Comparative Transcriptomics of Flammulina filiformis Suggests a High CO2 Concentration Inhibits Early Pileus Expansion by Decreasing Cell Division Control Pathways

 , ,
, ,
Abstract
:
{kind=link}
{kind=link}
{kind=link}
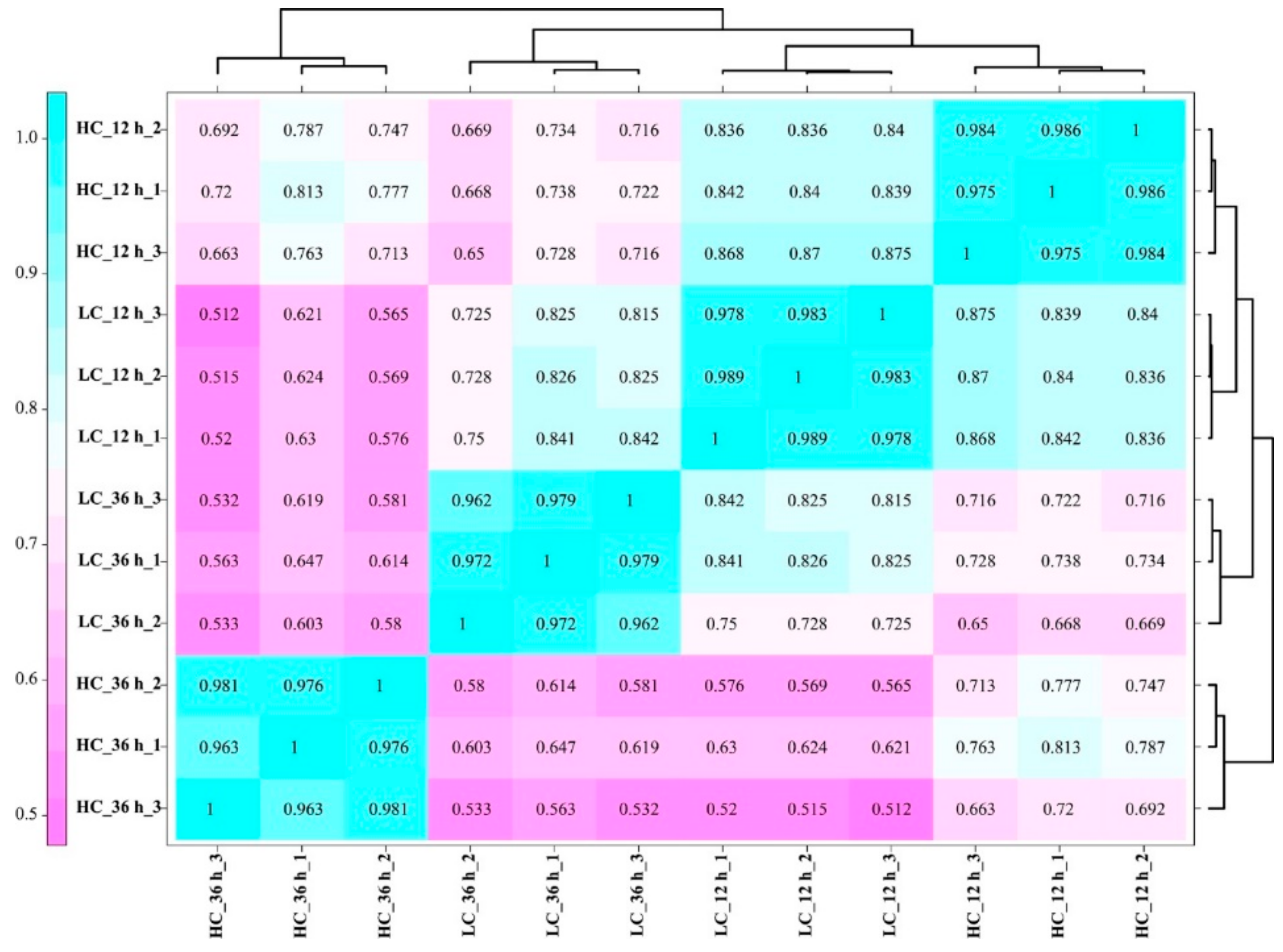{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
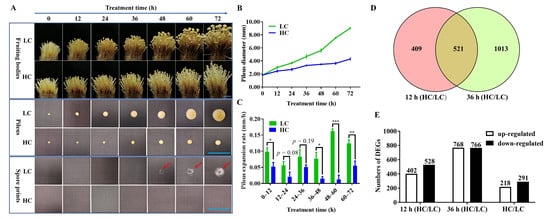
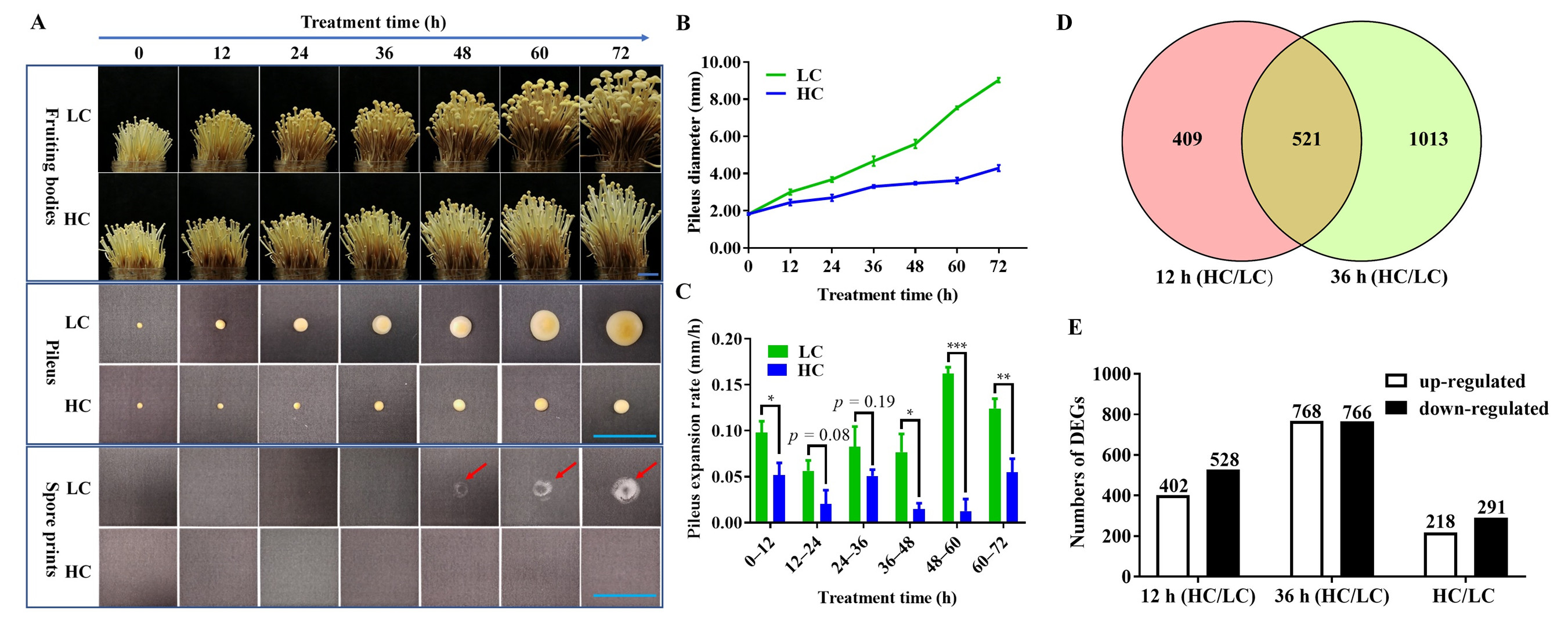
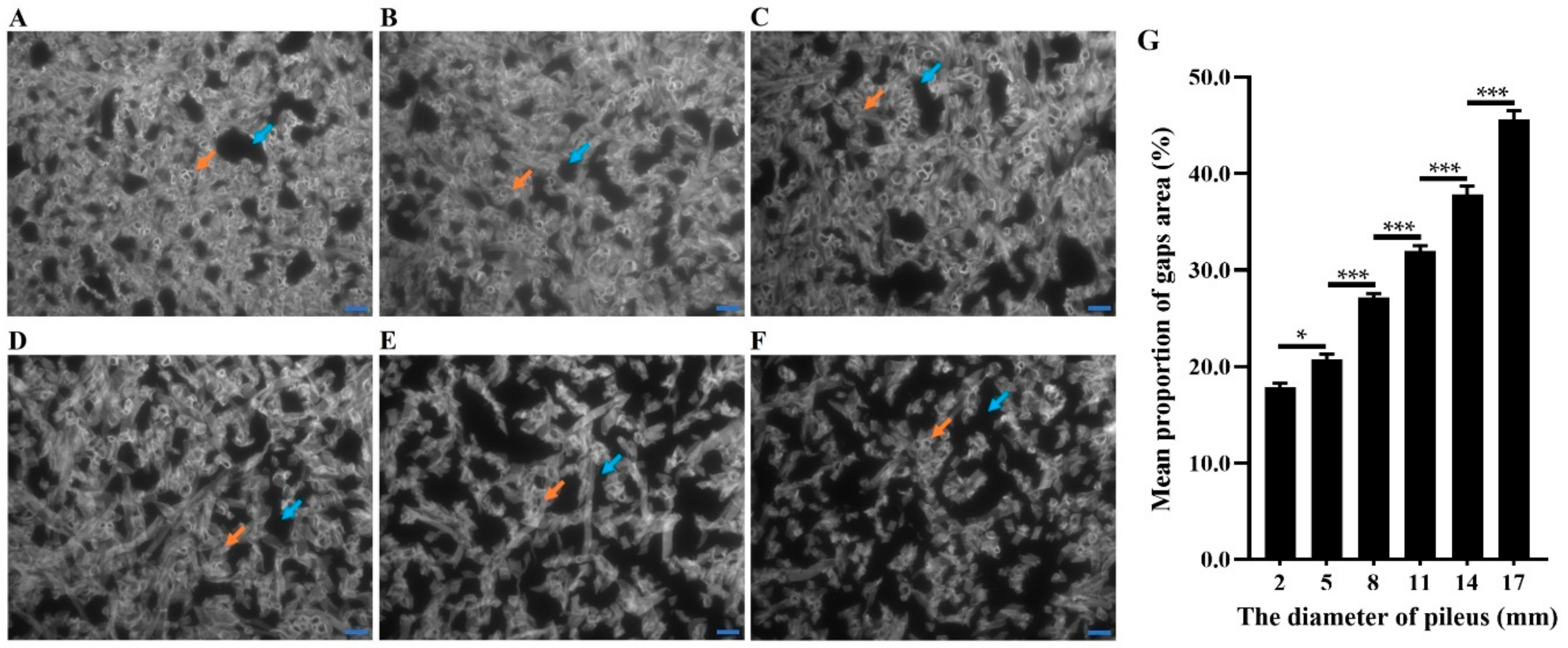
