Endogenous Retroviruses Activity as a Molecular Signature of Neurodevelopmental Disorders


 , , ,
, , , 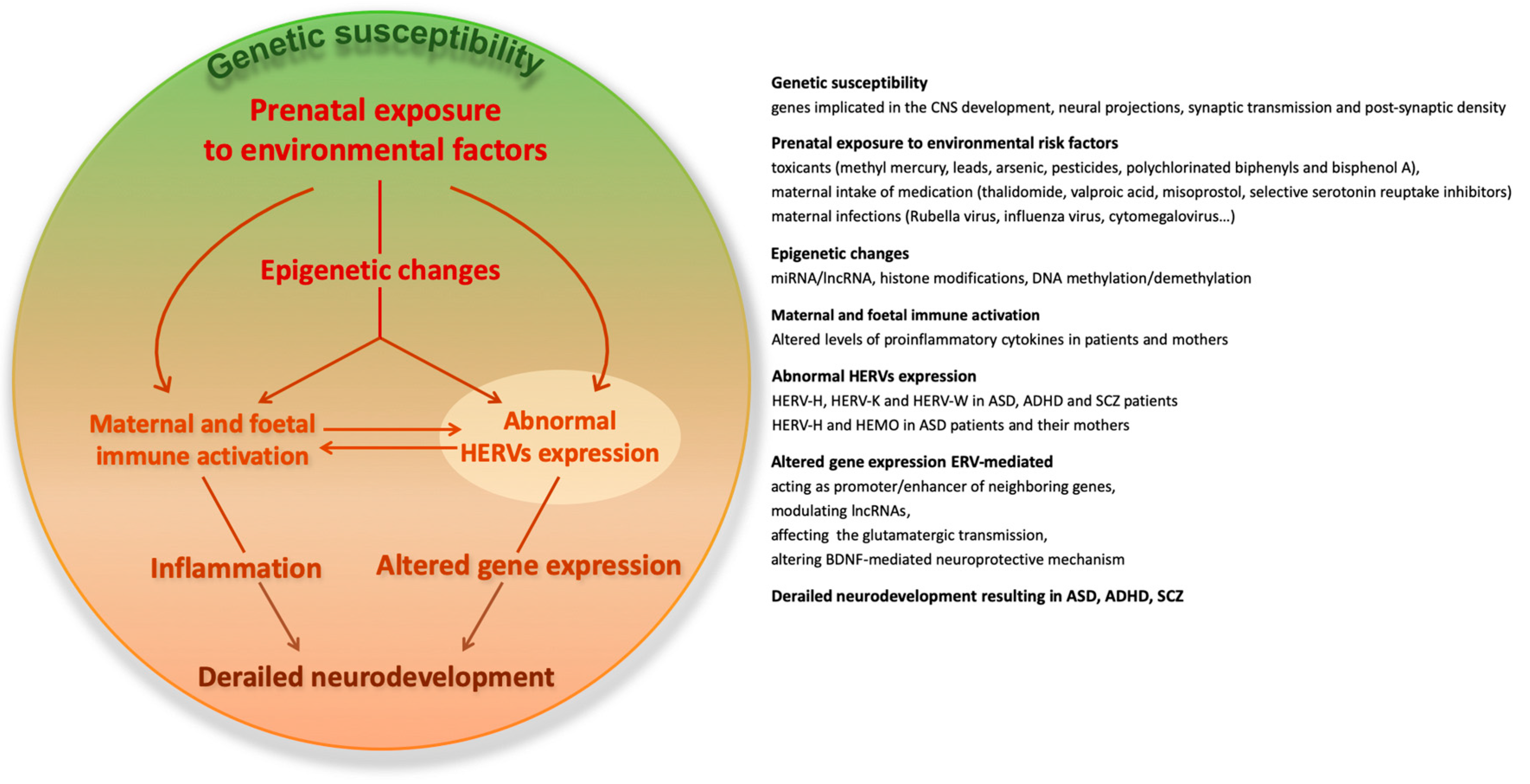{kind=link}
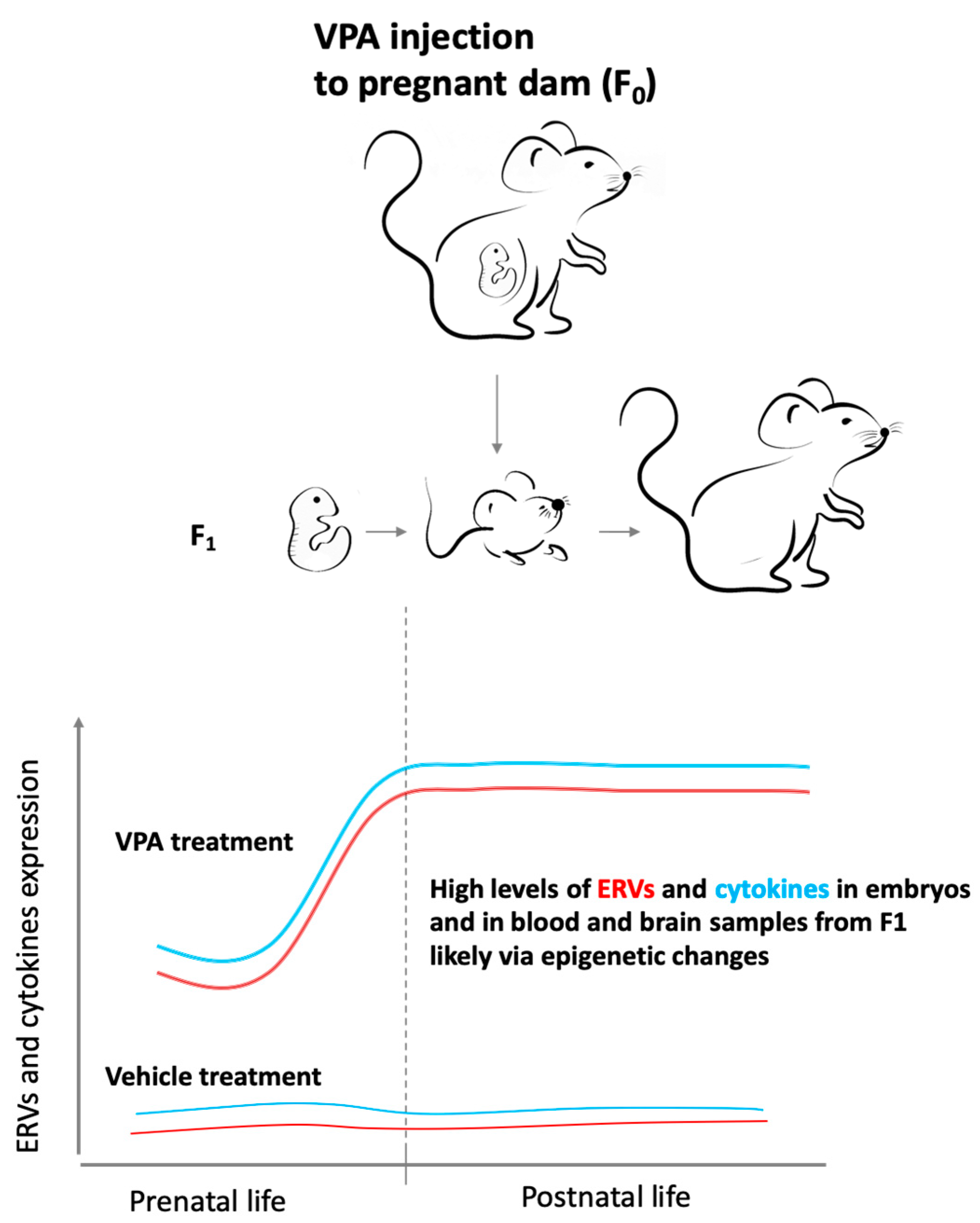{kind=link}
{kind=link}
Abstract
:1. Human Endogenous Retroviruses
1.1. The Origin of the Human Endogenous Retroviruses
1.2. Physiological Functions of HERVs
1.3. HERVs Responsiveness to Environmental Stimuli and their Deregulation in Human Diseases
2. Neurodevelopmental Disorders
2.1. Main Clinical and Genetic Features of Neurodevelopmental Disorders
2.2. Drugs and Infections Exposure in Sensitive Time-Windows of the Pregnancy as Risk Factors for NDDs
2.3. Inflammatory Mechanisms in NDDs
3. HERVs in Neurodevelopmental Disorders
3.1. Evidence from Human Studies on the Involvement of HERVs in Neurodevelopmental Disorders
3.2. Mechanisms by Which HERVsCouldDerail Neurodevelopment
4. Endogenous Retroviruses Activity in Mouse Models of Autism
4.1. Mouse Endogenous Retroviruses
4.2. Animal Models of Autism
4.3. Evidence of Abnormal ERVs Activity in Mouse Models of Autism
4.4. Recent Findings on the Transgenerational Inheritance of the Abnormal ERVs Expression in a Mouse Model of Autism: the Conceivable Underpinning Mechanisms
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| HERVs | Human Endogenous Retroviruses |
| LTRs | Long Terminal Repeats |
| LINEs | Long interspersed nuclear elements |
| RT | Reverse transcriptase |
| ncRNAs | Non-coding RNA |
| HEMO | Human endogenous MER34 (medium reiteration-frequency-family-34) ORF |
| HSV | Herpesviruses |
| HERV-W/MSRV | Human endogenous retrovirus W/ Multiple Sclerosis-associated retrovirus |
| EBV | Epstein Barr virus |
| HHV-6 | Human herpesvirus 6 |
| MS | Multiple sclerosis |
| HTLV-1 | Human T-lymphotropic virus-1 |
| HIV-1 | Human immunodeficiency virus-1 |
| AIDS | Acquired immune deficiency syndrome |
| PBMCs | Peripheral blood mononuclear cells |
| LPS | Lipopolysaccharide |
| IFN-γ | Interferon-γ |
| ALS | Amyotrophic lateral sclerosis |
| TNF-α | Tumor necrosis factor-alpha |
| IL-1α | Interleukin-1alpha |
| IL-1β | Interleukin-1β |
| PHA | Phytohemagglutinin |
| PMA | Horbol-12-myristate-13-acetate |
| HDACi | Inhibitors of the histone deacetylase |
| VPA | Valproic acid |
| UV | Ultraviolet irradiations |
| SLE | Systemic lupus erythematosus |
| RA | Rheumatoid arthritis |
| TLR4 | Toll-Like Receptor 4 |
| ASD | Autism spectrum disorders |
| ADHD | Attention deficit hyperactivity disorder |
| SCZ | Schizophrenia |
| NDDs | Neurodevelopmental disorders |
| BD | Bipolar disease |
| CNV | Copy number variation |
| HDAC1 | Inhibitor of histone deacetylase of class I |
| HDAC2 | Inhibitor of histone deacetylase of class II |
| MIA | Maternal immune activation |
| MPH | Methylphenidate |
| BDNF | Brain-derived neurotrophic factor |
| NTRK2 | Neurotrophic tyrosine kinase receptor type 2 |
| DRD3 | Dopamine receptor D3 |
| PRRs | Pattern recognition receptors |
| PAMPs | Pathogen associated molecular patterns |
| ERVs | Endogenous retroviruses |
| IAP | Intracisternal A-particle |
| ETn | Early transposon |
| MusD | Mouse endogenous proviruses |
| BTBR | BTBR T+tf/J inbred |
| F1 | First generation |
| F2 | Second generation |
| F3 | Third generation |
References
- Lander, E.S.; Linton, L.M.; Birren, B.; Nusbaum, C.; Zody, M.C.; Baldwin, J.; Devon, K.; Dewar, K.; Doyle, M.; FitzHugh, W.; et al. International Human Genome Sequencing Consortium. Initial sequencing and analysis of the human genome. Nature 2001, 409, 860–921. [Google Scholar] [CrossRef] [PubMed]
- Feschotte, C.; Gilbert, C. Endogenous viruses: Insights into viral evolution and impact on host biology. Nat. Rev. Genet. 2012, 13, 283–296. [Google Scholar] [CrossRef] [PubMed]
- Kazazian, H.H., Jr.; Moran, J.V. Mobile DNA in Health and Disease. N. Engl. J. Med. 2017, 377, 361–370. [Google Scholar] [CrossRef] [PubMed]
- Campos-Sánchez, R.; Kapusta, A.; Feschotte, C.; Chiaromonte, F.; Makova, K.D. Genomic landscape of human, bat, and ex vivo DNA transposon integrations. Mol. Biol. Evol. 2014, 31, 1816–1832. [Google Scholar] [CrossRef] [PubMed]
- Hancks, D.C.; Kazazian, H.H., Jr. Roles for retrotransposon insertions in human disease. Mob. DNA 2016, 7, 9. [Google Scholar] [CrossRef]
- Kassiotis, G.; Stoye, J.P. Making a virtue of necessity: The pleiotropic role of human endogenous retroviruses in cancer. Philos. Trans. R. Soc. Lond B Biol. Sci. 2017, 372, 20160277. [Google Scholar] [CrossRef]
- Ohshima, K. RNA-Mediated Gene Duplication and Retroposons: Retrogenes, LINEs, SINEs, and Sequence Specificity. Int. J. Evol. Biol. 2013, 2013, 424726. [Google Scholar] [CrossRef]
- Burns, K.H.; Boeke, J.D. Human transposon tectonics. Cell 2012, 149, 740–752. [Google Scholar] [CrossRef]
- Belshaw, R.; Pereira, V.; Katzourakis, A.; Talbot, G.; Paces, J.; Burt, A.; Tristem, M. Long-term reinfection of the human genome by endogenous retroviruses. Proc. Natl. Acad. Sci. USA 2004, 101, 4894–4899. [Google Scholar] [CrossRef]
- Bannert, N.; Kurth, R. The evolutionary dynamics of human endogenous retroviral families. Annu. Rev. Genomics Hum. Genet. 2006, 7, 149–173. [Google Scholar] [CrossRef]
- Bock, M.; Stoye, J.P. Endogenous retroviruses and the human germline. Curr. Opin. Genet. Dev. 2000, 10, 651–655. [Google Scholar] [CrossRef]
- Consortium, M.G.S. Initial sequencing and comparative analysis of the mouse genome. Nature 2002, 420, 520–562. [Google Scholar] [CrossRef] [PubMed]
- Herniou, E.; Martin, J.; Miller, K.; Cook, J.; Wilkinson, M.; Tristem, M. Retroviral diversity and distribution in vertebrates. J. Virol. 1998, 72, 5955–5966. [Google Scholar] [PubMed]
- Jern, P.; Sperber, G.O.; Blomberg, J. Divergent patterns of recent retroviral integrations in the human and chimpanzee genomes: Probable transmissions between other primates and chimpanzees. J. Virol. 2006, 80, 1367–1375. [Google Scholar] [CrossRef] [PubMed]
- Nellåker, C.; Keane, T.M.; Yalcin, B.; Wong, K.; Agam, A.; Belgard, T.G.; Flint, J.; Adams, D.J.; Frankel, W.N.; Ponting, C.P. The genomic landscape shaped by selection on transposable elements across 18 mouse strains. Genome Biol. 2012, 13, R45. [Google Scholar] [CrossRef]
- Paces, J.; Pavlícek, A.; Paces, V. HERVd: Database of human endogenous retroviruses. Nucleic Acids Res. 2002, 30, 205–206. [Google Scholar] [CrossRef]
- Mayer, J.; Blomberg, J.; Seal, R.L. A revised nomenclature for transcribed human endogenous retroviral loci. Mob. DNA 2011, 2, 7. [Google Scholar] [CrossRef]
- Tristem, M. Identification and characterization of novel human endogenous retrovirus families by phylogenetic screening of the human genome mapping project database. J. Virol. 2000, 74, 3715–3730. [Google Scholar] [CrossRef]
- Bénit, L.; Dessen, P.; Heidmann, T. Identification, phylogeny, and evolution of retroviral elements based on their envelope genes. J. Virol. 2001, 75, 11709–17719. [Google Scholar] [CrossRef]
- Thomas, J.; Perron, H.; Feschotte, C. Variation in proviral content among human genomes mediated by LTR recombination. Mob. DNA 2018, 9, 36. [Google Scholar] [CrossRef]
- Belshaw, R.; Katzourakis, A.; Paces, J.; Burt, A.; Tristem, M. High copy number in human endogenous retrovirus families is associated with copying mechanisms in addition to reinfection. Mol. Biol. Evol. 2005, 22, 814–817. [Google Scholar] [CrossRef] [PubMed]
- Wildschutte, J.H.; Williams, Z.H.; Montesion, M.; Subramanian, R.P.; Kidd, J.M.; Coffin, J.M. Discovery of unfixed endogenous retrovirus insertions in diverse human populations. Proc. Natl. Acad. Sci. USA 2016, 113, E2326–E2334. [Google Scholar] [CrossRef] [PubMed]
- Marchi, E.; Kanapin, A.; Magiorkinis, G.; Belshaw, R. Unfixed endogenous retroviral insertions in the human population. J. Virol. 2014, 88, 9529–9537. [Google Scholar] [CrossRef] [PubMed]
- Coffin, J.M.; Hughes, S.H.; Varmus, H.E. The Interactions of Retroviruses and their Hosts. In Retroviruses; Coffin, J.M., Hughes, S.H., Varmus, H.E., Eds.; Cold Spring Harbor Laboratory Press: Cold Spring Harbor, NY, USA, 1997. [Google Scholar]
- Dewannieux, M.; Heidmann, T. Endogenous retroviruses: Acquisition, amplification and taming of genome invaders. Curr. Opin. Virol. 2013, 3, 646–656. [Google Scholar] [CrossRef] [PubMed]
