Protofibrils of Amyloid-β are Important Targets of a Disease-Modifying Approach for Alzheimer’s Disease


1
Department of Internal Medicine, Division of Neurology, School of Medicine, Showa University, Tokyo 142-8666, Japan
2
Department of Pharmacology, School of Medicine, Showa University, Tokyo 142-8666, Japan
*
Author to whom correspondence should be addressed.
Int. J. Mol. Sci. 2020, 21(3), 952; https://doi.org/10.3390/ijms21030952
Submission received: 31 December 2019
/
Revised: 20 January 2020
/
Accepted: 29 January 2020
/
Published: 31 January 2020
(This article belongs to the Special Issue Amyloid-β: Structure, Function, and Pathophysiological Significance in Neurodegenerative Diseases)
{kind=link}
Abstract
:Worldwide, Alzheimer’s disease (AD) is the most common age-related neurodegenerative disease and is characterized by unique pathological hallmarks in the brain, including plaques composed of amyloid β-protein (Aβ) and neurofibrillary tangles of tau protein. Genetic studies, biochemical data, and animal models have suggested that Aβ is responsible for the pathogenesis of AD (i.e., the amyloid hypothesis). Indeed, Aβ molecules tend to aggregate, forming oligomers, protofibrils, and mature fibrils. However, while these Aβ species form amyloid plaques of the type implicated in AD neurodegeneration, recent clinical trials designed to reduce the production of Aβ and/or the plaque burden have not demonstrated clinical efficacy. In addition, recent studies using synthetic Aβ peptides, cell culture models, Arctic transgenic mice, and human samples of AD brain tissues have suggested that the pre-fibrillar forms of Aβ, particularly Aβ protofibrils, may be the most critical species, compared with extracellular fibrillar forms. We recently reported that protofibrils of Aβ1-42 disturbed membrane integrity by inducing reactive oxygen species generation and lipid peroxidation, resulting in decreased membrane fluidity, intracellular calcium dysregulation, depolarization, and synaptic toxicity. Therefore, the therapeutic reduction of protofibrils may prevent the progression of AD by ameliorating neuronal damage and cognitive dysfunction through multiple mechanisms.
1. Introduction
Neurodegenerative diseases, such as Alzheimer’s disease (AD), Parkinson’s disease, and spinocerebellar ataxia, have characteristic abnormal protein aggregates in the brain. In AD, the two neuropathological characteristics are amyloid plaques composed of amyloid β-protein (Aβ) and neurofibrillary tangles of hyperphosphorylated tau protein [1].
Human genetic association studies, biochemical analyses of AD plaque content, and various animal models with altered Aβ or tau expression have strongly implicated Aβ and tau in AD pathogenesis [1]. Furthermore, many in vivo and in vitro studies have demonstrated the neurotoxicity of these amyloidogenic proteins. However, amyloid neurotoxicity depends strongly on Aβ’s primary structure and aggregation state. For example, two predominant Aβ forms are produced in humans and are comprised of either 40 (Aβ1-40) or 42 (Aβ1-42) amino acid residues. The relative proportion of Aβ1-42 appears to be particularly crucial for AD progression, as this longer form is more prone to aggregation and is inherently more toxic than Aβ1-40 [2]. Aβ molecules form low molecular weight (LMW) oligomers, high molecular weight (HMW) oligomers such as protofibrils (PFs), and mature fibrils, which have been suggested to be primary agents of neuronal dysfunction in AD [3]. Although these Aβ aggregates may directly cause neuronal injury by acting on synapses or indirectly by activating astrocytes and microglia [2], evidence also supports the hypothesis that soluble oligomeric Aβ plays an important role in AD pathogenesis (i.e., the oligomer hypothesis) [1,3,4].
Many types of oligomeric Aβ species have been demonstrated in vitro, with PFs being commonly described. Aβ PFs are defined as curved linear structures >100 kDa that remain soluble upon centrifugation at 16,000–18,000× g [3,5,6,7]. The neurotoxicity of these Aβ PFs formed in vitro, as well as their ability to induce electrophysiological effects on neurons, has been demonstrated by several groups [8,9,10,11]. Arctic Aβ is the result of a mutation in the gene that encodes the amyloid precursor protein (APP) and leads to the production of a particular Aβ species, [Glu22Gly]Aβ, with a high propensity to form PFs [12]. We recently reported that PFs disturb membrane integrity by inducing reactive oxygen species’ (ROS) generation and lipid peroxidation, resulting in decreased membrane fluidity, intracellular calcium dysregulation, depolarization, and impaired long-term potentiation (LTP). In addition, the damaging effects of PFs were found to be significantly greater than those of LMW-Aβ1-42 [13].
Current treatments for AD are primarily aimed at mitigating symptoms, while disease-modifying approaches are aimed at halting or attenuating the progression of the disease, such as inhibiting Aβ production and aggregation or promoting Aβ1-42 clearance [14]. However, despite many long and expensive trials, no disease-modifying drug for AD has been approved [15,16]. A recent failure in phase 3 involved the investigation of a β secretase in patients with mild-to-moderate AD [17]. Other large, phase 3 trials using anti-amyloid approaches including semagacestat [18], bapineuzumab [19], and solanezumab [20], have yielded disappointing results. However, it has been recently reported that BAN2401 (mAb158), an antibody developed for early AD with a unique target binding profile selective for Aβ PFs, significantly slowed cognitive decline by 30%, with a concomitant reduction in amyloid plaques, compared with placebo at 18 months [21].
In this review, we focus on recent developments from basic and clinical studies of PFs, including research findings from our laboratory.
2. PFs Are Primary Toxins in AD
2.1. The Discovery of PFs and Their Role in AD Pathogenesis
PFs were first described by Teplow and colleagues in 1997 [6]. Using a size exclusion chromatography (SEC) system and the synthetic Aβ1-42 peptide, they found a peak representing a large (>100 kDa) soluble species before the peak of the LMW-Aβ (mainly monomer) [6]. Using electron microscopy (EM), they further revealed that this peak contained predominantly curved fibrils, with a diameter of ~5 nm and a length of up to 200 nm, which they termed PFs [6]. Subsequently, the authors elucidated that the PFs were composed primarily of β-sheets and partially random coils and α-helices in a secondary structure [6]. In the same year, using atomic force microscopy (AFM), Lansbury’s group found the existence of a metastable intermediate species, which was termed Aβ PF [22]. Many data have shown that LMW-Aβ oligomers are on-pathway to fibril formation, while HMW-Aβ oligomers such as PFs are off-pathway [22,23,24,25]. Although the PF-to-fibril transition, characterized by PF elongation, was very slow, preformed fibrillar seeds greatly accelerated this conversion [22]. Recently, using a combination of high-speed AFM with thioflavin T assay, EM, and re-injection assays by SEC, we demonstrated that fibril formation from PFs is more difficult than that from LMW-Aβ, suggesting that mature fibrils of Aβ1-42 are primarily formed from LMW-Aβ1-42 and not from PFs [24]. Furthermore, we determined that PFs instead supplied precursors to LMW-Aβ1-42 by their dissociation, suggesting that PFs may not always represent the “on-pathway” of Aβ1-42 aggregation from the monomer to the mature fibrils [24]. Kodali and Wetzel mentioned that, although Aβ1-40 PFs can grow by monomer addition, their rate of growth is lower than that of mature fibrils. Additionally, while Aβ1-40 monomer was able to support the extension of mature fibrils at low concentrations of, Aβ1-40 PFs exhibited no extension [23]. They suggested another terminology, “curvilinear fibrils”, for the description of off-pathway PFs instead of PFs as on-pathway precursors of fibrils [23]. It was recently revealed that curvilinear fibrils inhibit fibril formation not only by slowing fibril nucleation and elongation, but also by actively disrupting either process based on combined thioflavin kinetics and AFM imaging data [26]. On the other hand, Iwatsubo’s group showed that Aβ1-42 PF injection induced Aβ deposition in the brains of A7 mice overexpressing human APP695 and harboring the K670N, M671L, and T714I familial AD neuronal mutations, suggesting that Aβ PFs may act as a seed for Aβ aggregation in vivo [27]. The injection of Aβ PFs mixed with apoE3 significantly attenuated Aβ deposition, whereas apoE4 did not, suggesting that the suppressive effect of apoE3 on the structural conversion of Aβ PFs to fibrils is stronger than that of apoE4, thereby impeding Aβ deposition in vivo [27].