2.1. Phenotype of F. filiformis Fruiting Bodies under Different CO2 Concentrations
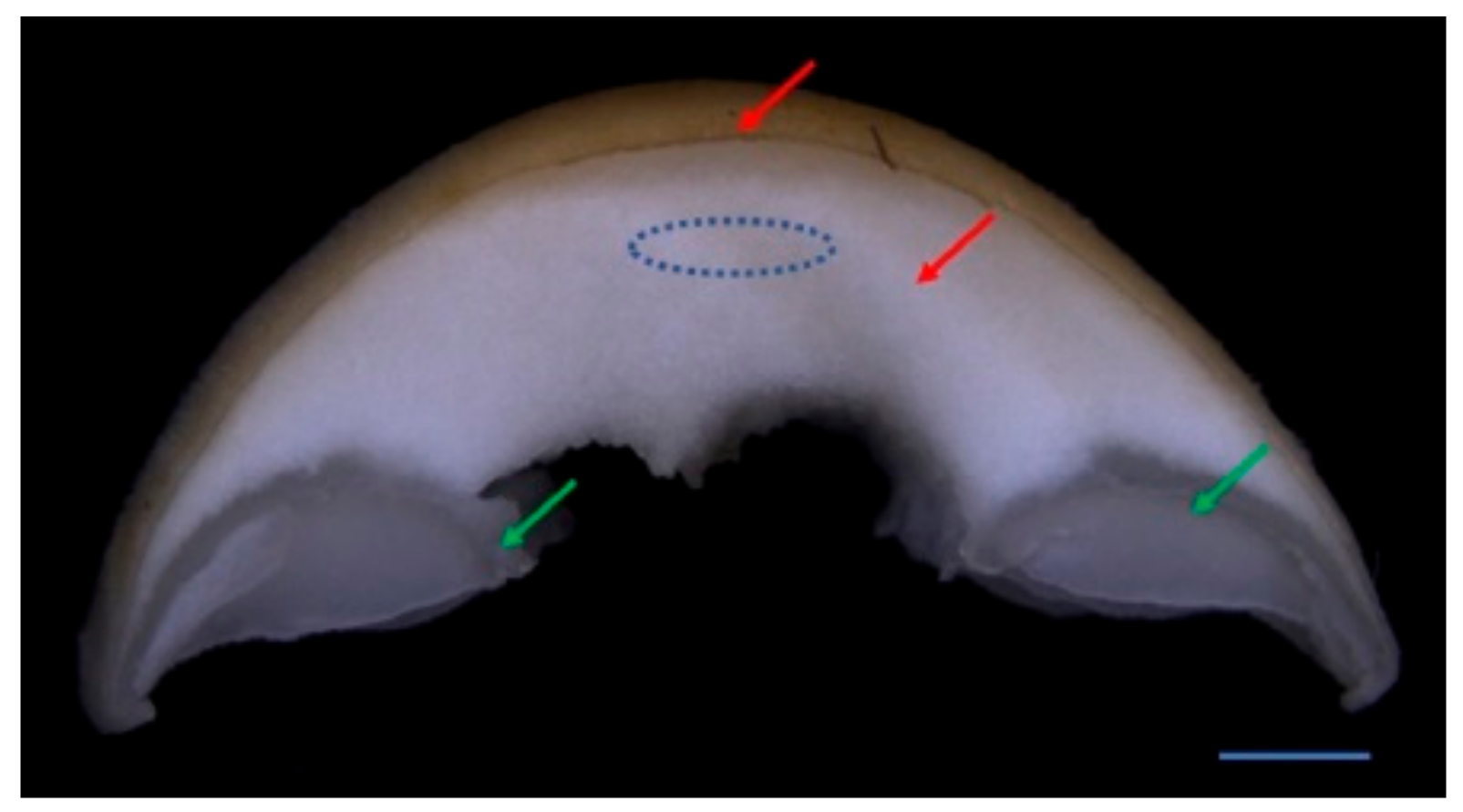
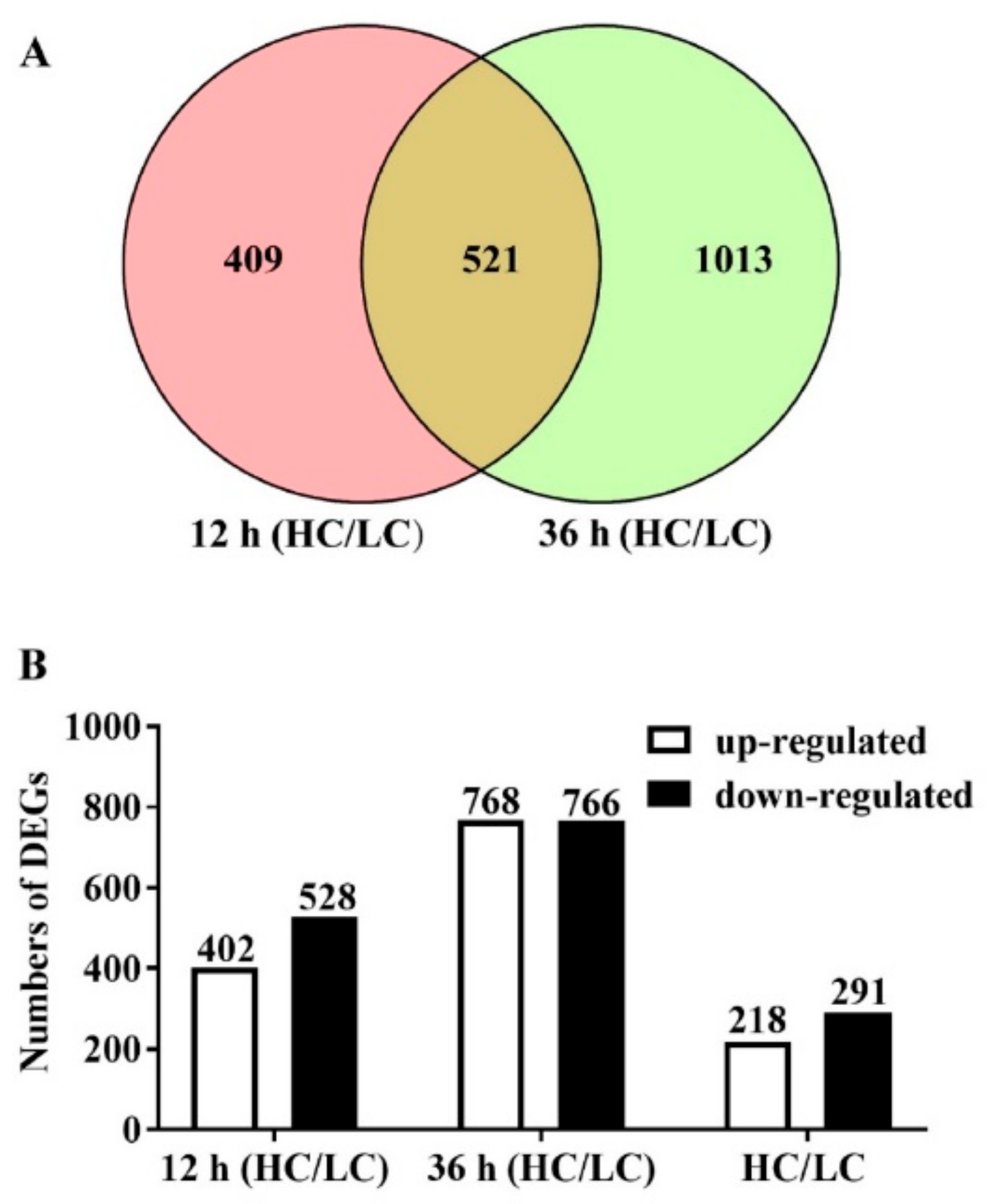
