PD-L1/PD-1-mediated CD8 T-cell dysfunction in the context of aging-related immune defects in the Emicro-TCL1 CLL mouse model. Blood 2015, 126, 212–221. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bichi, R.; Shinton, S.A.; Martin, E.S.; Koval, A.; Calin, G.A.; Cesari, R.; Russo, G.; Hardy, R.R.; Croce, C.M. Human chronic lymphocytic leukemia modeled in mouse by targeted TCL1 expression. Proc. Natl. Acad. Sci. USA 2002, 99, 6955–6960. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, C.; Thudium, K.B.; Han, M.; Wang, X.T.; Huang, H.; Feingersh, D.; Garcia, C.; Wu, Y.; Kuhne, M.; Srinivasan, M.; et al. In vitro characterization of the anti-PD-1 antibody nivolumab, BMS-936558, and in vivo toxicology in non-human primates. Cancer Immunol. Res. 2014, 2, 846–856. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jain, N.; Basu, S.; Thompson, P.A.; Ohanian, M.; Ferrajoli, A.; Pemmaraju, N.; Cortes, J.E.; Estrov, Z.; Burger, J.A.; Neelapu, S.S.; et al. Nivolumab Combined with Ibrutinib for CLL and Richter Transformation: A Phase II Trial. Blood 2016, 128. [Google Scholar] [CrossRef]
- Armand, P. Immune checkpoint blockade in hematologic malignancies. Blood 2015, 125, 3393–3400. [Google Scholar] [CrossRef]
- Yu, X.; Harden, K.; Gonzalez, L.C.; Francesco, M.; Chiang, E.; Irving, B.; Tom, I.; Ivelja, S.; Refino, C.J.; Clark, H.; et al. The surface protein TIGIT suppresses T cell activation by promoting the generation of mature immunoregulatory dendritic cells. Nat. Immunol. 2009, 10, 48–57. [Google Scholar] [CrossRef]
- Li, M.; Xia, P.; Du, Y.; Liu, S.; Huang, G.; Chen, J.; Zhang, H.; Hou, N.; Cheng, X.; Zhou, L.; et al. T-cell immunoglobulin and ITIM domain (TIGIT) receptor/poliovirus receptor (PVR) ligand engagement suppresses interferon-gamma production of natural killer cells via beta-arrestin 2-mediated negative signaling. J. Biol. Chem. 2014, 289, 17647–17657. [Google Scholar] [CrossRef] [Green Version]
- Liu, S.; Zhang, H.; Li, M.; Hu, D.; Li, C.; Ge, B.; Jin, B.; Fan, Z. Recruitment of Grb2 and SHIP1 by the ITT-like motif of TIGIT suppresses granule polarization and cytotoxicity of NK cells. Cell Death Differ. 2013, 20, 456–464. [Google Scholar] [CrossRef]
- Le Mercier, I.; Lines, J.L.; Noelle, R.J. Beyond CTLA-4 and PD-1, the Generation Z of Negative Checkpoint Regulators. Front. Immunol. 2015, 6, 418. [Google Scholar] [CrossRef]
- Dardalhon, V.; Schubart, A.S.; Reddy, J.; Meyers, J.H.; Monney, L.; Sabatos, C.A.; Ahuja, R.; Nguyen, K.; Freeman, G.J.; Greenfield, E.A.; et al. CD226 is specifically expressed on the surface of Th1 cells and regulates their expansion and effector functions. J. Immunol. 2005, 175, 1558–1565. [Google Scholar] [CrossRef]
