Topical MTII Therapy Suppresses Melanoma Through PTEN Upregulation and Cyclooxygenase II Inhibition

 and
and 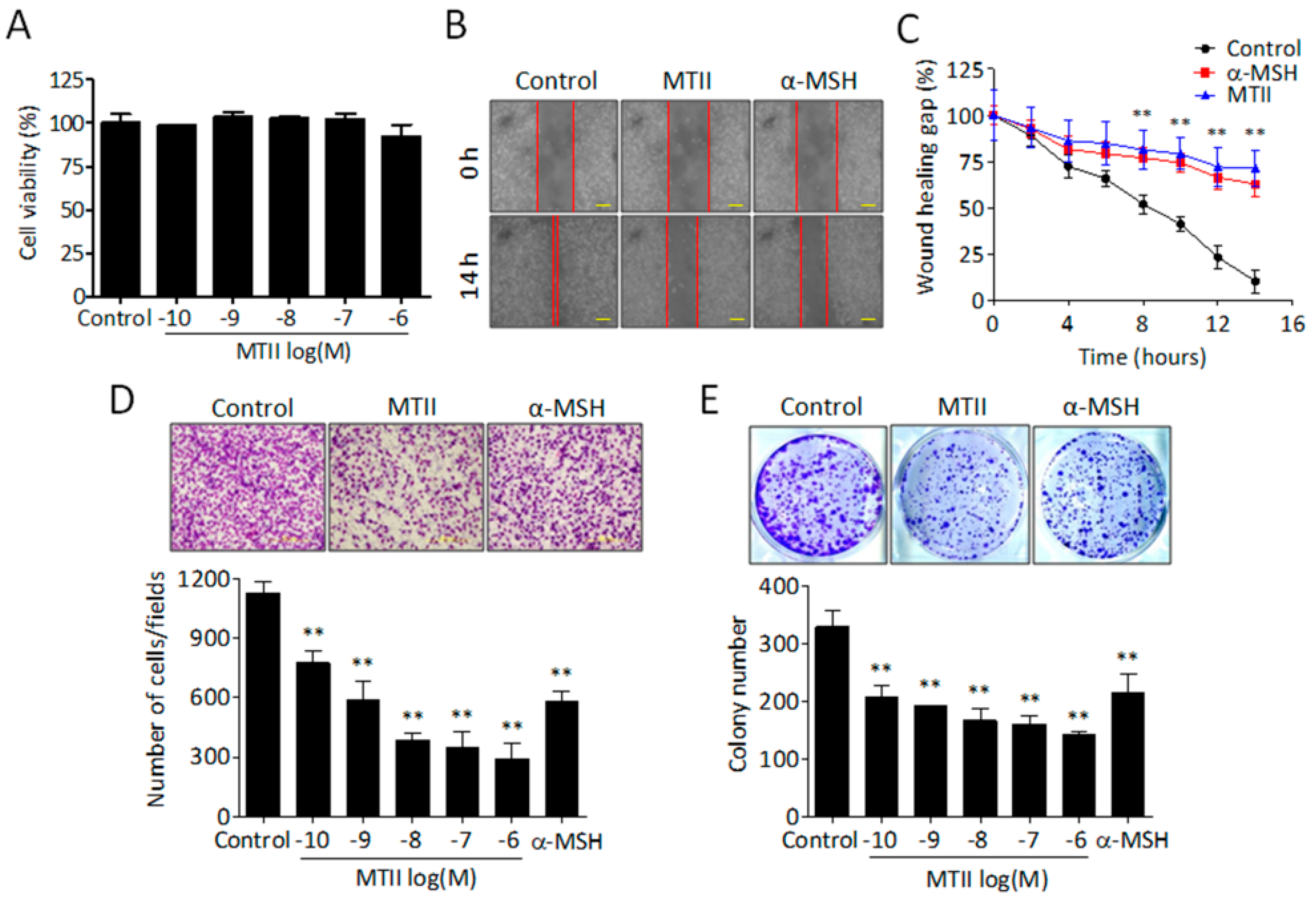{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Cultures and Reagents
2.2. Cell Proliferation Assay
2.3. Colony Formation Assay
2.4. Cell Invasion Assay
2.5. Scratch Migration Assay
2.6. Primary Melanoma Model
2.7. Immunohistochemistry Analysis
2.8. Immunofluorescence Staining of Fixed Tumor Sections
2.9. Terminal Deoxynucleotidyl Transferase (TdT) Mediated dUTP Nick End Labeling (TUNEL) Assay
2.10. NFκB Luciferase Assay
2.11. Secreted PGE2 Measurement
2.12. Western Blot Analysis
2.13. Quantitative Real-Time PCR
2.14. Statistical Analysis
3. Results
3.1. MTII Potently Suppressed the Invasiveness and Colony-Forming Capabilities of B16-F10 Melanoma Cells
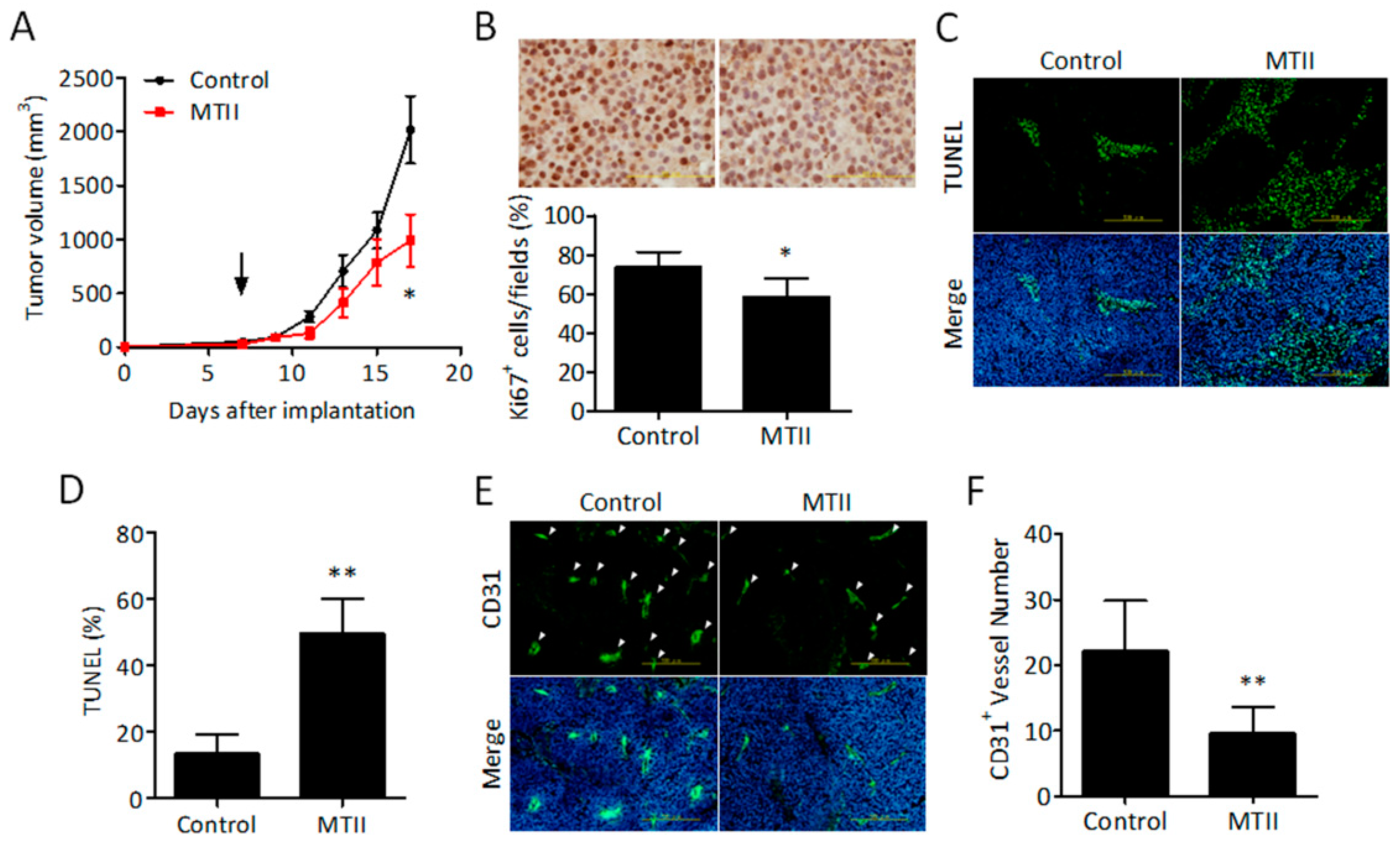
3.2. Topical MTII Application Attenuated the Progression of Established Melanoma in Mice
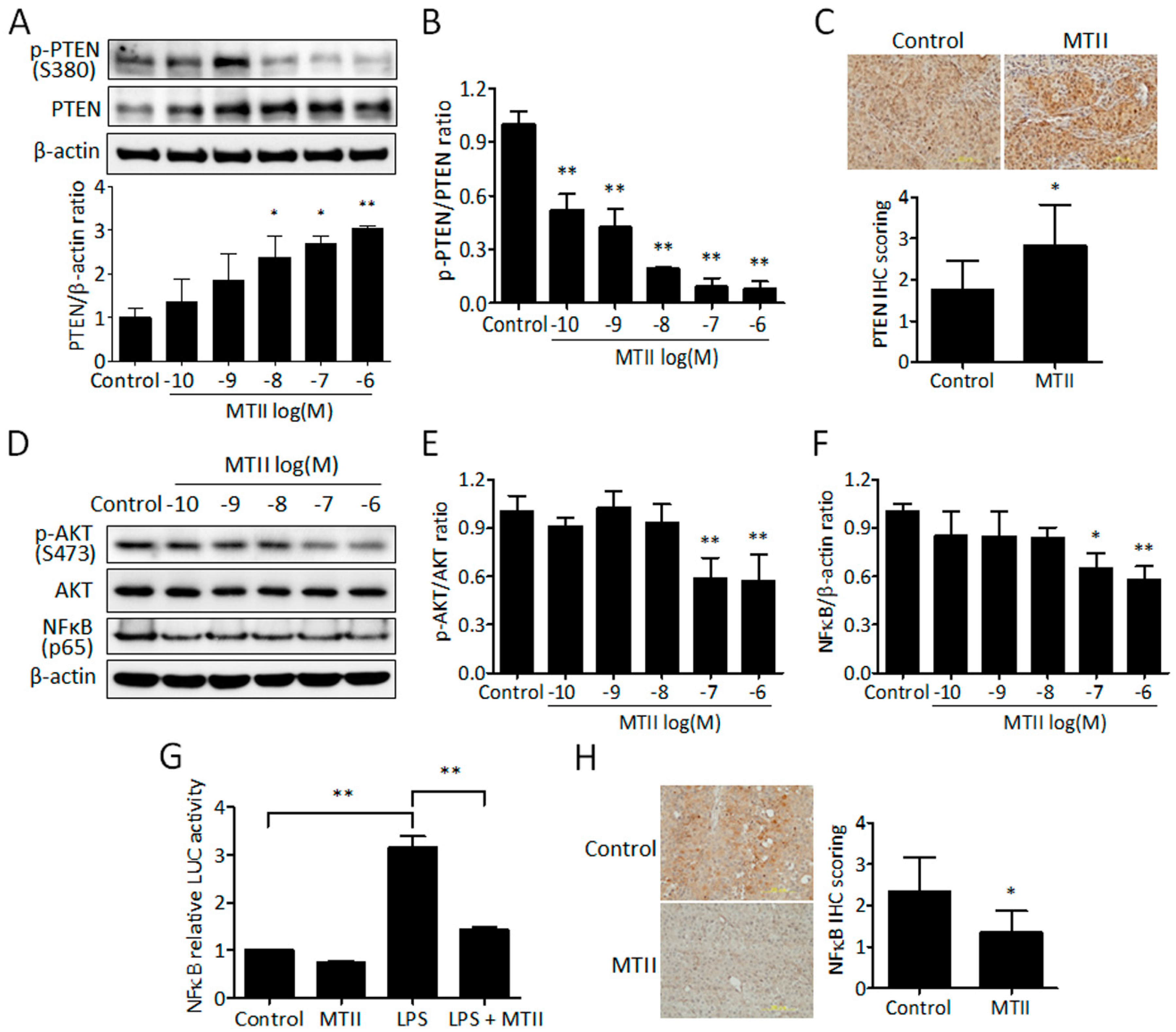
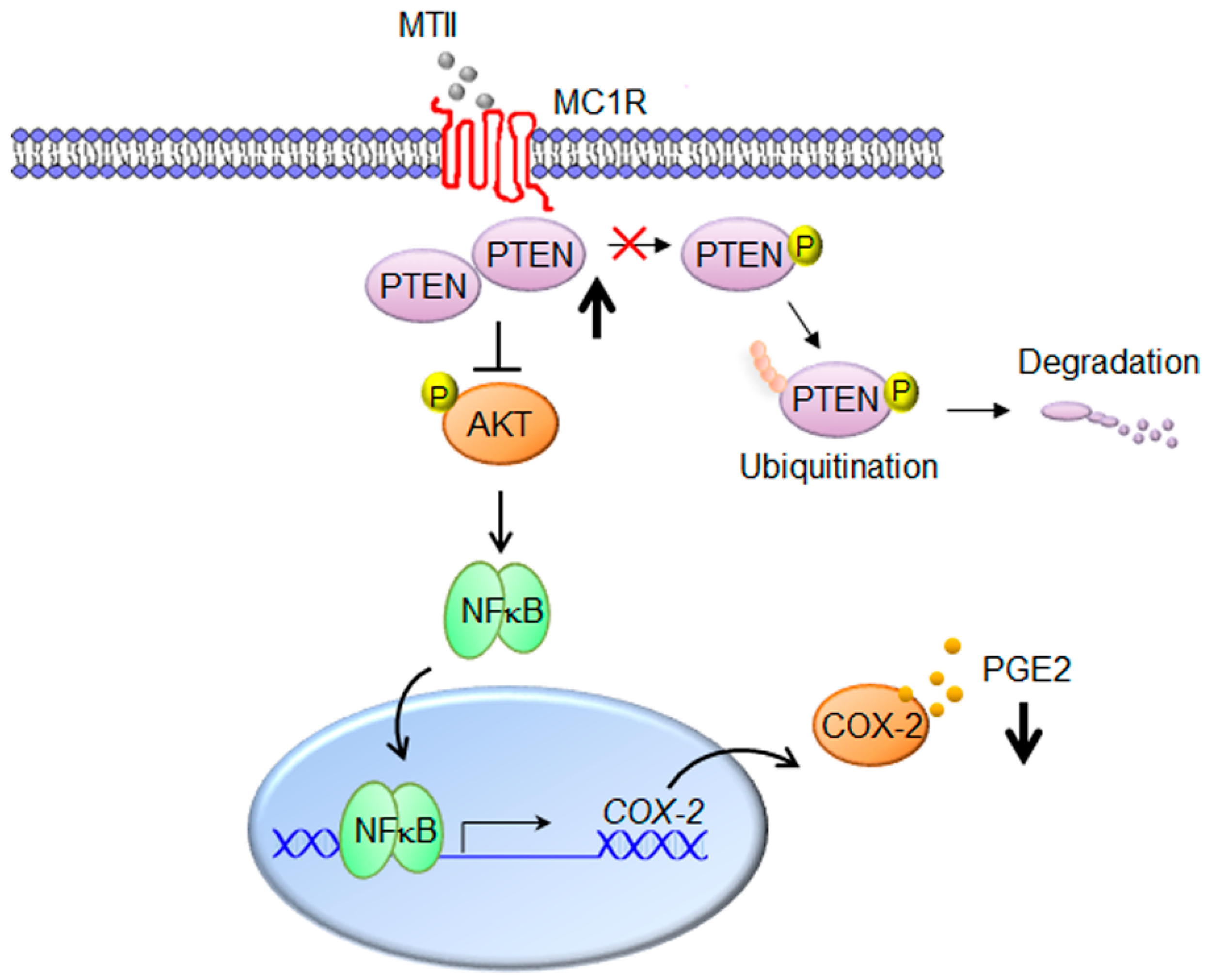
3.3. MTII Augmented the PTEN Expression and Repressed the Akt/NFκB Signaling in B16-F10 Melanoma Cells
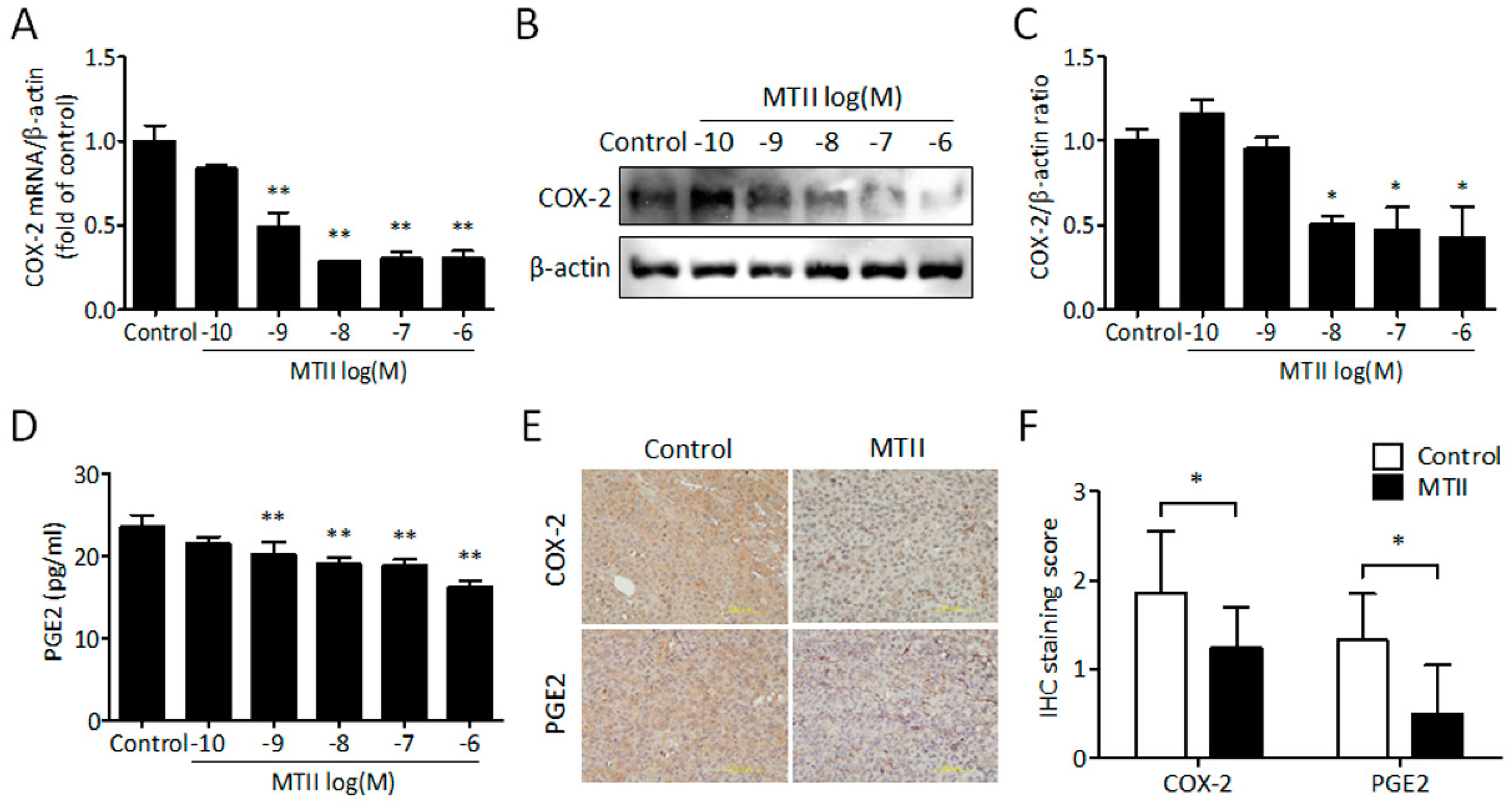
3.4. MTII Inhibited COX-2 Expression and PGE2 Production in Melanoma Cells
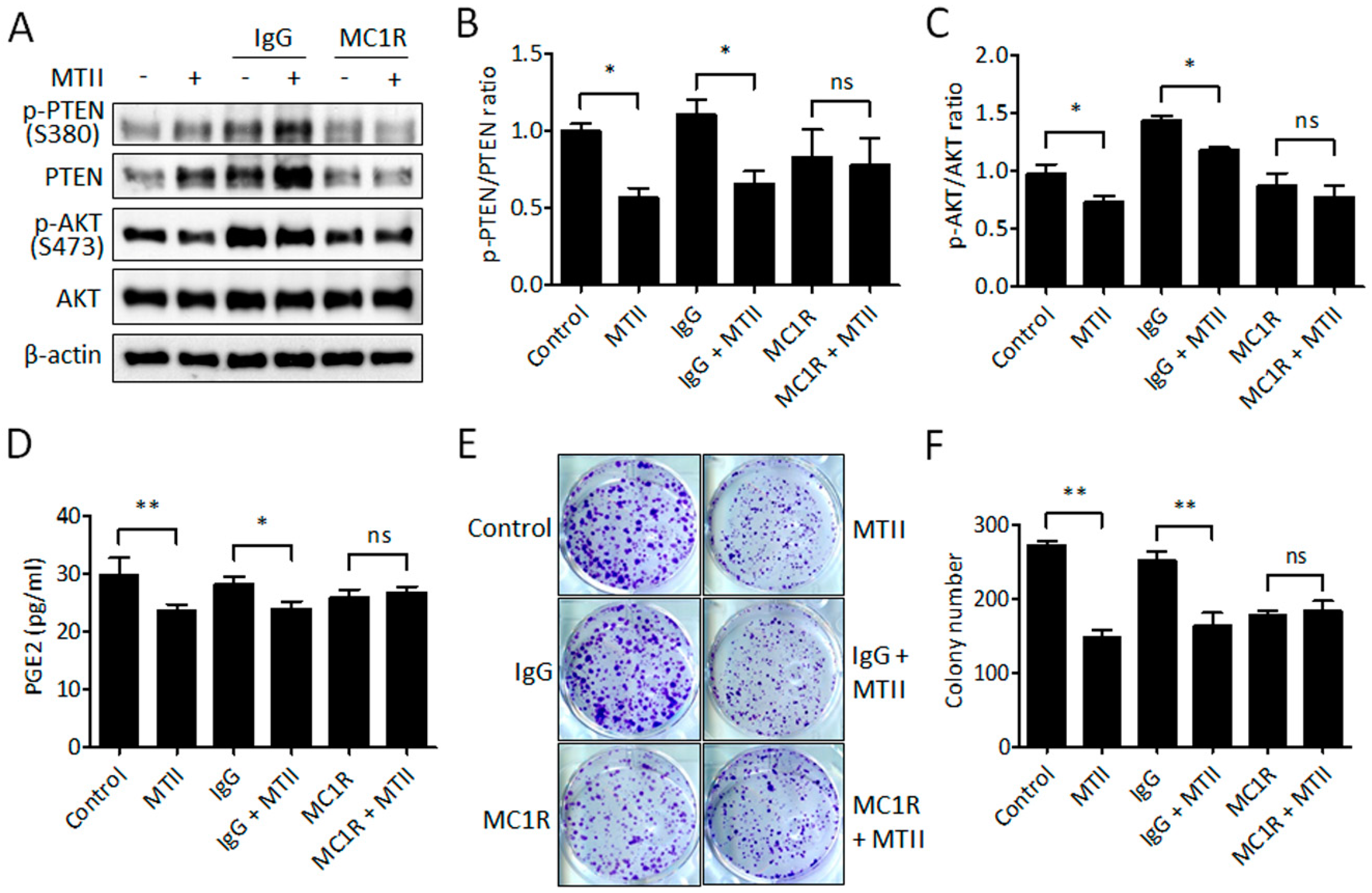
3.5. Antibody Neutralization of MC1R Abolished the MTII-Induced Melanoma Suppression
4. Discussion
Author Contributions
Funding
Conflicts of Interest
References
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2018. CA Cancer J. Clin. 2018, 68, 7–30. [Google Scholar] [CrossRef]