2.2. PFs Are Primary Toxins in AD
The solubility and diffusible nature of soluble oligomers may render them more effective in terms of intra- and extra-cellular interactions and engaging microglial receptors compared with mature insoluble fibrils. Indeed, it has been demonstrated that astrocytes engulf large amounts of accumulated, rather than digested, Aβ1-42 PFs. This intracellular storage of Aβ1-42 results in severe astrocytic endosomal/lysosomal defects and the secretion of extracellular vesicles containing N-truncated, neurotoxic Aβ [28]. Aβ1-42 PFs have also been shown to induce an inflammatory process through microglial activation [29] and initiate Toll-like receptor (TLR) signaling (Figure 1) [30]. In addition, these PFs are preferentially internalized by microglia [31]. Furthermore, it has been reported that Aβ1-42 PFs are more effective at inducing microglial tumor necrosis factor α (TNFα) production in BV-2 and primary murine microglia in vitro than monomers and mature fibrils. Moreover, PFs of Aβ1-40 exhibit significantly less activity than concentration-matched Aβ1-42 [29]. Aβ1-42 PFs also have been shown to trigger a time- and myeloid differentiation protein (MyD) 88-dependent process that generates TNFα and interleukin-1β (IL-1β) mRNA, along with pro and mature forms of the intracellular IL-1β protein [30]. The accumulation of both IL-1β forms has indicated that Aβ1-42 PFs are able to prime and activate the Nod-like receptor (NLR) P3 inflammasome. In this process, Aβ has been shown to elicit a quantized burst of secreted IL-1β which occurs prior to the Aβ priming of the microglia. The IL-1β secretion burst appears to be rapid and not sustained, yet it may be re-initiated with additional Aβ stimulation. These findings indicate multiple modes of IL-1β regulation by Aβ1-42 PFs, including TLR/MyD88-mediated priming, NLRP3 inflammasome activation, and modulation of the IL-1β secretory process, suggesting wide-ranging effects of Aβ on the innate immune response [30].
Recent evidence has suggested that the neuronal cell membrane is the chief site of oligomer-mediated neuronal damage. We recently studied the cellular response to short exposures to PFs using multiple indices of membrane integrity, cytolysis, oxidative stress, and synaptic function. We found that cellular membrane and metabolic integrity were more severely disrupted by PFs of Aβ1-42 than LMW-Aβ1-42, as evidenced by various experimental systems, including cell viability and leakage assays, fluorometric measures of ROS generation, lipid peroxidation assays, and electrophysiological recordings [13]. While our results for lactate dehydrogenase (LDH) and calcein and ethidium homodimer-1 assays reflected cellular membrane damage by PFs of Aβ1-42 to a greater extent than LMW-Aβ1-42, 3-[4,5-dimethylthiazol-2-yl]-2,5-diphenyltetrazolium bromide metabolism (MTT) and water soluble tetrazolium (WST) assays reflecting mitochondrial enzyme activity, they demonstrated only small differences between the Aβs in different cellular models, including SH-SY5Y cells and a healthy, human-induced pluripotent stem line [13]. From these results, in terms of short-term Aβ1-42 PF treatment, Aβ1-42 PFs may first attack the cell membrane, followed by subsequent damage to the mitochondria, although Aβ1-42 dimers might not be removed clearly in LMW-Aβ1-42 preparation using the above-mentioned SEC method [6]. Next, we found that exposure to PFs of Aβ1-42 in SH-SY5Y cells induces more severe oxidative stress, including greater levels of ROS production and membrane lipid peroxidation, than LMW-Aβ1-42. Indeed, many studies have reported that oxidative stress, which occurs in the presence of a physiological imbalance between ROS generation and antioxidant capacity, is a critical pathogenic mechanism in AD progression [32]. Along with the direct destruction/modification of lipids, DNA, and proteins, the byproducts of lipid peroxidation produced during oxidative stress cause damage to the mitochondria and upregulate tau phosphorylation, which appears essential for NFT formation [33]. In addition, the generation of superoxide by Aβ aggregates may lead to mitochondrial impairment and further induce ROS generation, thereby establishing a positive feedback pathway that ultimately results in cell death [34]. Moreover, Aβ aggregates may directly interact with the mitochondrial respiratory chain, causing metabolic dysfunction and increased ROS production [35]. In our study, the PFs of Aβ1-42 also reduced neuronal membrane fluidity to a significantly greater extent than LMW-Aβ1-42. Thus, we consider the possibility that the effects on membrane fluidity, and the resulting neuronal damage, depend on the specific Aβ conformation [13].
We further demonstrated that short exposures to Aβ1-42 PFs induces higher concentrations of [Ca2+]i than LMW-Aβ1-42, whereas a reduced depolarization-induced [Ca2+]i influx through voltage-dependent Ca2+ channels was observed following longer exposures to Aβ1-42 PFs [13]. These results suggested that PFs may not only directly damage voltage-gated calcium channels for a short time, but also alter the cell membrane environment required for proper channel insertion or gating for longer periods, as evidenced by lipid peroxidation and membrane fluidity measurements [13].
Consistent with the changes observed in [Ca2+]i and the loss of membrane integrity, the application of PFs of Aβ1-42, but not those of LMW-Aβ1-42, also has been shown to depolarize SH-SY5Y cells and significantly reduce membrane input resistance [13]. Bode et al. monitored transmembrane currents during Aβ exposure at the extracellular face of excised membranes from HEK293 cells, and found that annular Aβ1-42 oligomers formed ion channels, whereas Aβ1-40 oligomers and mature fibrils and monomers did not [36]. Drolle et al. used multi-component lipid models to mimic healthy and AD states of neuronal membranes and posited that Aβ1-42 increases lipid membrane roughness and membrane conductance, possibly through pore formation [37]. Taken together, the [Ca2+]i increase evoked by PFs may be due to pore formation and oxidative damage, as well as the suppression of calcium egress and sequestration pathways secondary to metabolic disruption.
We also demonstrated that Aβ1-42 PFs significantly inhibit LTP formation in the mouse hippocampal CA1 subfield [13]. Similarly, it has been reported that PFs induce electrophysiological changes, including rapid increases in the excitatory post synaptic and action potentials, membrane depolarizations in rat cortical neurons [8], and the inhibition of LTP in the rat hippocampus [38]. Excessive ROS accumulation and decreased membrane fluidity are associated with effects on LTP and learning [39,40]. Furthermore, membrane pore formation may also impair cellular and synaptic functions (Figure 1) [41,42].
The small (35kDa) and highly dispersible protein, secreted-frizzled-related protein 1 (SFRP1), regulates transmembrane metalloprotease ADAM10 activity and is essential for the development of tissue homeostasis and constitutive levels of α-secretase in the brain [43]. As a novel player in AD pathogenesis, SFRP1 has been shown to be significantly increased in the brain and cerebrospinal fluid of patients with AD. In addition, SFRP1 has been demonstrated in human AD cases and mouse models to prevent Aβ PF formation by binding to Aβ, suggesting it may be a promising AD therapeutic target [44].