- Montesion, M.; Williams, Z.H.; Subramanian, R.P.; Kuperwasser, C.; Coffin, J.M. Promoter expression of HERV-K (HML-2) provirus-derived sequences is related to LTR sequence variation and polymorphic transcription factor binding sites. Retrovirology 2018, 15, 57. [Google Scholar] [CrossRef] [PubMed]
- Jern, P.; Coffin, J.M. Effects of retroviruses on host genome function. Annu. Rev. Genet. 2008, 42, 709–732. [Google Scholar] [CrossRef]
- Rebollo, R.; Romanish, M.T.; Mager, D.L. Transposable elements: An abundant and natural source of regulatory sequences for host genes. Annu. Rev. Genet. 2012, 46, 21–42. [Google Scholar] [CrossRef]
- Frank, J.A.; Feschotte, C. Co-option of endogenous viral sequences for host cell function. Curr. Opin. Virol. 2017, 25, 81–89. [Google Scholar] [CrossRef]
- Chuong, E.B.; Elde, N.C.; Feschotte, C. Regulatory activities of transposable elements: From conflicts to benefits. Nat. Rev. Genet. 2017, 18, 71–86. [Google Scholar] [CrossRef]
- Leboyer, M.; Tamouza, R.; Charron, D.; Faucard, R.; Perron, H. Human endogenous retrovirus type W (HERV-W) in schizophrenia: A new avenue of research at the gene-environment interface. World J. Biol. Psychiatry 2013, 14, 80–90. [Google Scholar] [CrossRef]
- Matteucci, C.; Balestrieri, E.; Argaw-Denboba, A.; Sinibaldi-Vallebona, P. Human endogenous retroviruses role in cancer cell stemness. Semin. Cancer Biol. 2018, 53, 17–30. [Google Scholar] [CrossRef] [PubMed]
- Küry, P.; Nath, A.; Créange, A.; Dolei, A.; Marche, P.; Gold, J.; Giovannoni, G.; Hartung, H.P.; Perron, H. Human Endogenous Retroviruses in Neurological Diseases. Trends Mol. Med. 2018, 24, 379–394. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ehlhardt, S.; Seifert, M.; Schneider, J.; Ojak, A.; Zang, K.D.; Mehraein, Y. Human endogenous retrovirus HERV-K(HML-2) Rec expression and transcriptional activities in normal and rheumatoid arthritis synovia. J. Rheumatol. 2006, 33, 16–23. [Google Scholar]
- Schmitt, K.; Heyne, K.; Roemer, K.; Meese, E.; Mayer, J. HERV-K(HML-2) rec and np9 transcripts not restricted to disease but present in many normal human tissues. Mob. DNA 2015, 6, 4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Balestrieri, E.; Pica, F.; Matteucci, C.; Zenobi, R.; Sorrentino, R.; Argaw-Denboba, A.; Cipriani, C.; Bucci, I.; Sinibaldi-Vallebona, P. Transcriptional activity of human endogenous retroviruses in human peripheral blood mononuclear cells. Biomed. Res. Int. 2015, 2015, 164529. [Google Scholar] [CrossRef]
- Buzdin, A.; Kovalskaya-Alexandrova, E.; Gogvadze, E.; Sverdlov, E. At least 50% of human-specific HERV-K (HML-2) long terminal repeats serve in vivo as active promoters for host nonrepetitive DNA transcription. J. Virol 2006, 80, 10752–10762. [Google Scholar] [CrossRef] [Green Version]
- Chuong, E.B.; Rumi, M.A.; Soares, M.J.; Baker, J.C. Endogenous retroviruses function as species-specific enhancer elements in the placenta. Nat. Genet. 2013, 45, 325–329. [Google Scholar] [CrossRef] [Green Version]
- Suntsova, M.; Garazha, A.; Ivanova, A.; Kaminsky, D.; Zhavoronkov, A.; Buzdin, A. Molecular functions of human endogenous retroviruses in health and disease. Cell. Mol. Life Sci. 2015, 72, 3653–3675. [Google Scholar] [CrossRef]
- Schumann, G.G.; Gogvadze, E.V.; Osanai-Futahashi, M.; Kuroki, A.; Münk, C.; Fujiwara, H.; Ivics, Z.; Buzdin, A.A. Unique functions of repetitive transcriptomes. Int. Rev. Cell Mol. Biol. 2010, 285, 115–188. [Google Scholar] [CrossRef]
- Wang, J.; Xie, G.; Singh, M.; Ghanbarian, A.T.; Raskó, T.; Szvetnik, A.; Cai, H.; Besser, D.; Prigione, A.; Fuchs, N.V.; et al. Primate-specific endogenous retrovirus-driven transcription defines naive-like stem cells. Nature 2014, 516, 405–409. [Google Scholar] [CrossRef] [Green Version]
- Hughes, J.F.; Coffin, J.M. Human endogenous retrovirus K solo-LTR formation and insertional polymorphisms: Implications for human and viral evolution. Proc. Natl. Acad. Sci. USA 2004, 101, 1668–1672. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trombetta, B.; Fantini, G.; D’Atanasio, E.; Sellitto, D.; Cruciani, F. Evidence of extensive non-allelic gene conversion among LTR elements in the human genome. Sci. Rep. 2016, 6, 28710. [Google Scholar] [CrossRef] [PubMed]
- Dunn, C.A.; Medstrand, P.; Mager, D.L. An endogenous retroviral long terminal repeat is the dominant promoter for human beta1,3-galactosyltransferase 5 in the colon. Proc. Natl. Acad. Sci. USA 2003, 100, 12841–12846. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bièche, I.; Laurent, A.; Laurendeau, I.; Duret, L.; Giovangrandi, Y.; Frendo, J.L.; Olivi, M.; Fausser, J.L.; Evain-Brion, D.; Vidaud, M. Placenta-specific INSL4 expression is mediated by a human endogenous retrovirus element. Biol. Reprod. 2003, 68, 1422–1429. [Google Scholar] [CrossRef] [PubMed]
- Ting, C.N.; Rosenberg, M.P.; Snow, C.M.; Samuelson, L.C.; Meisler, M.H. Endogenous retroviral sequences are required for tissue-specific expression of a human salivary amylase gene. Genes Dev. 1992, 6, 1457–1465. [Google Scholar] [CrossRef] [Green Version]
- Grow, E.J.; Flynn, R.A.; Chavez, S.L.; Bayless, N.L.; Wossidlo, M.; Wesche, D.J.; Martin, L.; Ware, C.B.; Blish, C.A.; Chang, H.Y.; et al. Intrinsic retroviral reactivation in human preimplantation embryos and pluripotent cells. Nature 2015, 522, 221–225. [Google Scholar] [CrossRef] [Green Version]
- Glinsky, G.V. Transposable Elements and DNA Methylation Create in Embryonic Stem Cells Human-Specific Regulatory Sequences Associated with Distal Enhancers and Noncoding RNAs. Genome Biol. Evol. 2015, 7, 1432–1454. [Google Scholar] [CrossRef] [Green Version]
- Gemmell, P.; Hein, J.; Katzourakis, A. Phylogenetic Analysis Reveals That ERVs “Die Young” but HERV-H Is Unusually Conserved. PLoSComput. Biol. 2016, 12, e1004964. [Google Scholar] [CrossRef]
- Heidmann, O.; Béguin, A.; Paternina, J.; Berthier, R.; Deloger, M.; Bawa, O.; Heidmann, T. HEMO, an ancestral endogenous retroviral envelope protein shed in the blood of pregnant women and expressed in pluripotent stem cells and tumors. Proc. Natl. Acad. Sci. USA 2017, 114, E6642–E6651. [Google Scholar] [CrossRef] [Green Version]
- Mi, S.; Lee, X.; Li, X.; Veldman, G.M.; Finnerty, H.; Racie, L.; LaVallie, E.; Tang, X.Y.; Edouard, P.; Howes, S.; et al. Syncytin is a captive retroviral envelope protein involved in human placental morphogenesis. Nature 2000, 403, 785–789. [Google Scholar] [CrossRef]
- Chuong, E.B.; Elde, N.C.; Feschotte, C. Regulatory evolution of innate immunity through co-option of endogenous retroviruses. Science 2016, 351, 1083–1087. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blaise, S.; de Parseval, N.; Bénit, L.; Heidmann, T. Genomewide screening for fusogenic human endogenous retrovirus envelopes identifies syncytin 2, a gene conserved on primate evolution. Proc. Natl. Acad. Sci. USA 2003, 100, 13013–13018. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frendo, J.L.; Olivier, D.; Cheynet, V.; Blond, J.L.; Bouton, O.; Vidaud, M.; Rabreau, M.; Evain-Brion, D.; Mallet, F. Direct involvement of HERV-W Env glycoprotein in human trophoblast cell fusion and differentiation. Mol. Cell. Biol. 2003, 23, 3566–3574. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dunlap, K.A.; Palmarini, M.; Varela, M.; Burghardt, R.C.; Hayashi, K.; Farmer, J.L.; Spencer, T.E. Endogenous retroviruses regulate periimplantation placental growth and differentiation. Proc. Natl. Acad. Sci. USA 2006, 103, 14390–14395. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Engel, M.E.; Hiebert, S.W. The enemy within: Dormant retroviruses awaken. Nat. Med. 2010, 16, 517–518. [Google Scholar] [CrossRef]
- Gröger, V.; Cynis, H. Human Endogenous Retroviruses and Their Putative Role in the Development of Autoimmune Disorders Such as Multiple Sclerosis. Front. Microbiol. 2018, 9, 265. [Google Scholar] [CrossRef]
- Grandi, N.; Tramontano, E. HERV Envelope Proteins: Physiological Role and Pathogenic Potential in Cancer and Autoimmunity. Front. Microbiol. 2018, 9, 462. [Google Scholar] [CrossRef]
