Genome-Wide Detection of Key Genes and Epigenetic Markers for Chicken Fatty Liver

 , ,
, ,
Abstract
:1. Introduction
2. Results
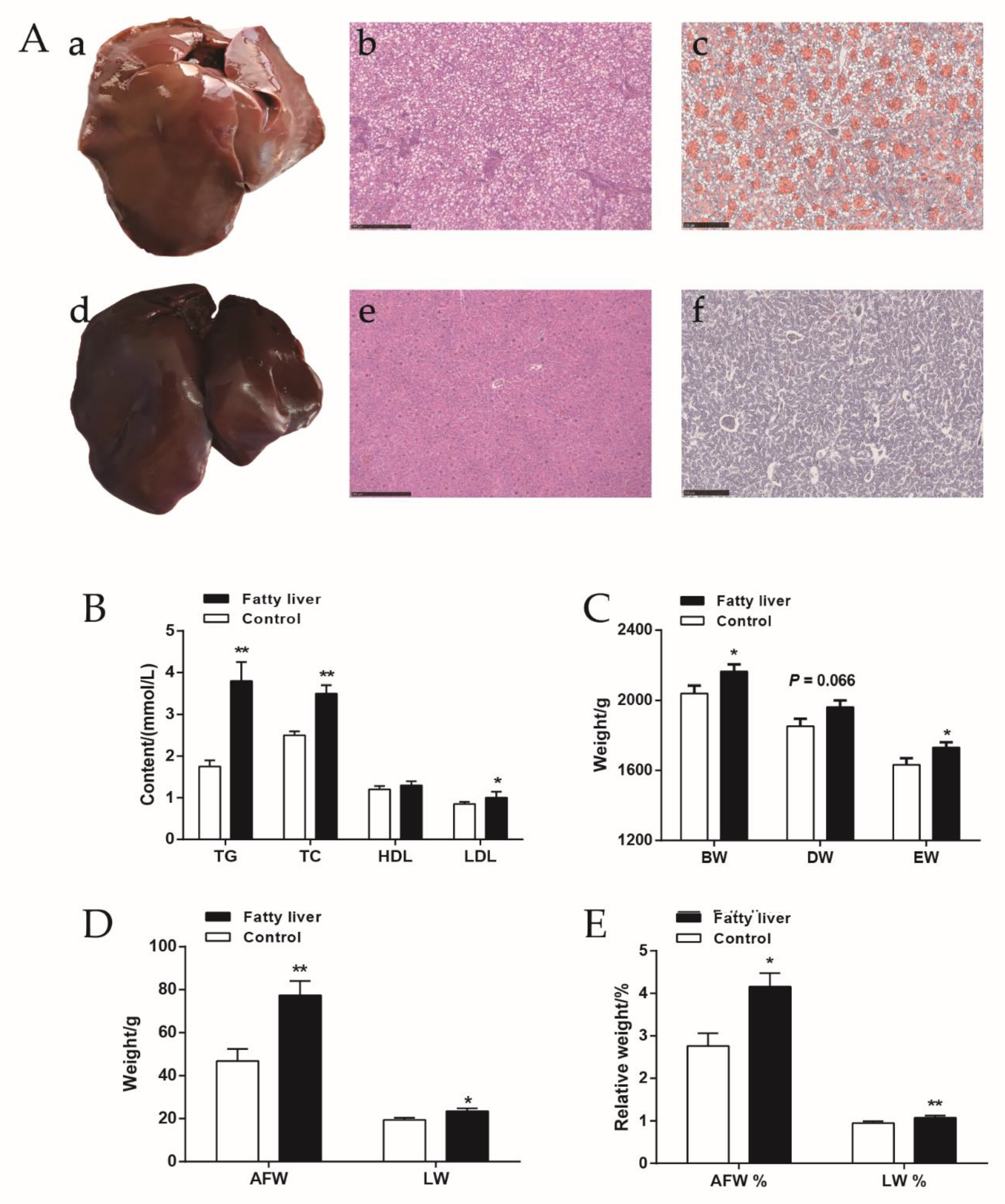
2.1. The Slaughter Performance and Serum Biochemical Indices of Chickens with Fatty Liver
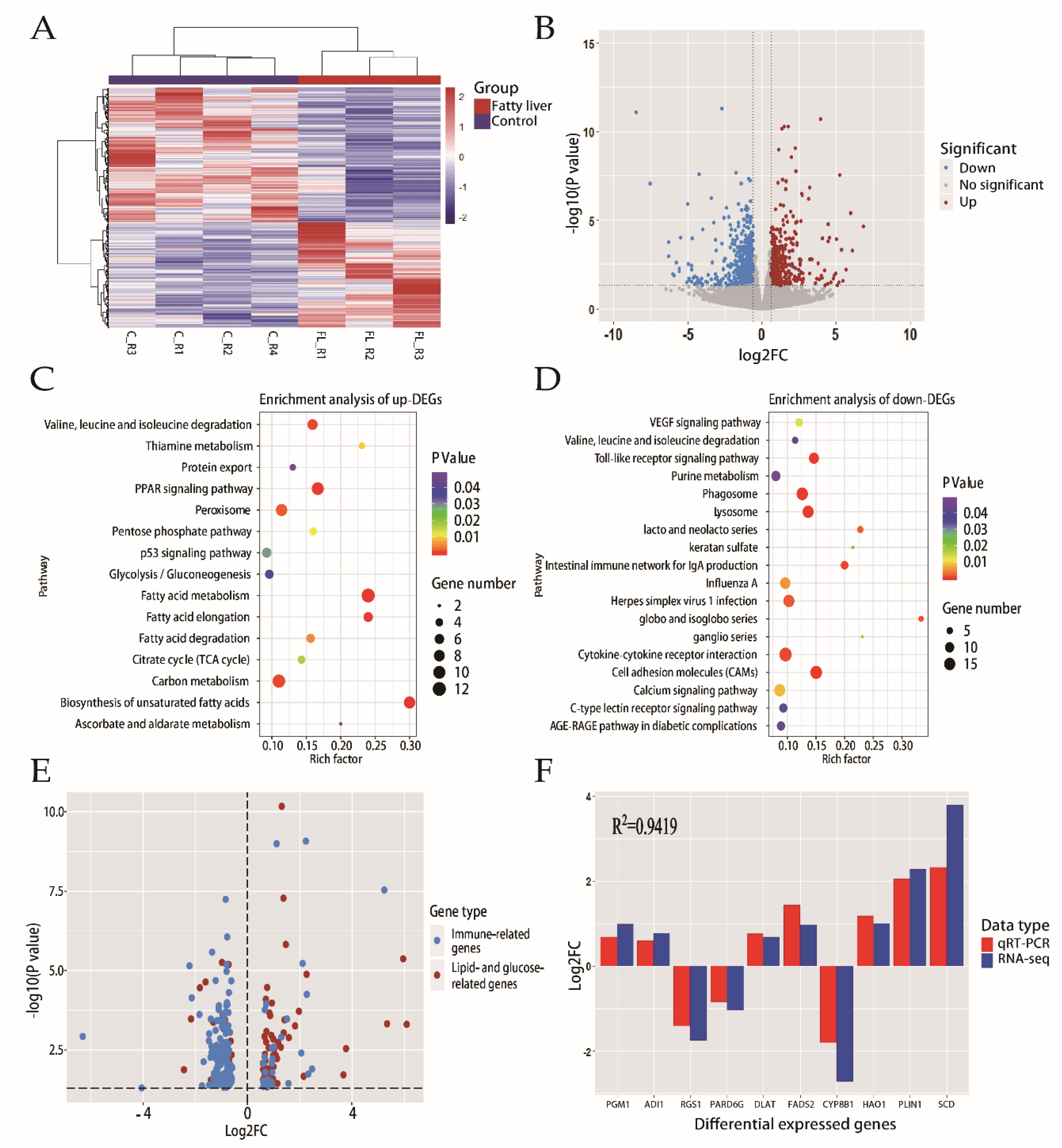
2.2. Transcriptome Profiling Analysis of Liver
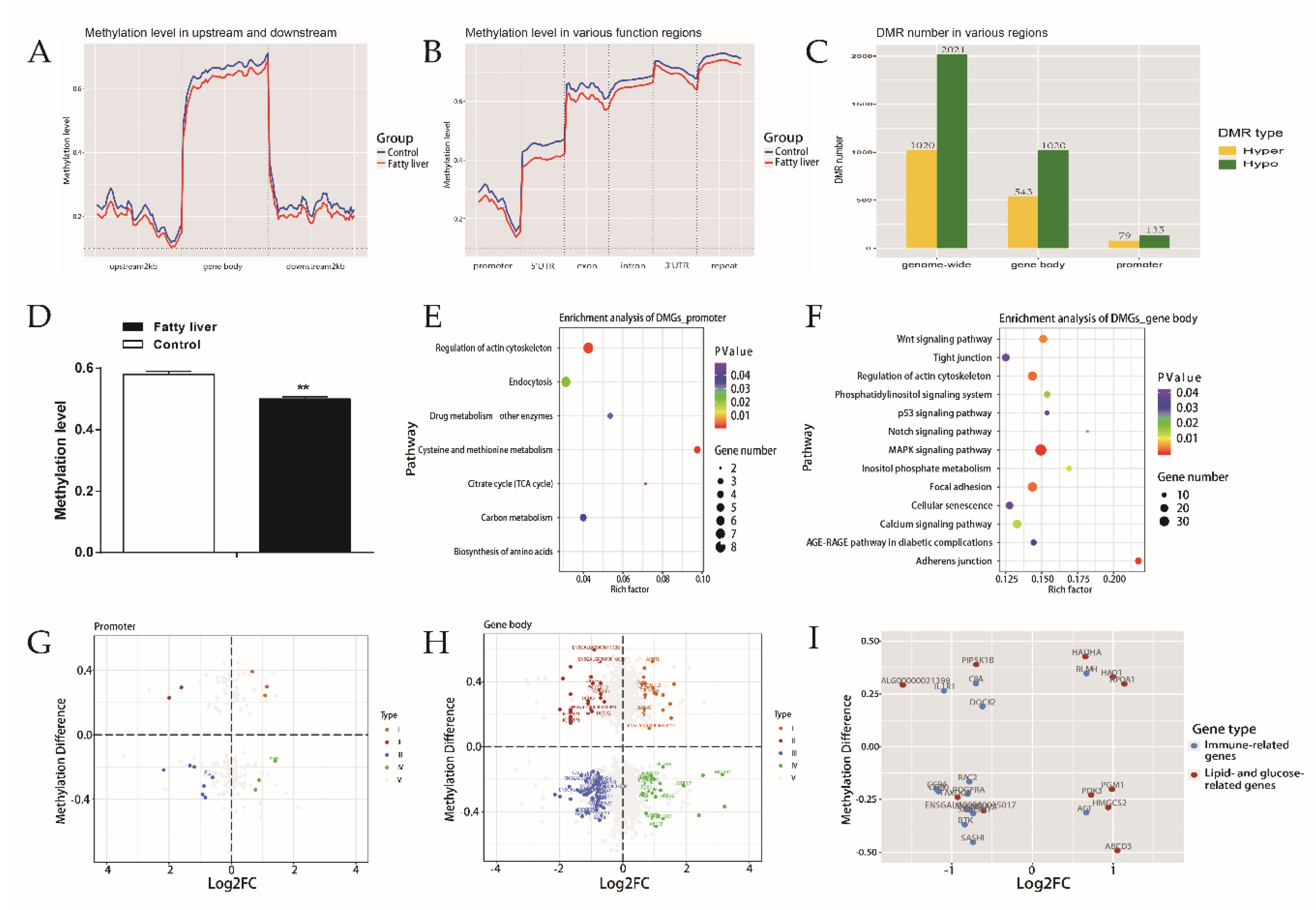
2.3. Integration Analysis of Methylome and Transcriptome
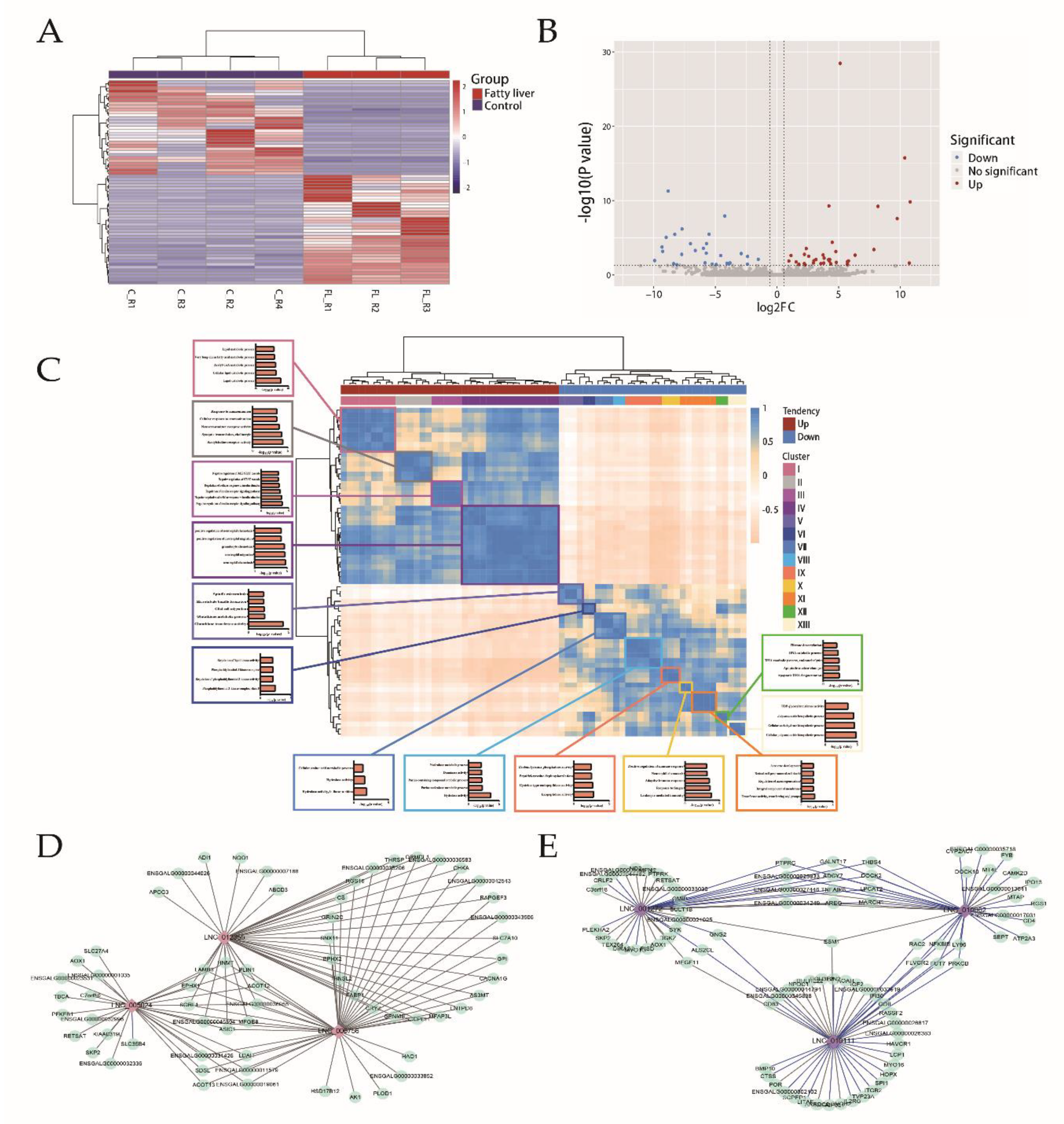
2.4. Integration Analysis of the LncRNA and the mRNA Profiles
