Attenuation of Hypertrophy in Human MSCs via Treatment with a Retinoic Acid Receptor Inverse Agonist

 ,
,  , ,
, ,  ,
,
Abstract
:1. Introduction
1.1. Cell Fate of Limb Chondrocytes
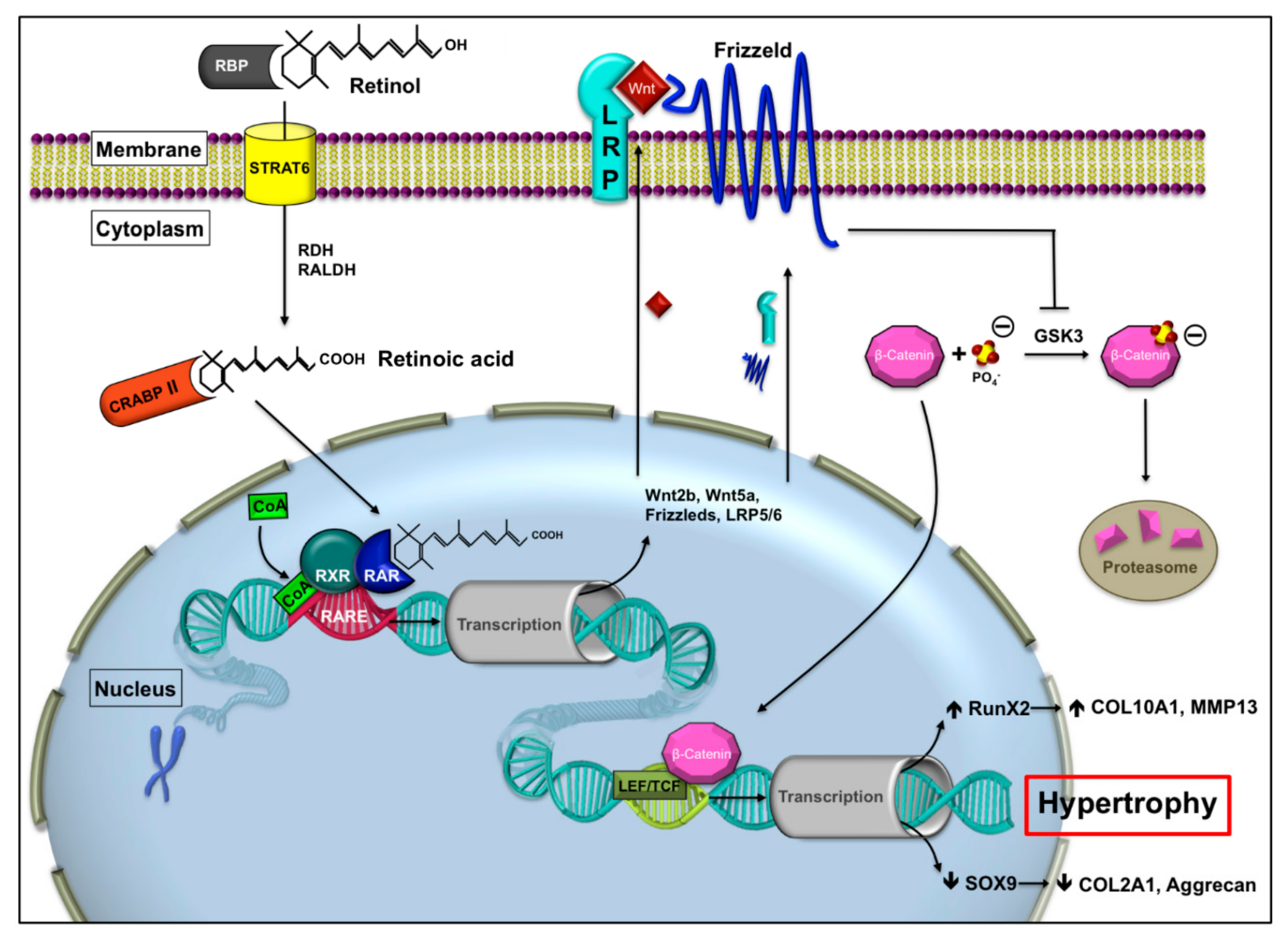
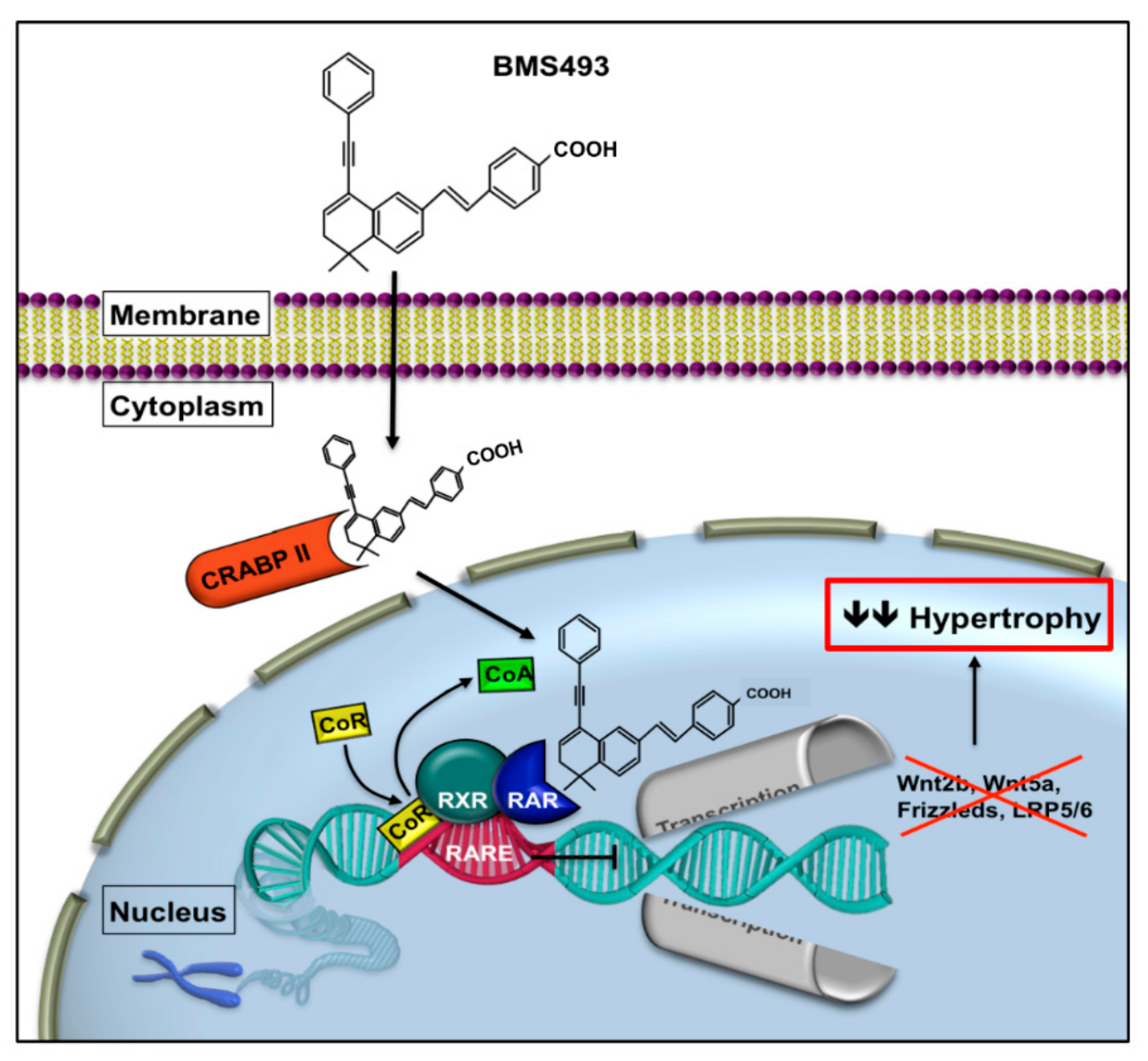
1.2. Retinoic Acid Receptor Signaling
2. Results
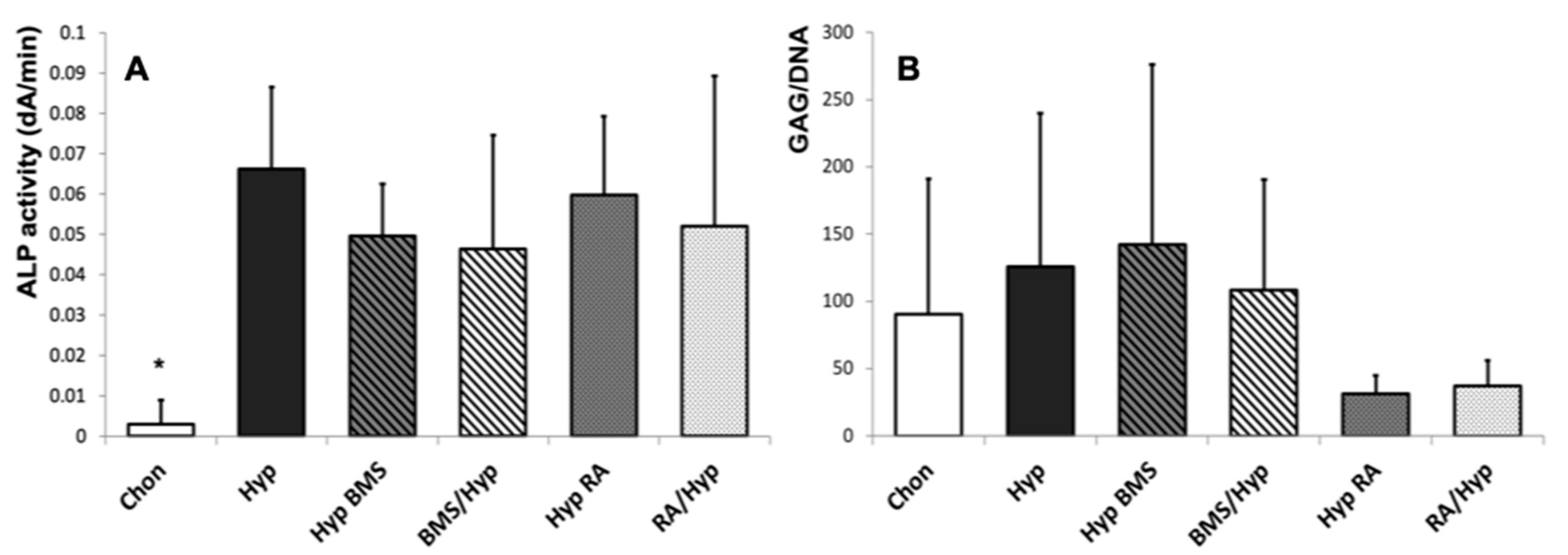
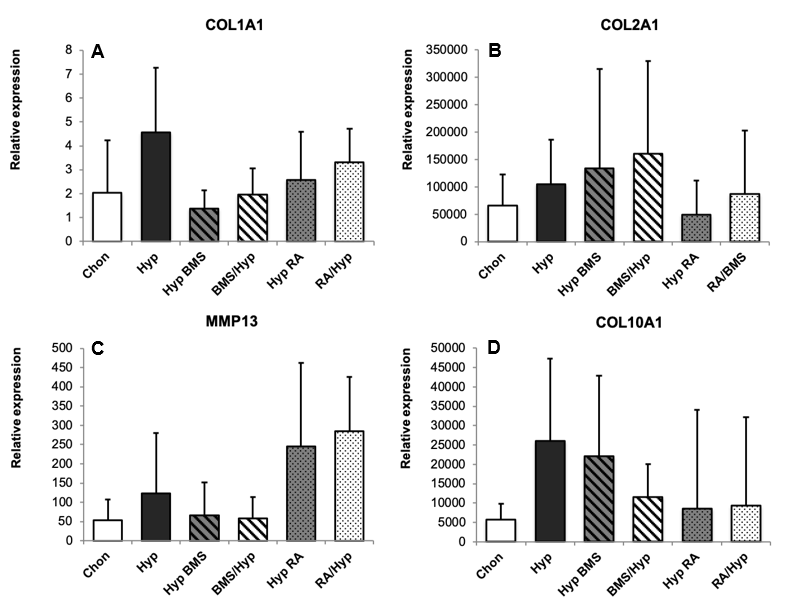
2.1. Induction of Hypertrophy
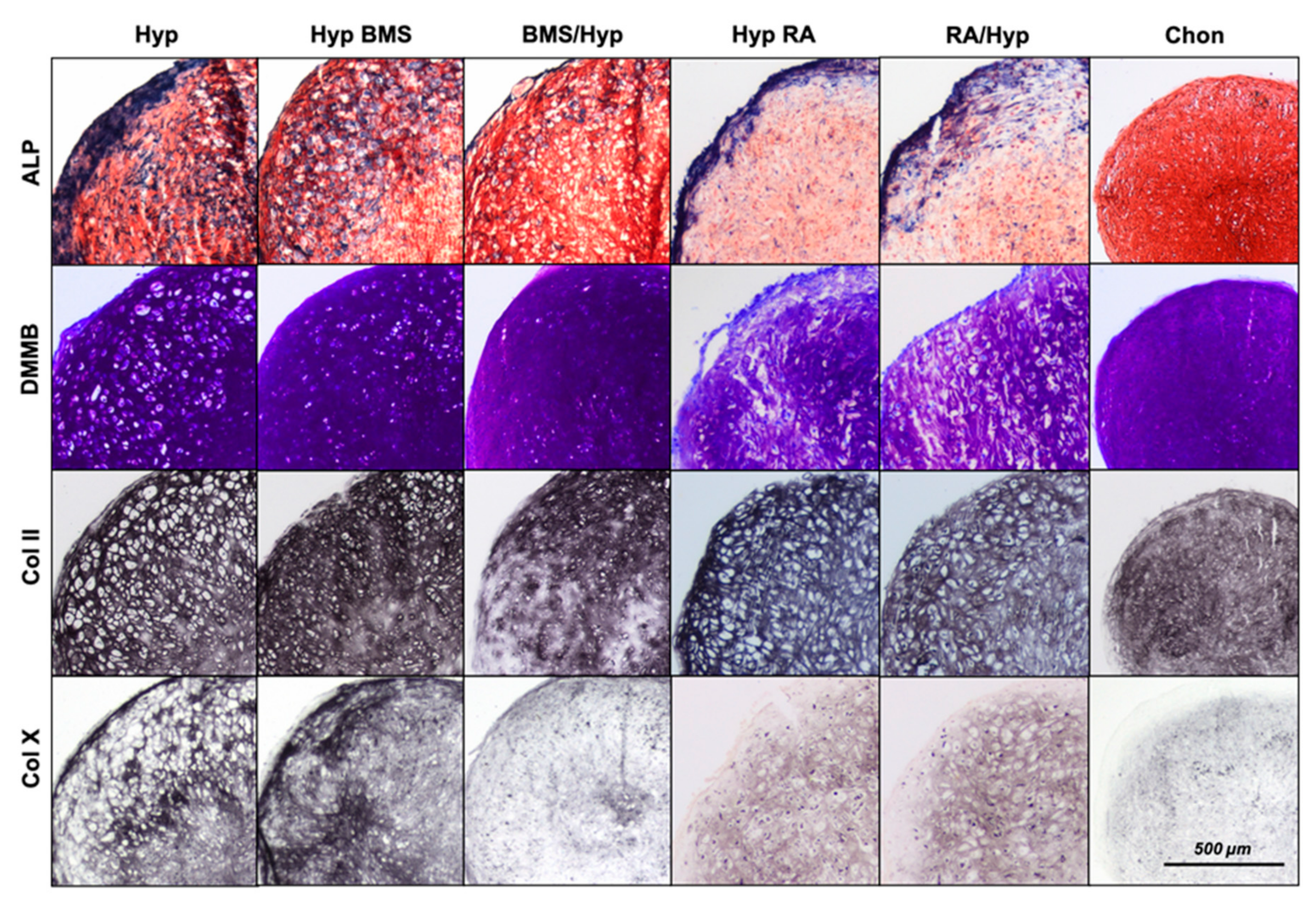
2.2. Enhancement of Hypertrophy by RA Treatment
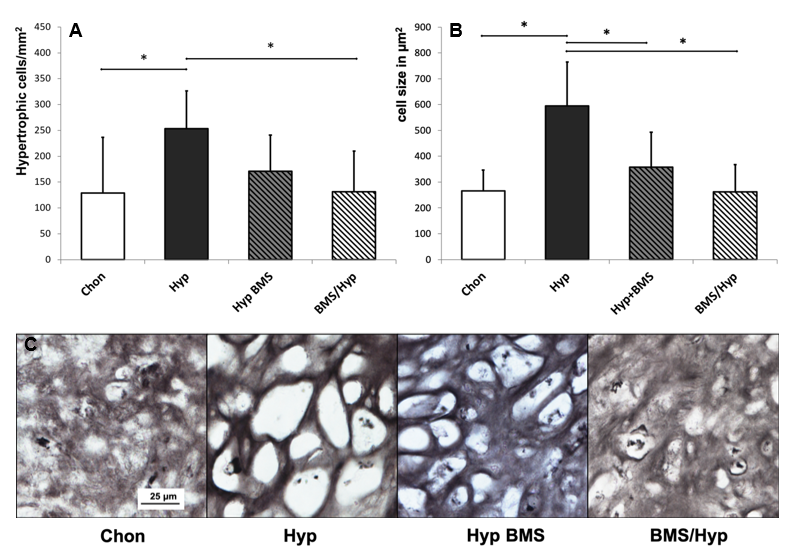
2.3. Attenuation of Hypertrophy by BMS Treatment
3. Discussion
4. Material and Methods
4.1. Isolation of MSCs
4.2. Chondrogenic Differentiation and Hypertrophic Conversion upon Treatment with BMS493 and RA
4.3. Histological and Immunohistochemical Analysis
4.4. RNA Isolation, cDNA Synthesis, and Gene Expression Analysis
4.5. ALP Activity
4.6. GAG Analysis
4.7. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Pittenger, M.F.; Mackay, A.M.; Beck, S.C.; Jaiswal, R.K.; Douglas, R.; Mosca, J.D.; Marshak, D.R. Multilineage potential of adult human mesenchymal stem cells. Science 1999, 284, 143–147. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zuk, P.A.; Zhu, M.I.N.; Mizuno, H.; Huang, J.; Futrell, J.W.; Katz, A.J.; Hedrick, M.H. Multilineage cells from human adipose tissue: Implications for cell-based therapies. Tissue Eng. 2001, 7, 211–228. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ashton, B.A.; Allen, T.D.; Howlett, C.R.; Eaglesom, C.C.; Hattori, A.; Owen, M. Formation of bone and cartilage by marrow stromal cells in diffusion chambers in vivo. Clin. Orthop. Relat. Res. 1980, 151, 294–307. [Google Scholar] [CrossRef]
- Johnstone, B.; Hering, T.M.; Caplan, A.I.; Goldberg, V.M.; Yoo, J.U. In vitro chondrogenesis of bone marrow-derived mesenchymal progenitor cells. Exp. Cell Res. 1998, 238, 265–272. [Google Scholar] [CrossRef]
- Yoo, J.U.; Barthel, T.S.; Nishimura, K.; Solchaga, L.; Caplan, A.I.; Goldberg, V.M.; Johnstone, B. The chondrogenic potential of human bone-marrow-derived mesenchymal progenitor cells. J. Bone Jt. Surg. Am. 1998, 80, 1745–1757. [Google Scholar] [CrossRef]