- Stanietsky, N.; Simic, H.; Arapovic, J.; Toporik, A.; Levy, O.; Novik, A.; Levine, Z.; Beiman, M.; Dassa, L.; Achdout, H.; et al. The interaction of TIGIT with PVR and PVRL2 inhibits human NK cell cytotoxicity. Proc. Natl. Acad. Sci. USA 2009, 106, 17858–17863. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kong, Y.; Zhu, L.; Schell, T.D.; Zhang, J.; Claxton, D.F.; Ehmann, W.C.; Rybka, W.B.; George, M.R.; Zeng, H.; Zheng, H. T-Cell Immunoglobulin and ITIM Domain (TIGIT) Associates with CD8+ T-Cell Exhaustion and Poor Clinical Outcome in AML Patients. Clin. Cancer Res. 2016, 22, 3057–3066. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hirota, T.; Irie, K.; Okamoto, R.; Ikeda, W.; Takai, Y. Transcriptional activation of the mouse Necl-5/Tage4/PVR/CD155 gene by fibroblast growth factor or oncogenic Ras through the Raf-MEK-ERK-AP-1 pathway. Oncogene 2005, 24, 2229–2235. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Soriani, A.; Zingoni, A.; Cerboni, C.; Iannitto, M.L.; Ricciardi, M.R.; Di Gialleonardo, V.; Cippitelli, M.; Fionda, C.; Petrucci, M.T.; Guarini, A.; et al. ATM-ATR-dependent up-regulation of DNAM-1 and NKG2D ligands on multiple myeloma cells by therapeutic agents results in enhanced NK-cell susceptibility and is associated with a senescent phenotype. Blood 2009, 113, 3503–3511. [Google Scholar] [CrossRef] [Green Version]
- Kamran, N.; Takai, Y.; Miyoshi, J.; Biswas, S.K.; Wong, J.S.; Gasser, S. Toll-like receptor ligands induce expression of the costimulatory molecule CD155 on antigen-presenting cells. PLoS ONE 2013, 8, e54406. [Google Scholar] [CrossRef] [Green Version]
- Chan, C.J.; Andrews, D.M.; McLaughlin, N.M.; Yagita, H.; Gilfillan, S.; Colonna, M.; Smyth, M.J. DNAM-1/CD155 interactions promote cytokine and NK cell-mediated suppression of poorly immunogenic melanoma metastases. J. Immunol. 2010, 184, 902–911. [Google Scholar] [CrossRef]
- Shirakawa, J.; Wang, Y.; Tahara-Hanaoka, S.; Honda, S.; Shibuya, K.; Shibuya, A. LFA-1-dependent lipid raft recruitment of DNAM-1 (CD226) in CD4+ T cell. Int. Immunol. 2006, 18, 951–957. [Google Scholar] [CrossRef]
- Shibuya, K.; Shirakawa, J.; Kameyama, T.; Honda, S.; Tahara-Hanaoka, S.; Miyamoto, A.; Onodera, M.; Sumida, T.; Nakauchi, H.; Miyoshi, H.; et al. CD226 (DNAM-1) is involved in lymphocyte function-associated antigen 1 costimulatory signal for naive T cell differentiation and proliferation. J. Exp. Med. 2003, 198, 1829–1839. [Google Scholar] [CrossRef] [Green Version]
- Johnston, R.J.; Comps-Agrar, L.; Hackney, J.; Yu, X.; Huseni, M.; Yang, Y.; Park, S.; Javinal, V.; Chiu, H.; Irving, B.; et al. The immunoreceptor TIGIT regulates antitumor and antiviral CD8(+) T cell effector function. Cancer Cell 2014, 26, 923–937. [Google Scholar] [CrossRef] [Green Version]
- Zhang, T.; Wang, J.; Zhou, X.; Liang, R.; Bai, Q.; Yang, L.; Gu, H.; Gao, G.; Dong, B.; Zhu, H.; et al. Increased expression of TIGIT on CD4+ T cells ameliorates immune-mediated bone marrow failure of aplastic anemia. J. Cell Biochem. 2014, 115, 1918–1927. [Google Scholar] [CrossRef]
- Cella, M.; Presti, R.; Vermi, W.; Lavender, K.; Turnbull, E.; Ochsenbauer-Jambor, C.; Kappes, J.C.; Ferrari, G.; Kessels, L.; Williams, I.; et al. Loss of DNAM-1 contributes to CD8+ T-cell exhaustion in chronic HIV-1 infection. Eur. J. Immunol. 2010, 40, 949–954. [Google Scholar] [CrossRef] [Green Version]