- Liu, Q.; Das, M.; Liu, Y.; Huang, L. Targeted drug delivery to melanoma. Adv. Drug Deliv. Rev. 2018, 127, 208–221. [Google Scholar] [CrossRef] [PubMed]
- Miller, A.J.; Mihm, M.C., Jr. Melanoma. N. Engl. J. Med. 2006, 355, 51–65. [Google Scholar] [CrossRef] [PubMed]
- Sheen, Y.S.; Tan, K.T.; Tse, K.P.; Liao, Y.H.; Lin, M.H.; Chen, J.S.; Liau, J.Y.; Tseng, Y.J.; Lee, C.H.; Hong, C.H.; et al. Genetic alterations in primary melanoma in Taiwan. Br. J. Dermatol. 2019. [Google Scholar] [CrossRef] [PubMed]
- Gao, H.W.; Tsai, W.C.; Perng, C.L.; Wang, W.M.; Chiang, C.P. Distinct MAPK and PI3K pathway mutations in different melanoma types in Taiwanese individuals. Eur. J. Dermatol. 2018, 28, 509–518. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.H.; Kim, J.E.; Jang, H.S.; Park, K.H.; Oh, B.H.; Shin, S.J.; Chung, K.Y.; Roh, M.R.; Rha, S.Y. Genetic Alterations among Korean Melanoma Patients Showing Tumor Heterogeneity: A Comparison between Primary Tumors and Corresponding Metastatic Lesions. Cancer Res. Treat. 2018, 50, 1378–1387. [Google Scholar] [CrossRef]
- Chamcheu, J.C.; Roy, T.; Uddin, M.B.; Banang-Mbeumi, S.; Chamcheu, R.N.; Walker, A.L.; Liu, Y.Y.; Huang, S. Role and Therapeutic Targeting of the PI3K/Akt/mTOR Signaling Pathway in Skin Cancer: A Review of Current Status and Future Trends on Natural and Synthetic Agents Therapy. Cells 2019, 8, 803. [Google Scholar] [CrossRef] [Green Version]