- Ichinose, S.; Tagami, M.; Muneta, T.; Sekiya, I. Morphological examination during in vitro cartilage formation by human mesenchymal stem cells. Cell Tissue Res. 2005, 322, 217–226. [Google Scholar] [CrossRef]
- Barry, F.; Boynton, R.E.; Liu, B.; Murphy, J.M. Chondrogenic differentiation of mesenchymal stem cells from bone marrow: Differentiation-dependent gene expression of matrix components. Exp. Cell Res. 2001, 268, 189–200. [Google Scholar] [CrossRef]
- Mueller, M.B.; Fischer, M.; Zellner, J.; Berner, A.; Dienstknecht, T.; Prantl, L.; Angele, P. Hypertrophy in mesenchymal stem cell chondrogenesis: Effect of TGF-beta isoforms and chondrogenic conditioning. Cells Tissues Organs 2010, 192, 158–166. [Google Scholar] [CrossRef] [Green Version]
- Pelttari, K.; Winter, A.; Steck, E.; Goetzke, K.; Hennig, T.; Ochs, B.G.; Richter, W. Premature induction of hypertrophy during in vitro chondrogenesis of human mesenchymal stem cells correlates with calcification and vascular invasion after ectopic transplantation in SCID mice. Arthr. Rheum. 2006, 54, 3254–3266. [Google Scholar] [CrossRef]
- Wu, L.; Bluguermann, C.; Kyupelyan, L.; Latour, B.; Gonzalez, S.; Shah, S.; Nsair, A. Human developmental chondrogenesis as a basis for engineering chondrocytes from pluripotent stem cells. Stem Cell Rep. 2013, 1, 575–589. [Google Scholar] [CrossRef] [Green Version]
- Goldring, M.B.; Tsuchimochi, K.; Ijiri, K. The control of chondrogenesis. J. Cell. Biochem. 2006, 97, 33–44. [Google Scholar] [CrossRef] [PubMed]
- Decker, R.S.; Koyama, E.; Pacifici, M. Genesis and morphogenesis of limb synovial joints and articular cartilage. Matrix Biol. J. Int. Soc. Matrix Biol. 2014, 39, 5–10. [Google Scholar] [CrossRef] [PubMed]
- Pacifici, M.; Koyama, E.; Shibukawa, Y.; Wu, C.; Tamamura, Y.; Enomoto-Iwamoto, M.; Iwamoto, M. Cellular and molecular mechanisms of synovial joint and articular cartilage formation. Ann. N. Y. Acad. Sci. 2006, 1068, 74–86. [Google Scholar] [CrossRef] [PubMed]