2.2. RNA-Seq Data Analysis of F. filiformis Grown under Different CO2 Conditions
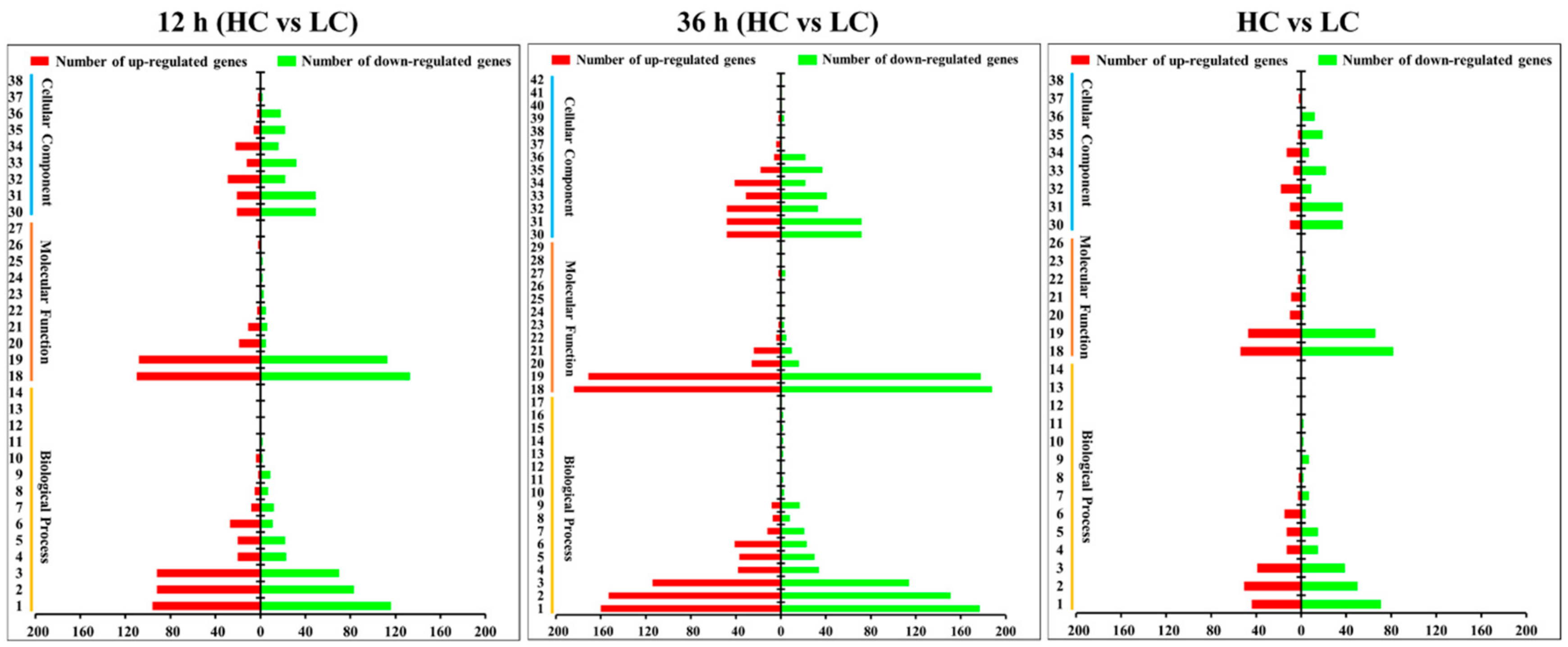
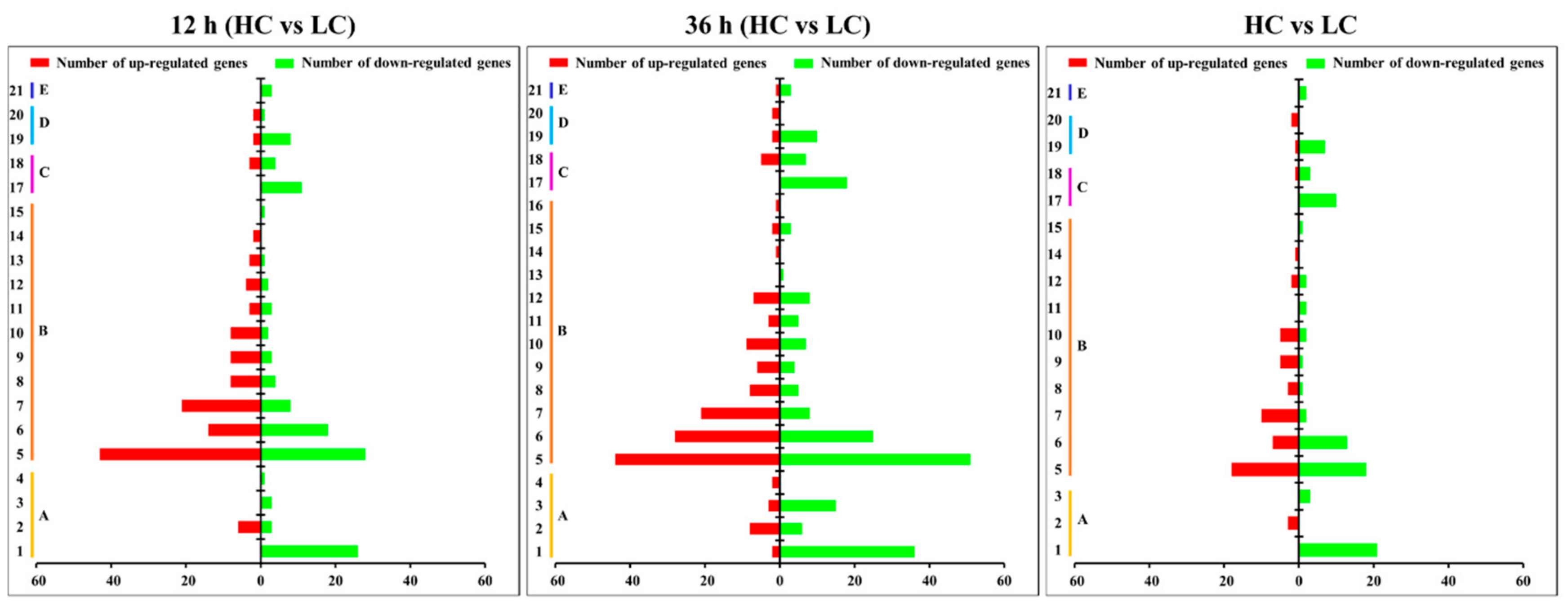
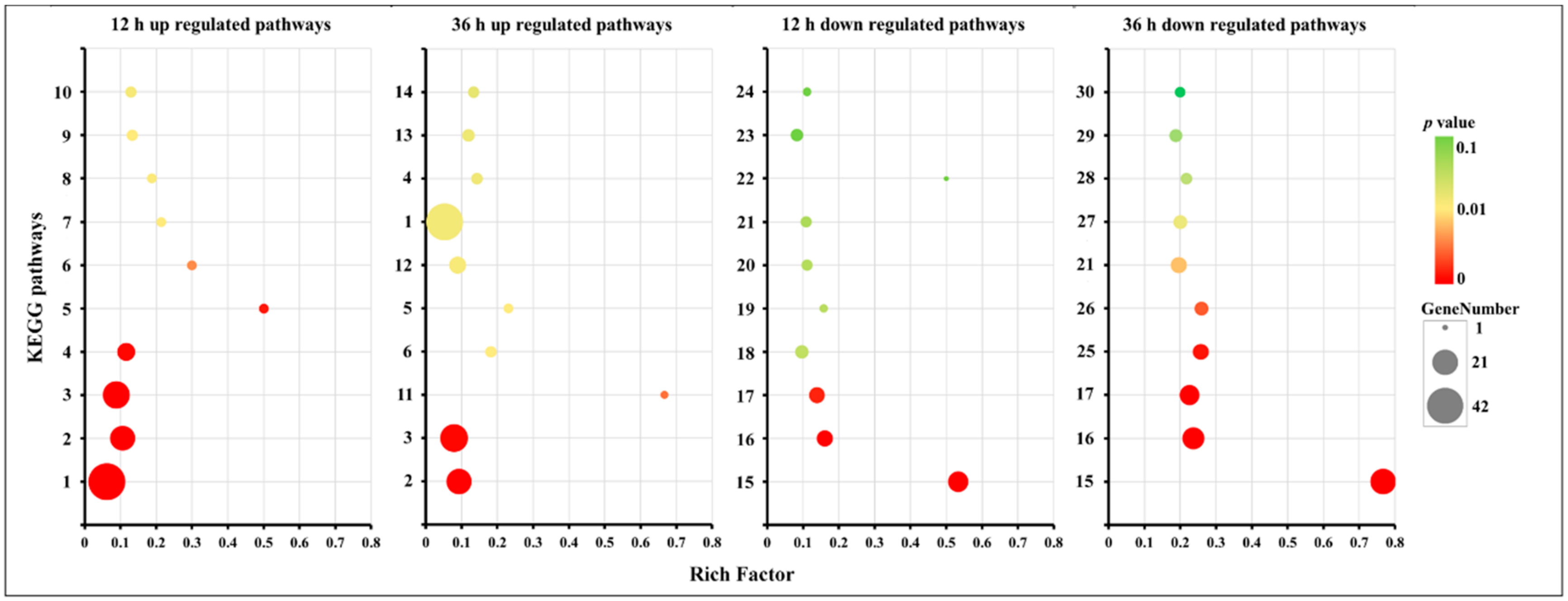
2.3. Gene Ontology (GO) Enrichment and KEGG Pathway Analysis for DEGs