- Madhunapantula, S.V.; Robertson, G.P. The PTEN-AKT3 signaling cascade as a therapeutic target in melanoma. Pigment Cell Melanoma Res. 2009, 22, 400–419. [Google Scholar] [CrossRef]
- Vazquez, F.; Grossman, S.R.; Takahashi, Y.; Rokas, M.V.; Nakamura, N.; Sellers, W.R. Phosphorylation of the PTEN tail acts as an inhibitory switch by preventing its recruitment into a protein complex. J. Biol. Chem. 2001, 276, 48627–48630. [Google Scholar] [CrossRef] [Green Version]
- Vazquez, F.; Ramaswamy, S.; Nakamura, N.; Sellers, W.R. Phosphorylation of the PTEN tail regulates protein stability and function. Mol. Cell Biol. 2000, 20, 5010–5018. [Google Scholar] [CrossRef] [Green Version]
- Maddika, S.; Kavela, S.; Rani, N.; Palicharla, V.R.; Pokorny, J.L.; Sarkaria, J.N.; Chen, J. WWP2 is an E3 ubiquitin ligase for PTEN. Nat. Cell Biol. 2011, 13, 728–733. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gupta, A.; Leslie, N.R. Controlling PTEN (Phosphatase and Tensin Homolog) Stability: A DOMINANT ROLE FOR LYSINE 66. J. Biol. Chem. 2016, 291, 18465–18473. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, Y.R.; Chen, M.; Lee, J.D.; Zhang, J.; Lin, S.Y.; Fu, T.M.; Chen, H.; Ishikawa, T.; Chiang, S.Y.; Katon, J.; et al. Reactivation of PTEN tumor suppressor for cancer treatment through inhibition of a MYC-WWP1 inhibitory pathway. Science 2019, 364, eaau0159. [Google Scholar] [CrossRef] [PubMed]