- Koyama, E.; Shibukawa, Y.; Nagayama, M.; Sugito, H.; Young, B.; Yuasa, T.; Okabe, T.; Ochiai, T.; Kamiya, N.; Rountree, R.B.; et al. A distinct cohort of progenitor cells participates in synovial joint and articular cartilage formation during mouse limb skeletogenesis. Dev. Biol. 2008, 316, 62–73. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hyde, G.; Dover, S.; Aszodi, A.; Wallis, G.A.; Boot-Handford, R.P. Lineage tracing using matrilin-1 gene expression reveals that articular chondrocytes exist as the joint interzone forms. Dev. Biol. 2007, 304, 825–833. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoffman, L.M.; Garcha, K.; Karamboulas, K.; Cowan, M.F.; Drysdale, L.M.; Horton, W.A.; Underhill, T.M. BMP action in skeletogenesis involves attenuation of retinoid signaling. J. Cell Biol. 2006, 174, 101–113. [Google Scholar] [CrossRef]
- Weston, A.D.; Chandraratna, R.A.S.; Torchia, J.; Underhill, T.M. Requirement for RAR-mediated gene repression in skeletal progenitor differentiation. J. Cell Biol. 2002, 158, 39–51. [Google Scholar] [CrossRef]
- Weston, A.D.; Rosen, V.; Chandraratna, R.A.; Underhill, T.M. Regulation of skeletal progenitor differentiation by the BMP and retinoid signaling pathways. J. Cell Biol. 2000, 148, 679–690. [Google Scholar] [CrossRef]
- Pacifici, M. Retinoid roles and action in skeletal development and growth provide the rationale for an ongoing heterotopic ossification prevention trial. Bone 2018, 109, 267–275. [Google Scholar] [CrossRef]
- Hoffman, L.M.; Weston, A.D.; Underhill, T.M. Molecular mechanisms regulating chondroblast differentiation. J. Bone Jt. Surg. Am. 2003, 85, 124–132. [Google Scholar] [CrossRef]
- Koyama, E.; Golden, E.B.; Kirsch, T.; Adams, S.L.; Chandraratna, R.A.; Michaille, J.J.; Pacifici, M. Retinoid signaling is required for chondrocyte maturation and endochondral bone formation during limb skeletogenesis. Dev. Biol. 1999, 208, 375–391. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shimono, K.; Tung, W.E.; Macolino, C.; Chi, A.H.T.; Didizian, J.H.; Mundy, C.; Iwamoto, M. Potent inhibition of heterotopic ossification by nuclear retinoic acid receptor-γ agonists. Nat. Med. 2011, 17, 454–460. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cohen, A.J.; Lassová, L.; Golden, E.B.; Niu, Z.; Adams, S.L. Retinoids directly activate the collagen X promoter in prehypertrophic chondrocytes through a distal retinoic acid response element. J. Cell. Biochem. 2006, 99, 269–278. [Google Scholar] [CrossRef] [PubMed]