2.3. Arctic Mutation Causes Aβ PF Formation
Arctic mutation is a pathogenic APP mutation located within the Aβ sequence at codon 693, at which point glutamic acid is substituted for glycine (E693G). In 2001, Lannfelt’s group named the mutation the ‘Arctic’ mutation because the family in which it was detected was from northern Sweden [12]. Affected subjects have clinical features of early AD and plasma levels of both Aβ1-40 and Aβ1-42 are lower in mutation carriers compared with healthy family members. In addition, concentrations of Aβ1-42 were found to be reduced in media from cells transfected with APPE693G [12]. Furthermore, the authors reported that the Arctic Aβ mutation (Aβ1-40 Arc) causes enhanced the formation of Aβ1-40 PFs in vitro [12]. Subsequently, Lannfelt’s group found that the Arctic mutation significantly accelerated Aβ1-42 PF formation, as well as PF fibrillization [7].
It has been reported that Aβ1-40 Arc inhibits LTP ~100-fold more potently than wild-type Aβ1-40 when wild-type and Aβ1-40 Arc peptides are injected into the CA1 area in rats intracerebroventricularly. In this study, the isolated soluble fraction that included the PFs of Aβ1-40 Arc after high-speed centrifugation was shown to still retain full LTP inhibitory activity [38]. In a later study, Lord et al. demonstrated that the Arc mutation accelerates early intraneuronal Aβ aggregation and PF formation, followed by plaque formation, in APP transgenic mice with both the Arctic (E693G) and Swedish (K670N, M671L) mutations (tg-APPArcSwe) [45,46]. In addition, cognitive deficits have been shown to occur concomitantly with the formation of intracellular Aβ deposits, but before plaque formation, in transgenic mice [45]. In addition, the levels of PFs in the brain, but not those of total Aβ, have been correlated with spatial learning, which adds further evidence to the theory that soluble PFs are the toxic species [47]. The pool of toxic Aβ species reportedly consists of molecules in the size range of 80 to 500 kDa [48].
3. Therapeutic Approaches Targeting Aβ PFs
3.1. Small Molecules Inhibit the Formation of Aβ PFs
Small molecules with the potential to mitigate toxic AD species such as Aβ1-42 PFs are promising preventive and therapeutic candidates. We previously demonstrated that a grape-seed-derived polyphenol was able to inhibit Aβ1-42 aggregation by preventing PF formation, pre-protofibrillar oligomerization, and random coil-aggregation-prone α-helix/β-sheet secondary structure transitions using various analyses, including circular dichroism spectroscopy, thioflavin T fluorescence, SEC, and EM [49]. Importantly, this polyphenol demonstrated protective effects in cytotoxicity assays, in which it was mixed with Aβ1-42 aggregates and exposed to cells [49]. Furthermore, our in vivo studies using the Tg2576 AD mouse model showed that this grape seed polyphenolic extract significantly attenuated AD-type cognitive deterioration and reduced cerebral amyloid deposition [50].
Using multiple molecular dynamics (MD) simulations, Jin et al. reported that dihydrochalcone, a compound extracted from the daemonorops draco tree, could effectively inhibit Aβ1-42 fibrillization and reduce Aβ-induced cytotoxicity by destabilizing the Aβ PFs. In this process, dihydrochalcone was shown to bind to the cavity of the Aβ1-40/Aβ1-42 PFs themselves and disrupt the D23-K28 salt bridge and inter-peptide β–sheet in the β1 region [51]. In addition, Zhou et al. reported that 1,2-(dimethoxymethano)fullerene (DMF), a water-soluble fullerene derivative, strongly inhibited Aβ1-42 aggregation by binding with Aβ PFs on three dominant binding sites, namely, the central hydrophobic core (17LVFFA21), the turn site (27NKGAI31), and the C-terminal β-sheet site comprised of glycine and hydrophobic residues (31IIGLMVGGVVI41), by MD stimulations [52]. In addition, the binding of DMF to the turn region served to disrupt the D23-K28 salt-bridge critical for PF Aβ fibril formation [52]. Another series of MD stimulations showed that wgx-50, a compound extracted from the Sichuan pepper (Zanthoxylum bungeanum), can destabilize Aβ1-42 PFs through three possible stable binding sites, including two sites in the hydrophobic grooves on the surface of the Aβ PFs, which resulted in no significant changes in Aβ structure, and one site in the interior that caused PF destabilization. At this site, wgx-50 was observed to be packed against the side chains of I32 and L34, disrupting the D23-K28 salt bridge and partially opening the two tightly compacted β-sheets [53]. Recently, Saini et al. reported that a resveratrol and clioquinol hybrid compound, (E)-5-(4-hydroxystyryl)quinolone-8-ol, inhibits Aβ1-42 aggregation by preventing the conformational transition of the Aβ1-42 monomer and causing destablization of the Aβ1-42 PF structure using MD simulation [54]. The destabilizing mechanisms of the Aβ1-42 PF structure may be due to the increasing interchain distance between chains A–B, disrupting the salt-bridge interaction between D23-K28 and decreasing the number of backbone hydrogen bonds between the chains [54]. In the same year, it was reported that β-sheet breaker peptides, particularly PPFFE pentapeptides, display strong destablizing effects that shift the energy minima toward the lowest value of sheet content and the lowest number of hydrogen bonds in Aβ1-42 PFs, using in silico methodologies including the molecular mechanics Poisson–Bolzmann surface area method and MD simulations [55].
3.2. Aβ PF-Selective Antibody
PFs have been identified in the human brain and the APP transgenic mouse brain [48,56]. mAb158 is a murine monoclonal antibody developed to selectively target HMW-Aβ1-42 assemblies [56]. Using an enzyme-linked immunosorbent assay (ELISA), it has been elucidated that mAb158 has an at least 1000-fold higher selectivity for PFs than monomeric Aβ and 10-15 times better binding affinity to PFs than to mature fibrils, thereby targeting the more toxic species of the peptide [57]. In immunohistochemistry, mAb158 also detects Aβ in plaques and the vasculature of AD brains because of the massive amount of Aβ in these structures [58]. In addition, Lord et al. reported that mAb158 inhibits in vitro Aβ1-42 fibril formation and protected cells from Aβ PF-induced cytotoxicity [59]. A co-culture study of astrocytes, neurons, and oligodendrocytes exposed to Aβ1-42 PFs in the presence or absence of mAb158 demonstrated that the presence of mAb158 almost entirely abolished Aβ accumulation in astrocytes, indicating an effect towards Aβ PF degradation. Consequently, mAb158 treatment was shown to rescue neurons from Aβ-induced cell death [60].
The treatment of tg-APPArcSwe mice with mAb158 resulted in the prevention of plaque formation if the antibody was administered before the appearance of plaques in young mice. If the treatment was started later in this mouse model, levels of insoluble Aβ were unaffected in the brains of plaque-bearing older mice. However, in both cases, soluble Aβ PF levels were diminished, supporting the notion that mAb158 can selectively reduce PF levels [59]. Similarly, the authors found that PF levels were elevated in young tg-APPArcSwe mice compared with several transgenic models lacking the Arctic mutation. In older tg-APPArcSwe mice with plaque deposition, the levels of Aβ PFs were approximately 50% higher than in younger mice, whereas levels of total Aβ were exponentially increased. Young tg-APPArcSwe mice showed deficits in spatial learning, and individual performances in the Morris water maze were inversely correlated with levels of Aβ PF, but not with total Aβ levels. These findings indicated that Aβ PFs accumulated in an age-dependent manner, and increased levels of Aβ PFs may result in spatial learning impairments in tg-APPArcSwe mice [47]. Lannfelt et al. reported that the murine version of mAb158 reached the brain and reduced brain PF levels by 42% in an exposure-dependent manner both after long-term (13 weeks) and short-term (4 weeks) treatment in tg-APPArcSwe mice [14]. Notably, a 53% reduction in PFs/oligomers in the cerebrospinal fluid (CSF), found to be correlated with reduced brain PF levels, was observed after long-term treatment, suggesting that CSF PFs/oligomers may be used as potential biomarkers of AD [14].