- Fuhrman, C.A.; Yeh, W.I.; Seay, H.R.; Saikumar Lakshmi, P.; Chopra, G.; Zhang, L.; Perry, D.J.; McClymont, S.A.; Yadav, M.; Lopez, M.C.; et al. Divergent Phenotypes of Human Regulatory T Cells Expressing the Receptors TIGIT and CD226. J. Immunol. 2015, 195, 145–155. [Google Scholar] [CrossRef] [Green Version]
- Chauvin, J.M.; Pagliano, O.; Fourcade, J.; Sun, Z.; Wang, H.; Sander, C.; Kirkwood, J.M.; Chen, T.H.; Maurer, M.; Korman, A.J.; et al. TIGIT and PD-1 impair tumor antigen-specific CD8(+) T cells in melanoma patients. J. Clin. Invest. 2015, 125, 2046–2058. [Google Scholar] [CrossRef]
- Inozume, T.; Yaguchi, T.; Furuta, J.; Harada, K.; Kawakami, Y.; Shimada, S. Melanoma Cells Control Antimelanoma CTL Responses via Interaction between TIGIT and CD155 in the Effector Phase. J. Invest. Dermatol. 2016, 136, 255–263. [Google Scholar] [CrossRef] [Green Version]
- Catakovic, K.; Gassner, F.J.; Ratswohl, C.; Zaborsky, N.; Rebhandl, S.; Schubert, M.; Steiner, M.; Gutjahr, J.C.; Pleyer, L.; Egle, A.; et al. TIGIT expressing CD4+T cells represent a tumor-supportive T cell subset in chronic lymphocytic leukemia. Oncoimmunology 2017, 7, e1371399. [Google Scholar] [CrossRef]
- Rouas-Freiss, N.; Goncalves, R.M.; Menier, C.; Dausset, J.; Carosella, E.D. Direct evidence to support the role of HLA-G in protecting the fetus from maternal uterine natural killer cytolysis. Proc. Natl. Acad. Sci. USA 1997, 94, 11520–11525. [Google Scholar] [CrossRef] [Green Version]
- Rouas-Freiss, N.; Moreau, P.; Menier, C.; Carosella, E.D. HLA-G in cancer: A way to turn off the immune system. Semin. Cancer Biol. 2003, 13, 325–336. [Google Scholar] [CrossRef]
- Rouas-Freiss, N.; Moreau, P.; Ferrone, S.; Carosella, E.D. HLA-G proteins in cancer: Do they provide tumor cells with an escape mechanism? Cancer Res. 2005, 65, 10139–10144. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sebti, Y.; Le Friec, G.; Pangault, C.; Gros, F.; Drenou, B.; Guilloux, V.; Bernard, M.; Lamy, T.; Fauchet, R.; Amiot, L. Soluble HLA-G molecules are increased in lymphoproliferative disorders. Hum. Immunol. 2003, 64, 1093–1101. [Google Scholar] [CrossRef] [PubMed]
- Giannopoulos, K.; Schmitt, M.; Kowal, M.; Wlasiuk, P.; Bojarska-Junak, A.; Rolinski, J.; Dmoszynska, A. The significance of soluble HLA-G plasma levels as well as messenger HLA-G for B-cell chronic lymphocytic leukemia (B-CLL). Leuk. Res. 2008, 32, 1815–1819. [Google Scholar] [CrossRef] [PubMed]
- Nuckel, H.; Rebmann, V.; Durig, J.; Duhrsen, U.; Grosse-Wilde, H. HLA-G expression is associated with an unfavorable outcome and immunodeficiency in chronic lymphocytic leukemia. Blood 2005, 105, 1694–1698. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Erikci, A.A.; Karagoz, B.; Ozyurt, M.; Ozturk, A.; Kilic, S.; Bilgi, O. HLA-G expression in B chronic lymphocytic leukemia: A new prognostic marker? Hematology 2009, 14, 101–105. [Google Scholar] [CrossRef] [PubMed]