- Lu, J.; Pallante, K.; Niu, Z.; Leboy, P.; Cohen, A.; Adam, S. Induction of type-X collagen gene transcription by retinoids occurs in part through the BMP-signaling pathway. Penn Dent. J. 2002, 102, 5–31. [Google Scholar] [PubMed]
- Ballock, R.T.; Heydemann, A.; Wakefield, L.M.; Flanders, K.C.; Roberts, A.B.; Sporn, M.B. Inhibition of the chondrocyte phenotype by retinoic acid involves upregulation of metalloprotease genes independent of TGF-beta. J. Cell. Physiol. 1994, 159, 340–346. [Google Scholar] [CrossRef] [PubMed]
- Cash, D.; Bock, C.; Schughart, K.; Linney, E.; Underhill, T. Retinoic acid receptor alpha function in vertebrate limb skeletogenesis: A modulator of chondrogenesis. J. Cell Biol. 1997, 136, 445–457. [Google Scholar] [CrossRef] [Green Version]
- Hill, D.S.; Ragsdale, C.W.; Brockes, J.P. Isoform-specific immunological detection of newt retinoic acid receptor delta 1 in normal and regenerating limbs. Development 1993, 117, 937–945. [Google Scholar]
- Ragsdale, C.W.; Gates, P.B.; Hill, D.S.; Brockes, J.P. Delta retinoic acid receptor isoform delta 1 is distinguished by its exceptional N-terminal sequence and abundance in the limb regeneration blastema. Mech. Dev. 1993, 40, 99–112. [Google Scholar] [CrossRef]
- Germain, P.; Gaudon, C.; Pogenberg, V.; Sanglier, S.; Van Dorsselaer, A.; Royer, C.A.; Gronemeyer, H. Differential action on coregulator interaction defines inverse retinoid agonists and neutral antagonists. Chem. Biol. 2009, 16, 479–489. [Google Scholar] [CrossRef]
- Chambon, P. A decade of molecular biology of retinoic acid receptors. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 1996, 10, 940–954. [Google Scholar] [CrossRef]
- Janesick, A.; Nguyen, T.T.; Aisaki, K.I.; Igarashi, K.; Kitajima, S.; Chandraratna, R.A.; Blumberg, B. Active repression by RARγ signaling is required for vertebrate axial elongation. Development 2014, 141, 2260–2270. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koide, T.; Downes, M.; Chandraratna, R.A.; Blumberg, B.; Umesono, K. Active repression of RAR signaling is required for head formation. Genes Dev. 2001, 15, 2111–2121. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gaur, T.; Lengner, C.J.; Hovhannisyan, H.; Bhat, R.A.; Bodine, P.V.; Komm, B.S.; Lian, J.B. Canonical WNT signaling promotes osteogenesis by directly stimulating Runx2 gene expression. J. Biol. Chem. 2005, 280, 33132–33140. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dong, Y.-F.; Soung, D.Y.; Schwarz, E.M.; O’Keefe, R.J.; Drissi, H. Wnt induction of chondrocyte hypertrophy through the Runx2 transcription factor. J. Cell. Physiol. 2006, 208, 77–86. [Google Scholar] [CrossRef] [PubMed]
- Hill, T.; Später, D.; Taketo, M.; Birchmeier, W.; Hartmann, C. Canonical Wnt/beta-catenin signaling prevents osteoblasts from differentiating into chondrocytes. Dev. Cell 2005, 8, 727–738. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sassi, N.; Laadhar, L.; Allouche, M.; Achek, A.; Kallel-Sellami, M.; Makni, S.; Sellami, S. WNT signaling and chondrocytes: From cell fate determination to osteoarthritis physiopathology. J. Recept. Signal Transduct. Res. 2014, 34, 73–80. [Google Scholar] [CrossRef]
- Kawaguchi, R.; Yu, J.; Honda, J.; Hu, J.; Whitelegge, J.; Ping, P.; Sun, H. A membrane receptor for retinol binding protein mediates cellular uptake of vitamin A. Science 2007, 315, 820–825. [Google Scholar] [CrossRef]
- Kedishvili, N.Y. Enzymology of retinoic acid biosynthesis and degradation. J. Lipid Res. 2013, 54, 1744–1760. [Google Scholar] [CrossRef] [Green Version]
- Dong, D.; Ruuska, S.E.; Levinthal, D.J.; Noy, N. Distinct Roles for Cellular Retinoic Acid-binding Proteins I and II in Regulating Signaling by Retinoic Acid. J. Biol. Chem. 1999, 274, 23695–23698. [Google Scholar] [CrossRef] [Green Version]
- Napoli, J.L. Cellular retinoid binding-proteins, CRBP, CRABP, FABP5: Effects on retinoid metabolism, function and related diseases. Pharmacol. Ther. 2017, 173, 19–33. [Google Scholar] [CrossRef] [Green Version]