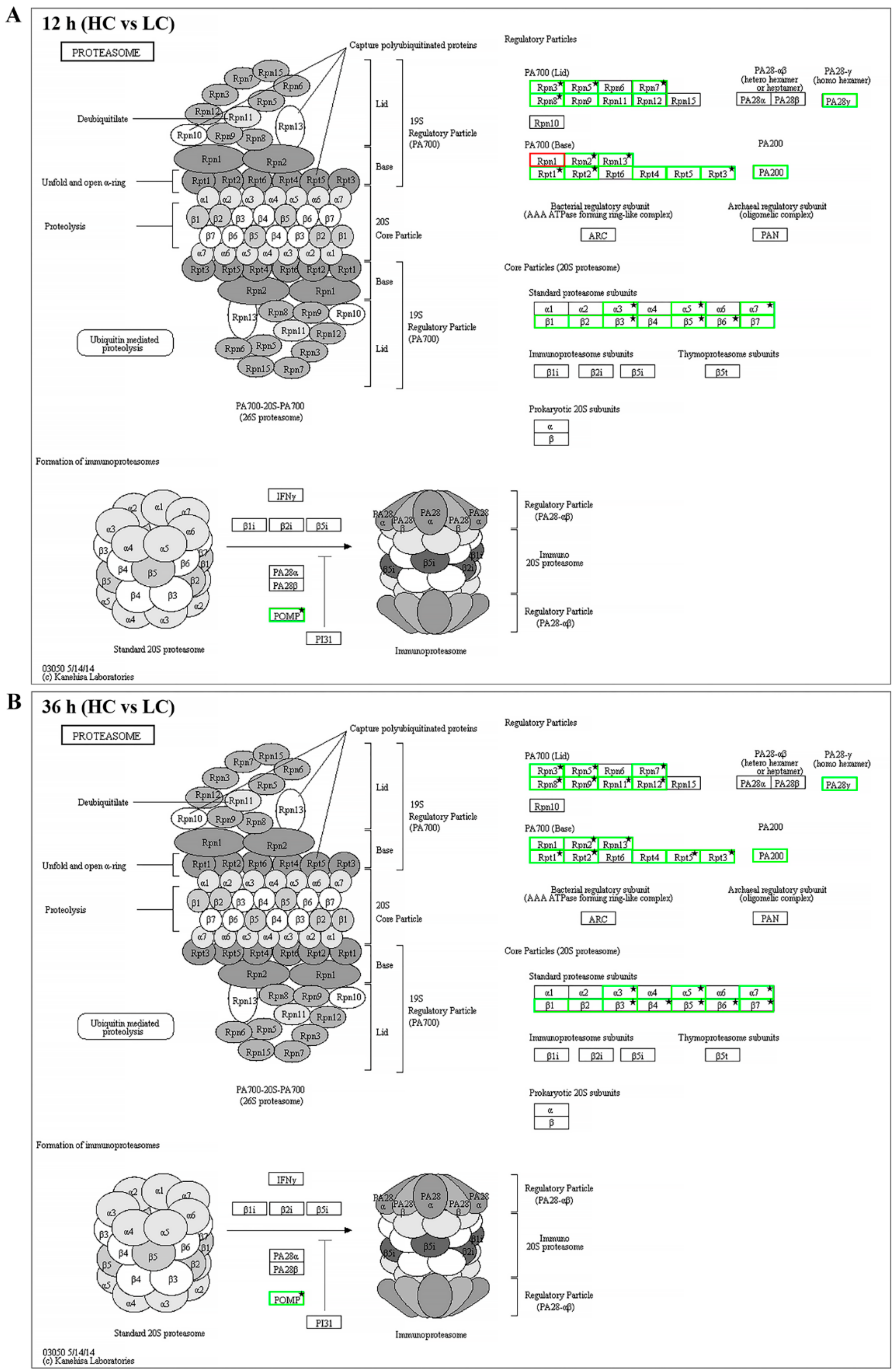
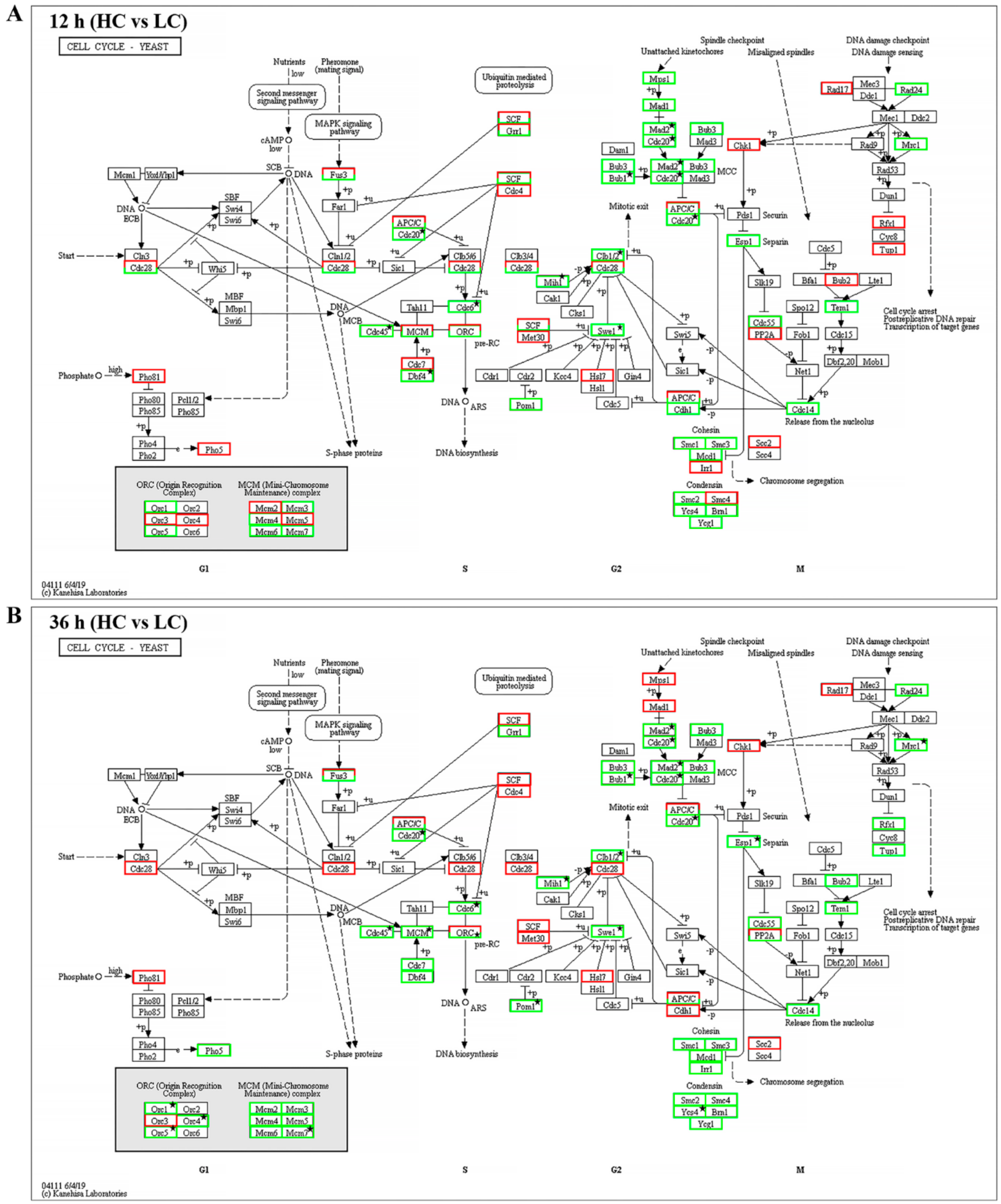
2.4. Proteasome and Cell Cycle Pathways were Down-Regulated under High CO2 Conditions