Recently, Sehlin’s group succeeded in facilitating the brain uptake of mAb158 by using transferrin receptor-mediated transcytosis across the blood–brain barrier in tg-APPArcSwe mice [61]. ELISA analysis of the brain extracts demonstrated a 40% reduction in soluble Aβ PFs in both ten-fold lower-dose modified mAb158 and high-dose mAb158-treated mice, whereas there was no Aβ PF reduction in mice treated with a low dose of mAb158 [61]. Furthermore, ex vivo autoradiography and PET imaging have revealed different brain distribution patterns of modified mAb158 (brain parenchyma) and mAb158 (central periventricular areas), suggesting that these antibodies may affect Aβ levels by different mechanisms. This strategy may allow for decreased antibody doses, thereby reducing the side effects and treatment costs [61].
3.3. Clinical Application of mAb158
BAN2401, a humanized IgG1 monoclonal form of mAb158, exhibits a strong binding preference for soluble Aβ PFs compared with monomers [14]. In addition, it has been confirmed that both mAb158 and BAN2401 efficiently immunoprecipitate soluble Aβ aggregates in human AD brain extracts.
The first clinical study of BAN2401 demonstrated that the compound was safe and well tolerated in mild to moderate AD [62]. The incidence of amyloid-related imaging abnormalities (ARIA-E for edema /H for hemorrhage) on brain MRI scans was comparable to that of the placebo. BAN2401 exposure was approximately dose-proportional, with a serum terminal elimination half-life of approximately seven days. Only a slight increase in plasma Aβ1-40 was observed, but there were no measurable effects of BAN2401 on CSF biomarkers such as Aβ1-42, total-tau, and phosphorylayed-tau (p-tau) [62]. A recent phase 2 randomized trial reported that BAN2401’s highest dose (10 mg/kg) significantly slowed cognitive decline in early AD, with a concomitant reduction in amyloid plaques, as measured by amyloid PET compared with placebo at 18 months [21]. BAN2401 significantly reduced amyloid plaques in the brain at all five treatment doses used in the trial, which involved 856 patients with mild cognitive impairment. The 30% slowing of cognitive decline at 18 months was based on the Alzheimer’s Disease Composite Score (ADCOMS) created by Eisai. On the more widely used Alzheimer’s Disease Assessment Scale cognitive subscale (ADAS-Cog), the highest dose of BAN2401 slowed a cognitive decline of 47% compared with placebo. However, the trial was not large enough to definitively demonstrate efficacy in improving cognitive function according to an overall optimistic statement from the Alzheimer Association. The drug also did not achieve its primary efficacy endpoint, namely, a change from baseline on the ADCOMS at 12 months [21]. Currently, BAN2401 is a part of an ongoing phase 3 clinical trial. In contrast, other clinical trials of monoclonal antibodies targeting fibrillar Aβ, such as bapineuzumab [63], or soluble monomeric Aβ, such as solanezumab [20], have failed to produce clinical effects.
In the fall of 2019, after trials of the drug EMERGE (aducanumab; BIIB037) were previously discontinued following a phase III futility analysis, Biogen, the company that developed the drug, announced that subsequent analysis of a larger dataset instead showed that EMERGE had met its primary endpoint. Patients on the highest dose, 10 mg/kg, had a significant reduction in decline in terms of the primary endpoint using the Clinical Dementia Rating Scale-Sum of Boxes (CDR-SB). This group also declined less in terms of secondary endpoints, including the Mini-Mental State Examination (MMSE), ADAS-Cog, and the Alzheimer’s Disease Cooperative Study/Activities of Daily Living scale adapted for patients with mild cognitive impairment (ADCS-ADL-MCI). In a parallel clinical trial of aducanumab, termed the ENGAGE trial, aducanumab did not meet the primary endpoint; however, an exploratory analysis suggested that a subgroup of people who had received 10 or more 10 mg/kg doses declined more slowly, which is consistent with the EMERGE participants. In both trials, aducanumab caused a dose-dependent reduction in brain Aβ and CSF p-tau. Based on the updated data analysis, Biogen announced plans to apply for regulatory approval of aducanumab in the US in early 2020 [64]. Since aducanumab may also bind aggregates such as oligomers of Aβ [65], these results may be important for interpreting data from the phase 3 clinical trial of BAN2401.
4. PFs Are Present in Other Neurodegenerative Diseases
PFs are formed from proteins implicated in other neurodegenerative diseases, including tauopathy [66], Parkinson’s disease [67,68], familial amyloid polyneuropathy [69], and Huntington’s disease [70], indicating a common mechanism. Similar to Aβ, tau and α-synuclein (αS) also form PFs with annular, pore-like structures, thereby exerting membrane permeabilization activity [66,67]. Analyses of annular tau PFs in brain tissue from patients with progressive supranuclear palsy, as well as that from the P301L mouse model, indicated that the annular PFs of tau are preceded by tau oligomers and do not go on to form neurofibrillarly tangles (mature fibrils) [66]. In addition, it was recently reported that the αS oligomer and PFs interconvert during polymerization reactions, using the thioflavin T assay combined with SEC and EM [68]. Similarly, Groenning et al. described a dynamic transthyretin (TTR) protofibril structure that exchanges protomers with highly unfolded monomers in solution, using a combination of primarily small-angle X-ray scattering and hydrogen exchange mass spectrometry analysis. The TTR PFs were shown to only grow to an approximate final size of 2900 kDa and a length of 70 nm [69]. In a recent micro electron diffraction study at 0.75Å resolution, ultrahigh-resolution cryo-EM revealed that prion PFs are stabilized by a dense three-dimensional network of stabilizing hydrogen bonds that link residues between and within its β strands through polar clasps [71].
5. Conclusions and Future Perspectives
Unlike current therapies limited to the treatment of AD symptoms, research on Aβ aggregation has rapidly advanced, with growing evidence that soluble pre-fibrillar aggregates (i.e., oligomers of Aβ) are proximate neurotoxins. Indeed, recent data from both in vitro and in vivo studies have suggested that HMW oligomers as PFs induce neuronal injury and cognitive deficits via multiple mechanisms, including not only increasing Aβ plaque accumulation but also increasing direct membrane and synaptic damage. Furthermore, additional projects to fully characterize the PFs actually present in the human brain have been undertaken. Aβ PFs may be the primary pathogenic species of Aβ-related cognitive deficits, particularly in the early stage of AD, although it remains to be established how Aβ PFs, alone or together with other soluble oligomeric Aβ species, cause the neurodegeneration leading to AD. Disease-modifying therapies targeting toxic PFs will reach the clinical stage in the near future, and may have the potential to delay or even halt the further progression of AD. Further clarification of the toxic PFs of brain Aβ should aid in the development of more effective and safe drugs, as well as in novel diagnostic assays.
Author Contributions
K.O. and M.T. wrote the paper. Authorship must be limited to those who have contributed substantially to the work reported. All authors have read and agreed to the published version of the manuscript.
Funding
This work was supported by a Grant-in-Aid for Scientific Research (C) (26461266) (K.O.) from the Ministry of Education, Culture, Sports, Science and Technology, Japan, and a Grant for Research and Development Grants for Dementia from the Japan Agency for Medical Research and Development (16dk0207021h0001) (K.O.).
Conflicts of Interest
The authors declare no conflicts of interest.