- Ming, M.; Feng, L.; Shea, C.R.; Soltani, K.; Zhao, B.; Han, W.; Smart, R.C.; Trempus, C.S.; He, Y.Y. PTEN positively regulates UVB-induced DNA damage repair. Cancer Res. 2011, 71, 5287–5295. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shen, W.H.; Balajee, A.S.; Wang, J.; Wu, H.; Eng, C.; Pandolfi, P.P.; Yin, Y. Essential role for nuclear PTEN in maintaining chromosomal integrity. Cell 2007, 128, 157–170. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chang, C.J.; Mulholland, D.J.; Valamehr, B.; Mosessian, S.; Sellers, W.R.; Wu, H. PTEN nuclear localization is regulated by oxidative stress and mediates p53-dependent tumor suppression. Mol. Cell Biol. 2008, 28, 3281–3289. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cao, J.; Wan, L.; Hacker, E.; Dai, X.; Lenna, S.; Jimenez-Cervantes, C.; Wang, Y.; Leslie, N.R.; Xu, G.X.; Widlund, H.R.; et al. MC1R is a potent regulator of PTEN after UV exposure in melanocytes. Mol. Cell 2013, 51, 409–422. [Google Scholar] [CrossRef] [Green Version]
- Sobolewski, C.; Cerella, C.; Dicato, M.; Ghibelli, L.; Diederich, M. The role of cyclooxygenase-2 in cell proliferation and cell death in human malignancies. Int. J. Cell Biol. 2010, 2010, 215158. [Google Scholar] [CrossRef] [Green Version]
- Lee, C.; Ramirez, J.A.; Guitart, J.; Diaz, L.K. Expression of cyclooxygenase-2 and peroxisome proliferator-activated receptor gamma during malignant melanoma progression. J. Cutan Pathol. 2008, 35, 989–994. [Google Scholar] [CrossRef] [Green Version]
- Elmets, C.A.; Ledet, J.J.; Athar, M. Cyclooxygenases: Mediators of UV-induced skin cancer and potential targets for prevention. J. Investig. Dermatol. 2014, 134, 2497–2502. [Google Scholar] [CrossRef] [Green Version]