- Yasuhara, R.; Yuasa, T.; Williams, J.A.; Byers, S.W.; Shah, S.; Pacifici, M.; Enomoto-Iwamoto, M. Wnt/beta-catenin and retinoic acid receptor signaling pathways interact to regulate chondrocyte function and matrix turnover. J. Biol. Chem. 2010, 285, 317–327. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kafienah, W.; Mistry, S.; Perry, M.J.; Politopoulou, G.; Hollander, A.P. Pharmacological regulation of adult stem cells: Chondrogenesis can be induced using a synthetic inhibitor of the retinoic acid receptor. Stem Cells 2007, 25, 2460–2468. [Google Scholar] [CrossRef] [PubMed]
- Klein, E.S.; Pino, M.E.; Johnson, A.T.; Davies, P.J.; Nagpal, S.; Thacher, S.M.; Chandraratna, R.A. Identification and functional separation of retinoic acid receptor neutral antagonists and inverse agonists. J. Biol. Chem. 1996, 271, 22692–22696. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klein, E.S.; Wang, J.W.; Khalifa, B.; Gavigan, S.A.; Chandraratna, R.A. Recruitment of nuclear receptor corepressor and coactivator to the retinoic acid receptor by retinoid ligands. Influence of DNA-heterodimer interactions. J. Biol. Chem. 2000, 275, 19401–19408. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Castagnola, P.; Dozin, B.; Moro, G.; Cancedda, R. Changes in the expression of collagen genes show two stages in chondrocyte differentiation in vitro. J. Cell Biol. 1988, 106, 461–467. [Google Scholar] [CrossRef] [PubMed]
- Kosher, R.A.; Kulyk, W.M.; Gay, S.W. Collagen gene expression during limb cartilage differentiation. J. Cell Biol. 1986, 102, 1151–1156. [Google Scholar] [CrossRef] [Green Version]
- Focht, R.J.; Adams, S.L. Tissue specificity of type I collagen gene expression is determined at both transcriptional and post-transcriptional levels. Mol. Cell. Biol. 1984, 4, 1843–1852. [Google Scholar] [CrossRef] [Green Version]
- Karl, A.; Olbrich, N.; Pfeifer, C.; Berner, A.; Zellner, J.; Kujat, R.; Mueller, M.B. Thyroid hormone-induced hypertrophy in mesenchymal stem cell chondrogenesis is mediated by bone morphogenetic protein-4. Tissue Eng. Part A 2014, 20, 178–188. [Google Scholar] [CrossRef] [Green Version]
- Mueller, M.B.; Fischer, M.; Zellner, J.; Berner, A.; Dienstknecht, T.; Kujat, R.; Angele, P. Effect of parathyroid hormone-related protein in an in vitro hypertrophy model for mesenchymal stem cell chondrogenesis. Int. Orthop. 2013, 37, 945–951. [Google Scholar] [CrossRef] [Green Version]