- Chen, J.; Foroozesh, M.; Qin, Z. Transactivation of human endogenous retroviruses by tumor viruses and their functions in virus-associated malignancies. Oncogenesis 2019, 8, 6. [Google Scholar] [CrossRef] [Green Version]
- Nellåker, C.; Yao, Y.; Jones-Brando, L.; Mallet, F.; Yolken, R.H.; Karlsson, H. Transactivation of elements in the human endogenous retrovirus W family by viral infection. Retrovirology 2006, 3, 44. [Google Scholar] [CrossRef]
- Ruprecht, K.; Obojes, K.; Wengel, V.; Gronen, F.; Kim, K.S.; Perron, H.; Schneider-Schaulies, J.; Rieckmann, P. Regulation of human endogenous retrovirus W protein expression by herpes simplex virus type 1: Implications for multiple sclerosis. J. Neurovirol. 2006, 12, 65–71. [Google Scholar] [CrossRef]
- Mameli, G.; Poddighe, L.; Mei, A.; Uleri, E.; Sotgiu, S.; Serra, C.; Manetti, R.; Dolei, A. Expression and activation by Epstein Barr virus of human endogenous Retroviruses-W in blood cells and astrocytes: Inference for multiple sclerosis. PLoS ONE 2012, 7, e44991. [Google Scholar] [CrossRef] [PubMed]
- Hsiao, F.C.; Lin, M.; Tai, A.; Chen, G.; Huber, B.T. Cutting edge: Epstein-Barr virus transactivates the HERV-K18 superantigen by docking to the human complement receptor 2 (CD21) on primary B cells. J. Immunol. 2006, 177, 2056–2060. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leung, A.; Trac, C.; Kato, H.; Costello, K.R.; Chen, Z.; Natarajan, R.; Schones, D.E. LTRs activated by Epstein-Barr virus-induced transformation of B cells alter the transcriptome. Genome Res. 2018, 28, 1791–1798. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dai, L.; Del Valle, L.; Miley, W.; Whitby, D.; Ochoa, A.C.; Flemington, E.K.; Qin, Z. Transactivation of human endogenous retrovirus K (HERV-K) by KSHV promotes Kaposi’s sarcoma development. Oncogene 2018, 37, 4534–4545. [Google Scholar] [CrossRef] [PubMed]
- Charvet, B.; Reynaud, J.M.; Gourru-Lesimple, G.; Perron, H.; Marche, P.N.; Horvat, B. Induction of Proinflammatory Multiple Sclerosis-Associated Retrovirus Envelope Protein by Human Herpesvirus-6A and CD46 Receptor Engagement. Front. Immunol. 2018, 9, 2803. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Jovel, J.; Halloran, B.; Wine, E.; Patterson, J.; Ford, G.; O’Keefe, S.; Meng, B.; Song, D.; Zhang, Y.; et al. Metagenomic analysis of microbiome in colon tissue from subjects with inflammatory bowel diseases reveals interplay of viruses and bacteria. Inflamm. Bowel Dis. 2015, 21, 1419–1427. [Google Scholar] [CrossRef]
- Liu, C.; Liu, L.; Wang, X.; Liu, Y.; Wang, M.; Zhu, F. HBV X Protein induces overexpression of HERV-W env through NF-κB in HepG2 cells. Virus Genes 2017, 53, 797–806. [Google Scholar] [CrossRef]
- Toufaily, C.; Landry, S.; Leib-Mosch, C.; Rassart, E.; Barbeau, B. Activation of LTRs from different human endogenous retrovirus (HERV) families by the HTLV-1 tax protein and T-cell activators. Viruses 2011, 3, 2146–2159. [Google Scholar] [CrossRef]
- Contreras-Galindo, R.; Kaplan, M.H.; Markovitz, D.M.; Lorenzo, E.; Yamamura, Y. Detection of HERV-K(HML-2) viral RNA in plasma of HIV type 1-infected individuals. AIDS Res. Hum. Retrovir. 2006, 22, 979–984. [Google Scholar] [CrossRef] [Green Version]
- Young, G.R.; Terry, S.N.; Manganaro, L.; Cuesta-Dominguez, A.; Deikus, G.; Bernal-Rubio, D.; Campisi, L.; Fernandez-Sesma, A.; Sebra, R.; Simon, V.; et al. HIV-1 Infection of Primary CD4+ T Cells Regulates the Expression of Specific Human Endogenous Retrovirus HERV-K (HML-2) Elements. J. Virol. 2017, 92, e01507-17. [Google Scholar] [CrossRef] [Green Version]
- Gonzalez-Hernandez, M.J.; Cavalcoli, J.D.; Sartor, M.A.; Contreras-Galindo, R.; Meng, F.; Dai, M.; Dube, D.; Saha, A.K.; Gitlin, S.D.; Omenn, G.S.; et al. Regulation of the human endogenous retrovirus K (HML-2) transcriptome by the HIV-1 Tat protein. J. Virol. 2014, 88, 8924–8935. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Uleri, E.; Mei, A.; Mameli, G.; Poddighe, L.; Serra, C.; Dolei, A. HIV Tat acts on endogenous retroviruses of the W family and this occurs via Toll-like receptor 4: Inference for neuroAIDS. AIDS 2014, 28, 2659–2670. [Google Scholar] [CrossRef] [PubMed]
- Li, F.; Nellåker, C.; Sabunciyan, S.; Yolken, R.H.; Jones-Brando, L.; Johansson, A.S.; Owe-Larsson, B.; Karlsson, H. Transcriptional derepression of the ERVWE1 locus following influenza A virus infection. J. Virol. 2014, 88, 4328–4337. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frank, O.; Jones-Brando, L.; Leib-Mosch, C.; Yolken, R.; Seifarth, W. Altered transcriptional activity of human endogenous retroviruses in neuroepithelial cells after infection with Toxoplasma gondii. J. Infect. Dis. 2006, 194, 1447–1449. [Google Scholar] [CrossRef] [Green Version]
- Young, G.R.; Mavrommatis, B.; Kassiotis, G. Microarray analysis reveals global modulation of endogenous retroelement transcription by microbes. Retrovirology 2014, 11, 59. [Google Scholar] [CrossRef] [Green Version]
- Mommert, M.; Tabone, O.; Oriol, G.; Cerrato, E.; Guichard, A.; Naville, M.; Fournier, P.; Volff, J.N.; Pachot, A.; Monneret, G.; et al. LTR-retrotransposon transcriptome modulation in response to endotoxin-induced stress in PBMCs. BMC Genomics 2018, 19, 522. [Google Scholar] [CrossRef]
- Manghera, M.; Ferguson-Parry, J.; Lin, R.; Douville, R.N. NF-κB and IRF1 Induce Endogenous Retrovirus K Expression via Interferon-Stimulated Response Elements in Its 5’ Long Terminal Repeat. J. Virol. 2016, 90, 9338–9349. [Google Scholar] [CrossRef] [Green Version]
- Katsumata, K.; Ikeda, H.; Sato, M.; Ishizu, A.; Kawarada, Y.; Kato, H.; Wakisaka, A.; Koike, T.; Yoshiki, T. Cytokine regulation of env gene expression of human endogenous Retrovirus-R in human vascular endothelial cells. Clin. Immunol. 1999, 93, 75–80. [Google Scholar] [CrossRef]
- Mueller, O.; Moore, D.W.; Giovannucci, J.; Etter, A.R.; Peterson, E.M.; Mudge, A.; Liu, Y. Expression of Human Endogenous Retroviruses in Peripheral Leukocytes During the Menstrual Cycle Suggests Coordinated Hormonal Regulation. AIDS Res. Hum. Retrovir. 2018, 34, 909–911. [Google Scholar] [CrossRef]
- Wang-Johanning, F.; Frost, A.R.; Jian, B.; Epp, L.; Lu, D.W.; Johanning, G.L. Quantitation of HERV-K env gene expression and splicing in human breast cancer. Oncogene 2003, 22, 1528–1535. [Google Scholar] [CrossRef] [Green Version]
- Buslei, R.; Strissel, P.L.; Henke, C.; Schey, R.; Lang, N.; Ruebner, M.; Stolt, C.C.; Fabry, B.; Buchfelder, M.; Strick, R. Activation and regulation of endogenous retroviral genes in the human pituitary gland and related endocrine tumours. Neuropathol. Appl. Neurobiol. 2015, 41, 180–200. [Google Scholar] [CrossRef] [PubMed]
- Ogasawara, H.; Naito, T.; Kaneko, H.; Hishikawa, T.; Sekigawa, I.; Hashimoto, H.; Kaneko, Y.; Yamamoto, N.; Maruyama, N.; Yamamoto, N. Quantitative analyses of messenger RNA of human endogenous retrovirus in patients with systemic lupus erythematosus. J. Rheumatol. 2001, 28, 533–538. [Google Scholar] [PubMed]
- Kelleher, C.A.; Wilkinson, D.A.; Freeman, J.D.; Mager, D.L.; Gelfand, E.W. Expression of novel-transposon-containing mRNAs in human T cells. J. Gen. Virol. 1996, 77, 1101–1110. [Google Scholar] [CrossRef] [PubMed]
- Johnston, J.B.; Silva, C.; Holden, J.; Warren, K.G.; Clark, A.W.; Power, C. Monocyte activation and differentiation augment human endogenous retrovirus expression: Implications for inflammatory brain diseases. Ann. Neurol. 2001, 50, 434–442. [Google Scholar] [CrossRef]
- Bollati, V.; Favero, C.; Albetti, B.; Tarantini, L.; Moroni, A.; Byun, H.M.; Motta, V.; Conti, D.M.; Tirelli, A.S.; Vigna, L.; et al. Nutrients intake is associated with DNA methylation of candidate inflammatory genes in a population of obese subjects. Nutrients 2014, 6, 4625–4639. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, M.; Ohtani, H.; Zhou, W.; Ørskov, A.D.; Charlet, J.; Zhang, Y.W.; Shen, H.; Baylin, S.B.; Liang, G.; Grønbæk, K.; et al. Vitamin C increases viral mimicry induced by 5-aza-2′-deoxycytidine. Proc. Natl. Acad. Sci. USA 2016, 113, 10238–10244. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Serafino, A.; Balestrieri, E.; Pierimarchi, P.; Matteucci, C.; Moroni, G.; Oricchio, E.; Rasi, G.; Mastino, A.; Spadafora, C.; Garaci, E.; et al. The activation of human endogenous retrovirus K (HERV-K) is implicated in melanoma cell malignant transformation. Exp. Cell. Res. 2009, 315, 849–862. [Google Scholar] [CrossRef]