2.5. Integration Analysis of the Methylation, the LncRNA, and the mRNA Profiles
3. Discussion
4. Materials and Methods
4.1. Ethics Statement
4.2. Animal Model and Environment
4.3. Sample Collection
4.4. Serum Biochemical Analysis and Liver Histology
4.5. Evaluation of fatty liver
4.6. Sequencing and Identification of Differentially Expressed LncRNAs and mRNAs
4.7. Quantitative Real-Time PCR
4.8. Construction and Analysis of lncRNA-mRNA Network
4.9. Whole-Genome Bisulfite Sequencing and DMGs Identification
4.10. KEGG Pathways Analysis
4.11. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| DEG | Differentially Expressed Gene |
| DMG | Differentially Methylated Gene |
| DMR | Differentially Methylated Region |
| FC | Fold Change |
| FLS | Fatty Liver Syndrome |
| HFD | High Fat Diet |
| LncRNA | Long Noncoding RNA |
| NAFLD | Non-Alcoholic Fatty Liver Disease |
| PCC | Pearson Correlation Coefficient |
| HDL | High-Density Lipoprotein |
| LDL | Low-Density lipoprotein |
| TC | Total Cholesterol |
| TG | Triglyceride |
References
- Faostat. Available online: http://www.fao.org/faostat/en/#home (accessed on 20 February 2020).