Abbreviations
| Aβ | amyloid β-protein |
| AD | Alzheimer’s disease |
| ADAS-Cog | Alzheimer’s Disease Assessment Scale cognitive subscale |
| ADCOMS | Alzheimer’s Disease Composite Score |
| AFM | atomic force microscopy |
| APP | amyloid precursor protein |
| ARIA | amyloid-related imaging abnormalities |
| αS | α-synuclein |
| CSF | cerebrospinal fluid |
| DMF | 1,2-(dimethoxymethano)fullerene |
| ELISA | enzyme-linked immunosorbent assay |
| EM | electron microscopy |
| HMW | high molecular weight |
| IL-1β | interleukin-1β |
| LDH | lactate dehydrogenase |
| LMW | low molecular weight |
| LTPs | long-term potentiation |
| MD | molecular dynamics |
| MTT | 3-[4,5-dimethylthiazol-2-yl]-2,5-diphenyltetrazolium bromide metabolism |
| MyD | myeloid differentiation protein |
| NLR | Nod-like receptor |
| PFs | protofibrils |
| p-tau | phosphorylayed-tau |
| ROS | reactive oxygen species |
| SEC | size exclusion chromatography |
| SFRP1 | secreted-frizzled-related protein 1 |
| TLR | Toll-like receptor |
| TNFα | tumor necrosis factor α |
| TTR | transthyretin |
| WST | water soluble tetrazolium |
References
- Selkoe, D.J.; Hardy, J. The amyloid hypothesis of Alzheimer’s disease at 25 years. EMBO Mol. Med. 2016, 8, 595–608. [Google Scholar] [CrossRef] [PubMed]
- Hardy, J.; Selkoe, D.J. The amyloid hypothesis of Alzheimer’s disease: Progress and problems on the road to therapeutics. Science 2002, 297, 353–356. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ono, K. Alzheimer’s disease as oligomeropathy. Neurochem. Int. 2018, 119, 57–70. [Google Scholar] [CrossRef] [PubMed]
- Ono, K.; Condron, M.M.; Teplow, D.B. Structure-neurotoxicity relationships of amyloid β-protein oligomers. Proc. Natl. Acad. Sci. USA 2009, 106, 14745–14750. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harper, J.D.; Lansbury, P.T., Jr. Models of amyloid seeding in Alzheimer’s disease and scrapie: Mechanistic truths and physiological consequences of the time-dependent solubility of amyloid proteins. Annu. Rev. Biochem. 1997, 66, 385–407. [Google Scholar] [CrossRef]
- Walsh, D.M.; Lomakin, A.; Benedek, G.B.; Condron, M.M.; Teplow, D.B. Amyloid β-protein fibrillogenesis. Detection of a protofibrillar intermediate. J. Biol. Chem. 1997, 272, 22364–22372. [Google Scholar] [CrossRef] [Green Version]
- Johansson, A.S.; Berglind-Dehlin, F.; Karlsson, G.; Edwards, K.; Gellerfors, P.; Lannfelt, L. Physiochemical characterization of the Alzheimer’s disease-related peptides Aβ 1-42Arctic and Aβ 1-42wt. FEBS J. 2006, 273, 2618–2630. [Google Scholar] [CrossRef]
- Hartley, D.M.; Walsh, D.M.; Ye, C.P.; Diehl, T.; Vasquez, S.; Vassilev, P.M.; Teplow, D.B.; Selkoe, D.J. Protofibrillar intermediates of amyloid β-protein induce acute electrophysiological changes and progressive neurotoxicity in cortical neurons. J. Neurosci. 1999, 19, 8876–8884. [Google Scholar] [CrossRef] [Green Version]
- Walsh, D.M.; Hartley, D.M.; Kusumoto, Y.; Fezoui, Y.; Condron, M.M.; Lomakin, A.; Benedek, G.B.; Selkoe, D.J.; Teplow, D.B. Amyloid β-protein fibrillogenesis. Structure and biological activity of protofibrillar intermediates. J. Biol. Chem. 1999, 274, 25945–25952. [Google Scholar] [CrossRef] [Green Version]
- Ward, R.V.; Jennings, K.H.; Jepras, R.; Neville, W.; Owen, D.E.; Hawkins, J.; Christie, G.; Davis, J.B.; George, A.; Karran, E.H.; et al. Fractionation and characterization of oligomeric, protofibrillar and fibrillar forms of β-amyloid peptide. Biochem. J. 2000, 348, 137–144. [Google Scholar] [CrossRef]
- Johansson, A.S.; Garlind, A.; Berglind-Dehlin, F.; Karlsson, G.; Edwards, K.; Gellerfors, P.; Ekholm-Pettersson, F.; Palmblad, J.; Lannfelt, L. Docosahexaenoic acid stabilizes soluble amyloid-β protofibrils and sustains amyloid-β-induced neurotoxicity in vitro. FEBS J. 2007, 274, 990–1000. [Google Scholar] [CrossRef] [PubMed]
- Nilsberth, C.; Westlind-Danielsson, A.; Eckman, C.B.; Condron, M.M.; Axelman, K.; Forsell, C.; Stenh, C.; Luthman, J.; Teplow, D.B.; Younkin, S.G.; et al. The ‘Arctic’ APP mutation (E693G) causes Alzheimer’s disease by enhanced Aβ protofibril formation. Nat. Neurosci. 2001, 4, 887–893. [Google Scholar] [CrossRef] [PubMed]
- Yasumoto, T.; Takamura, Y.; Tsuji, M.; Watanabe-Nakayama, T.; Imamura, K.; Inoue, H.; Nakamura, S.; Inoue, T.; Kimura, A.; Yano, S.; et al. High molecular weight amyloid β1-42 oligomers induce neurotoxicity via plasma membrane damage. FASEB J. 2019, 33, 9220–9234. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tucker, S.; Moller, C.; Tegerstedt, K.; Lord, A.; Laudon, H.; Sjodahl, J.; Soderberg, L.; Spens, E.; Sahlin, C.; Waara, E.R.; et al. The murine version of BAN2401 (mAb158) selectively reduces amyloid-β protofibrils in brain and cerebrospinal fluid of tg-ArcSwe mice. J. Alzheimers Dis. 2015, 43, 575–588. [Google Scholar] [CrossRef]
- Gauthier, S.; Albert, M.; Fox, N.; Goedert, M.; Kivipelto, M.; Mestre-Ferrandiz, J.; Middleton, L.T. Why has therapy development for dementia failed in the last two decades? Alzheimers Dement. 2016, 12, 60–64. [Google Scholar] [CrossRef]
- Cummings, J.; Aisen, P.S.; DuBois, B.; Frolich, L.; Jack, C.R., Jr.; Jones, R.W.; Morris, J.C.; Raskin, J.; Dowsett, S.A.; Scheltens, P. Drug development in Alzheimer’s disease: The path to 2025. Alzheimers Res. Ther. 2016, 8, 39. [Google Scholar] [CrossRef] [Green Version]
- Burki, T. Alzheimer’s disease research: The future of BACE inhibitors. Lancet 2018, 391, 2486. [Google Scholar] [CrossRef]
- Doody, R.S.; Raman, R.; Farlow, M.; Iwatsubo, T.; Vellas, B.; Joffe, S.; Kieburtz, K.; He, F.; Sun, X.; Thomas, R.G.; et al. A phase 3 trial of semagacestat for treatment of Alzheimer’s disease. N. Engl. J. Med. 2013, 369, 341–350. [Google Scholar] [CrossRef]