- Argaw-Denboba, A.; Balestrieri, E.; Serafino, A.; Cipriani, C.; Bucci, I.; Sorrentino, R.; Sciamanna, I.; Gambacurta, A.; Sinibaldi-Vallebona, P.; Matteucci, C. HERV-K activation is strictly required to sustain CD133+ melanoma cells with stemness features. J. Exp. Clin. Cancer Res. 2017, 36, 20. [Google Scholar] [CrossRef] [Green Version]
- Balestrieri, E.; Argaw-Denboba, A.; Gambacurta, A.; Cipriani, C.; Bei, R.; Serafino, A.; Sinibaldi-Vallebona, P.; Matteucci, C. Human Endogenous Retrovirus K in the Crosstalk Between Cancer Cells Microenvironment and Plasticity: A New Perspective for Combination Therapy. Front. Microbiol. 2018, 9, 1448. [Google Scholar] [CrossRef]
- Díaz-Carballo, D.; Acikelli, A.H.; Klein, J.; Jastrow, H.; Dammann, P.; Wyganowski, T.; Guemues, C.; Gustmann, S.; Bardenheuer, W.; Malak, S.; et al. Therapeutic potential of antiviral drugs targeting chemorefractory colorectal adenocarcinoma cells overexpressing endogenous retroviral elements. J. Exp. Clin. Cancer Res. 2015, 34, 81. [Google Scholar] [CrossRef] [Green Version]
- Diem, O.; Schäffner, M.; Seifarth, W.; Leib-Mösch, C. Influence of antipsychotic drugs on human endogenous retrovirus (HERV) transcription in brain cells. PLoS ONE 2012, 7, e30054. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- White, C.H.; Beliakova-Bethell, N.; Lada, S.M.; Breen, M.S.; Hurst, T.P.; Spina, C.A.; Richman, D.D.; Frater, J.; Magiorkinis, G.; Woelk, C.H. Transcriptional Modulation of Human Endogenous Retroviruses in Primary CD4+ T Cells Following Vorinostat Treatment. Front. Immunol. 2018, 9, 603. [Google Scholar] [CrossRef] [PubMed]
- Conti, A.; Rota, F.; Ragni, E.; Favero, C.; Motta, V.; Lazzari, L.; Bollati, V.; Fustinoni, S.; Dieci, G. Hydroquinoneinduces DNA hypomethylation-independentoverexpression of retroelements in human leukemia and hematopoieticstemcells. Biochem. Biophys. Res. Commun. 2016, 474, 691–695. [Google Scholar] [CrossRef] [PubMed]
- Schanab, O.; Humer, J.; Gleiss, A.; Mikula, M.; Sturlan, S.; Grunt, S.; Okamoto, I.; Muster, T.; Pehamberger, H.; Waltenberger, A. Expression of human endogenous retrovirus K is stimulated by ultraviolet radiation in melanoma. Pigment Cell Melanoma Res. 2011, 24, 656–665. [Google Scholar] [CrossRef]
- Wu, Z.; Mei, X.; Zhao, D.; Sun, Y.; Song, J.; Pan, W.; Shi, W. DNA methylation modulates HERV-E expression in CD4+ T cells from systemic lupus erythematosus patients. J. Dermatol. Sci. 2015, 77, 110–116. [Google Scholar] [CrossRef]
- Sauter, M.; Schommer, S.; Kremmer, E.; Remberger, K.; Dölken, G.; Lemm, I.; Buck, M.; Best, B.; Neumann-Haefelin, D.; Mueller-Lantzsch, N. Human endogenous retrovirus K10: Expression of Gag protein and detection of antibodies in patients with seminomas. J. Virol. 1995, 69, 414–421. [Google Scholar]
- Wang-Johanning, F.; Li, M.; Esteva, F.J.; Hess, K.R.; Yin, B.; Rycaj, K.; Plummer, J.B.; Garza, J.G.; Ambs, S.; Johanning, G.L. Human endogenous retrovirus type K antibodies and mRNA as serum biomarkers of early-stage breast cancer. Int. J. Cancer 2014, 134, 587–595. [Google Scholar] [CrossRef]
- Giovinazzo, A.; Balestrieri, E.; Petrone, V.; Argaw-Denboba, A.; Cipriani, C.; Miele, M.T.; Grelli, S.; Sinibaldi-Vallebona, P.; Matteucci, C. The concomitant expression of human endogenous retroviruses and embryonic genes in cancer cells under microenvironmental changes is a potential target for antiretroviral drugs. Cancer Microenviron. 2019, 1–14. [Google Scholar] [CrossRef]
- Perron, H.; Dougier-Reynaud, H.L.; Lomparski, C.; Popa, I.; Firouzi, R.; Bertrand, J.B.; Marusic, S.; Portoukalian, J.; Jouvin-Marche, E.; Villiers, C.L.; et al. Human endogenous retrovirus protein activates innate immunity and promotes experimental allergic encephalomyelitis in mice. PLoS ONE 2013, 8, e80128. [Google Scholar] [CrossRef]
- Curtin, F.; Perron, H.; Kromminga, A.; Porchet, H.; Lang, A.B. Preclinical and early clinical development of GNbAC1, a humanized IgG4 monoclonal antibody targeting endogenous retroviral MSRV-Env protein. mAbs 2015, 7, 265–275. [Google Scholar] [CrossRef] [Green Version]
- Nelson, P.N.; Roden, D.; Nevill, A.; Freimanis, G.L.; Trela, M.; Ejtehadi, H.D.; Bowman, S.; Axford, J.; Veitch, A.M.; Tugnet, N.; et al. Rheumatoid arthritis is associated with IgG antibodies to human endogenous retrovirus gag matrix: A potential pathogenic mechanism of disease? J. Rheumatol. 2014, 41, 1952–1960. [Google Scholar] [CrossRef] [PubMed]
- Bendiksen, S.; Martinez-Zubiavrra, I.; Tümmler, C.; Knutsen, G.; Elvenes, J.; Olsen, E.; Olsen, R.; Moens, U. Human endogenous retrovirus W activity in cartilage of osteoarthritis patients. Biomed. Res. Int. 2014, 2014, 698609. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marguerat, S.; Wang, W.Y.; Todd, J.A.; Conrad, B. Association of human endogenous retrovirus K-18 polymorphisms with type 1 diabetes. Diabetes 2004, 53, 852–854. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Levet, S.; Medina, J.; Joanou, J.; Demolder, A.; Queruel, N.; Réant, K.; Normand, M.; Seffals, M.; Dimier, J.; Germi, R.; et al. An ancestral retroviral protein identified as a therapeutic target in type-1 diabetes. JCI Insight 2017, 2, 94387. [Google Scholar] [CrossRef] [Green Version]
- Moreno-De-Luca, A.; Myers, S.M.; Challman, T.D.; Moreno-De-Luca, D.; Evans, D.W.; Ledbetter, D.H. Developmental brain dysfunction: Revival and expansion of old concepts based on new genetic evidence. Lancet Neurol. 2013, 12, 406–414. [Google Scholar] [CrossRef] [Green Version]
- Ploeger, A.; Raijmakers, M.E.; van der Maas, H.L.; Galis, F. The association between autism and errors in early embryogenesis: What is the causal mechanism? Biol. Psychiatry 2010, 67, 602–607. [Google Scholar] [CrossRef]
- American Psychiatric Association. Diagnostic and Statistical Manual of Mental Disorders, 5th ed.; American Psychiatric Press: Washington, DC, USA, 2013. [Google Scholar]
- Cannon, T.D.; van Erp, T.G.; Bearden, C.E.; Loewy, R.; Thompson, P.; Toga, A.W.; Huttunen, M.O.; Keshavan, M.S.; Seidman, L.J.; Tsuang, M.T. Early and late neurodevelopmental influences in the prodrome to schizophrenia: Contributions of genes, environment, and their interactions. Schizophr. Bull. 2003, 29, 653–669. [Google Scholar] [CrossRef] [Green Version]
- Rapoport, J.; Chavez, A.; Greenstein, D.; Addington, A.; Gogtay, N. Autism spectrum disorders and childhood-onset schizophrenia: Clinical and biological contributions to a relation revisited. J. Am. Acad. Child Adolesc. Psychiatry 2009, 48, 10–18. [Google Scholar] [CrossRef] [Green Version]
- Park, M.T.M.; Raznahan, A.; Shaw, P.; Gogtay, N.; Lerch, J.P.; Chakravarty, M.M. Neuroanatomical phenotypes in mental illness: Identifying convergent and divergent cortical phenotypes across autism, ADHD and schizophrenia. J. Psychiatry Neurosci. 2018, 43, 201–212. [Google Scholar] [CrossRef] [Green Version]
- Septier, M.; Peyre, H.; Amsellem, F.; Beggiato, A.; Maruani, A.; Poumeyreau, M.; Amestoy, A.; Scheid, I.; Gaman, A.; Bolognani, F.; et al. Increased risk of ADHD in families with ASD. Eur. Child Adolesc. Psychiatry 2019, 28, 281–288. [Google Scholar] [CrossRef]
- Simonoff, E.; Pickles, A.; Charman, T.; Chandler, S.; Loucas, T.; Baird, G. Psychiatric disorders in children with autism spectrum disorders: Prevalence, comorbidity, and associated factors in a population-derived sample. J. Am. Acad. Child Adolesc. Psychiatry 2008, 47, 921–929. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jokiranta-Olkoniemi, E.; Cheslack-Postava, K.; Sucksdorff, D.; Suominen, A.; Gyllenberg, D.; Chudal, R.; Leivonen, S.; Gissler, M.; Brown, A.S.; Sourander, A. Risk of Psychiatric and Neurodevelopmental Disorders Among Siblings of Probands with Autism Spectrum Disorders. JAMA Psychiatry 2016, 73, 622–629. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guilmatre, A.; Dubourg, C.; Mosca, A.L.; Legallic, S.; Goldenberg, A.; Drouin-Garraud, V.; Layet, V.; Rosier, A.; Briault, S.; Bonnet-Brilhault, F.; et al. Recurrent rearrangements in synaptic and neurodevelopmental genes and shared biologic pathways in schizophrenia, autism, and mental retardation. Arch. Gen. Psychiatry 2009, 66, 947–956. [Google Scholar] [CrossRef] [PubMed]