- Hermier, D. Lipoprotein metabolism and fattening in poultry. J. Nutr. 1997, 127, 805s–808s. [Google Scholar] [CrossRef] [PubMed]
- Saadoun, A.; Leclercq, B. In vivo lipogenesis of genetically lean and fat chickens: Effects of nutritional state and dietary fat. J. Nutr. 1987, 117, 428–435. [Google Scholar] [CrossRef] [PubMed]
- Burdge, G.C.; Lillycrop, K.A. Fatty acids and epigenetics. Curr. Opin. Clin. Nutr. Metab. Care 2014, 17, 156–161. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, D.M.; Simao, A.L.; Rodrigues, C.M.; Castro, R.E. Revisiting the metabolic syndrome and paving the way for microRNAs in non-alcoholic fatty liver disease. FEBS J. 2014, 281, 2503–2524. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wolford, J.H.; Polin, D. Lipid accumulation and hemorrhage in livers of laying chickens. A study on fatty liver-hemorrhagic syndrome (FLHS). Poult. Sci. 1972, 51, 1707–1713. [Google Scholar] [CrossRef]
- Deacon, L. The fatty liver syndrome—History and early observations. In Proceedings of the 23rd Annual Texas Nutrition Conference, College Park, MD, USA, October 1968; pp. 124–126. [Google Scholar]
- Reedy, L.M. Some clinical observations on the fatty liver syndrome (FLS) in laying hens. Proc. Tex. Nutr. Conf. 1968, 23, 1046. [Google Scholar]
- Grimes, T.M. Causes of disease in two commercial flocks of laying hens. Aust. Vet. J. 1975, 51, 337–343. [Google Scholar] [CrossRef]
- Zhang, Y.; Liu, Z.; Liu, R.; Wang, J.; Zheng, M.; Li, Q.; Cui, H.; Zhao, G.; Wen, J. Alteration of Hepatic Gene Expression along with the Inherited Phenotype of Acquired Fatty Liver in Chicken. Genes 2018, 9, 199. [Google Scholar] [CrossRef] [Green Version]
- Sherriff, J.L.; O’Sullivan, T.A.; Properzi, C.; Oddo, J.L.; Adams, L.A. Choline, Its Potential Role in Nonalcoholic Fatty Liver Disease, and the Case for Human and Bacterial Genes. Adv. Nutr. 2016, 7, 5–13. [Google Scholar] [CrossRef] [Green Version]
- Veskovic, M.; Mladenovic, D.; Milenkovic, M.; Tosic, J.; Borozan, S.; Gopcevic, K.; Labudovic-Borovic, M.; Dragutinovic, V.; Vucevic, D.; Jorgacevic, B.; et al. Betaine modulates oxidative stress, inflammation, apoptosis, autophagy, and Akt/mTOR signaling in methionine-choline deficiency-induced fatty liver disease. Eur. J. Pharmacol. 2019, 848, 39–48. [Google Scholar] [CrossRef]
- Dongiovanni, P.; Valenti, L. A nutrigenomic approach to non-alcoholic fatty liver disease. Int. J. Mol. Sci. 2017, 18, 1534. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, J.H.; Kim, Y.; Kim, Y.J.; Park, Y. Conjugated Linoleic Acid: Potential Health Benefits as a Functional Food Ingredient. Annu. Rev. Food Sci. Technol. 2016, 7, 221–244. [Google Scholar] [CrossRef] [PubMed]
- McCarty, R. Cross-fostering: Elucidating the effects of genexenvironment interactions on phenotypic development. Neurosci. Biobehav. Rev. 2017, 73, 219–254. [Google Scholar] [CrossRef] [PubMed]
- Huang, T.; Hu, F.B. Gene-environment interactions and obesity: Recent developments and future directions. BMC Med. Genom. 2015, 8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gluckman, P.D. Epigenetics and metabolism in 2011: Epigenetics, the life-course and metabolic disease. Nat. Rev. Endocrinol. 2011, 8, 74. [Google Scholar] [CrossRef] [PubMed]
- Jiang, M.; Zhang, Y.; Liu, M.; Lan, M.S.; Fei, J.; Fan, W.; Gao, X.; Lu, D. Hypermethylation of hepatic glucokinase and L-type pyruvate kinase promoters in high-fat diet–induced obese rats. Endocrinology 2011, 152, 1284–1289. [Google Scholar] [CrossRef] [Green Version]
- Sookoian, S.; Rosselli, M.S.; Gemma, C.; Burgueno, A.L.; Fernandez Gianotti, T.; Castano, G.O.; Pirola, C.J. Epigenetic regulation of insulin resistance in nonalcoholic fatty liver disease: Impact of liver methylation of the peroxisome proliferator-activated receptor gamma coactivator 1alpha promoter. Hepatology 2010, 52, 1992–2000. [Google Scholar] [CrossRef]
- Cordero, P.; Gomez-Uriz, A.M.; Campion, J.; Milagro, F.I.; Martinez, J.A. Dietary supplementation with methyl donors reduces fatty liver and modifies the fatty acid synthase DNA methylation profile in rats fed an obesogenic diet. Genes Nutr. 2013, 8, 105–113. [Google Scholar] [CrossRef] [Green Version]