- Perez-Chacon, G.; Rosado, S.; Rebolleda, N.; Losada-Fernandez, I.; Vargas, J.A.; Morado, M.; Jorda, J.; Perez-Aciego, P. Prognostic irrelevance of HLA-G in B-cell chronic lymphocytic leukemia. Int. J. Lab. Hematol. 2009, 31, 327–337. [Google Scholar] [CrossRef]
- Hviid, T.V.; Rizzo, R.; Christiansen, O.B.; Melchiorri, L.; Lindhard, A.; Baricordi, O.R. HLA-G and IL-10 in serum in relation to HLA-G genotype and polymorphisms. Immunogenetics 2004, 56, 135–141. [Google Scholar] [CrossRef] [PubMed]
- Rizzo, R.; Audrito, V.; Vacca, P.; Rossi, D.; Brusa, D.; Stignani, M.; Bortolotti, D.; D’Arena, G.; Coscia, M.; Laurenti, L.; et al. HLA-G is a component of the chronic lymphocytic leukemia escape repertoire to generate immune suppression: Impact of the HLA-G 14 base pair (rs66554220) polymorphism. Haematologica 2014, 99, 888–896. [Google Scholar] [CrossRef] [PubMed]
- Zhong, Y.; Byrd, J.C.; Dubovsky, J.A. The B-cell receptor pathway: A critical component of healthy and malignant immune biology. Semin. Hematol. 2014, 51, 206–218. [Google Scholar] [CrossRef]
- Stamatopoulos, K.; Agathangelidis, A.; Rosenquist, R.; Ghia, P. Antigen receptor stereotypy in chronic lymphocytic leukemia. Leukemia 2017, 31, 282–291. [Google Scholar] [CrossRef]
- Ghia, P.; Scielzo, C.; Frenquelli, M.; Muzio, M.; Caligaris-Cappio, F. From normal to clonal B cells: Chronic lymphocytic leukemia (CLL) at the crossroad between neoplasia and autoimmunity. Autoimmun. Rev. 2007, 7, 127–131. [Google Scholar] [CrossRef]
- Duhren-von Minden, M.; Ubelhart, R.; Schneider, D.; Wossning, T.; Bach, M.P.; Buchner, M.; Hofmann, D.; Surova, E.; Follo, M.; Kohler, F.; et al. Chronic lymphocytic leukaemia is driven by antigen-independent cell-autonomous signalling. Nature 2012, 489, 309–312. [Google Scholar] [CrossRef]
- Herishanu, Y.; Perez-Galan, P.; Liu, D.; Biancotto, A.; Pittaluga, S.; Vire, B.; Gibellini, F.; Njuguna, N.; Lee, E.; Stennett, L.; et al. The lymph node microenvironment promotes B-cell receptor signaling, NF-kappaB activation, and tumor proliferation in chronic lymphocytic leukemia. Blood 2011, 117, 563–574. [Google Scholar] [CrossRef]
- Petrova, V.; Annicchiarico-Petruzzelli, M.; Melino, G.; Amelio, I. The hypoxic tumour microenvironment. Oncogenesis 2018, 7, 10. [Google Scholar] [CrossRef] [PubMed]
- Semenza, G.L. HIF-1 mediates metabolic responses to intratumoral hypoxia and oncogenic mutations. J. Clin. Invest. 2013, 123, 3664–3671. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pavlova, N.N.; Thompson, C.B. The Emerging Hallmarks of Cancer Metabolism. Cell Metab. 2016, 23, 27–47. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gottfried, E.; Kunz-Schughart, L.A.; Ebner, S.; Mueller-Klieser, W.; Hoves, S.; Andreesen, R.; Mackensen, A.; Kreutz, M. Tumor-derived lactic acid modulates dendritic cell activation and antigen expression. Blood 2006, 107, 2013–2021. [Google Scholar] [CrossRef]