- Cross-Disorder Group of the Psychiatric Genomics Consortium. Identification of risk loci with shared effects on five major psychiatric disorders: A genome-wide analysis. Lancet 2013, 381, 1371–1379. [Google Scholar] [CrossRef]
- Brainstorm Consortium; Anttila, V.; Bulik-Sullivan, B.; Finucane, H.K.; Walters, R.K.; Bras, J.; Duncan, L.; Escott-Price, V.; Falcone, G.J.; Gormley, P. Analysis of shared heritability in common disorders of the brain. Science 2018, 360, eaap8757. [Google Scholar] [CrossRef] [Green Version]
- Lotan, A.; Fenckova, M.; Bralten, J.; Alttoa, A.; Dixson, L.; Williams, R.W.; van der Voet, M. Neuroinformatic analyses of common and distinct genetic components associated with major neuropsychiatric disorders. Front. Neurosci. 2014, 8, 331. [Google Scholar] [CrossRef] [Green Version]
- Liu, X.; Li, Z.; Fan, C.; Zhang, D.; Chen, J. Genetics implicate common mechanisms in autism and schizophrenia: Synaptic activity and immunity. J. Med. Genet. 2017, 54, 511–520. [Google Scholar] [CrossRef] [Green Version]
- Glessner, J.T.; Li, J.; Wang, D.; March, M.; Lima, L.; Desai, A.; Hadley, D.; Kao, C.; Gur, R.E.; Cohen, N.; et al. Copy number variation meta-analysis reveals a novel duplication at 9p24 associated with multiple neurodevelopmental disorders. Genome Med. 2017, 9, 106. [Google Scholar] [CrossRef] [Green Version]
- Brown, A.S.; Derkits, E.J. Prenatal infection and schizophrenia: A review of epidemiologic and translational studies. Am. J. Psychiatry 2010, 167, 261–280. [Google Scholar] [CrossRef] [Green Version]
- Heyer, D.B.; Meredith, R.M. Environmental toxicology: Sensitive periods of development and neurodevelopmental disorders. Neurotoxicology 2017, 58, 23–41. [Google Scholar] [CrossRef]
- Miodovnik, A.; Engel, S.M.; Zhu, C.; Ye, X.; Soorya, L.V.; Silva, M.J.; Calafat, A.M.; Wolff, M.S. Endocrine disruptors and childhood social impairment. Neurotoxicology 2011, 32, 261–267. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Christensen, J.; Grønborg, T.K.; Sørensen, M.J.; Schendel, D.; Parner, E.T.; Pedersen, L.H.; Vestergaard, M. Prenatal valproate exposure and risk of autism spectrum disorders and childhood autism. JAMA 2013, 309, 1696–1703. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bromley, R.L.; Mawer, G.E.; Briggs, M.; Cheyne, C.; Clayton-Smith, J.; García-Fiñana, M.; Kneen, R.; Lucas, S.B.; Shallcross, R.; Baker, G.A.; et al. The prevalence of neurodevelopmental disorders in children prenatally exposed to antiepileptic drugs. J. Neurol. Neurosurg Psychiatry 2013, 84, 637–643. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Soontornpun, A.; Choovanichvong, T.; Tongsong, T. Pregnancy outcomes among women with epilepsy: A retrospective cohort study. Epilepsy Behav. 2018, 82, 52–56. [Google Scholar] [CrossRef]
- Ornoy, A. Valproic acid in pregnancy: How much are we endangering the embryo and fetus? Reprod. Toxicol. 2009, 28, 1–10. [Google Scholar] [CrossRef]
- Kolozsi, E.; Mackenzie, R.N.; Roullet, F.; deCatanzaro, D.; Foster, J.A. Prenatal exposure to valproic acid leads to reduced expression of synaptic adhesion molecule neuroligin 3 in mice. Neuroscience 2009, 163, 1201–1210. [Google Scholar] [CrossRef]
- Kazlauskas, N.; Campolongo, M.; Lucchina, L.; Zappala, C.; Depino, A.M. Postnatal behavioral and inflammatory alterations in female pups prenatally exposed to valproic acid. Psychoneuroendocrinology. 2016, 72, 11–21. [Google Scholar] [CrossRef]
- Manent, J.B.; Jorquera, I.; Mazzucchelli, I.; Depaulis, A.; Perucca, E.; Ben-Ari, Y.; Represa, A. Fetal exposure to GABA-acting antiepileptic drugs generates hippocampal and cortical dysplasias. Epilepsia 2007, 48, 684–693. [Google Scholar] [CrossRef]
- Schneider, T.; Roman, A.; Basta-Kaim, A.; Kubera, M.; Budziszewska, B.; Schneider, K.; Przewłocki, R. Gender-specific behavioral and immunological alterations in an animal model of autism induced by prenatal exposure to valproic acid. Psychoneuroendocrinology 2008, 33, 728–740. [Google Scholar] [CrossRef]
- Fukuchi, M.; Nii, T.; Ishimaru, N.; Minamino, A.; Hara, D.; Takasaki, I.; Tabuchi, A.; Tsuda, M. Valproic acid induces up- or down-regulation of gene expression responsible for the neuronal excitation and inhibition in rat cortical neurons through its epigenetic actions. Neurosci. Res. 2009, 65, 35–43. [Google Scholar] [CrossRef]
- Barrett, C.E.; Hennessey, T.M.; Gordon, K.M.; Ryan, S.J.; McNair, M.L.; Ressler, K.J.; Rainnie, D.G. Developmental disruption of amygdala transcriptome and socioemotional behavior in rats exposed to valproic acid prenatally. Mol. Autism 2017, 8, 42. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kawanai, T.; Ago, Y.; Watanabe, R.; Inoue, A.; Taruta, A.; Onaka, Y.; Hasebe, S.; Hashimoto, H.; Matsuda, T.; Takuma, K. Prenatal Exposure to Histone Deacetylase Inhibitors Affects Gene Expression of Autism-Related Molecules and Delays Neuronal Maturation. Neurochem. Res. 2016, 41, 2574–2584. [Google Scholar] [CrossRef] [PubMed]
- Choi, C.S.; Gonzales, E.L.; Kim, K.C.; Yang, S.M.; Kim, J.W.; Mabunga, D.F.; Cheong, J.H.; Han, S.H.; Bahn, G.H.; Shin, C.Y. The transgenerational inheritance of autism-like phenotypes in mice exposed to valproic acid during pregnancy. Sci. Rep. 2016, 6, 36250. [Google Scholar] [CrossRef] [PubMed]
- Chess, S. Autism in children with congenital rubella. J. Autism Child. Schizophr. 1971, 1, 33–47. [Google Scholar] [CrossRef] [PubMed]
- Lundstrom, R.; Ahnsjo, S. Mental development following maternal rubella:A follow-up study of children born in 1951–1952. Acta Paediatr. 1962, 51, 153–159. [Google Scholar] [CrossRef]
- Brown, A.S.; Susser, E.S. In utero infection and adult schizophrenia. Ment. Retard. Dev. Disabil. Res. Rev. 2002, 8, 51–57. [Google Scholar] [CrossRef]
- Brown, A.S.; Begg, M.D.; Gravenstein, S.; Schaefer, C.A.; Wyatt, R.J.; Bresnahan, M.; Babulas, V.P.; Susser, E.S. Serologic evidence of prenatal influenza in the etiology of schizophrenia. Arch. Gen. Psychiatry 2004, 61, 774–778. [Google Scholar] [CrossRef] [Green Version]
- Mann, J.R.; McDermott, S. Are maternal genitourinary infection and pre-eclampsia associated with ADHD in school-aged children? J. Atten. Disord. 2011, 15, 667–673. [Google Scholar] [CrossRef]
- Werenberg Dreier, J.; Nybo Andersen, A.M.; Hvolby, A.; Garne, E.; Kragh Andersen, P.; Berg-Beckhoff, G. Fever and infections in pregnancy and risk of attention deficit/hyperactivity disorder in the offspring. J. Child Psychol. Psychiatry 2016, 57, 540–548. [Google Scholar] [CrossRef]
- Atladóttir, H.O.; Thorsen, P.; Østergaard, L.; Schendel, D.E.; Lemcke, S.; Abdallah, M.; Parner, E.T. Maternal infection requiring hospitalization during pregnancy and autism spectrum disorders. J. Autism Dev. Disord. 2010, 40, 1423–1430. [Google Scholar] [CrossRef]
- Maeyama, K.; Tomioka, K.; Nagase, H.; Yoshioka, M.; Takagi, Y.; Kato, T.; Mizobuchi, M.; Kitayama, S.; Takada, S.; Nagai, M.; et al. Congenital Cytomegalovirus Infection in Children with Autism Spectrum Disorder: Systematic Review and Meta-Analysis. J. Autism Dev. Disord. 2018, 48, 1483–1491. [Google Scholar] [CrossRef] [PubMed]
- Lintas, C.; Altieri, L.; Lombardi, F.; Sacco, R.; Persico, A.M. Association of autism with polyomavirus infection in postmortem brains. J. Neurovirol. 2010, 16, 141–149. [Google Scholar] [CrossRef] [PubMed]
- Atladóttir, H.Ó.; Henriksen, T.B.; Schendel, D.E.; Parner, E.T. Autism after infection, febrile episodes, and antibiotic use during pregnancy: An exploratory study. Pediatrics 2012, 130, e1447–e1454. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shi, L.; Fatemi, S.H.; Sidwell, R.W.; Patterson, P.H. Maternal influenza infection causes marked behavioral and pharmacological changes in the offspring. J. Neurosci. 2003, 23, 297–302. [Google Scholar] [CrossRef]
- Knuesel, I.; Chicha, L.; Britschgi, M.; Schobel, S.A.; Bodmer, M.; Hellings, J.A.; Toovey, S.; Prinssen, E.P. Maternal immune activation and abnormal brain development across CNS disorders. Nat. Rev. Neurol. 2014, 10, 643–660. [Google Scholar] [CrossRef]