- Hatakeyama, Y.; Tuan, R.S.; Shum, L. Distinct functions of BMP4 and GDF5 in the regulation of chondrogenesis. J. Cell. Biochem. 2004, 91, 1204–1217. [Google Scholar] [CrossRef]
- Kramer, J.; Hegert, C.; Guan, K.; Wobus, A.M.; Müller, P.K.; Rohwedel, J. Embryonic stem cell-derived chondrogenic differentiation in vitro: Activation by BMP-2 and BMP-4. Mech. Dev. 2000, 92, 193–205. [Google Scholar] [CrossRef]
- Semba, I.; Nonaka, K.; Takahashi, I.; Takahashi, K.; Dashner, R.; Shum, L.; Slavkin, H. Positionally-dependent chondrogenesis induced by BMP4 is co-regulated by Sox9 and Msx2. Dev. Dyn. Off. Publ. Am. Assoc. Anat. 2000, 217, 401–414. [Google Scholar] [CrossRef]
- Müller, P.; Doliva, R.; Busch, M.; Philippeit, C.; Stephan, H.; Dünker, N. Additive Effects of Retinoic Acid (RA) and Bone Morphogenetic Protein 4 (BMP-4) Apoptosis Signaling in Retinoblastoma Cell Lines. PLoS ONE 2015, 10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McLeod, C.M.; Mauck, R.L. On the origin and impact of mesenchymal stem cell heterogeneity: New insights and emerging tools for single cell analysis. Eur. Cells Mater. 2017, 34, 217–231. [Google Scholar] [CrossRef] [PubMed]
- Siegel, G.; Kluba, T.; Kluba, T.; Hermanutz-Klein, U.; Bieback, K.; Northoff, H.; Schäfer, R. Phenotype, donor age and gender affect function of human bone marrow-derived mesenchymal stromal cells. BMC Med. 2013, 11, 146. [Google Scholar] [CrossRef] [Green Version]
- Yin, L.; Agustinus, A.S.; Yuvienco, C.; Minashima, T.; Schnabel, N.L.; Kirsch, T.; Montclare, J.K. Engineered Coiled-Coil Protein for Delivery of Inverse Agonist for Osteoarthritis. Biomacromolecules 2018, 19, 1614–1624. [Google Scholar] [CrossRef]
- Busby, S.A.; Kumar, N.; Kuruvilla, D.S.; Istrate, M.A.; Conkright, J.J.; Wang, Y.; Kamenecka, T.M.; Cameron, M.D.; Roush, W.R.; Burris, T.P.; et al. Identification of a novel non-retinoid pan inverse agonist of the retinoic acid receptors. ACS Chem. Biol. 2011, 6, 618–627. [Google Scholar] [CrossRef]
- Weber, M.; Steinert, A.; Jork, A.; Dimmler, A.; Thürmer, F.; Schütze, N.; Hendrich, C.; Zimmerman, U. Formation of cartilage matrix proteins by BMP-transfected murine mesenchymal stem cells encapsulated in a novel class of alginates. Biomaterials 2002, 23, 2003–2013. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Group | Week 1+2 | d14 | Week 3+4 | d28 |