- Sulaiman, S.A.; Muhsin, N.I.A.; Jamal, R. Regulatory Non-coding RNAs Network in Non-alcoholic Fatty Liver Disease. Front. Physiol. 2019, 10, 279. [Google Scholar] [CrossRef]
- Chen, Y.; Huang, H.; Xu, C.; Yu, C.; Li, Y. Long Non-Coding RNA Profiling in a Non-Alcoholic Fatty Liver Disease Rodent Model: New Insight into Pathogenesis. Int. J. Mol. Sci. 2017, 18, 21. [Google Scholar] [CrossRef] [Green Version]
- Sun, C.; Liu, X.; Yi, Z.; Xiao, X.; Yang, M.; Hu, G.; Liu, H.; Liao, L.; Huang, F. Genome-wide analysis of long noncoding RNA expression profiles in patients with non-alcoholic fatty liver disease. IUBMB Life 2015, 67, 847–852. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Gu, Z.; Yang, L.; Tian, Y.; Kang, X.; Liu, X. Transcriptome Profile Analysis Reveals an Estrogen Induced LncRNA Associated with Lipid Metabolism and Carcass Traits in Chickens (Gallus Gallus). Cell. Physiol. Biochem. Int. J. Exp. Cell. Physiol. Biochem. Pharmacol. 2018, 50, 1638–1658. [Google Scholar] [CrossRef] [PubMed]
- Wei, Y.; Yang, C.R.; Wei, Y.P.; Zhao, Z.A.; Hou, Y.; Schatten, H.; Sun, Q.Y. Paternally induced transgenerational inheritance of susceptibility to diabetes in mammals. Proc. Natl. Acad. Sci. USA 2014, 111, 1873–1878. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dhana, K.; Haines, J.; Liu, G.; Zhang, C.; Wang, X.; Field, A.E.; Chavarro, J.E.; Sun, Q. Association between maternal adherence to healthy lifestyle practices and risk of obesity in offspring: Results from two prospective cohort studies of mother-child pairs in the United States. BMJ 2018, 362, k2486. [Google Scholar] [CrossRef] [Green Version]
- Liu, Z.; Li, Q.; Liu, R.; Zhao, G.; Zhang, Y.; Zheng, M.; Cui, H.; Li, P.; Cui, X.; Liu, J.; et al. Expression and methylation of microsomal triglyceride transfer protein and acetyl-CoA carboxylase are associated with fatty liver syndrome in chicken. Poult. Sci. 2016, 95, 1387–1395. [Google Scholar] [CrossRef]
- Pan, X.; Gong, D.; Gao, F.; Sangild, P.T. Diet-dependent changes in the intestinal DNA methylome after introduction of enteral feeding in preterm pigs. Epigenomics 2018, 10, 395–408. [Google Scholar] [CrossRef]
- Hardy, T.; Zeybel, M.; Day, C.P.; Dipper, C.; Masson, S.; McPherson, S.; Henderson, E.; Tiniakos, D.; White, S.; French, J.; et al. Plasma DNA methylation: A potential biomarker for stratification of liver fibrosis in non-alcoholic fatty liver disease. Gut 2017, 66, 1321–1328. [Google Scholar] [CrossRef]
- Jung, G.S.; Jeon, J.H.; Choi, Y.K.; Jang, S.Y.; Park, S.Y.; Kim, S.W.; Byun, J.K.; Kim, M.K.; Lee, S.; Shin, E.C.; et al. Pyruvate dehydrogenase kinase regulates hepatitis C virus replication. Sci. Rep. 2016, 6, 30846. [Google Scholar] [CrossRef] [Green Version]
- Degenhardt, T.; Saramaki, A.; Malinen, M.; Rieck, M.; Vaisanen, S.; Huotari, A.; Herzig, K.H.; Muller, R.; Carlberg, C. Three members of the human pyruvate dehydrogenase kinase gene family are direct targets of the peroxisome proliferator-activated receptor beta/delta. J. Mol. Biol. 2007, 372, 341–355. [Google Scholar] [CrossRef]
- Paschos, G.K.; Ibrahim, S.; Song, W.L.; Kunieda, T.; Grant, G.; Reyes, T.M.; Bradfield, C.A.; Vaughan, C.H.; Eiden, M.; Masoodi, M.; et al. Obesity in mice with adipocyte-specific deletion of clock component Arntl. Nat. Med. 2012, 18, 1768–1777. [Google Scholar] [CrossRef] [Green Version]
- Shimba, S.; Ogawa, T.; Hitosugi, S.; Ichihashi, Y.; Nakadaira, Y.; Kobayashi, M.; Tezuka, M.; Kosuge, Y.; Ishige, K.; Ito, Y.; et al. Deficient of a clock gene, brain and muscle Arnt-like protein-1 (BMAL1), induces dyslipidemia and ectopic fat formation. PLoS ONE 2011, 6, e25231. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Satapati, S.; Sunny, N.E.; Kucejova, B.; Fu, X.; He, T.T.; Mendez-Lucas, A.; Shelton, J.M.; Perales, J.C.; Browning, J.D.; Burgess, S.C. Elevated TCA cycle function in the pathology of diet-induced hepatic insulin resistance and fatty liver. J. Lipid Res. 2012, 53, 1080–1092. [Google Scholar] [CrossRef] [Green Version]
- White, H.M. The Role of TCA Cycle Anaplerosis in Ketosis and Fatty Liver in Periparturient Dairy Cows. Animals 2015, 5, 793–802. [Google Scholar] [CrossRef] [PubMed]
