Novel Diels–Alder Type Adducts from Morus alba Root Bark Targeting Human Monoamine Oxidase and Dopaminergic Receptors for the Management of Neurodegenerative Diseases



Abstract
:1. Introduction
2. Results
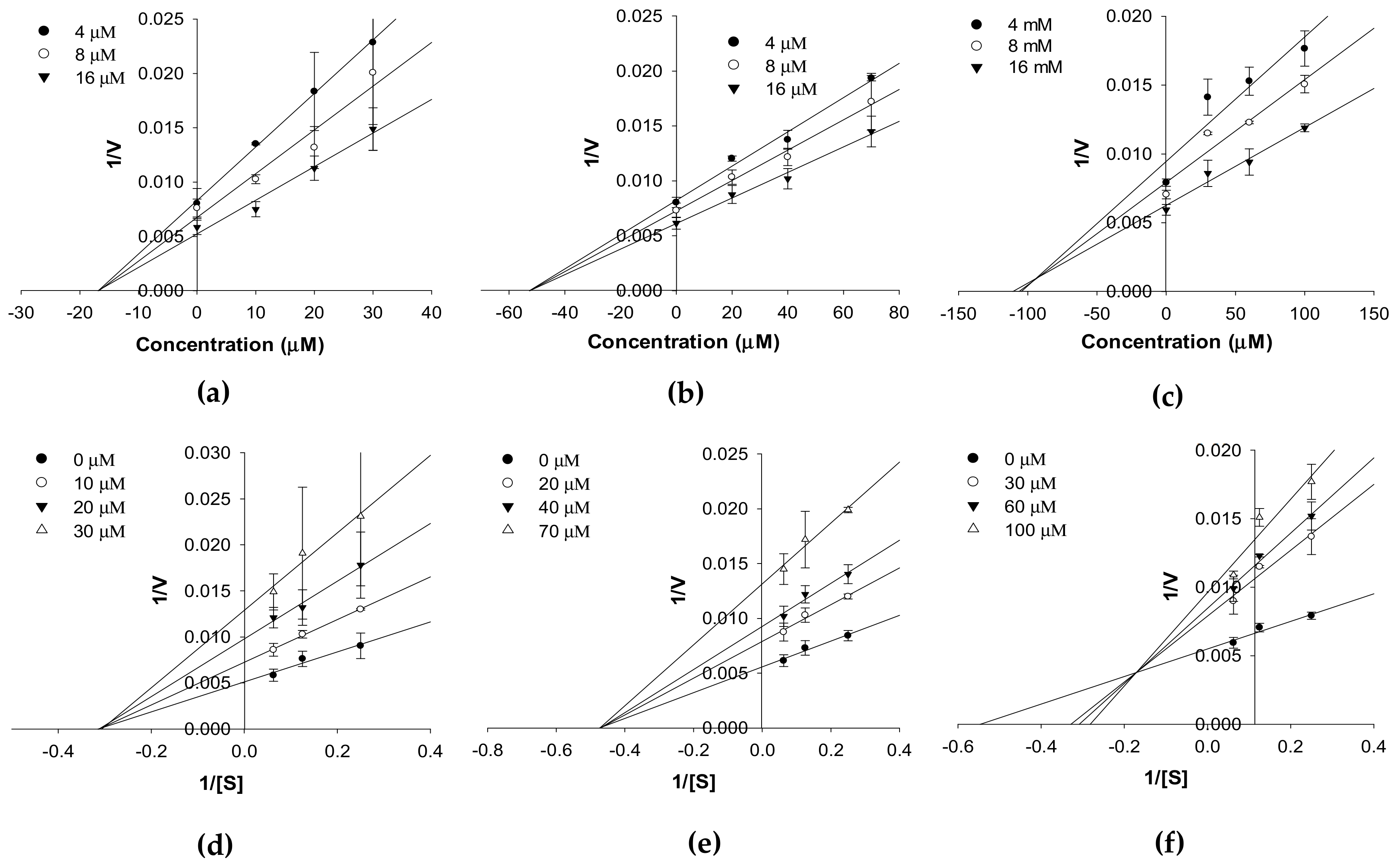
2.1. In Vitro hMAO Inhibition and Enzyme Kinetics
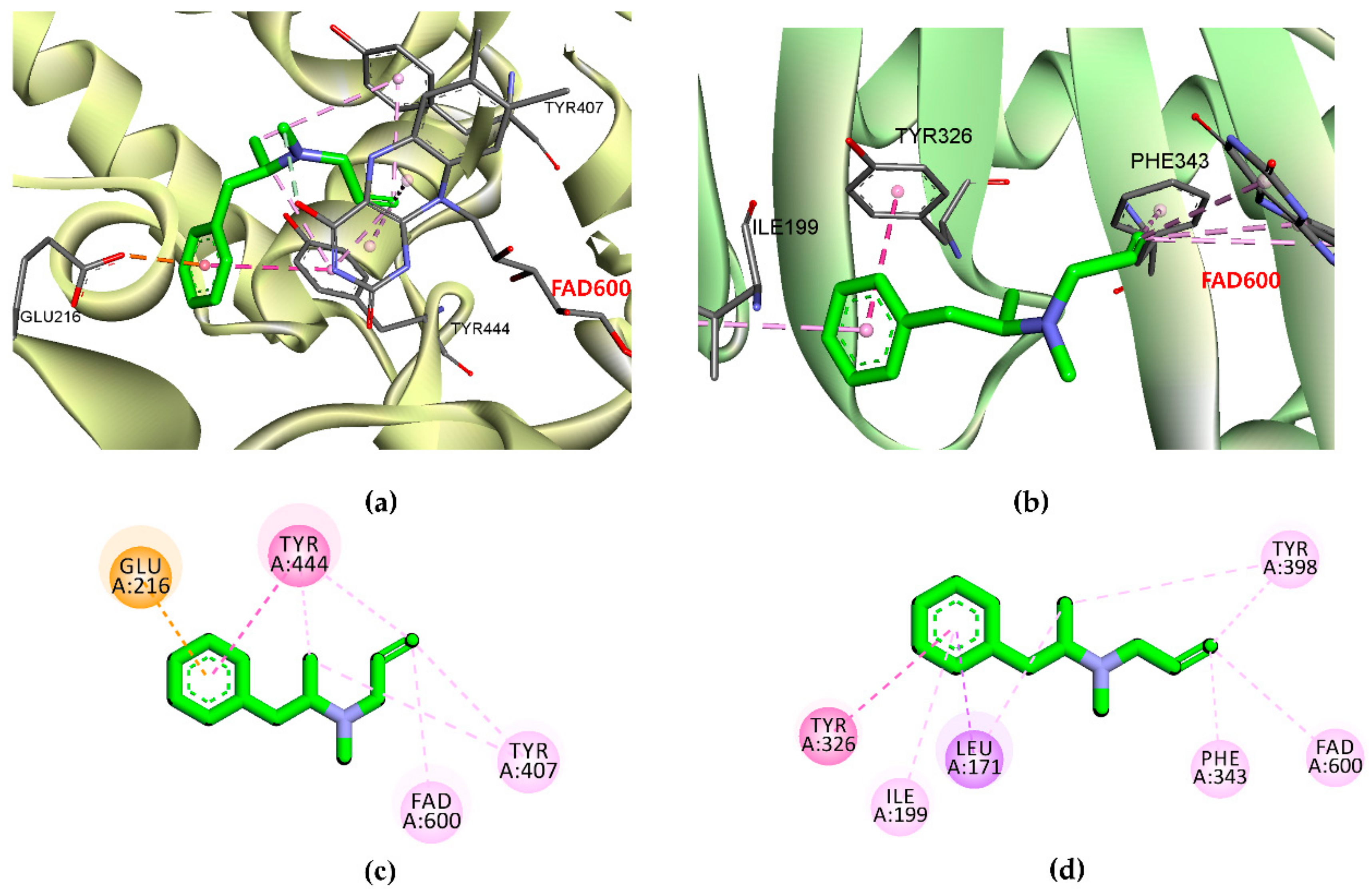
2.2. In Silico Docking Simulation of hMAO
2.3. Cell-Based Functional GPCR Assays
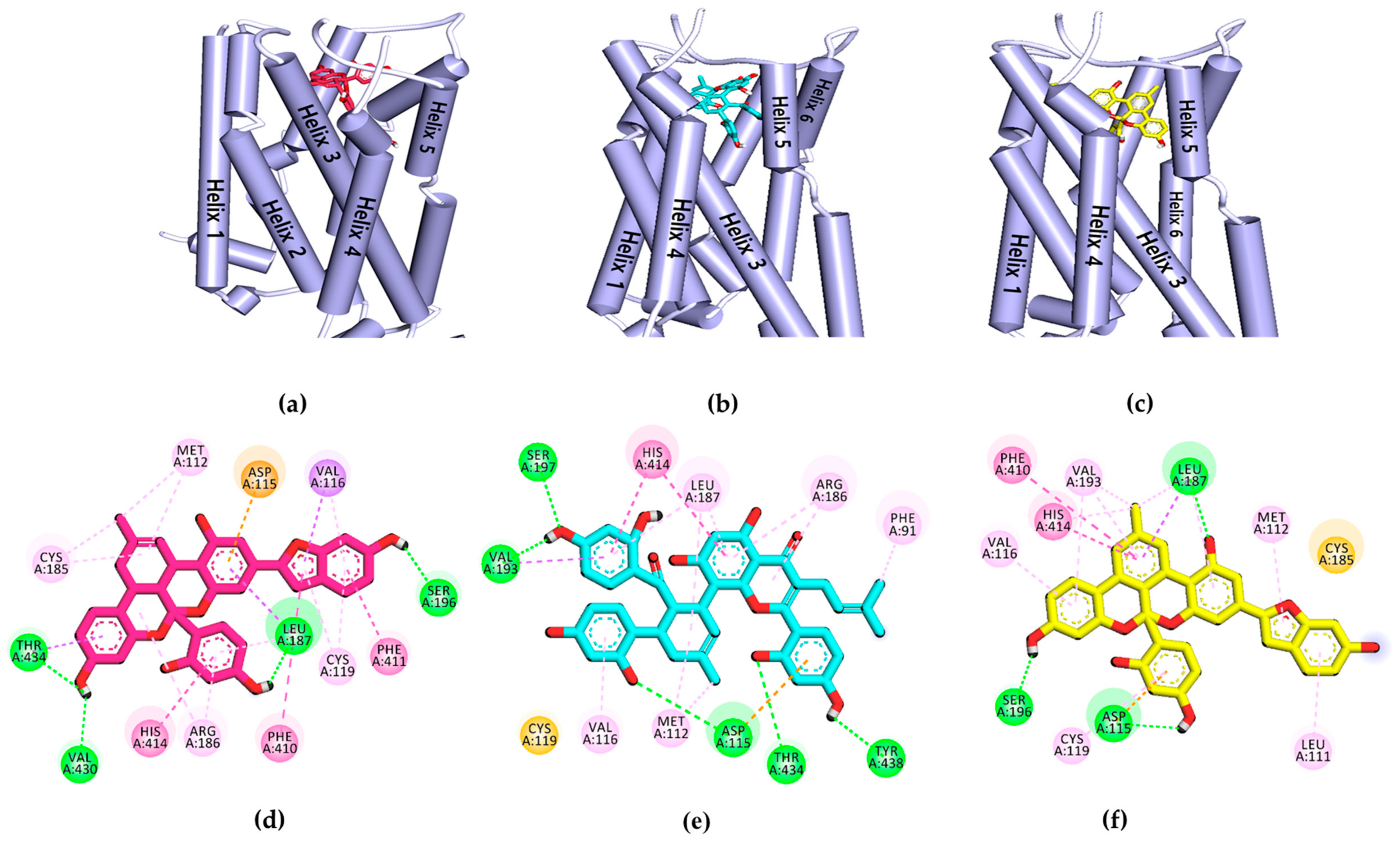
2.4. In Silico Docking Simulation of Dopamine Receptors
3. Discussion
4. Materials and Methods
4.1. Chemicals and Reagents
4.2. In Vitro Human MAO Inhibition and Enzyme Kinetics
4.3. Cell-Based Functional GPCR Assay
4.4. Measurement of cAMP Level
4.5. Measurement of Intracellular [Ca2+] Levels
4.6. Homology Modeling
4.7. In Silico Molecular Docking Simulation
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Adachi, N.; Yoshimura, A.; Chiba, S.; Ogawa, S.; Kunugi, H. Rotigotine, a dopamine receptor agonist, increased BDNF protein levels in the rat cortex and hippocampus. Neurosci. Lett. 2018, 662, 44–50. [Google Scholar] [CrossRef] [PubMed]
- Naoi, M.; Maruyama, W. Monoamine oxidase inhibitors as neuroprotective agents in age-dependent neurodegenerative disorders. Curr. Pharm. Des. 2010, 16, 2799–2817. [Google Scholar] [CrossRef] [PubMed]
- Riederer, P.; Konradi, C.; Schay, V.; Kienzl, E.; Birkmayer, G.; Danielczyk, W.; Sofic, E.; Youdim, M.B. Localization of MAO-A and MAO-B in human brain: A step in understanding the therapeutic action of l-deprenyl. Adv. Neurol. 1987, 45, 111–118. [Google Scholar] [PubMed]
- Bortolato, M.; Chen, K.; Shih, J.C. Monoamine oxidase inactivation: From pathophysiology to therapeutics. Adv. Drug Deliv. Rev. 2008, 60, 1527–1533. [Google Scholar] [CrossRef] [Green Version]
- Brunner, H.G.; Nelen, M.; Breakefield, X.O.; Ropers, H.H.; van Oost, B.A. Abnormal behavior associated with a point mutation in the structural gene for monoamine oxidase A. Science 1993, 262, 578–580. [Google Scholar] [CrossRef] [Green Version]
- Meyer, J.H.; Ginovart, N.; Boovariwala, A.; Sagrati, S.; Hussey, D.; Garcia, A.; Young, T.; Praschak-Rieder, N.; Wilson, A.A.; Houle, S. Elevated monoamine oxidase A levels in the brain: An explanation for the monoamine imbalance of major depression. Arch. Gen. Psychiatry 2006, 63, 1209–1216. [Google Scholar] [CrossRef] [Green Version]
- Finberg, J.P.M.; Rabey, J.M. Inhibitors of MAO-A and MAO-B in psychiatry and neurology. Front. Pharmacol. 2016, 7, 340. [Google Scholar] [CrossRef] [Green Version]
- Winner, B.M.; Zhang, H.; Farthing, M.M.; Karchalla, L.M.; Lookingland, K.J.; Goudreau, J.L. Metabolism of dopamine in nucleus accumbens astrocytes is preserved in aged mice exposed to MPTP. Front. Aging Neurosci. 2017, 9, 410. [Google Scholar] [CrossRef] [Green Version]
- Colzi, A.; d’Agostini, F.; Kettler, R.; Borroni, E.; Da Prada, M. Effect of selective and reversible MAO inhibitors on dopamine outflow in rat striatum: A microdialysis study. J. Neural. Transm. Suppl. 1990, 32, 79–84. [Google Scholar]
- Hauser, A.S.; Attwood, M.M.; Rask-Andersen, M.; Schiöth, H.B.; Gloriam, D.E. Trends in GPCR drug discovery: New agents, targets and indications. Nat. Rev. Drug Discov. 2017, 16, 829–842. [Google Scholar] [CrossRef]
- Bräuner-Osborne, H.; Rosenkilde, M.M.; Gether, U.; Gloriam, D.E. G protein-coupled receptor pharmacology—The next generation. Basic Clin. Pharmacol. Toxicol. 2019. [Google Scholar] [CrossRef] [PubMed]