- Fischer, K.; Hoffmann, P.; Voelkl, S.; Meidenbauer, N.; Ammer, J.; Edinger, M.; Gottfried, E.; Schwarz, S.; Rothe, G.; Hoves, S.; et al. Inhibitory effect of tumor cell-derived lactic acid on human T cells. Blood 2007, 109, 3812–3819. [Google Scholar] [CrossRef]
- Goetze, K.; Walenta, S.; Ksiazkiewicz, M.; Kunz-Schughart, L.A.; Mueller-Klieser, W. Lactate enhances motility of tumor cells and inhibits monocyte migration and cytokine release. Int. J. Oncol. 2011, 39, 453–463. [Google Scholar] [CrossRef]
- Colegio, O.R.; Chu, N.Q.; Szabo, A.L.; Chu, T.; Rhebergen, A.M.; Jairam, V.; Cyrus, N.; Brokowski, C.E.; Eisenbarth, S.C.; Phillips, G.M.; et al. Functional polarization of tumour-associated macrophages by tumour-derived lactic acid. Nature 2014, 513, 559–563. [Google Scholar] [CrossRef]
- Serra, S.; Vaisitti, T.; Audrito, V.; Bologna, C.; Buonincontri, R.; Chen, S.S.; Arruga, F.; Brusa, D.; Coscia, M.; Jaksic, O.; et al. Adenosine signaling mediates hypoxic responses in the chronic lymphocytic leukemia microenvironment. Blood Adv. 2016, 1, 47–61. [Google Scholar] [CrossRef] [Green Version]
- Koczula, K.M.; Ludwig, C.; Hayden, R.; Cronin, L.; Pratt, G.; Parry, H.; Tennant, D.; Drayson, M.; Bunce, C.M.; Khanim, F.L.; et al. Metabolic plasticity in CLL: Adaptation to the hypoxic niche. Leukemia 2016, 30, 65–73. [Google Scholar] [CrossRef] [Green Version]
- De Rosa, V.; Galgani, M.; Porcellini, A.; Colamatteo, A.; Santopaolo, M.; Zuchegna, C.; Romano, A.; De Simone, S.; Procaccini, C.; La Rocca, C.; et al. Glycolysis controls the induction of human regulatory T cells by modulating the expression of FOXP3 exon 2 splicing variants. Nat. Immunol. 2015, 16, 1174–1184. [Google Scholar] [CrossRef] [Green Version]
- Kepp, O.; Loos, F.; Liu, P.; Kroemer, G. Extracellular nucleosides and nucleotides as immunomodulators. Immunol. Rev. 2017, 280, 83–92. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vaisitti, T.; Arruga, F.; Guerra, G.; Deaglio, S. Ectonucleotidases in Blood Malignancies: A Tale of Surface Markers and Therapeutic Targets. Front. Immunol. 2019, 10, 2301. [Google Scholar] [CrossRef] [PubMed]
- Chiarugi, A.; Dolle, C.; Felici, R.; Ziegler, M. The NAD metabolome--a key determinant of cancer cell biology. Nat. Rev. Cancer 2012, 12, 741–752. [Google Scholar] [CrossRef] [PubMed]
- Audrito, V.; Manago, A.; Gaudino, F.; Sorci, L.; Messana, V.G.; Raffaelli, N.; Deaglio, S. NAD-Biosynthetic and Consuming Enzymes as Central Players of Metabolic Regulation of Innate and Adaptive Immune Responses in Cancer. Front. Immunol. 2019, 10, 1720. [Google Scholar] [CrossRef] [Green Version]
- Samal, B.; Sun, Y.; Stearns, G.; Xie, C.; Suggs, S.; McNiece, I. Cloning and characterization of the cDNA encoding a novel human pre-B-cell colony-enhancing factor. Mol. Cell Biol. 1994, 14, 1431–1437. [Google Scholar] [CrossRef]