- Zhang, K.; Shi, Z.M.; Chang, Y.N.; Hu, Z.M.; Qi, H.X.; Hong, W. The ways of action of long non-coding RNAs in cytoplasm and nucleus. Gene 2014, 547, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Grimaldi, B.; Bellet, M.M.; Katada, S.; Astarita, G.; Hirayama, J.; Amin, R.H.; Granneman, J.G.; Piomelli, D.; Leff, T.; Sassone-Corsi, P. PER2 controls lipid metabolism by direct regulation of PPARgamma. Cell Metab. 2010, 12, 509–520. [Google Scholar] [CrossRef] [Green Version]
- Chen, L.L.; Carmichael, G.G. Long noncoding RNAs in mammalian cells: What, where, and why? Wiley Interdiscip. Rev. RNA 2010, 1, 2–21. [Google Scholar] [CrossRef]
- Guo, J.; Zhou, Y.; Cheng, Y.; Fang, W.; Hu, G.; Wei, J.; Lin, Y.; Man, Y.; Guo, L.; Sun, M.; et al. Metformin-Induced Changes of the Coding Transcriptome and Non-Coding RNAs in the Livers of Non-Alcoholic Fatty Liver Disease Mice. Cell. Physiol. Biochem. Int. J. Exp. Cell. Physiol. Biochem. Pharmacol. 2018, 45, 1487–1505. [Google Scholar] [CrossRef]
- Yan, P.; Luo, S.; Lu, J.Y.; Shen, X. Cis- and trans-acting lncRNAs in pluripotency and reprogramming. Curr. Opin. Genet. Dev. 2017, 46, 170–178. [Google Scholar] [CrossRef]
- Wang, L.; Zhao, Y.; Bao, X.; Zhu, X.; Kwok, Y.K.; Sun, K.; Chen, X.; Huang, Y.; Jauch, R.; Esteban, M.A.; et al. LncRNA Dum interacts with Dnmts to regulate Dppa2 expression during myogenic differentiation and muscle regeneration. Cell Res. 2015, 25, 335–350. [Google Scholar] [CrossRef] [Green Version]
- O’Leary, V.B.; Ovsepian, S.V.; Carrascosa, L.G.; Buske, F.A.; Radulovic, V.; Niyazi, M.; Moertl, S.; Trau, M.; Atkinson, M.J.; Anastasov, N. PARTICLE, a Triplex-Forming Long ncRNA, Regulates Locus-Specific Methylation in Response to Low-Dose Irradiation. Cell Rep. 2015, 11, 474–485. [Google Scholar] [CrossRef] [Green Version]
- Morita, M.; Imanaka, T. Peroxisomal ABC transporters: Structure, function and role in disease. Biochim. Biophys. Acta 2012, 1822, 1387–1396. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Imanaka, T.; Aihara, K.; Takano, T.; Yamashita, A.; Sato, R.; Suzuki, Y.; Yokota, S.; Osumi, T. Characterization of the 70-kDa peroxisomal membrane protein, an ATP binding cassette transporter. J. Biol. Chem. 1999, 274, 11968–11976. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schwam, H.; Michelson, S.; Randall, W.C.; Sondey, J.M.; Hirschmann, R. Purification and characterization of human liver glycolate oxidase. Molecular weight, subunit, and kinetic properties. Biochemistry 1979, 18, 2828–2833. [Google Scholar] [CrossRef] [PubMed]
- Recalcati, S.; Tacchini, L.; Alberghini, A.; Conte, D.; Cairo, G. Oxidative stress-mediated down-regulation of rat hydroxyacid oxidase 1, a liver-specific peroxisomal enzyme. Hepatology 2003, 38, 1159–1166. [Google Scholar] [CrossRef]
- De Craemer, D.; Pauwels, M.; Van den Branden, C. Alterations of peroxisomes in steatosis of the human liver: A quantitative study. Hepatology 1995, 22, 744–752. [Google Scholar] [CrossRef]
- Suszynska-Zajczyk, J.; Wroblewski, J.; Utyro, O.; Luczak, M.; Marczak, L.; Jakubowski, H. Bleomycin hydrolase and hyperhomocysteinemia modulate the expression of mouse proteins involved in liver homeostasis. Amino Acids 2014, 46, 1471–1480. [Google Scholar] [CrossRef]
- Okamura, Y.; Nomoto, S.; Hayashi, M.; Hishida, M.; Nishikawa, Y.; Yamada, S.; Fujii, T.; Sugimoto, H.; Takeda, S.; Kodera, Y.; et al. Identification of the bleomycin hydrolase gene as a methylated tumor suppressor gene in hepatocellular carcinoma using a novel triple-combination array method. Cancer Lett. 2011, 312, 150–157. [Google Scholar] [CrossRef]
- Xie, L.; Li, M.; Liu, D.; Wang, X.; Wang, P.; Dai, H.; Yang, W.; Liu, W.; Hu, X.; Zhao, M. Secalonic Acid-F, a Novel Mycotoxin, Represses the Progression of Hepatocellular Carcinoma via MARCH1 Regulation of the PI3K/AKT/beta-catenin Signaling Pathway. Molecules 2019, 24, 393. [Google Scholar] [CrossRef] [Green Version]
- Bhagwandin, C.; Ashbeck, E.L.; Whalen, M.; Bandola-Simon, J.; Roche, P.A.; Szajman, A.; Truong, S.M.; Wertheim, B.C.; Klimentidis, Y.C.; Ishido, S.; et al. The E3 ubiquitin ligase MARCH1 regulates glucose-tolerance and lipid storage in a sex-specific manner. PLoS ONE 2018, 13, e0204898. [Google Scholar] [CrossRef]