- Zhai, S.; Tanimura, A.; Graves, S.M.; Shen, W.; Surmeier, D.J. Striatal synapses, circuits, and Parkinson’s disease. Curr. Opin. Neurobiol. 2018, 48, 9–16. [Google Scholar] [CrossRef] [PubMed]
- Levite, M. Neurotransmitters activate T-cells and elicit crucial functions via neurotransmitter receptors. Curr. Opin. Pharmacol. 2008, 8, 460–471. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.; Wang, B.L.; Li, Y. Advances in the pharmacological study of Morus alba L. Yao Xue Xue Bao 2014, 49, 824–831. [Google Scholar] [PubMed]
- Wei, H.; Zhu, J.-J.; Liu, X.-Q.; Feng, W.-H.; Wang, Z.-M.; Yan, L.-H. Review of bioactive compounds from root barks of Morus plants (Sang-Bai-Pi) and their pharmacological effects. Cogent Chem. 2016, 2, 1212320. [Google Scholar] [CrossRef]
- Kim, H.G.; Ju, M.S.; Shim, J.S.; Kim, M.C.; Lee, S.-H.; Huh, Y.; Kim, S.Y.; Oh, M.S. Mulberry fruit protects dopaminergic neurons in toxin-induced Parkinson’s disease models. Br. J. Nutr. 2010, 104, 8–16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- El-Sayyad, H.I.H.; El-Sherbiny, M.A.; Sobh, M.A.; Abou-El-Naga, A.M.; Ibrahim, M.A.N.; Mousa, S.A. Protective effects of Morus alba leaves extract on ocular functions of pups from diabetic and hypercholesterolemic mother rats. Int. J. Biol. Sci. 2011, 7, 715. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gurukar, M.S.A.; Chilkunda, N.D. Morus alba leaf bioactives modulate peroxisome proliferator activated receptor γ in the kidney of diabetic rat and impart beneficial effect. J. Agric. Food Chem. 2018, 66, 7923–7934. [Google Scholar] [CrossRef]
- Yadav, A.; Kawale, L.; Nade, V. Effect of Morus alba L. (mulberry) leaves on anxiety in mice. Indian J. Pharmacol. 2008, 40, 32. [Google Scholar] [CrossRef] [Green Version]
- Paudel, P.; Yu, T.; Seong, S.H.; Kuk, E.B.; Jung, H.A.; Choi, J.S. Protein tyrosine phosphatase 1B inhibition and glucose uptake potentials of mulberrofuran G, albanol B, and kuwanon G from root bark of Morus alba L. in insulin-resistant HepG2 cells: An in vitro and in silico study. Int. J. Mol. Sci. 2018, 19, 1542. [Google Scholar] [CrossRef] [Green Version]
- Ha, M.T.; Seong, S.H.; Nguyen, T.D.; Cho, W.-K.; Ah, K.J.; Ma, J.Y.; Woo, M.H.; Choi, J.S.; Min, B.S. Chalcone derivatives from the root bark of Morus alba L. act as inhibitors of PTP1B and α-glucosidase. Phytochemistry 2018, 155, 114–125. [Google Scholar] [CrossRef] [PubMed]
- Paudel, P.; Seong, S.H.; Zhou, Y.; Ha, M.T.; Min, B.S.; Jung, H.A.; Choi, J.S. Arylbenzofurans from the root bark of Morus alba as triple inhibitors of cholinesterase, β-site amyloid precursor protein cleaving enzyme 1, and glycogen synthase kinase-3β: Relevance to Alzheimer’s disease. ACS Omega 2019, 4, 6283–6294. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seong, S.H.; Ha, M.T.; Min, B.S.; Jung, H.A.; Choi, J.S. Moracin derivatives from Morus Radix as dual BACE1 and cholinesterase inhibitors with antioxidant and anti-glycation capacities. Life Sci. 2018, 210, 20–28. [Google Scholar] [CrossRef] [PubMed]
- Paudel, P.; Seong, S.H.; Wagle, A.; Min, B.S.; Jung, H.A.; Choi, J.S. Antioxidant and anti-browning property of 2-arylbenzofuran derivatives from Morus alba Linn root bark. Food Chem. 2019, 125739. [Google Scholar] [CrossRef] [PubMed]
- Yimam, M.; Jiao, P.; Hong, M.; Brownell, L.; Lee, Y.-C.; Kim, H.-J.; Nam, J.-B.; Kim, M.-R.; Jia, Q. Morus alba, a medicinal plant for appetite suppression and weight loss. J. Med. Food. 2019, 22, 741–751. [Google Scholar] [CrossRef] [PubMed]