- Audrito, V.; Serra, S.; Brusa, D.; Mazzola, F.; Arruga, F.; Vaisitti, T.; Coscia, M.; Maffei, R.; Rossi, D.; Wang, T.; et al. Extracellular nicotinamide phosphoribosyltransferase (NAMPT) promotes M2 macrophage polarization in chronic lymphocytic leukemia. Blood 2015, 125, 111–123. [Google Scholar] [CrossRef]
- Camp, S.M.; Ceco, E.; Evenoski, C.L.; Danilov, S.M.; Zhou, T.; Chiang, E.T.; Moreno-Vinasco, L.; Mapes, B.; Zhao, J.; Gursoy, G.; et al. Unique Toll-Like Receptor 4 Activation by NAMPT/PBEF Induces NFkappaB Signaling and Inflammatory Lung Injury. Sci. Rep. 2015, 5, 13135. [Google Scholar] [CrossRef] [Green Version]
- Antonioli, L.; Blandizzi, C.; Pacher, P.; Hasko, G. Immunity, inflammation and cancer: A leading role for adenosine. Nat. Rev. Cancer 2013, 13, 842–857. [Google Scholar] [CrossRef] [PubMed]
- Longhi, M.S.; Robson, S.C.; Bernstein, S.H.; Serra, S.; Deaglio, S. Biological functions of ecto-enzymes in regulating extracellular adenosine levels in neoplastic and inflammatory disease states. J. Mol. Med. 2013, 91, 165–172. [Google Scholar] [CrossRef] [Green Version]
- Gessi, S.; Merighi, S.; Sacchetto, V.; Simioni, C.; Borea, P.A. Adenosine receptors and cancer. Biochim. Biophys. Acta 2011, 1808, 1400–1412. [Google Scholar] [CrossRef] [Green Version]
- Ohta, A.; Sitkovsky, M. Role of G-protein-coupled adenosine receptors in downregulation of inflammation and protection from tissue damage. Nature 2001, 414, 916–920. [Google Scholar] [CrossRef]
- Vijayan, D.; Young, A.; Teng, M.W.L.; Smyth, M.J. Targeting immunosuppressive adenosine in cancer. Nat. Rev. Cancer 2017, 17, 709–724. [Google Scholar] [CrossRef] [PubMed]
- Serra, S.; Horenstein, A.L.; Vaisitti, T.; Brusa, D.; Rossi, D.; Laurenti, L.; D’Arena, G.; Coscia, M.; Tripodo, C.; Inghirami, G.; et al. CD73-generated extracellular adenosine in chronic lymphocytic leukemia creates local conditions counteracting drug-induced cell death. Blood 2011, 118, 6141–6152. [Google Scholar] [CrossRef]
- Sitkovsky, M.V.; Kjaergaard, J.; Lukashev, D.; Ohta, A. Hypoxia-adenosinergic immunosuppression: Tumor protection by T regulatory cells and cancerous tissue hypoxia. Clin. Cancer Res. 2008, 14, 5947–5952. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Viardot, A.; Goebeler, M.E.; Hess, G.; Neumann, S.; Pfreundschuh, M.; Adrian, N.; Zettl, F.; Libicher, M.; Sayehli, C.; Stieglmaier, J.; et al. Phase 2 study of the bispecific T-cell engager (BiTE) antibody blinatumomab in relapsed/refractory diffuse large B-cell lymphoma. Blood 2016, 127, 1410–1416. [Google Scholar] [CrossRef] [PubMed]
- Cao, J.X.; Gao, W.J.; You, J.; Wu, L.H.; Liu, J.L.; Wang, Z.X. The efficacy of anti-CD19 chimeric antigen receptor T cells for B-cell malignancies. Cytotherapy 2019, 21, 769–781. [Google Scholar] [CrossRef] [PubMed]
- Ding, W.; LaPlant, B.R.; Call, T.G.; Parikh, S.A.; Leis, J.F.; He, R.; Shanafelt, T.D.; Sinha, S.; Le-Rademacher, J.; Feldman, A.L.; et al. Pembrolizumab in patients with CLL and Richter transformation or with relapsed CLL. Blood 2017, 129, 3419–3427. [Google Scholar] [CrossRef] [PubMed]