- De Cicco, P.; Panza, E.; Ercolano, G.; Armogida, C.; Sessa, G.; Pirozzi, G.; Cirino, G.; Wallace, J.L.; Ianaro, A. ATB-346, a novel hydrogen sulfide-releasing anti-inflammatory drug, induces apoptosis of human melanoma cells and inhibits melanoma development in vivo. Pharmacol. Res. 2016, 114, 67–73. [Google Scholar] [CrossRef] [PubMed]
- Zhou, P.; Qin, J.; Li, Y.; Li, G.; Wang, Y.; Zhang, N.; Chen, P.; Li, C. Combination therapy of PKCzeta and COX-2 inhibitors synergistically suppress melanoma metastasis. J. Exp. Clin. Cancer Res. 2017, 36, 115. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumar, D.; Rahman, H.; Tyagi, E.; Liu, T.; Li, C.; Lu, R.; Lum, D.; Holmen, S.L.; Maschek, J.A.; Cox, J.E.; et al. Aspirin Suppresses PGE2 and Activates AMP Kinase to Inhibit Melanoma Cell Motility, Pigmentation, and Selective Tumor Growth in Vivo. Cancer Prev. Res. 2018, 11, 629–642. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, G.S.; Liu, L.F.; Lin, C.J.; Tseng, J.C.; Chuang, M.J.; Lam, H.C.; Lee, J.K.; Yang, L.C.; Chan, J.H.; Howng, S.L.; et al. Gene transfer of pro-opiomelanocortin prohormone suppressed the growth and metastasis of melanoma: Involvement of alpha-melanocyte-stimulating hormone-mediated inhibition of the nuclear factor kappaB/cyclooxygenase-2 pathway. Mol. Pharmacol. 2006, 69, 440–451. [Google Scholar] [CrossRef] [PubMed]
- Weng, W.T.; Huang, S.C.; Ma, Y.L.; Chan, H.H.; Lin, S.W.; Wu, J.C.; Wu, C.Y.; Wen, Z.H.; Wang, E.M.; Wu, C.L.; et al. Alpha-Melanocyte-stimulating hormone inhibits angiogenesis through attenuation of VEGF/VEGFR2 signaling pathway. Biochim. Biophys. Acta 2014, 1840, 1850–1860. [Google Scholar] [CrossRef]