- Wagmann, L.; Brandt, S.D.; Kavanagh, P.V.; Maurer, H.H.; Meyer, M.R. In vitro monoamine oxidase inhibition potential of alpha-methyltryptamine analog new psychoactive substances for assessing possible toxic risks. Toxicol. Lett. 2017, 272, 84–93. [Google Scholar] [CrossRef] [Green Version]
- Tzvetkov, N.T.; Hinz, S.; Küppers, P.; Gastreich, M.; Müller, C.E. Indazole-and indole-5-carboxamides: Selective and reversible monoamine oxidase B inhibitors with subnanomolar potency. J. Med. Chem. 2014, 57, 6679–6703. [Google Scholar] [CrossRef]
- Kołaczkowski, M.; Bucki, A.; Feder, M.; Pawłowski, M. Ligand-optimized homology models of D1 and D2 dopamine receptors: Application for virtual screening. J. Chem. Inf. Model. 2013, 53, 638–648. [Google Scholar] [CrossRef]
- Gerlach, M.; Double, K.; Arzberger, T.; Leblhuber, F.; Tatschner, T.; Riederer, P. Dopamine receptor agonists in current clinical use: Comparative dopamine receptor binding profiles defined in the human striatum. J. Neural. Transm. 2003, 110, 1119–1127. [Google Scholar] [CrossRef]
- Jaber, M.; Robinson, S.W.; Missale, C.; Caron, M.G. Dopamine receptors and brain function. Neuropharmacology 1996, 35, 1503–1519. [Google Scholar] [CrossRef]
- Ettenberg, A.; MacConell, L.A.; Geist, T.D. Effects of haloperidol in a response-reinstatement model of heroin relapse. Psychopharmacology 1996, 124, 205–210. [Google Scholar] [CrossRef] [PubMed]
- Shaham, Y.; Stewart, J. Effects of opioid and dopamine receptor antagonists on relapse induced by stress and re-exposure to heroin in rats. Psychopharmacology 1996, 125, 385–391. [Google Scholar] [CrossRef] [PubMed]
- Ashby, C.R.; Paul, M.; Gardner, E.L.; Heidbreder, C.A.; Hagan, J.J. Acute administration of the selective D3 receptor antagonist SB-277011A blocks the acquisition and expression of the conditioned place preference response to heroin in male rats. Synapse 2003, 48, 154–156. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.B.; Mantsch, J.R. l-tetrahydropalamatine: A potential new medication for the treatment of cocaine addiction. Future Med. Chem. 2012, 4, 177–186. [Google Scholar] [CrossRef] [Green Version]
- Lapish, C.C.; Belardetti, F.; Ashby, D.M.; Ahn, S.; Butts, K.A.; So, K.; Macrae, C.M.; Hynd, J.J.; Miller, J.J.; Phillips, A.G. A preclinical assessment of dl-govadine as a potential antipsychotic and cognitive enhancer. Int. J. Neuropsychopharmacol. 2012, 15, 1441–1455. [Google Scholar] [CrossRef] [Green Version]
- Masri, B.; Salahpour, A.; Didriksen, M.; Ghisi, V.; Beaulieu, J.-M.; Gainetdinov, R.R.; Caron, M.G. Antagonism of dopamine D2 receptor/β-arrestin 2 interaction is a common property of clinically effective antipsychotics. Proc. Natl. Acad. Sci. USA 2008, 105, 13656–13661. [Google Scholar] [CrossRef] [Green Version]
- Kim, K.H.; Chung, W.S.; Kim, Y.; Kim, K.S.; Lee, I.S.; Park, J.Y.; Jeong, H.S.; Na, Y.C.; Lee, C.H.; Jang, H.J. Transcriptomic analysis reveals wound healing of Morus alba root extract by up-regulating keratin filament and CXCL12/CXCR4 signaling. Phytother. Res. 2015, 29, 1251–1258. [Google Scholar] [CrossRef]
- Shoichet, B.K. Interpreting steep dose-response curves in early inhibitor discovery. J. Med. Chem. 2006, 49, 7274–7277. [Google Scholar] [CrossRef]
- McGovern, S.L.; Caselli, E.; Grigorieff, N.; Shoichet, B.K. A common mechanism underlying promiscuous inhibitors from virtual and high-throughput screening. J. Med. Chem. 2002, 45, 1712–1722. [Google Scholar] [CrossRef]