- Estes, M.L.; McAllister, A.K. Maternal immune activation: Implications for neuropsychiatric disorders. Science 2016, 353, 772–777. [Google Scholar] [CrossRef] [Green Version]
- Gilmore, J.; Jarskog, L.F.; Vadlamudi, S. Maternal poly I:C exposure during pregnancy regulates TNF alpha, BDNF, and NGF expression in neonatal brain and the maternal-fetal unit of the rat. J. Neuroimmunol. 2005, 159, 106–112. [Google Scholar] [CrossRef]
- Urakubo, A.; Jarskog, L.F.; Lieberman, J.A.; Gilmore, J.H. Prenatal exposure to maternal infection alters cytokine expression in the placenta, amniotic fluid, and fetal brain. Schizophr. Res. 2001, 47, 27–36. [Google Scholar] [CrossRef]
- Zuckerman, L.; Weiner, I. Maternal immune activation leads to behavioral and pharmacological changes in the adult offspring. J. Psychiatr Res. 2005, 39, 311–323. [Google Scholar] [CrossRef]
- Meyer, U.; Feldon, J. Neural basis of psychosis-related behaviour in the infection model of schizophrenia. Behav. Brain Res. 2009, 204, 322–334. [Google Scholar] [CrossRef]
- Shi, L.; Smith, S.E.; Malkova, N.; Tse, D.; Su, Y.; Patterson, P.H. Activation of the maternal immune system alters cerebellar development in the offspring. Brain Behav. Immun. 2009, 23, 116–123. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goines, P.E.; Croen, L.A.; Braunschweig, D.; Yoshida, C.K.; Grether, J.; Hansen, R.; Kharrazi, M.; Ashwood, P.; Van de Water, J. Increased midgestational IFN-γ, IL-4 and IL-5 in women bearing a child with autism: A case-control study. Mol. Autism 2011, 2, 13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abdallah, M.W.; Larsen, N.; Grove, J.; Nørgaard-Pedersen, B.; Thorsen, P.; Mortensen, E.L.; Hougaard, D.M. Amniotic fluid inflammatory cytokines: Potential markers of immunologic dysfunction in autism spectrum disorders. World J. Biol. Psychiatry 2013, 14, 528–538. [Google Scholar] [CrossRef] [PubMed]
- Jones, K.L.; Croen, L.A.; Yoshida, C.K.; Heuer, L.; Hansen, R.; Zerbo, O.; DeLorenze, G.N.; Kharrazi, M.; Yolken, R.; Ashwood, P.; et al. Autism with intellectual disability is associated with increased levels of maternal cytokines and chemokines during gestation. Mol. Psychiatry 2017, 22, 273–279. [Google Scholar] [CrossRef] [Green Version]
- Meyer, U.; Feldon, J.; Dammann, O. Schizophrenia and autism: Both shared and disorder-specific pathogenesis via perinatal inflammation? Pediatr. Res. 2011, 69, 26R–33R. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hsiao, E.Y.; McBride, S.W.; Chow, J.; Mazmanian, S.K.; Patterson, P.H. Modeling an autism risk factor in mice leads to permanent immune dysregulation. Proc. Natl. Acad. Sci. USA 2012, 109, 12776–12781. [Google Scholar] [CrossRef] [Green Version]
- Masi, A.; Glozier, N.; Dale, R.; Guastella, A.J. The Immune System, Cytokines, and Biomarkers in Autism Spectrum Disorder. Neurosci. Bull. 2017, 33, 194–204. [Google Scholar] [CrossRef] [Green Version]
- Balestrieri, E.; Cipriani, C.; Matteucci, C.; Benvenuto, A.; Coniglio, A.; Argaw-Denboba, A.; Toschi, N.; Bucci, I.; Miele, M.T.; Grelli, S.; et al. Children with Autism Spectrum Disorder and Their Mothers Share Abnormal Expression of Selected Endogenous Retroviruses Families and Cytokines. Front. Immunol. 2019, 10, 2244. [Google Scholar] [CrossRef] [Green Version]
- Verlaet, A.A.; Noriega, D.B.; Hermans, N.; Savelkoul, H.F. Nutrition, immunological mechanisms and dietary immunomodulation in ADHD. Eur. Child Adolesc. Psychiatry 2014, 23, 519–529. [Google Scholar] [CrossRef]
- Instanes, J.T.; Halmøy, A.; Engeland, A.; Haavik, J.; Furu, K.; Klungsøyr, K. Attention-Deficit/Hyperactivity Disorder in Offspring of Mothers with Inflammatory and Immune System Diseases. Biol. Psychiatry 2017, 81, 452–459. [Google Scholar] [CrossRef] [Green Version]
- Miller, B.J.; Buckley, P.; Seabolt, W.; Mellor, A.; Kirkpatrick, B. Meta-analysis of cytokine alterations in schizophrenia: Clinical status and antipsychotic effects. Biol. Psychiatry 2011, 70, 663–671. [Google Scholar] [CrossRef]
- Upthegrove, R.; Manzanares-Teson, N.; Barnes, N.M. Cytokine function in medication-naive first episode psychosis: A systematic review and meta-analysis. Schizophr. Res. 2014, 155, 101–108. [Google Scholar] [CrossRef] [PubMed]
- Khoury, R.; Nasrallah, H.A. Inflammatory biomarkers in individuals at clinical high risk for psychosis (CHR-P): State or trait? Schizophr. Res. 2018, 199, 31–38. [Google Scholar] [CrossRef] [PubMed]
- Goldsmith, D.R.; Rapaport, M.H.; Miller, B.J. A meta-analysis of blood cytokine network alterations in psychiatric patients: Comparisons between schizophrenia, bipolar disorder and depression. Mol. Psychiatry 2016, 21, 1696–1709. [Google Scholar] [CrossRef] [PubMed]
- Balestrieri, E.; Arpino, C.; Matteucci, C.; Sorrentino, R.; Pica, F.; Alessandrelli, R.; Coniglio, A.; Curatolo, P.; Rezza, G.; Macciardi, F.; et al. HERVs expression in Autism Spectrum Disorders. PLoS ONE 2012, 7, e48831. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Balestrieri, E.; Pitzianti, M.; Matteucci, C.; D’Agati, E.; Sorrentino, R.; Baratta, A.; Caterina, R.; Zenobi, R.; Curatolo, P.; Garaci, E.; et al. Human endogenous retroviruses and ADHD. World J. Biol. Psychiatry 2014, 15, 499–504. [Google Scholar] [CrossRef] [PubMed]
- Balestrieri, E.; Cipriani, C.; Matteucci, C.; Capodicasa, N.; Pilika, A.; Korca, I.; Sorrentino, R.; Argaw-Denboba, A.; Bucci, I.; Miele, M.T.; et al. Transcriptional activity of human endogenous retrovirus in Albanian children with autism spectrum disorders. New Microbiol. 2016, 39, 228–231. [Google Scholar]
- D’Agati, E.; Pitzianti, M.; Balestrieri, E.; Matteucci, C.; SinibaldiVallebona, P.; Pasini, A. First evidence of HERV-H transcriptional activity reduction after methylphenidate treatment in a young boy with ADHD. New Microbiol. 2016, 39, 237–239. [Google Scholar]
- Cipriani, C.; Pitzianti, M.B.; Matteucci, C.; D’Agati, E.; Miele, M.T.; Rapaccini, V.; Grelli, S.; Curatolo, P.; Sinibaldi-Vallebona, P.; Pasini, A.; et al. The Decrease in Human Endogenous Retrovirus-H Activity Runs in Parallel with Improvement in ADHD Symptoms in Patients Undergoing Methylphenidate Therapy. Int. J. Mol. Sci. 2018, 19, 3286. [Google Scholar] [CrossRef] [Green Version]
- Huang, W.J.; Liu, Z.C.; Wei, W.; Wang, G.H.; Wu, J.G.; Zhu, F. Human endogenous retroviral pol RNA and protein detected and identified in the blood of individuals with schizophrenia. Schizophr. Res. 2006, 83, 193–199. [Google Scholar] [CrossRef]
- Yao, Y.; Schröder, J.; Nellåker, C.; Bottmer, C.; Bachmann, S.; Yolken, R.H.; Karlsson, H. Elevated levels of human endogenous Retrovirus-W transcripts in blood cells from patients with first episode schizophrenia. Genes Brain Behav. 2008, 7, 103–112. [Google Scholar] [CrossRef] [PubMed]
- Perron, H.; Hamdani, N.; Faucard, R.; Lajnef, M.; Jamain, S.; Daban-Huard, C.; Sarrazin, S.; LeGuen, E.; Houenou, J.; Delavest, M.; et al. Molecular characteristics of Human Endogenous Retrovirus Type-W in schizophrenia and bipolar disorder. Transl. Psychiatry 2012, 2, 201. [Google Scholar] [CrossRef] [PubMed]
- Huang, W.; Li, S.; Hu, Y.; Yu, H.; Luo, F.; Zhang, Q.; Zhu, F. Implication of the env gene of the human endogenous retrovirus W family in the expression of BDNF and DRD3 and development of recent-onset schizophrenia. Schizophr. Bull. 2011, 37, 988–1000. [Google Scholar] [CrossRef] [PubMed]
- Frank, O.; Giehl, M.; Zheng, C.; Hehlmann, R.; Leib-Mösch, C.; Seifarth, W. Human endogenous retrovirus expression profiles in samples from brains of patients with schizophrenia and bipolar disorders. J. Virol. 2005, 79, 10890–10901. [Google Scholar] [CrossRef] [Green Version]
- Karlsson, H.; Bachmann, S.; Schröder, J.; McArthur, J.; Torrey, E.F.; Yolken, R.H. Retroviral RNA identified in the cerebrospinal fluids and brains of individuals with schizophrenia. Proc. Natl. Acad. Sci. USA 2001, 98, 4634–4639. [Google Scholar] [CrossRef] [Green Version]
- Weis, S.; Llenos, I.C.; Sabunciyan, S.; Dulay, J.R.; Isler, L.; Yolken, R.; Perron, H. Reduced expression of human endogenous retrovirus (HERV)-W GAG protein in the cingulate gyrus and hippocampus in schizophrenia, bipolar disorder, and depression. J. Neural Transm. 2007, 114, 645–655. [Google Scholar] [CrossRef]