- Vandenberghe, R.; Rinne, J.O.; Boada, M.; Katayama, S.; Scheltens, P.; Vellas, B.; Tuchman, M.; Gass, A.; Fiebach, J.B.; Hill, D.; et al. Bapineuzumab for mild to moderate Alzheimer’s disease in two global, randomized, phase 3 trials. Alzheimers Res. Ther. 2016, 8, 18. [Google Scholar] [CrossRef] [Green Version]
- Honig, L.S.; Vellas, B.; Woodward, M.; Boada, M.; Bullock, R.; Borrie, M.; Hager, K.; Andreasen, N.; Scarpini, E.; Liu-Seifert, H.; et al. Trial of Solanezumab for Mild Dementia Due to Alzheimer’s Disease. N. Engl. J. Med. 2018, 378, 321–330. [Google Scholar] [CrossRef]
- Abbasi, J. Promising Results in 18-Month Analysis of Alzheimer Drug Candidate. JAMA 2018, 320, 965. [Google Scholar] [CrossRef] [PubMed]
- Harper, J.D.; Wong, S.S.; Lieber, C.M.; Lansbury, P.T. Observation of metastable Aβ amyloid protofibrils by atomic force microscopy. Chem. Biol. 1997, 4, 119–125. [Google Scholar] [CrossRef] [Green Version]
- Kodali, R.; Wetzel, R. Polymorphism in the intermediates and products of amyloid assembly. Curr. Opin. Struct. Biol. 2007, 17, 48–57. [Google Scholar] [CrossRef] [PubMed]
- Watanabe-Nakayama, T.; Ono, K.; Itami, M.; Takahashi, R.; Teplow, D.B.; Yamada, M. High-speed atomic force microscopy reveals structural dynamics of amyloid β1-42 aggregates. Proc. Natl. Acad. Sci. USA 2016, 113, 5835–5840. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cline, E.N.; Bicca, M.A.; Viola, K.L.; Klein, W.L. The Amyloid-β Oligomer Hypothesis: Beginning of the Third Decade. J. Alzheimers Dis. 2018, 64, S567–S610. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hasecke, F.; Miti, T.; Perez, C.; Barton, J.; Scholzel, D.; Gremer, L.; Gruning, C.S.R.; Matthews, G.; Meisl, G.; Knowles, T.P.J.; et al. Origin of metastable oligomers and their effects on amyloid fibril self-assembly. Chem. Sci. 2018, 9, 5937–5948. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hori, Y.; Hashimoto, T.; Nomoto, H.; Hyman, B.T.; Iwatsubo, T. Role of Apolipoprotein E in β-Amyloidogenesis: Isoform-Specific Effects On Protofibril To Fibril Conversion Of Aβ In Vitro And Brain Aβ Deposition In Vivo. J. Biol. Chem. 2015, 290, 15163–15174. [Google Scholar] [CrossRef] [Green Version]
- Sollvander, S.; Nikitidou, E.; Brolin, R.; Soderberg, L.; Sehlin, D.; Lannfelt, L.; Erlandsson, A. Accumulation of amyloid-β by astrocytes result in enlarged endosomes and microvesicle-induced apoptosis of neurons. Mol. Neurodegener. 2016, 11, 38. [Google Scholar] [CrossRef]
- Paranjape, G.S.; Gouwens, L.K.; Osborn, D.C.; Nichols, M.R. Isolated amyloid-β(1-42) protofibrils, but not isolated fibrils, are robust stimulators of microglia. ACS Chem. Neurosci. 2012, 3, 302–311. [Google Scholar] [CrossRef]
- Terrill-Usery, S.E.; Mohan, M.J.; Nichols, M.R. Amyloid-β(1-42) protofibrils stimulate a quantum of secreted IL-1β despite significant intracellular IL-1β accumulation in microglia. Biochim. Biophys. Acta 2014, 1842, 2276–2285. [Google Scholar] [CrossRef] [Green Version]
- Gouwens, L.K.; Makoni, N.J.; Rogers, V.A.; Nichols, M.R. Amyloid-β42 protofibrils are internalized by microglia more extensively than monomers. Brain Res. 2016, (Pt A), 485–495. [Google Scholar] [CrossRef] [Green Version]
- Rosini, M.; Simoni, E.; Milelli, A.; Minarini, A.; Melchiorre, C. Oxidative stress in Alzheimer’s disease: Are we connecting the dots? J. Med. Chem. 2014, 57, 2821–2831. [Google Scholar] [CrossRef] [PubMed]
- Ansari, M.A.; Scheff, S.W. Oxidative stress in the progression of Alzheimer disease in the frontal cortex. J. Neuropathol. Exp. Neurol. 2010, 69, 155–167. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pohanka, M. Alzheimer’s disease and oxidative stress: A review. Curr. Med. Chem. 2014, 21, 356–364. [Google Scholar] [CrossRef] [PubMed]
- Hernandez-Zimbron, L.F.; Luna-Munoz, J.; Mena, R.; Vazquez-Ramirez, R.; Kubli-Garfias, C.; Cribbs, D.H.; Manoutcharian, K.; Gevorkian, G. Amyloid-β peptide binds to cytochrome C oxidase subunit 1. PLoS ONE 2012, 7, e42344. [Google Scholar] [CrossRef] [Green Version]
- Bode, D.C.; Baker, M.D.; Viles, J.H. Ion Channel Formation by Amyloid-β42 Oligomers but Not Amyloid-β40 in Cellular Membranes. J. Biol. Chem. 2017, 292, 1404–1413. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Drolle, E.; Negoda, A.; Hammond, K.; Pavlov, E.; Leonenko, Z. Changes in lipid membranes may trigger amyloid toxicity in Alzheimer’s disease. PLoS ONE 2017, 12, e0182194. [Google Scholar] [CrossRef]
- Klyubin, I.; Walsh, D.M.; Cullen, W.K.; Fadeeva, J.V.; Anwyl, R.; Selkoe, D.J.; Rowan, M.J. Soluble Arctic amyloid β protein inhibits hippocampal long-term potentiation in vivo. Eur. J. Neurosci. 2004, 19, 2839–2846. [Google Scholar] [CrossRef]
- Massaad, C.A.; Klann, E. Reactive oxygen species in the regulation of synaptic plasticity and memory. Antioxid Redox Signal. 2011, 14, 2013–2054. [Google Scholar] [CrossRef] [Green Version]
- Schaeffer, E.L.; Bassi, F., Jr.; Gattaz, W.F. Inhibition of phospholipase A2 activity reduces membrane fluidity in rat hippocampus. J. Neural. Transm. (Vienna) 2005, 112, 641–647. [Google Scholar] [CrossRef]
- Alzheimer’s Association Calcium Hypothesis Workgroup. Calcium Hypothesis of Alzheimer’s disease and brain aging: A framework for integrating new evidence into a comprehensive theory of pathogenesis. Alzheimers Dement. 2017, 13, 178–182. [Google Scholar] [CrossRef] [PubMed]
- Berridge, M.J. Calcium signalling in health and disease. Biochem. Biophys. Res. Commun. 2017, 485, 5. [Google Scholar] [CrossRef] [PubMed]
- Jorissen, E.; Prox, J.; Bernreuther, C.; Weber, S.; Schwanbeck, R.; Serneels, L.; Snellinx, A.; Craessaerts, K.; Thathiah, A.; Tesseur, I.; et al. The disintegrin/metalloproteinase ADAM10 is essential for the establishment of the brain cortex. J. Neurosci. 2010, 30, 4833–4844. [Google Scholar] [CrossRef]