- Coan, K.E.D.; Shoichet, B.K. Stoichiometry and physical chemistry of promiscuous aggregate-based inhibitors. J. Am. Chem. Soc. 2008, 130, 9606–9612. [Google Scholar] [CrossRef] [Green Version]
- Jadhav, A.; Ferreira, R.S.; Klumpp, C.; Mott, B.T.; Austin, C.P.; Inglese, J.; Thomas, C.J.; Maloney, D.J.; Shoichet, B.K.; Simeonov, A. Quantitative analyses of aggregation, autofluorescence, and reactivity artifacts in a screen for inhibitors of a thiol protease. J. Med. Chem. 2010, 53, 37–51. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, C.; Li, Q.; Meng, L.; Ren, Y. Design of novel dopamine D2 and serotonin 5-HT2A receptors dual antagonists toward schizophrenia: An integrated study with QSAR, molecular docking, virtual screening and molecular dynamics simulations. J. Biomol. Struct. Dyn. 2019, 1–26. [Google Scholar] [CrossRef]
- Kalani, M.Y.S.; Vaidehi, N.; Hall, S.E.; Trabanino, R.J.; Freddolino, P.L.; Kalani, M.A.; Floriano, W.B.; Kam, V.W.T.; Goddard, W.A. The predicted 3D structure of the human D2 dopamine receptor and the binding site and binding affinities for agonists and antagonists. Proc. Natl. Acad. Sci. USA 2004, 101, 3815–3820. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ramdani, E.D.; Yanuar, A.; Tjandrawinata, R.R. Comparison of dopamine D2 receptor (homology model and X-ray structure) and virtual screening protocol validation for the antagonism mechanism. J. Appl. Pharm. Sci. 2019, 9, 17–22. [Google Scholar]
- Salmas, R.E.; Yurtsever, M.; Stein, M.; Durdagi, S. Modeling and protein engineering studies of active and inactive states of human dopamine D2 receptor (D2R) and investigation of drug/receptor interactions. Mol. Divers. 2015, 19, 321–332. [Google Scholar] [CrossRef]
- Yadav, A.V.; Nade, V.S. Anti-dopaminergic effect of the methanolic extract of Morus alba L. leaves. Indian J. Pharmacol. 2008, 40, 221. [Google Scholar] [CrossRef] [Green Version]
- Gurevich, E.V.; Joyce, J.N. Distribution of dopamine D3 receptor expressing neurons in the human forebrain: Comparison with D2 receptor expressing neurons. Neuropsychopharmacology 1999, 20, 60–80. [Google Scholar] [CrossRef] [Green Version]
- Levant, B.; Ling, Z.D.; Carvey, P.M. Dopamine D3 Receptors. CNS Drugs 1999, 12, 391–402. [Google Scholar] [CrossRef]
- Scheller, D.; Ullmer, C.; Berkels, R.; Gwarek, M.; Lübbert, H. The in vitro receptor profile of rotigotine: A new agent for the treatment of Parkinson’s disease. Naunyn-Schmiedeberg’s Arch. Pharmacol. 2009, 379, 73–86. [Google Scholar] [CrossRef]
- Sokoloff, P.; Leriche, L.; Diaz, J.; Louvel, J.; Pumain, R. Direct and indirect interactions of the dopamine D3 receptor with glutamate pathways: Implications for the treatment of schizophrenia. Naunyn-Schmiedeberg’s Arch. Pharmacol. 2013, 386, 107–124. [Google Scholar] [CrossRef] [Green Version]
- Leggio, G.M.; Bucolo, C.; Platania, C.B.M.; Salomone, S.; Drago, F. Current drug treatments targeting dopamine D3 receptor. Pharmacol. Ther. 2016, 165, 164–177. [Google Scholar] [CrossRef] [PubMed]
- Huang, M.; Kwon, S.; He, W.; Meltzer, H.Y. Neurochemical arguments for the use of dopamine D4 receptor stimulation to improve cognitive impairment associated with schizophrenia. Pharmacol. Biochem. Behav. 2017, 157, 16–23. [Google Scholar] [CrossRef] [PubMed]
- Tomlinson, A.; Grayson, B.; Marsh, S.; Hayward, A.; Marshall, K.M.; Neill, J.C. Putative therapeutic targets for symptom subtypes of adult ADHD: D4 receptor agonism and COMT inhibition improve attention and response inhibition in a novel translational animal model. Eur. Neuropsychopharmacol. 2015, 25, 454–467. [Google Scholar] [CrossRef] [PubMed]