- Younes, A.; Brody, J.; Carpio, C.; Lopez-Guillermo, A.; Ben-Yehuda, D.; Ferhanoglu, B.; Nagler, A.; Ozcan, M.; Avivi, I.; Bosch, F.; et al. Safety and activity of ibrutinib in combination with nivolumab in patients with relapsed non-Hodgkin lymphoma or chronic lymphocytic leukaemia: A phase 1/2a study. Lancet Haematol. 2019, 6, e67–e78. [Google Scholar] [CrossRef]
- Middleton, O.; Cosimo, E.; Dobbin, E.; McCaig, A.M.; Clarke, C.; Brant, A.M.; Leach, M.T.; Michie, A.M.; Wheadon, H. Complement deficiencies limit CD20 monoclonal antibody treatment efficacy in CLL. Leukemia 2015, 29, 107–114. [Google Scholar] [CrossRef] [Green Version]
- Evers, M.; Jak, M.; Leusen, J.H.W. The latest developments with anti-CD20 monoclonal antibodies in chronic lymphocytic leukemia. Expert Opin. Biol. Ther. 2018, 18, 973–982. [Google Scholar] [CrossRef]
- June, C.H.; O’Connor, R.S.; Kawalekar, O.U.; Ghassemi, S.; Milone, M.C. CAR T cell immunotherapy for human cancer. Science 2018, 359, 1361–1365. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Porter, D.L.; Hwang, W.T.; Frey, N.V.; Lacey, S.F.; Shaw, P.A.; Loren, A.W.; Bagg, A.; Marcucci, K.T.; Shen, A.; Gonzalez, V.; et al. Chimeric antigen receptor T cells persist and induce sustained remissions in relapsed refractory chronic lymphocytic leukemia. Sci. Transl. Med. 2015, 7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bair, S.M.; Porter, D.L. Accelerating chimeric antigen receptor therapy in chronic lymphocytic leukemia: The development and challenges of chimeric antigen receptor T-cell therapy for chronic lymphocytic leukemia. Am. J. Hematol. 2019, 94, S10–S17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hofland, T.; Eldering, E.; Kater, A.P.; Tonino, S.H. Engaging Cytotoxic T and NK Cells for Immunotherapy in Chronic Lymphocytic Leukemia. Int. J. Mol. Sci. 2019, 20. [Google Scholar] [CrossRef] [PubMed] [Green Version]


© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Arruga, F.; Gyau, B.B.; Iannello, A.; Vitale, N.; Vaisitti, T.; Deaglio, S. Immune Response Dysfunction in Chronic Lymphocytic Leukemia: Dissecting Molecular Mechanisms and Microenvironmental Conditions. Int. J. Mol. Sci. 2020, 21, 1825. https://doi.org/10.3390/ijms21051825
Arruga F, Gyau BB, Iannello A, Vitale N, Vaisitti T, Deaglio S. Immune Response Dysfunction in Chronic Lymphocytic Leukemia: Dissecting Molecular Mechanisms and Microenvironmental Conditions. International Journal of Molecular Sciences. 2020; 21(5):1825. https://doi.org/10.3390/ijms21051825
Chicago/Turabian StyleArruga, Francesca, Benjamin Baffour Gyau, Andrea Iannello, Nicoletta Vitale, Tiziana Vaisitti, and Silvia Deaglio. 2020. "Immune Response Dysfunction in Chronic Lymphocytic Leukemia: Dissecting Molecular Mechanisms and Microenvironmental Conditions" International Journal of Molecular Sciences 21, no. 5: 1825. https://doi.org/10.3390/ijms21051825