- Weng, W.T.; Wu, C.S.; Wang, F.S.; Wu, C.Y.; Ma, Y.L.; Chan, H.H.; Wu, D.C.; Wu, J.C.; Chu, T.H.; Huang, S.C.; et al. Alpha-Melanocyte-Stimulating Hormone Attenuates Neovascularization by Inducing Nitric Oxide Deficiency via MC-Rs/PKA/NF-kappaB Signaling. Int. J. Mol. Sci. 2018, 19, 3823. [Google Scholar] [CrossRef] [Green Version]
- Liu, G.S.; Wu, J.C.; Tsai, H.E.; Dusting, G.J.; Chan, E.C.; Wu, C.S.; Tai, M.H. Proopiomelanocortin gene delivery induces apoptosis in melanoma through NADPH oxidase 4-mediated ROS generation. Free Radic. Biol. Med. 2014, 70, 14–22. [Google Scholar] [CrossRef] [Green Version]
- Langan, E.A.; Nie, Z.; Rhodes, L.E. Melanotropic peptides: More than just ‘Barbie drugs’ and ‘sun-tan jabs’? Br. J. Dermatol. 2010, 163, 451–455. [Google Scholar] [CrossRef]
- Uckert, S.; Bannowsky, A.; Albrecht, K.; Kuczyk, M.A. Melanocortin receptor agonists in the treatment of male and female sexual dysfunctions: Results from basic research and clinical studies. Expert Opin. Investig. Drugs 2014, 23, 1477–1483. [Google Scholar] [CrossRef]
- Habbema, L.; Halk, A.B.; Neumann, M.; Bergman, W. Risks of unregulated use of alpha-melanocyte-stimulating hormone analogues: A review. Int. J. Dermatol. 2017, 56, 975–980. [Google Scholar] [CrossRef]
- Lam, H.C.; Kuo, S.M.; Chuang, M.J.; Keng, H.M.; Lin, P.R.; Liu, G.S.; Hsu, C.M.; Howng, S.L.; Tai, M.H. Blockade of endothelin-1 release contributes to the anti-angiogenic effect by pro-opiomelanocortin overexpression in endothelial cells. Exp. Biol. Med. 2006, 231, 782–788. [Google Scholar]