- Negrete-Díaz, J.V.; Sumilov, K.; Real, M.Á.; Medina-Luque, J.; Valderrama-Carvajal, A.; Flores, G.; Rodríguez-Moreno, A.; Rivera, A. Pharmacological activation of dopamine D4 receptor modulates morphine-induced changes in the expression of GAD65/67 and GABAB receptors in the basal ganglia. Neuropharmacology 2019, 152, 22–29. [Google Scholar] [CrossRef]
- Chien, E.Y.; Liu, W.; Zhao, Q.; Katritch, V.; Han, G.W.; Hanson, M.A.; Shi, L.; Newman, A.H.; Javitch, J.A.; Cherezov, V. Structure of the human dopamine D3 receptor in complex with a D2/D3 selective antagonist. Science 2010, 330, 1091–1095. [Google Scholar] [CrossRef] [Green Version]
- Neve, K.A.; Seamans, J.K.; Trantham-Davidson, H. Dopamine receptor signaling. J. Recept. Signal. Transduct. 2004, 24, 165–205. [Google Scholar] [CrossRef]
- Santini, E.; Heiman, M.; Greengard, P.; Valjent, E.; Fisone, G. Inhibition of mTOR signaling in Parkinson’s disease prevents l-DOPA–induced dyskinesia. Sci. Signal. 2009, 2, ra36. [Google Scholar] [CrossRef] [Green Version]
- Chen, J.; Rusnak, M.; Lombroso, P.J.; Sidhu, A. Dopamine promotes striatal neuronal apoptotic death via ERK signaling cascades. Eur. J. Neurosci. 2009, 29, 287–306. [Google Scholar] [CrossRef] [Green Version]
- Greengard, P. The neurobiology of slow synaptic transmission. Science 2001, 294, 1024–1030. [Google Scholar] [CrossRef] [Green Version]
- Nishi, A.; Bibb, J.A.; Snyder, G.L.; Higashi, H.; Nairn, A.C.; Greengard, P. Amplification of dopaminergic signaling by a positive feedback loop. Proc. Natl. Acad. Sci. USA 2000, 97, 12840–12845. [Google Scholar] [CrossRef] [Green Version]
- Nishi, A.; Snyder, G.L.; Greengard, P. Bidirectional regulation of DARPP-32 phosphorylation by dopamine. J. Neurosci. 1997, 17, 8147–8155. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klein, M.O.; Battagello, D.S.; Cardoso, A.R.; Hauser, D.N.; Bittencourt, J.C.; Correa, R.G. Dopamine: Functions, signaling, and association with neurological diseases. Cell. Mol. Neurobiol. 2019, 39, 31–59. [Google Scholar] [CrossRef] [PubMed]
- Kuk, E.B.; Jo, A.R.; Oh, S.I.; Sohn, H.S.; Seong, S.H.; Roy, A.; Choi, J.S.; Jung, H.A. Anti-Alzheimer’s disease activity of compounds from the root bark of Morus alba L. Arch. Pharm. Res. 2017, 40, 338–349. [Google Scholar] [CrossRef] [PubMed]
- Paudel, P.; Seong, S.H.; Jung, H.A.; Choi, J.S. Rubrofusarin as a dual protein tyrosine phosphate 1B and human monoamine oxidase-A inhibitor: An in vitro and in silico study. ACS Omega 2019, 4, 11621–11630. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paudel, P.; Seong, S.H.; Shrestha, S.; Jung, H.A.; Choi, J.S. In vitro and in silico human monoamine oxidase inhibitory potential of anthraquinones, naphthopyrones, and naphthalenic lactones from Cassia obtusifolia Linn seeds. ACS Omega 2019, 4, 16139–16152. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paudel, P.; Seong, S.H.; Wu, S.; Park, S.; Jung, H.A.; Choi, J.S. Eckol as a potential therapeutic against neurodegenerative diseases targeting dopamine D3/D4 receptors. Mar. Drugs 2019, 17, 108. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paudel, P.; Seong, S.H.; Jung, H.A.; Choi, J.S. Characterizing fucoxanthin as a selective dopamine D3/D4 receptor agonist: Relevance to Parkinson’s disease. Chem. Biol. Interact. 2019, 310, 108757. [Google Scholar] [CrossRef]