- Esteve, P.; Rueda-Carrasco, J.; Ines Mateo, M.; Martin-Bermejo, M.J.; Draffin, J.; Pereyra, G.; Sandonis, A.; Crespo, I.; Moreno, I.; Aso, E.; et al. Elevated levels of Secreted-Frizzled-Related-Protein 1 contribute to Alzheimer’s disease pathogenesis. Nat. Neurosci. 2019, 22, 1258–1268. [Google Scholar] [CrossRef] [PubMed]
- Knobloch, M.; Konietzko, U.; Krebs, D.C.; Nitsch, R.M. Intracellular Aβ and cognitive deficits precede β-amyloid deposition in transgenic arcAβ mice. Neurobiol. Aging 2007, 28, 1297–1306. [Google Scholar] [CrossRef]
- Lord, A.; Kalimo, H.; Eckman, C.; Zhang, X.Q.; Lannfelt, L.; Nilsson, L.N. The Arctic Alzheimer mutation facilitates early intraneuronal Aβ aggregation and senile plaque formation in transgenic mice. Neurobiol. Aging 2006, 27, 67–77. [Google Scholar] [CrossRef]
- Lord, A.; Englund, H.; Soderberg, L.; Tucker, S.; Clausen, F.; Hillered, L.; Gordon, M.; Morgan, D.; Lannfelt, L.; Pettersson, F.E.; et al. Amyloid-β protofibril levels correlate with spatial learning in Arctic Alzheimer’s disease transgenic mice. FEBS J. 2009, 276, 995–1006. [Google Scholar] [CrossRef] [PubMed]
- Sehlin, D.; Englund, H.; Simu, B.; Karlsson, M.; Ingelsson, M.; Nikolajeff, F.; Lannfelt, L.; Pettersson, F.E. Large aggregates are the major soluble Aβ species in AD brain fractionated with density gradient ultracentrifugation. PLoS ONE 2012, 7, e32014. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ono, K.; Condron, M.M.; Ho, L.; Wang, J.; Zhao, W.; Pasinetti, G.M.; Teplow, D.B. Effects of grape seed-derived polyphenols on amyloid β-protein self-assembly and cytotoxicity. J. Biol. Chem. 2008, 283, 32176–32187. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, J.; Ho, L.; Zhao, W.; Ono, K.; Rosensweig, C.; Chen, L.; Humala, N.; Teplow, D.B.; Pasinetti, G.M. Grape-derived polyphenolics prevent Aβ oligomerization and attenuate cognitive deterioration in a mouse model of Alzheimer’s disease. J. Neurosci. 2008, 28, 6388–6392. [Google Scholar] [CrossRef]
- Jin, Y.; Sun, Y.; Lei, J.; Wei, G. Dihydrochalcone molecules destabilize Alzheimer’s amyloid-β protofibrils through binding to the protofibril cavity. Phys. Chem. Chem. Phys. 2018, 20, 17208–17217. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.; Xi, W.; Luo, Y.; Cao, S.; Wei, G. Interactions of a water-soluble fullerene derivative with amyloid-β protofibrils: Dynamics, binding mechanism, and the resulting salt-bridge disruption. J. Phys. Chem. B 2014, 118, 6733–6741. [Google Scholar] [CrossRef] [PubMed]
- Fan, H.M.; Gu, R.X.; Wang, Y.J.; Pi, Y.L.; Zhang, Y.H.; Xu, Q.; Wei, D.Q. Destabilization of Alzheimer’s Aβ42 Protofibrils with a Novel Drug Candidate wgx-50 by Molecular Dynamics Simulations. J. Phys. Chem. B 2015, 119, 11196–11202. [Google Scholar] [CrossRef] [PubMed]
- Saini, R.K.; Shuaib, S.; Goyal, D.; Goyal, B. Insights into the inhibitory mechanism of a resveratrol and clioquinol hybrid against Aβ42 aggregation and protofibril destabilization: A molecular dynamics simulation study. J. Biomol. Struct. Dyn. 2019, 37, 3183–3197. [Google Scholar] [CrossRef] [PubMed]
- Shuaib, S.; Narang, S.S.; Goyal, D.; Goyal, B. Computational design and evaluation of β-sheet breaker peptides for destabilizing Alzheimer’s amyloid-β42 protofibrils. J. Cell Biochem. 2019, 120, 17935–17950. [Google Scholar]
- Englund, H.; Sehlin, D.; Johansson, A.S.; Nilsson, L.N.; Gellerfors, P.; Paulie, S.; Lannfelt, L.; Pettersson, F.E. Sensitive ELISA detection of amyloid-β protofibrils in biological samples. J. Neurochem. 2007, 103, 334–345. [Google Scholar] [CrossRef]
- Sehlin, D.; Hedlund, M.; Lord, A.; Englund, H.; Gellerfors, P.; Paulie, S.; Lannfelt, L.; Pettersson, F.E. Heavy-chain complementarity-determining regions determine conformation selectivity of anti-Aβantibodies. Neurodegener. Dis. 2011, 8, 117–123. [Google Scholar] [CrossRef]
- Lannfelt, L.; Moller, C.; Basun, H.; Osswald, G.; Sehlin, D.; Satlin, A.; Logovinsky, V.; Gellerfors, P. Perspectives on future Alzheimer therapies: Amyloid-β protofibrils - a new target for immunotherapy with BAN2401 in Alzheimer’s disease. Alzheimers Res. Ther. 2014, 6, 16. [Google Scholar] [CrossRef] [Green Version]
- Lord, A.; Gumucio, A.; Englund, H.; Sehlin, D.; Sundquist, V.S.; Soderberg, L.; Moller, C.; Gellerfors, P.; Lannfelt, L.; Pettersson, F.E.; et al. An amyloid-β protofibril-selective antibody prevents amyloid formation in a mouse model of Alzheimer’s disease. Neurobiol. Dis. 2009, 36, 425–434. [Google Scholar] [CrossRef]
- Sollvander, S.; Nikitidou, E.; Gallasch, L.; Zysk, M.; Soderberg, L.; Sehlin, D.; Lannfelt, L.; Erlandsson, A. The Aβ protofibril selective antibody mAb158 prevents accumulation of Aβ in astrocytes and rescues neurons from Aβ-induced cell death. J. Neuroinflammation 2018, 15, 98. [Google Scholar] [CrossRef]
- Syvanen, S.; Hultqvist, G.; Gustavsson, T.; Gumucio, A.; Laudon, H.; Soderberg, L.; Ingelsson, M.; Lannfelt, L.; Sehlin, D. Efficient clearance of Aβ protofibrils in AβPP-transgenic mice treated with a brain-penetrating bifunctional antibody. Alzheimers Res. Ther. 2018, 10, 49. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Logovinsky, V.; Satlin, A.; Lai, R.; Swanson, C.; Kaplow, J.; Osswald, G.; Basun, H.; Lannfelt, L. Safety and tolerability of BAN2401--a clinical study in Alzheimer’s disease with a protofibril selective Aβ antibody. Alzheimers Res. Ther. 2016, 8, 14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Salloway, S.; Sperling, R.; Fox, N.C.; Blennow, K.; Klunk, W.; Raskind, M.; Sabbagh, M.; Honig, L.S.; Porsteinsson, A.P.; Ferris, S.; et al. Two phase 3 trials of bapineuzumab in mild-to-moderate Alzheimer’s disease. N. Engl. J. Med. 2014, 370, 322–333. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aducanumab. Available online: https://www.alzforum.org/therapeutics/aducanumab (accessed on 25 October 2019).