- Liu, L.; Geng, X.; Cai, Y.; Copple, B.; Yoshinaga, M.; Shen, J.; Nebert, D.W.; Wang, H.; Liu, Z. Hepatic ZIP8 deficiency is associated with disrupted selenium homeostasis, liver pathology, and tumor formation. Am. J. Physiol. Gastrointest. Liver Physiol. 2018, 315, G569–G579. [Google Scholar] [CrossRef]
- Kim, D.; Paggi, J.M.; Park, C.; Bennett, C.; Salzberg, S.L. Graph-based genome alignment and genotyping with HISAT2 and HISAT-genotype. Nat. Biotechnol. 2019, 37, 907–915. [Google Scholar] [CrossRef] [PubMed]
- Pertea, M.; Kim, D.; Pertea, G.M.; Leek, J.T.; Salzberg, S.L. Transcript-level expression analysis of RNA-seq experiments with HISAT, StringTie and Ballgown. Nat. Protoc. 2016, 11, 1650. [Google Scholar] [CrossRef] [PubMed]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, J.; Xiong, Q.; Chen, H.; Yang, C.; Fan, Y. Identification of the Spinal Expression Profile of Non-coding RNAs Involved in Neuropathic Pain Following Spared Nerve Injury by Sequence Analysis. Front. Mol. Neurosci. 2017, 10, 91. [Google Scholar] [CrossRef]
- Sun, L.; Luo, H.; Bu, D.; Zhao, G.; Yu, K.; Zhang, C.; Liu, Y.; Chen, R.; Zhao, Y. Utilizing sequence intrinsic composition to classify protein-coding and long non-coding transcripts. Nucleic Acids Res. 2013, 41, e166. [Google Scholar] [CrossRef]
- Punta, M.; Coggill, P.C.; Eberhardt, R.Y.; Mistry, J.; Tate, J.; Boursnell, C.; Pang, N.; Forslund, K.; Ceric, G.; Clements, J. The Pfam protein families database. Nucleic Acids Res. 2011, 40, D290–D301. [Google Scholar] [CrossRef] [PubMed]
- Kong, L.; Zhang, Y.; Ye, Z.-Q.; Liu, X.-Q.; Zhao, S.-Q.; Wei, L.; Gao, G. CPC: Assess the protein-coding potential of transcripts using sequence features and support vector machine. Nucleic Acids Res. 2007, 35, W345–W349. [Google Scholar] [CrossRef]
- Zerbino, D.R.; Achuthan, P.; Akanni, W.; Amode, M.R.; Barrell, D.; Bhai, J.; Billis, K.; Cummins, C.; Gall, A.; Giron, C.G.; et al. Ensembl 2018. Nucleic Acids Res. 2018, 46, D754–D761. [Google Scholar] [CrossRef]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef]
- Xie, C.; Mao, X.; Huang, J.; Ding, Y.; Wu, J.; Dong, S.; Kong, L.; Gao, G.; Li, C.Y.; Wei, L. KOBAS 2.0: A web server for annotation and identification of enriched pathways and diseases. Nucleic Acids Res. 2011, 39, W316–W322. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, S.; Qin, C.; Cao, G.; Guo, L.; Feng, C.; Zhang, W. Genome-wide analysis of DNA methylation profiles in a senescence-accelerated mouse prone 8 brain using whole-genome bisulfite sequencing. Bioinformatics 2017, 33, 1591–1595. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, C.; Li, Y.; Zhou, G.; Gao, Y.; Ma, S.; Chen, Y.; Song, J.; Wang, X. Whole-genome bisulfite sequencing of goat skins identifies signatures associated with hair cycling. BMC Genom. 2018, 19, 638. [Google Scholar] [CrossRef] [PubMed]
- Felix, K.; Andrews, S.R. Bismark: A flexible aligner and methylation caller for Bisulfite-Seq applications. Bioinformatics 2011, 27, 1571–1572. [Google Scholar]
- Hao, F.; Conneely, K.N.; Hao, W. A Bayesian hierarchical model to detect differentially methylated loci from single nucleotide resolution sequencing data. Nucleic Acids Res. 2014, 42, e69. [Google Scholar]
- Hao, W.; Tianlei, X.; Hao, F.; Li, C.; Ben, L.; Bing, Y.; Zhaohui, Q.; Peng, J.; Conneely, K.N. Detection of differentially methylated regions from whole-genome bisulfite sequencing data without replicates. Nucleic Acids Res. 2015, 43, e141. [Google Scholar]
- Park, Y.; Wu, H. Differential methylation analysis for BS-seq data under general experimental design. Bioinformatics 2016, 32, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Yu, G.; Wang, L.G.; Han, Y.; He, Q.Y. clusterProfiler: An R package for comparing biological themes among gene clusters. Omics A J. Integr. Biol. 2012, 16, 284–287. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| ID | Pathway | Tendency of DEG | p-Value 1 | DMR | p-Value 2 |
|---|---|---|---|---|---|
| gga01200 | Carbon metabolism | up | 8.53 × 10−4 | promoter | 3.69 × 10−2 |
| gga00020 | Citrate cycle (TCA cycle) | up | 1.70 × 10−2 | promoter | 4.82 ×10−2 |
| gga04115 | p53 signaling pathway | up | 2.88 ×10−2 | gene body | 3.76 × 10−2 |
| gga04020 | Calcium signaling pathway | down | 8.94 × 10−3 | gene body | 1.35 × 10−2 |
| gga04933 | AGE-RAGE signaling pathway in diabetic complications | down | 4.41 × 10−2 | gene body | 3.09 × 10−2 |