- Seong, S.H.; Paudel, P.; Choi, J.-W.; Ahn, D.H.; Nam, T.-J.; Jung, H.A.; Choi, J.S. Probing multi-target action of phlorotannins as new monoamine oxidase inhibitors and dopaminergic receptor modulators with the potential for treatment of neuronal disorders. Mar. Drugs 2019, 17, 377. [Google Scholar] [CrossRef] [Green Version]
- Xu, D.; Zhang, Y. Improving the physical realism and structural accuracy of protein models by a two-step atomic-level energy minimization. Biophys. J. 2011, 101, 2525–2534. [Google Scholar] [CrossRef] [Green Version]
- Goodsell, D.S.; Morris, G.M.; Olson, A.J. Automated docking of flexible ligands: Applications of AutoDock. J. Mol. Recognit. 1996, 9, 1–5. [Google Scholar] [CrossRef]













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compounds | Human Monoamine Oxidase A (hMAO-A) | ||
|---|---|---|---|
| IC50 (μM, Mean ± SD) a | Ki Value b | Inhibition Type c | |
| 1 | 54.79 ± 0.03 | 26.96 ± 3.98 | Competitive |
| 2 | 70.16 ± 2.60 | 28.29 ± 2.02 | Competitive |
| 3 | 114.31 ± 2.30 | 46.93 ± 4.12 | Competitive |
| Selegiline d | 12.51 ± 1.11 | NT | NT |
| Harmine d, e | 0.006 [26] | NT | NT |
| Human Monoamine Oxidase B (hMAO-B) | |||
| 1 | 18.14 ± 1.06 | 17.01 ± 3.31 | Noncompetitive |
| 2 | 57.71 ± 2.12 | 52.09 ± 5.56 | Noncompetitive |
| 3 | 90.59 ± 1.72 | 55.19 ± 7.79 f/186.2 ± 10.26 g | Mixed |
| Selegiline d | 0.30 ± 0.01 | NT | NT |
| Safinamide d,e | 0.00512 [27] | NT | NT |
| Compound | Binding Energy (kcal/mol) a | H-bond Interacting Residues b | Hydrophobic Interacting Residues b | Electrostatic Interacting Residues b |
|---|---|---|---|---|
| 1 | −9.54 | Gly110, Thr336, Ile207, Gly214, Ser209 | Val210 (Pi-Sigma, Pi-Alkyl), Ile325 (Pi-Sigma), Phe208 (Pi-Pi Stacked, Pi-Pi T-Shaped), Ile358 (Alkyl), Leu337 (Alkyl), Ile335 (Alkyl), Met350 (Alkyl), Val93 (Pi-Alkyl), | - |
| 2 | −6.74 | Met300, Leu298, Asp359, Gly404, Cys398, Trp397, Glu400 | Ala302 (Pi-Alkyl, Alkyl) | - |
| 3 | −8.62 | Gln296, Ile295, Gly404, Tyr410, Met300, Thr183, Ser184 | Pro299, Ala279, Ala302 (Pi-Alkyl) | Glu188 (Pi-Anion) |
| Selegiline | −6.54 | - | Ile335 (Pi-Sigma), Leu337 (Pi-Alkyl), FAD600 (Pi-Alkyl), Tyr407 (Pi-Alkyl), Tyr444 (Pi-Alkyl) | - |
| HRM c(Harmine) | −6.46 | FAD600 | Tyr444 (Pi-Sigma), FAD600 (Pi-Sigma, Pi-Pi T-shaped, Pi-Alkyl), Tyr444 (Pi-Pi Stacked), Phe352 (Pi-Pi T-shaped), Tyr407 (Pi-Alkyl), Ile335 (Pi-Alkyl) | - |
| Compound | Binding Energy (kcal/mol) a | H-bond Interacting Residues b | Hydrophobic Interacting Residues b | Electrostatic Interacting Residues b |
|---|---|---|---|---|
| 1 | −11.09 | His115, Pro476, Glu483 | Phe103 (Pi-Pi Stacked, Pi-Pi T-shaped, Pi-Alkyl), Val106 (Pi-Alkyl), Ile477 (Pi-Alkyl) | Glu483(Pi-Anion) |
| 2 | −12.65 | Pro104, Asn116, Glu483, Phe103, Thr478 | Tyr112 (Pi-Sigma), Phe103 (Pi-Pi Stacked), Val106 (Alkyl, Pi-Alkyl), Pro102 (Alkyl, Pi-Alkyl), Tyr112 (Pi-Alkyl), Trp119 (Pi-Alkyl), Pro104 (Pi-Alkyl), Leu164 (Pi-Alkyl) | Glu483(Pi-Anion) |
| 3 | −10.05 | Thr195, Pro104, Asn116, Thr478, Gly193 | Ile477 (Pi-Sigma), Trp119 (Pi-Pi Stacked), Phe103 (Pi-Pi T-shaped), Thr195 (Amide-Pi Stacked), Gly194 (Amide-Pi Stacked), Arg120 (Alkyl, Pi-Alkyl), Val106 (Pi-Alkyl) | Asp123(Pi-Anion), Glu483(Pi-Anion) |
| Selegiline c | −7.06 | Ile198 | Tyr398 (Pi-Pi Stacked), Tyr435 (Pi-Pi Stacked), FAD600 (Pi-Pi T-shaped), Leu171 (Alkyl), Cys172 (Alkyl), Phe188 (Pi-Alkyl) | - |
| Safinamide c | −9.86 | Cys172, Ile199, Tyr326, Thr201 | Leu171 (Pi-Sigma, Pi-Alkyl), Tyr398 (Pi-Pi Stacked), Tyr326 (Pi-Pi T-shaped), Ile199 (Pi-Alkyl) | - |