- Sevigny, J.; Chiao, P.; Bussiere, T.; Weinreb, P.H.; Williams, L.; Maier, M.; Dunstan, R.; Salloway, S.; Chen, T.; Ling, Y.; et al. The antibody aducanumab reduces Aβ plaques in Alzheimer’s disease. Nature 2016, 537, 50–56. [Google Scholar] [CrossRef] [PubMed]
- Lasagna-Reeves, C.A.; Sengupta, U.; Castillo-Carranza, D.; Gerson, J.E.; Guerrero-Munoz, M.; Troncoso, J.C.; Jackson, G.R.; Kayed, R. The formation of tau pore-like structures is prevalent and cell specific: Possible implications for the disease phenotypes. Acta Neuropathol. Commun. 2014, 2, 56. [Google Scholar] [CrossRef] [Green Version]
- Lashuel, H.A.; Petre, B.M.; Wall, J.; Simon, M.; Nowak, R.J.; Walz, T.; Lansbury, P.T., Jr. Alpha-synuclein, especially the Parkinson’s disease-associated mutants, forms pore-like annular and tubular protofibrils. J. Mol. Biol. 2002, 322, 1089–1102. [Google Scholar] [CrossRef] [Green Version]
- De Oliveira, G.A.P.; Silva, J.L. Alpha-synuclein stepwise aggregation reveals features of an early onset mutation in Parkinson’s disease. Commun. Biol. 2019, 2, 374. [Google Scholar] [CrossRef] [Green Version]
- Groenning, M.; Campos, R.I.; Hirschberg, D.; Hammarstrom, P.; Vestergaard, B. Considerably Unfolded Transthyretin Monomers Preceed and Exchange with Dynamically Structured Amyloid Protofibrils. Sci. Rep. 2015, 5, 11443. [Google Scholar] [CrossRef] [Green Version]
- Beasley, M.; Stonebraker, A.R.; Hasan, I.; Kapp, K.L.; Liang, B.J.; Agarwal, G.; Groover, S.; Sedighi, F.; Legleiter, J. Lipid Membranes Influence the Ability of Small Molecules To Inhibit Huntingtin Fibrillization. Biochemistry 2019, 58, 4361–4373. [Google Scholar] [CrossRef]
- Gallagher-Jones, M.; Glynn, C.; Boyer, D.R.; Martynowycz, M.W.; Hernandez, E.; Miao, J.; Zee, C.T.; Novikova, I.V.; Goldschmidt, L.; McFarlane, H.T.; et al. Sub-angstrom cryo-EM structure of a prion protofibril reveals a polar clasp. Nat. Struct. Mol. Biol. 2018, 25, 131–134. [Google Scholar] [CrossRef]
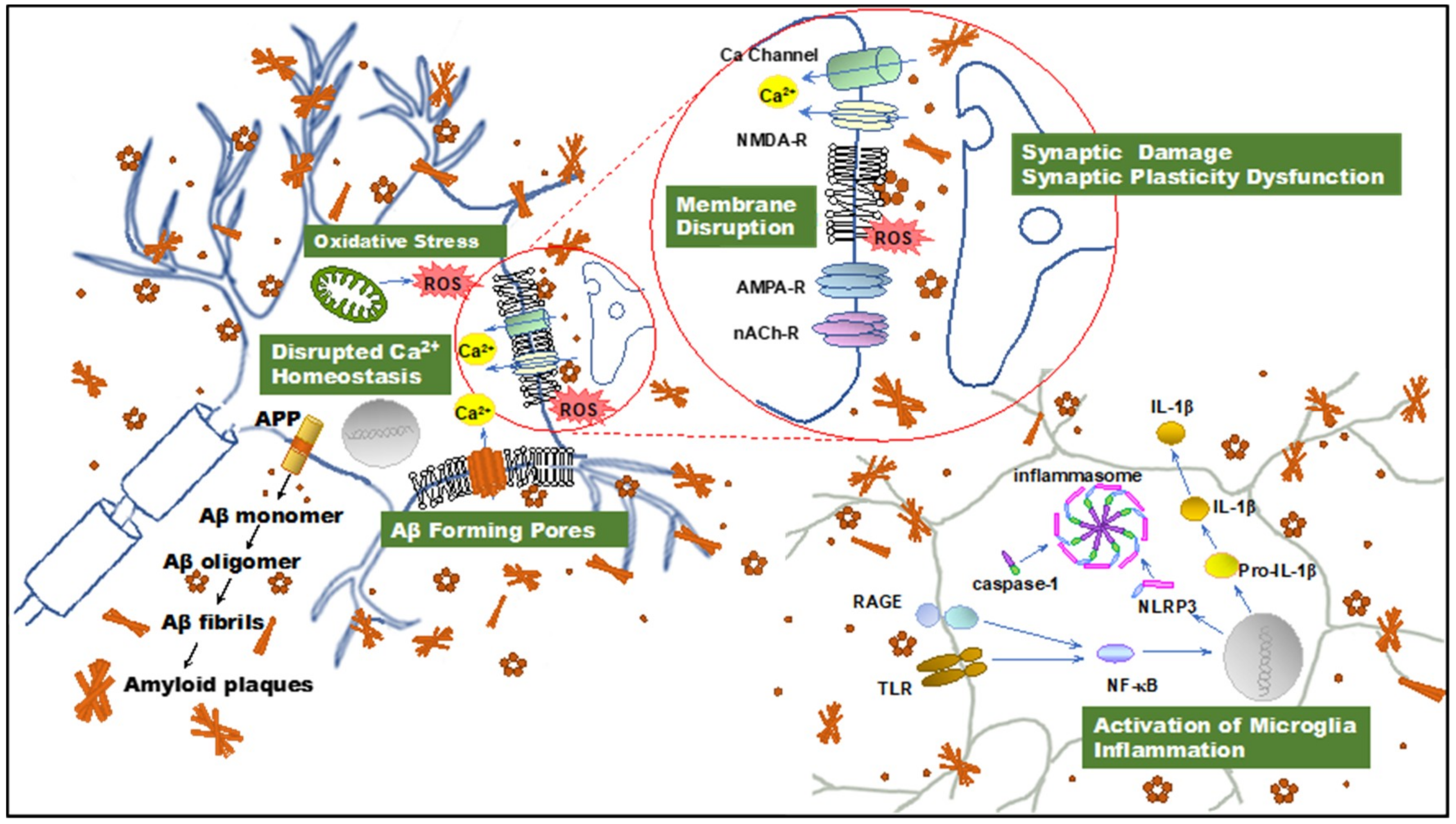
Figure 1.
Illustration summarizing amyloid β-protein (Aβ) neurotoxicity. Aβ aggregates induce disruption of cellular homeostasis, which may be the result of inducing or exacerbating membrane disruption, oxidative stress, calcium dysregulation, synaptic plasticity dysfunction, and inflammation. APP: amyloid precursor protein; Aβ: amyloid β-protein; ROS: reactive oxygen species; NMDAR, N-methyl-d-aspartate receptor. AMPAR, α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptor; nAChR, nicotinic acetylcholine receptor; TLR: toll-like receptor; RAGE: receptor for advanced glycation endproducts; NF-κB, nuclear factor κB; NLRP3: NOD-, LRR- and pyrin domain-containing protein 3; IL-1β: interleukin-1β.
Figure 1.
Illustration summarizing amyloid β-protein (Aβ) neurotoxicity. Aβ aggregates induce disruption of cellular homeostasis, which may be the result of inducing or exacerbating membrane disruption, oxidative stress, calcium dysregulation, synaptic plasticity dysfunction, and inflammation. APP: amyloid precursor protein; Aβ: amyloid β-protein; ROS: reactive oxygen species; NMDAR, N-methyl-d-aspartate receptor. AMPAR, α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptor; nAChR, nicotinic acetylcholine receptor; TLR: toll-like receptor; RAGE: receptor for advanced glycation endproducts; NF-κB, nuclear factor κB; NLRP3: NOD-, LRR- and pyrin domain-containing protein 3; IL-1β: interleukin-1β.

© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Ono, K.; Tsuji, M. Protofibrils of Amyloid-β are Important Targets of a Disease-Modifying Approach for Alzheimer’s Disease. Int. J. Mol. Sci. 2020, 21, 952. https://doi.org/10.3390/ijms21030952
AMA Style
Ono K, Tsuji M. Protofibrils of Amyloid-β are Important Targets of a Disease-Modifying Approach for Alzheimer’s Disease. International Journal of Molecular Sciences. 2020; 21(3):952. https://doi.org/10.3390/ijms21030952
Chicago/Turabian StyleOno, Kenjiro, and Mayumi Tsuji. 2020. "Protofibrils of Amyloid-β are Important Targets of a Disease-Modifying Approach for Alzheimer’s Disease" International Journal of Molecular Sciences 21, no. 3: 952. https://doi.org/10.3390/ijms21030952
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.