- Perron, H.; Mekaoui, L.; Bernard, C.; Veas, F.; Stefas, I.; Leboyer, M. Endogenous retrovirus type W GAG and envelope protein antigenemia in serum of schizophrenic patients. Biol. Psychiatry 2008, 64, 1019–1023. [Google Scholar] [CrossRef]
- Melbourne, J.K.; Chase, K.A.; Feiner, B.; Rosen, C.; Sharma, R.P. Long non-coding and endogenous retroviral RNA levels are associated with proinflammatory cytokine mRNA expression in peripheral blood cells: Implications for schizophrenia. Psychiatry Res. 2018, 262, 465–468. [Google Scholar] [CrossRef]
- Ibi, D.; González-Maeso, J. Epigeneticsignaling in schizophrenia. Cell. Signal. 2015, 27, 2131–2136. [Google Scholar] [CrossRef] [Green Version]
- Mak, M.; Samochowiec, J.; Frydecka, D.; Pełka-Wysiecka, J.; Szmida, E.; Karpiński, P.; Sąsiadek, M.M.; Piotrowski, P.; Samochowiec, A.; Misiak, B. First-episode schizophrenia is associated with a reduction of HERV-K methylation in peripheral blood. Psychiatry Res. 2019, 271, 459–463. [Google Scholar] [CrossRef]
- Perron, H.; Lang, A. The human endogenous retrovirus link between genes and environment in multiple sclerosis and in multifactorial diseases associating neuroinflammation. Clin. Rev. Allergy Immunol. 2010, 39, 51–61. [Google Scholar] [CrossRef]
- Robbez-Masson, L.; Rowe, H.M. Retrotransposons shape species-specific embryonic stem cell gene expression. Retrovirology 2015, 12, 45. [Google Scholar] [CrossRef] [Green Version]
- Magiorkinis, G.; Katzourakis, A.; Lagiou, P. Roles of Endogenous Retroviruses in Early Life Events. Trends Microbiol. 2017, 25, 876–877. [Google Scholar] [CrossRef] [PubMed]
- Gropman, A.L.; Batshaw, M.L. Epigenetics, copy number variation, and other molecular mechanisms underlying neurodevelopmental disabilities: New insights and diagnostic approaches. J. Dev. Behav. Pediatr. 2010, 31, 582–591. [Google Scholar] [CrossRef] [Green Version]
- LaSalle, J.M.; Powell, W.T.; Yasui, D.H. Epigenetic layers and players underlying neurodevelopment. Trends Neurosci. 2013, 36, 460–470. [Google Scholar] [CrossRef] [Green Version]
- Grafodatskaya, D.; Chung, B.; Szatmari, P.; Weksberg, R. Autism spectrum disorders and epigenetics. J. Am. Acad. Child Adolesc. Psychiatry 2010, 49, 794–809. [Google Scholar] [CrossRef] [PubMed]
- Kelley, D.; Rinn, J. Transposable elements reveal a stem cell-specific class of long noncoding RNAs. Genome Biol. 2012, 13, R107. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cogill, S.B.; Srivastava, A.K.; Yang, M.Q.; Wang, L. Co-expression of long non-coding RNAs and autism risk genes in the developing human brain. BMC Syst. Biol. 2018, 12, 91. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hakim, S.T.; Alsayari, M.; McLean, D.C.; Saleem, S.; Addanki, K.C.; Aggarwal, M.; Mahalingam, K.; Bagasra, O. A large number of the human microRNAs target lentiviruses, retroviruses, and endogenous retroviruses. Biochem. Biophys. Res. Commun. 2008, 369, 357–362. [Google Scholar] [CrossRef]
- Hadjiargyrou, M.; Delihas, N. The intertwining of transposable elements and non-coding RNAs. Int. J. Mol. Sci. 2013, 14, 13307–13328. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Xu, Z.; Jiang, J.; Xu, C.; Kang, J.; Xiao, L.; Wu, M.; Xiong, J.; Guo, X.; Liu, H. Endogenous miRNA sponge lincRNA-RoR regulates Oct4, Nanog, and Sox2 in human embryonic stem cell self-renewal. Dev. Cell 2013, 25, 69–80. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Durruthy-Durruthy, J.; Sebastiano, V.; Wossidlo, M.; Cepeda, D.; Cui, J.; Grow, E.J.; Davila, J.; Mall, M.; Wong, W.H.; Wysocka, J.; et al. The primate-specific noncoding RNA HPAT5 regulates pluripotency during human preimplantation development and nuclear reprogramming. Nat. Genet. 2016, 48, 44–52. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lavillette, D.; Marin, M.; Ruggieri, A.; Mallet, F.; Cosset, F.L.; Kabat, D. The envelope glycoprotein of human endogenous retrovirus type W uses a divergent family of amino acid transporters/cell surface receptors. J. Virol. 2002, 76, 6442–6452. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weis, S.; Llenos, I.C.; Dulay, J.R.; Verma, N.; Sabunciyan, S.; Yolken, R.H. Changes in region- and cell type-specific expression patterns of neutral amino acid transporter 1 (ASCT-1) in the anterior cingulate cortex and hippocampus in schizophrenia, bipolar disorder and major depression. J. Neural Transm 2007, 114, 261–271. [Google Scholar] [CrossRef] [PubMed]
- Rojas, D.C. The role of glutamate and its receptors in autism and the use of glutamate receptor antagonists in treatment. J. Neural Transm 2014, 121, 891–905. [Google Scholar] [CrossRef] [Green Version]
- Li, S.; Liu, Z.C.; Yin, S.J.; Chen, Y.T.; Yu, H.L.; Zeng, J.; Zhang, Q.; Zhu, F. Human endogenous retrovirus W family envelope gene activates the small conductance Ca2+-activated K+ channel in human neuroblastoma cells through CREB. Neuroscience 2013, 247, 164–174. [Google Scholar] [CrossRef]
- Angelucci, F.; Brenè, S.; Mathé, A.A. BDNF in schizophrenia, depression and corresponding animal models. Mol. Psychiatry 2005, 10, 345–352. [Google Scholar] [CrossRef] [Green Version]
- Kasarpalkar, N.J.; Kothari, S.T.; Dave, U.P. Brain-Derived Neurotrophic Factor in children with Autism Spectrum Disorder. Ann. Neurosci. 2014, 21, 129–133. [Google Scholar] [CrossRef] [Green Version]
- Grandi, N.; Tramontano, E. Human Endogenous Retroviruses Are Ancient Acquired Elements Still Shaping Innate Immune Responses. Front. Immunol. 2018, 9, 2039. [Google Scholar] [CrossRef] [Green Version]
- Hurst, T.P.; Magiorkinis, G. Activation of the innate immune response by endogenous retroviruses. J. Gen. Virol. 2015, 96, 1207–1218. [Google Scholar] [CrossRef]
- Hurst, T.P.; Magiorkinis, G. Epigenetic Control of Human Endogenous Retrovirus Expression: Focus on Regulation of Long-Terminal Repeats (LTRs). Viruses 2017, 9, 130. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aswad, A.; Katzourakis, A. Paleovirology and virally derived immunity. Trends Ecol. Evol. 2012, 27, 627–636. [Google Scholar] [CrossRef] [PubMed]
- Stocking, C.; Kozak, C.A. Murine endogenous retroviruses. Cell. Mol. Life Sci. 2008, 65, 3383–3398. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maksakova, I.A.; Romanish, M.T.; Gagnier, L.; Dunn, C.A.; van de Lagemaat, L.N.; Mager, D.L. Retroviral elements and their hosts: Insertional mutagenesis in the mouse germ line. PLoS Genet. 2006, 2, e2. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ribet, D.; Harper, F.; Dupressoir, A.; Dewannieux, M.; Pierron, G.; Heidmann, T. An infectious progenitor for the murine IAP retrotransposon: Emergence of an intracellular genetic parasite from an ancient retrovirus. Genome Res. 2008, 18, 597–609. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Magiorkinis, G.; Gifford, R.J.; Katzourakis, A.; De Ranter, J.; Belshaw, R. Env-less endogenous retroviruses are genomic superspreaders. Proc. Natl. Acad. Sci. USA 2012, 109, 7385–7390. [Google Scholar] [CrossRef] [Green Version]
- Rowe, H.M.; Trono, D. Dynamic control of endogenous retroviruses during development. Virology 2011, 411, 273–287. [Google Scholar] [CrossRef] [Green Version]
- Kuff, E.L.; Fewell, J.W. Intracisternal A-particle gene expression in normal mouse thymus tissue: Gene products and strain-related variability. Mol. Cell. Biol. 1985, 5, 474–483. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.Y.; Steelman, L.S.; McCubrey, J.A. Abnormal activation of cytokine gene expression by intracisternal type A particle transposition: Effects of mutations that result in autocrine growth stimulation and malignant transformation. Cytokines Cell. Mol. Ther. 1997, 3, 3–19. [Google Scholar]
- Howard, G.; Eiges, R.; Gaudet, F.; Jaenisch, R.; Eden, A. Activation and transposition of endogenous retroviral elements in hypomethylation induced tumors in mice. Oncogene 2008, 27, 404–408. [Google Scholar] [CrossRef] [Green Version]
- Sharif, J.; Shinkai, Y.; Koseki, H. Is there a role for endogenous retroviruses to mediate long-term adaptive phenotypic response upon environmental inputs? Philos. Trans. R. Soc. Lond B Biol. Sci. 2013, 368, 20110340. [Google Scholar] [CrossRef] [Green Version]