| Receptors | 1 | 2 | 3 | Reference Drugs |
|---|---|---|---|---|
| % Stimulation a (% Inhibition b) | % Stimulation a (% Inhibition b) | % Stimulation a (% Inhibition b) | EC50 c (IC50 d) | |
| D1 (h) | 17.2 ± 8.4 (87.65 ± 1.19) | 0.85 ± 0.24 (98.85 ± 1.79) | INTER (67.80 ± 9.05) | 28 (2.8) |
| D2L (h) | 7.10 ± 1.47 (101.30 ± 0.16) | NSI (99.15 ± 0.77) | 4.10 ± 1.06 (78.55 ± 3.61) | 12 (28) |
| D3 (h) | 119.9 ± 2.44 (−28.7 ± 11.15) | 124.3 ± 0.76 (−27.4 ± 7.79) | 102.8 ± 1.36 (−13.4 ± 1.87) | 4.1 (20) |
| D4 (h) | 86.30 ± 0.99 (−20.8 ± 6.93) | 90.45 ± 0.14 (−29.6 ± 7.21) | 46.10 ± 1.76 (26.9 ± 5.09) | 21 (150) |
| Target | Compounds | Binding Energy (kcal/mol) | H-bond Interaction Residues | Hydrophobic Interacting Residues | Electrostatic Interacting Residues |
|---|---|---|---|---|---|
| hD1R | Dopamine a (agonist) | −5.59 | Asp103 (Salt bridge), Ser202, Asn292, Ser199 | Phe289 (Pi-Pi T-shaped), Ile104 (Pi-Alkyl) | Phe288(Pi-Cation) |
| SCH23390 a (antagonist) | − 6.94 | Asp103 (Salt bridge), Ser199, Ser202 | Leu190 (Pi-sigma), Phe288 (Pi-Pi T-shaped), Ile104 (Pi-Alkyl), Ala195 (Pi-Alkyl) | - | |
| 1 | −9.22 | Lys81, Leu291, Asp314, Ser188 | Leu295 (Pi-sigma), Phe313 (Pi-Pi Stacked), Phe306 (Pi-Pi T-shaped), Ser188 (Amide-Pi Stacked), Leu295 (Pi-Alkyl), Leu291 (Pi-Alkyl) | Lys81(Pi-Cation), Asp314(Pi-Anion) | |
| 2 | −7.1 | Lys81, Ser107, Ser202, Asp187, Asp103, Ser198 | Val100 (Pi-sigma), Val317 (Pi-Sigma, Pi-Alkyl), Phe313 (Pi-Pi T-shaped), Leu190 (Alkyl), Cys186 (Alkyl), Phe288 (Pi-Alkyl), Ile104 (Pi-Alkyl) | Asp187 (Pi-Anion) | |
| 3 | −9.2 | Asp187, Ser188 | Asp187 (Pi-Sigma). Leu295 (Pi-Sigma, Pi-Alkyl), Phe30 6 (Pi-Pi T-shaped), Pro171 (Pi-Alkyl), Arg192 (Pi-Alkyl), Ala195 (Pi-Alkyl) | - |
| Target | Compounds | Binding Energy (kcal/mol) | H-bond Interaction Residues | Hydrophobic Interacting Residues | Electrostatic Interacting Residues |
|---|---|---|---|---|---|
| hD2LR | Dopamine a (agonist) | −6.98 | Asp114 (Salt bridge), Tyr416, Thr119 | Trp386 (Pi-Pi T-shaped), Val115 (Pi-Alkyl) | - |
| Risperidone a (agonist) | −12.7 | Asp114 (salt bridge), Thr119 | Trp100 (Pi-Pi T-shaped, Pi-Alkyl), Trp386 (Pi-Pi T-shaped), Val91(Alkyl), Leu94 (Alkyl), Val115 (Alkyl, Pi-Alkyl), Val111 (Alkyl), Ile184 (Alkyl), Phe110 (Pi-Alkyl), Phe389 (Pi-Alkyl), Cys118 (Pi-Alkyl), Ala122 (Pi-Alkyl) | - | |
| Butaclamol a (antagonist) | −6.9 | Asp114 (Salt bridge), Ser193 | Phe389 (Pi-Pi Stacked, Pi-Pi T-shaped, Pi-Alkyl), Tyr416 (Pi-Pi Stacked), Cys118 (Alkyl), Phe198 (Pi-Alkyl), Trp386 (Pi-Alkyl), Phe390 (Pi-Alkyl) | - | |
| 1 | −8.11 | Ser197, Asp114, Thr412, | Thr412 (Pi-Sigma), Phe110 (Pi-Sigma), Trp110 (Pi-Pi T-shaped, Pi-Alkyl), Trp386 (Pi-Pi T-shaped), Tyr416 (Pi-Pi- T-shaped), Val111 (Alkyl), Ile184 (Alkyl), | Asp114 (Pi-Anion) | |
| 2 | −8.23 | Asn396, Tyr408, Ile184 | Tyr408 (Pi-Pi Stacked), Tyr100 (Pi-Pi T-shaped), Phe389 (Pi-Alkyl), Tyr416 (Pi-Alkyl), Ile184 (Pi-Alkyl), Val190 (Pi-Alkyl) | - | |
| 3 | −10.45 | Trp100, Cys118, Ser193, Asp114 | Ile184 (Pi-Sigma, Alkyl), Trp100 (Pi-Pi T-shaped), Trp386 (Pi-Pi T-shaped), Val190 (Alkyl), Phe189 (Pi-Alkyl), Val115 (Pi-Alkyl) | Asp114 (Pi-Anion) |
| Target | Compounds | Binding Energy (kcal/mol) | H-bond Interaction Residues | Hydrophobic Interacting Residues | Electrostatic Interacting Residues |
|---|---|---|---|---|---|
| hD3R | Dopamine a (agonist) | −5.72 | Asp110 (Salt bridge), Tyr373, Val111, Thr115, Ser196 | Val111 (Pi-Alkyl), Cys114 (Pi-Alkyl) | |