- Wu, J.C.; Tsai, H.E.; Liu, G.S.; Wu, C.S.; Tai, M.H. Autophagic cell death participates in POMC-induced melanoma suppression. Cell Death Discov. 2018, 4, 11. [Google Scholar] [CrossRef] [PubMed]
- Kim, A.; Son, M.; Kim, K.I.; Yang, Y.; Song, E.Y.; Lee, H.G.; Lim, J.S. Elevation of intracellular cyclic AMP inhibits NF-kappaB-mediated thymosin beta4 expression in melanoma cells. Exp. Cell Res. 2009, 315, 3325–3335. [Google Scholar] [CrossRef] [PubMed]
- Liu, G.S.; Tsai, H.E.; Weng, W.T.; Liu, L.F.; Weng, C.H.; Chuang, M.R.; Lam, H.C.; Wu, C.S.; Tee, R.; Wen, Z.H.; et al. Systemic pro-opiomelanocortin expression induces melanogenic differentiation and inhibits tumor angiogenesis in established mouse melanoma. Hum. Gene Ther. 2011, 22, 325–335. [Google Scholar] [CrossRef]
- Song, M.S.; Salmena, L.; Pandolfi, P.P. The functions and regulation of the PTEN tumour suppressor. Nat. Rev. Mol. Cell Biol. 2012, 13, 283–296. [Google Scholar] [CrossRef]
- Eves, P.; Haycock, J.; Layton, C.; Wagner, M.; Kemp, H.; Szabo, M.; Morandini, R.; Ghanem, G.; Garcia-Borron, J.C.; Jimenez-Cervantes, C.; et al. Anti-inflammatory and anti-invasive effects of alpha-melanocyte-stimulating hormone in human melanoma cells. Br. J. Cancer 2003, 89, 2004–2015. [Google Scholar] [CrossRef]