| lncRNA | Regulation | Gene | Log2FC | DMR | Methylation Difference |
|---|---|---|---|---|---|
| LNC_008609, LNC_008671 | trans | LIMD2 | −0.83 | gene body | −0.39 |
| LNC_012679 | trans | BLMH1 | 0.67 | gene body | 0.35 |
| LNC_008303 | trans | ASPA | 1.41 | promoter | −0.16 |
| LNC_012355 | trans, cis | ABCD32 | 1.05 | gene body | −0.49 |
| LNC_010111 | trans, cis | CCDC18 | −0.74 | gene body | 0.35 |
| LNC_006756 | trans | HAO12 | 0.99 | gene body | 0.33 |
| LNC_010111, LNC_010862 | trans | FLVCR2 | −1.16 | gene body | −0.4 |
| LNC_010073, LNC_010240 | trans | FAM13A | 0.89 | gene body | −0.18 |
| LNC_002556 | trans, cis | ENSGALG00000010639 | −0.73 | gene body | 0.52 |
| LNC_009039 | trans, cis | ENSGALG00000011528 | −0.91 | gene body | 0.6 |
| LNC_000820 | trans | SLC39A8 | 0.73 | gene body | −0.2 |
| LNC_010111 | trans | MYO16 | 1.44 | gene body | −0.32 |
| LNC_000333 | trans | COTL1 | −0.84 | gene body | −0.36 |
| LNC_007320, LNC_007320 | trans | CELF2 | −0.72 | gene body | −0.24, −0.31 |
| LNC_005357, LNC_007350, LNC_010111, LNC_010862 | trans | RAC21 | −0.78 | gene body | −0.17 |
| LNC_001439, LNC_001531, LNC_007015, LNC_010098 | trans | JAM2 | 0.59 | gene body | 0.38 |
| LNC_001714, LNC_001742, LNC_006829, LNC_012722 | trans | WDPCP | 1.92 | gene body | −0.24 |
| LNC_001439, LNC_001531, LNC_005357, LNC_007015, LNC_010098, LNC_010111 | trans | ENSGALG00000033919 | 0.84 | gene body | 0.11 |
| LNC_001272, LNC_002705, LNC_003079, LNC_007151, LNC_010862, LNC_011070 | trans | DOCK21 | −0.62 | gene body | 0.19 |
| LNC_002705, LNC_003079, LNC_008608, LNC_012083, LNC_012722 | trans | DIP2C | 0.92 | gene body | −0.22 |
| LNC_001272, LNC_002705, LNC_003079, LNC_007151, LNC_008608, LNC_010862, LNC_011070 | trans | GALNT17 | −1.32 | gene body | −0.18 |
| ENSGALT00000085791, LNC_001272, LNC_007151, LNC_007350, LNC_010862, LNC_011070 | trans | MARCH11 | −0.78 | gene body | −0.27, −0.33 |
| LNC_001272, LNC_001439, LNC_002556, LNC_005357, LNC_007151, LNC_007350, LNC_008609, LNC_010111, LNC_010494 | trans | MEGF11 | 3.15 | gene body | −0.17 |
| Gene ID | Gene | Primer Sequence | Product Size (bp) |
|---|---|---|---|
| ENSGALG00000002549 | RGS1 | F:5′-AGGATTTACGAGGAGTTTGT-3′ | 105 |
| R:5′-TGTGTGAGTTGGGTCTTG-3′ | |||
| ENSGALG00000033511 | CYP8B1 | F:5′-GGATAAGTGAACAAGACCAGTA-3′ | 132 |
| R:5′-GATACAAGAGGAGCCAGAAG-3′ | |||
| ENSGALG00000007904 | DLAT | F:5′-TTGCTCTCCCTGCTCTGT-3′ | 127 |
| R:5′-CCTATTGTGGCTTTATCTGTCT-3′ | |||
| ENSGALG00000013594 | PARD6G | F:5′-GCCAACAGCCATAACCTT-3′ | 184 |
| R:5′-CCTCTTCGTCACTCTCCA-3′ | |||
| ENSGALG00000005739 | SCD | F:5′-GGCTGACAAAGTGGTGATG-3′ | 137 |
| R:5′-GGATGGCTGGAATGAAGA-3′ | |||
| ENSGALG00000007178 | FADS2 | F:5′-CTGAGGAAGACAGCAGAGGACAT-3′ | 153 |
| R:5′-GCAGGCAAGGATTAGAGTTGTG-3′ | |||
| ENSGALG00000025796 | ADI1 | F:5′-ACATGGACGAGTCCCAGGAG-3′ | 113 |
| R:5′-AGCATCCAATCTGCGGTAGG-3′ | |||
| ENSGALG00000011016 | PGM1 | F:5′-ACGGTGAAAACCAAGGCGT-3′ | 103 |
| R:5′-TGAAGTTCTCGGCGTAGTGG-3′ | |||
| ENSGALG00000023395 | PLIN1 | F:5′-GCAATCCAGGGCTTACAG-3′ | 171 |
| R:5′-ATCCAGACGACCAGTTCC-3′ | |||
| ENSGALG00000008845 | HAO1 | F:5′-CGGTTTGTGTTGCTGATTT-3′ | 116 |
| R:5′-TGCTGCTACATTATCTGCTA-3′ | |||
| ENSGALG00000015082 | RPS6 | F:5′-GAGCGCAACGTGAGAACATT-3′ | 92 |
| R:5′-CGGACAACATAGCCCTTCCA-3′ | |||
| ENSGALG00000009621 | ACTB | F:5′-GAGAAATTGTGCGTGACATCA-3′ | 152 |
| R:5′-CCTGAACCTCTCATTGCCA-3′ |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tan, X.; Liu, R.; Xing, S.; Zhang, Y.; Li, Q.; Zheng, M.; Zhao, G.; Wen, J. Genome-Wide Detection of Key Genes and Epigenetic Markers for Chicken Fatty Liver. Int. J. Mol. Sci. 2020, 21, 1800. https://doi.org/10.3390/ijms21051800
Tan X, Liu R, Xing S, Zhang Y, Li Q, Zheng M, Zhao G, Wen J. Genome-Wide Detection of Key Genes and Epigenetic Markers for Chicken Fatty Liver. International Journal of Molecular Sciences. 2020; 21(5):1800. https://doi.org/10.3390/ijms21051800
Chicago/Turabian StyleTan, Xiaodong, Ranran Liu, Siyuan Xing, Yonghong Zhang, Qinghe Li, Maiqing Zheng, Guiping Zhao, and Jie Wen. 2020. "Genome-Wide Detection of Key Genes and Epigenetic Markers for Chicken Fatty Liver" International Journal of Molecular Sciences 21, no. 5: 1800. https://doi.org/10.3390/ijms21051800