| Eticlopride a (antagonist) | −9.22 | Asp110 (Salt bridge), Tyr373 | Phe345 (Pi-Pi T-shaped), Ile183 (Alkyl, Pi-Alkyl), Val189 (Alkyl), VAl111 (Pi-Alkyl) | ||
| (+)-butaclamol a (antagonist) | −10.69 | Asp110(Salt bridge) | Val111 (Alkyl), Cys114 (Alkyl), Trp342 (Pi-Alkyl), Phe345 (Pi-Alkyl), Phe346 (Pi-Alkyl), Val86 (Pi-Alkyl) | ||
| 1 | −5.89 | Tyr365, Cys181, Ser366, Thr369 | Ile183 (Pi-Sigma), Phe345 (Pi-Pi T-shaped), His349 (Pi-Pi T-shaped), Tyr365 (Pi-Pi T-shaped), Val86 (Alkyl, Pi-Alkyl), Leu89 (Alkyl), Phe106 (PI-Alkyl), Val107 (Pi-Alkyl), Val111 (Pi-Alkyl) | Asp110 (Pi-Anion) | |
| 2 | −7.45 | Tyr365, Thr369, Cys181, | Thr369 (Pi-Sigma), Phe345(Pi-Pi Stacked, Pi-Alkyl), Phe106 (Pi-Pi T-shaped), Tyr365 (Pi-Pi T-shaped), Val86 (Alkyl), Leu89 (Alkyl), Phe346 (Pi-Alkyl), Val107 (PI-Alkyl) | Asp110 (Pi-Anion), | |
| 3 | −10.41 | Ile183, Val110, Thr115 | Leu89 (Pi-Sigma), Thr359 (Pi-Sigma), Phe345 (Pi-Pi Stacked), Tyr365 (PI-Pi T-shaped), Val86 (Alkyl, Pi-Alkyl), Tyr36 (Pi-Alkyl), Val111 (Pi-Alkyl), Cys114 (Pi-Alkyl), | Asp110 (Pi-Anion) |
| Target | Compounds | Binding Energy (kcal/mol) | H-bond Interaction Residues | Hydrophobic Interacting Residues | Electrostatic Interacting Residues |
|---|---|---|---|---|---|
| hD4R | Dopamine a (agonist) | −6.1 | Asp115(Salt bridge), Thr120, Ser196, Tyr438 | Cys119(Pi-Alkyl), Val116(Pi-Alkyl), Phe411(Pi-Pi T-shaped) | |
| Nemonapride a (agonist) | −13.08 | Asp115(Salt bridge), Tyr438, Ser196 | Val116 (Pi-Sigma), Phe91 (Pi-Pi T-shaped), Phe410 (Pi-Pi T-shaped), Leu90 (Amide-Pi Stacked), Val193 (Alkyl), Leu111 (Pi-Alkyl) | ||
| Clozapine a (antagonist) | −10.14 | Asp115(Salt bridge) | Leu187(Pi-Sigma), Phe410(Pi-PI T-shaped), His414(Pi-Pi T-shaped), Val116(Alkyl, Pi-Alkyl), Val193(Pi-Alkyl) | ||
| 1 | −9.67 | Ser196, Leu187, Val430, Thr434 | Val116 (Pi-Sigma, Pi-Alkyl), Leu187 (Pi-Sigma), Thr434 (Pi-Sigma), Phe411 (Pi-Pi T-shaped), His414 (PI-Pi T-shaped), Phe410 (Pi-Pi T-shaped), Met112 (Alkyl), Cys185 (Alkyl), Cys119 (Alkyl, Pi-Alkyl), Arg186 (Pi-Alkyl) | Asp115 (Pi-Anion) | |
| 2 | −10.34 | Ser197, Thr434, Asp115, Tyr438 | Val193 (Pi-sigma), His414 (Pi-Pi Stacked, Pi-Pi T-shaped), Met112 (Alkyl), Leu187 (Alkyl, Pi-Alkyl), Phe91 (Pi-Alkyl), Arg186 (Pi-Alkyl),Val116 (Pi-Alkyl) | Asp115 (Pi-Anion) | |
| 3 | −12.42 | Leu187, Asp115, Ser196 | Leu187 (Pi-Sigma, Alkyl, Pi-Alkyl), Phe410 (Pi-Pi T-shaped), His414 (Pi-Pi T-shaped, Pi-Alkyl), Val193 (Alkyl, Pi-Alkyl), Val116 (Pi-Alkyl) | Asp115 (Pi-Anion) |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Paudel, P.; Park, S.E.; Seong, S.H.; Jung, H.A.; Choi, J.S. Novel Diels–Alder Type Adducts from Morus alba Root Bark Targeting Human Monoamine Oxidase and Dopaminergic Receptors for the Management of Neurodegenerative Diseases. Int. J. Mol. Sci. 2019, 20, 6232. https://doi.org/10.3390/ijms20246232
Paudel P, Park SE, Seong SH, Jung HA, Choi JS. Novel Diels–Alder Type Adducts from Morus alba Root Bark Targeting Human Monoamine Oxidase and Dopaminergic Receptors for the Management of Neurodegenerative Diseases. International Journal of Molecular Sciences. 2019; 20(24):6232. https://doi.org/10.3390/ijms20246232
Chicago/Turabian StylePaudel, Pradeep, Se Eun Park, Su Hui Seong, Hyun Ah Jung, and Jae Sue Choi. 2019. "Novel Diels–Alder Type Adducts from Morus alba Root Bark Targeting Human Monoamine Oxidase and Dopaminergic Receptors for the Management of Neurodegenerative Diseases" International Journal of Molecular Sciences 20, no. 24: 6232. https://doi.org/10.3390/ijms20246232