- Castejon-Grinan, M.; Herraiz, C.; Olivares, C.; Jimenez-Cervantes, C.; Garcia-Borron, J.C. cAMP-independent non-pigmentary actions of variant melanocortin 1 receptor: AKT-mediated activation of protective responses to oxidative DNA damage. Oncogene 2018, 37, 3631–3646. [Google Scholar] [CrossRef]
- Sanz-Motilva, V.; Martorell-Calatayud, A.; Nagore, E. Non-steroidal anti-inflammatory drugs and melanoma. Curr. Pharm. Des. 2012, 18, 3966–3978. [Google Scholar] [CrossRef]
- Botti, G.; Fratangelo, F.; Cerrone, M.; Liguori, G.; Cantile, M.; Anniciello, A.M.; Scala, S.; D’Alterio, C.; Trimarco, C.; Ianaro, A.; et al. COX-2 expression positively correlates with PD-L1 expression in human melanoma cells. J. Transl. Med. 2017, 15, 46. [Google Scholar] [CrossRef] [Green Version]






© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wu, J.-C.; Tsai, H.-E.; Hsiao, Y.-H.; Wu, J.-S.; Wu, C.-S.; Tai, M.-H. Topical MTII Therapy Suppresses Melanoma Through PTEN Upregulation and Cyclooxygenase II Inhibition. Int. J. Mol. Sci. 2020, 21, 681. https://doi.org/10.3390/ijms21020681
Wu J-C, Tsai H-E, Hsiao Y-H, Wu J-S, Wu C-S, Tai M-H. Topical MTII Therapy Suppresses Melanoma Through PTEN Upregulation and Cyclooxygenase II Inhibition. International Journal of Molecular Sciences. 2020; 21(2):681. https://doi.org/10.3390/ijms21020681
Chicago/Turabian StyleWu, Jian-Ching, Han-En Tsai, Yi-Hsiang Hsiao, Ji-Syuan Wu, Chieh-Shan Wu, and Ming-Hong Tai. 2020. "Topical MTII Therapy Suppresses Melanoma Through PTEN Upregulation and Cyclooxygenase II Inhibition" International Journal of Molecular Sciences 21, no. 2: 681. https://doi.org/10.3390/ijms21020681