|---|---|---|---|---|
| 1 | Chondrogenic medium | Chondrogenic medium | Chon | |
| 2 | Chondrogenic medium | Hypertrophic medium | Hyp | |
| 3 | Chondrogenic medium | Hypertrophic medium + BMS | Hyp w BMS | |
| 4 | Chondrogenic medium + BMS | Hypertrophic medium | BMS/Hyp | |
| 5 | Chondrogenic medium | Hypertrophic medium + RA | Hyp w RA | |
| 6 | Chondrogenic medium + RA | Hypertrophic medium | RA/Hyp | |
| Gene | Sequence (Forward) | Sequence (Reverse) | Concentration |
|---|---|---|---|
| VPS29 | AGCTGGCAAACTGTTGCAC | GACGGTGGTGGTGACTGAG | 200 nM |
| PSMB4 | GCTTAGCACTGGCTGCTTCT | GGACATGCTTGGTGTAGCCT | 200 nM |
| REEP5 | AGGTCAGCCACTGGGTATCA | CCTCTCTCCTCTGCAACCTG | 200 nM |
| MMP13 | GACTGGTAATGGCATCAAGGGA | CACCGGCAAAAGCCACTTTA | 200 nM |
| COL1A1 | ACGTCCTGGTGAAGTTGGTC | ACCAGGGAAGCCTCTCTCTC | 200 nM |
| COL2A1 | GGGCAATAGCAGGTTCACGTA | TGTTTCGTGCAGCCATCCT | 200 nM |
| COL10A1 | CCCTCTTGTTAGTGCCAACC | AGATTCCCAGTCCTTGGGTCA | 200 nM |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Riedl, M.; Witzmann, C.; Koch, M.; Lang, S.; Kerschbaum, M.; Baumann, F.; Krutsch, W.; Docheva, D.; Alt, V.; Pfeifer, C. Attenuation of Hypertrophy in Human MSCs via Treatment with a Retinoic Acid Receptor Inverse Agonist. Int. J. Mol. Sci. 2020, 21, 1444. https://doi.org/10.3390/ijms21041444
Riedl M, Witzmann C, Koch M, Lang S, Kerschbaum M, Baumann F, Krutsch W, Docheva D, Alt V, Pfeifer C. Attenuation of Hypertrophy in Human MSCs via Treatment with a Retinoic Acid Receptor Inverse Agonist. International Journal of Molecular Sciences. 2020; 21(4):1444. https://doi.org/10.3390/ijms21041444
Chicago/Turabian StyleRiedl, Moritz, Christina Witzmann, Matthias Koch, Siegmund Lang, Maximilian Kerschbaum, Florian Baumann, Werner Krutsch, Denitsa Docheva, Volker Alt, and Christian Pfeifer. 2020. "Attenuation of Hypertrophy in Human MSCs via Treatment with a Retinoic Acid Receptor Inverse Agonist" International Journal of Molecular Sciences 21, no. 4: 1444. https://doi.org/10.3390/ijms21041444