- Ribet, D.; Dewannieux, M.; Heidmann, T. An active murine transposon family pair: Retrotransposition of “master” MusD copies and ETn trans-mobilization. Genome Res. 2004, 14, 2261–2267. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ribet, D.; Harper, F.; Dewannieux, M.; Pierron, G.; Heidmann, T. Murine MusD retrotransposon: Structure and molecular evolution of an “intracellularized” retrovirus. J. Virol. 2007, 81, 1888–1898. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maksakova, I.A.; Mager, D.L. Transcriptional regulation of early transposon elements, an active family of mouse long terminal repeat retrotransposons. J. Virol. 2005, 79, 13865–13874. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dupressoir, A.; Marceau, G.; Vernochet, C.; Bénit, L.; Kanellopoulos, C.; Sapin, V.; Heidmann, T. Syncytin-A and syncytin-B, two fusogenic placenta-specific murine envelope genes of retroviral origin conserved in Muridae. Proc. Natl. Acad. Sci. USA 2005, 102, 725–730. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dupressoir, A.; Vernochet, C.; Harper, F.; Guégan, J.; Dessen, P.; Pierron, G.; Heidmann, T. A pair of co-opted retroviral envelope syncytin genes is required for formation of the two-layered murine placental syncytiotrophoblast. Proc. Natl. Acad. Sci. USA 2011, 108, E1164–E1173. [Google Scholar] [CrossRef] [Green Version]
- Redelsperger, F.; Raddi, N.; Bacquin, A.; Vernochet, C.; Mariot, V.; Gache, V.; Blanchard-Gutton, N.; Charrin, S.; Tiret, L.; Dumonceaux, J.; et al. Genetic Evidence That Captured Retroviral Envelope syncytins Contribute to Myoblast Fusion and Muscle Sexual Dimorphism in Mice. PLoS Genet. 2016, 12, e1006289. [Google Scholar] [CrossRef]
- Søe, K.; Hobolt-Pedersen, A.S.; Delaisse, J.M. The elementary fusion modalities of osteoclasts. Bone 2015, 73, 181–189. [Google Scholar] [CrossRef]
- Ergaz, Z.; Weinstein-Fudim, L.; Ornoy, A. Genetic and non-genetic animal models for autism spectrum disorders (ASD). Reprod. Toxicol. 2016, 64, 116–140. [Google Scholar] [CrossRef]
- Scattoni, M.L.; Ricceri, L.; Crawley, J.N. Unusual repertoire of vocalizations in adult BTBR T+tf/J mice during three types of social encounters. Genes Brain Behav. 2011, 10, 44–56. [Google Scholar] [CrossRef] [Green Version]
- Hava, G.; Vered, L.; Yael, M.; Mordechai, H.; Mahoud, H. Alterations in behaviour in adult offspring mice following maternal inflammation during pregnancy. Dev. Psychobiol. 2006, 48, 162–168. [Google Scholar] [CrossRef] [PubMed]
- Malkova, N.V.; Yu, C.Z.; Hsiao, E.Y.; Moore, M.J.; Patterson, P.H. Maternal immune activation yields offspring displaying mouse versions of the three core symptoms of autism. Brain Behav. Immun. 2012, 26, 607–616. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smith, S.E.; Li, J.; Garbett, K.; Mirnics, K.; Patterson, P.H. Maternal immune activation alters fetal brain development through interleukin-6. J. Neurosci. 2007, 27, 10695–10702. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weber-Stadlbauer, U.; Richetto, J.; Labouesse, M.A.; Bohacek, J.; Mansuy, I.M.; Meyer, U. Transgenerational transmission and modification of pathological traits induced by prenatal immune activation. Mol. Psychiatry 2017, 22, 102–112. [Google Scholar] [CrossRef]
- Choi, G.B.; Yim, Y.S.; Wong, H.; Kim, S.; Kim, H.; Kim, S.V.; Hoeffer, C.A.; Littman, D.R.; Huh, J.R. The maternal interleukin-17a pathway in mice promotes autism-like phenotypes in offspring. Science 2016, 351, 933–939. [Google Scholar] [CrossRef] [Green Version]
- Deckmann, I.; Schwingel, G.B.; Fontes-Dutra, M.; Bambini-Junior, V.; Gottfried, C. Neuroimmune Alterations in Autism: A Translational Analysis Focusing on the Animal Model of Autism Induced by Prenatal Exposure to Valproic Acid. Neuroimmunomodulation 2018, 25, 285–299. [Google Scholar] [CrossRef]
- Wagner, G.C.; Reuhl, K.R.; Cheh, M.; McRae, P.; Halladay, A.K. A new neurobehavioral model of autism in mice: Pre- and postnatal exposure to sodium valproate. J. Autism Dev. Disord. 2006, 36, 779–793. [Google Scholar] [CrossRef]
- Nicolini, C.; Fahnestock, M. The valproic acid-induced rodent model of autism. Exp. Neurol. 2018, 299, 217–227. [Google Scholar] [CrossRef]
- Cipriani, C.; Ricceri, L.; Matteucci, C.; De Felice, A.; Tartaglione, A.M.; Argaw-Denboba, A.; Pica, F.; Grelli, S.; Calamandrei, G.; Sinibaldi Vallebona, P.; et al. High expression of Endogenous Retroviruses from intrauterine life to adulthood in two mouse models of Autism Spectrum Disorders. Sci. Rep. 2018, 8, 629. [Google Scholar] [CrossRef] [Green Version]
- Tartaglione, A.M.; Cipriani, C.; Chiarotti, F.; Perrone, B.; Balestrieri, E.; Matteucci, C.; Sinibaldi-Vallebona, P.; Calamandrei, G.; Ricceri, L. Early Behavioral Alterations and Increased Expression of Endogenous Retroviruses Are Inherited Across Generations in Mice Prenatally Exposed to Valproic Acid. Mol. Neurobiol. 2019, 56, 3736–3750. [Google Scholar] [CrossRef]
- Konopko, M.A.; Densmore, A.L.; Krueger, B.K. Sexually Dimorphic Epigenetic Regulation of Brain-Derived Neurotrophic Factor in Fetal Brain in the Valproic Acid Model of Autism Spectrum Disorder. Dev. Neurosci. 2017, 39, 507–518. [Google Scholar] [CrossRef] [PubMed]
- Tung, E.W.; Winn, L.M. Epigenetic modifications in valproic acid-induced teratogenesis. Toxicol. Appl. Pharmacol. 2010, 248, 201–209. [Google Scholar] [CrossRef] [PubMed]
- Holliday, R. The possibility of epigenetic transmission of defects induced by teratogens. Mutat. Res. 1998, 422, 203–205. [Google Scholar] [CrossRef]
- Gagnier, L.; Belancio, V.P.; Mager, D.L. Mouse germ line mutations due to retrotransposon insertions. Mob. DNA 2019, 10, 15. [Google Scholar] [CrossRef] [Green Version]
- Guliyev, M.; Yilmaz, S.; Sahin, K.; Marakli, S.; Gozukirmizi, N. Human endogenous Retrovirus-H insertion screening. Mol. Med. Rep. 2013, 7, 1305–1309. [Google Scholar] [CrossRef]
- CakmakGuner, B.; Karlik, E.; Marakli, S.; Gozukirmizi, N. Detection of HERV-K6 and HERV-K11 transpositions in the human genome. Biomed. Rep. 2018, 9, 53–59. [Google Scholar] [CrossRef] [Green Version]
- Garcia-Montojo, M.; Dominguez-Mozo, M.; Arias-Leal, A.; Garcia-Martinez, Á.; De lasHeras, V.; Casanova, I.; Faucard, R.; Gehin, N.; Madeira, A.; Arroyo, R.; et al. The DNA copy number of human endogenous Retrovirus-W (MSRV-type) is increased in multiple sclerosis patients and is influenced by gender and disease severity. PLoS ONE 2013, 8, e53623. [Google Scholar] [CrossRef]
- Bundo, M.; Toyoshima, M.; Okada, Y.; Akamatsu, W.; Ueda, J.; Nemoto-Miyauchi, T.; Sunaga, F.; Toritsuka, M.; Ikawa, D.; Kakita, A.; et al. Increased l1 retrotransposition in the neuronal genome in schizophrenia. Neuron 2014, 81, 306–313. [Google Scholar] [CrossRef] [Green Version]
- Nomura, J.; Takumi, T. Animal models of psychiatric disorders that reflect human copy number variation. Neural Plast. 2012, 2012, 589524. [Google Scholar] [CrossRef] [Green Version]
- Nishioka, M.; Bundo, M.; Iwamoto, K.; Kato, T. Somatic mutations in the human brain: Implications for psychiatric research. Mol. Psychiatry 2019, 24, 839–856. [Google Scholar] [CrossRef] [Green Version]



© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Balestrieri, E.; Matteucci, C.; Cipriani, C.; Grelli, S.; Ricceri, L.; Calamandrei, G.; Sinibaldi Vallebona, P. Endogenous Retroviruses Activity as a Molecular Signature of Neurodevelopmental Disorders. Int. J. Mol. Sci. 2019, 20, 6050. https://doi.org/10.3390/ijms20236050
Balestrieri E, Matteucci C, Cipriani C, Grelli S, Ricceri L, Calamandrei G, Sinibaldi Vallebona P. Endogenous Retroviruses Activity as a Molecular Signature of Neurodevelopmental Disorders. International Journal of Molecular Sciences. 2019; 20(23):6050. https://doi.org/10.3390/ijms20236050
Chicago/Turabian StyleBalestrieri, Emanuela, Claudia Matteucci, Chiara Cipriani, Sandro Grelli, Laura Ricceri, Gemma Calamandrei, and Paola Sinibaldi Vallebona. 2019. "Endogenous Retroviruses Activity as a Molecular Signature of Neurodevelopmental Disorders" International Journal of Molecular Sciences 20, no. 23: 6050. https://doi.org/10.3390/ijms20236050