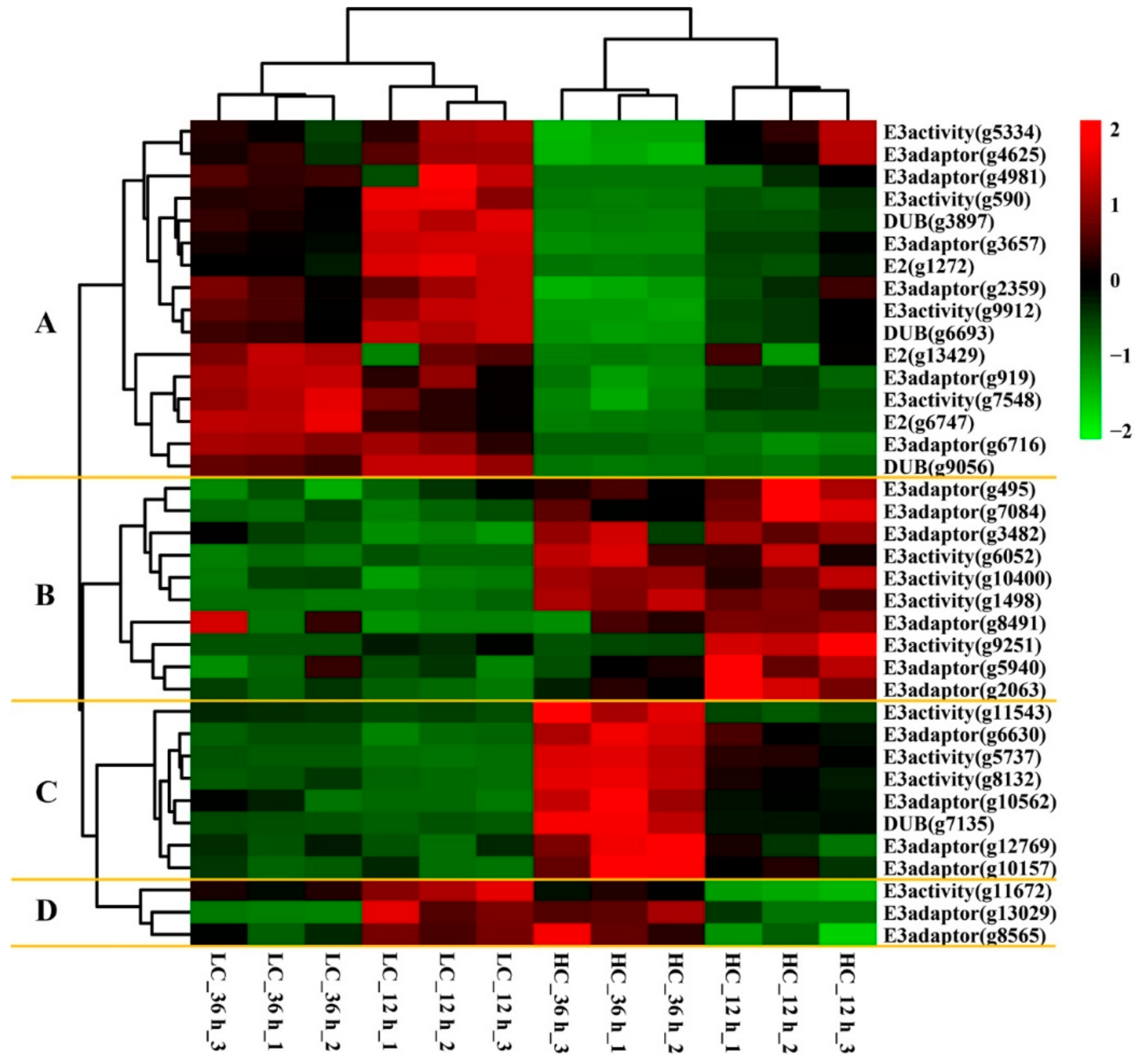
2.5. Ubiquitin-Conjugating Machinery Showed Differential Gene Expression under Different CO2 Conditions
3. Discussion
4. Materials and Methods
4.1. Strain Maintenance and Fruiting Body Cultivation Methods
4.2. Growth of Fruiting Bodies under Different CO2 Concentrations
4.3. Comparison of Fruiting Body Morphology, Pileus Size, and Basidiospore Ejection among Different CO2 Concentration
4.4. Sample Collection
4.5. RNA Extraction, Library Construction, and Sequencing
4.6. Sequence Read Mapping to the F. filiformis Reference Genome
4.7. Quantification and Differential Expression Analysis of Transcripts
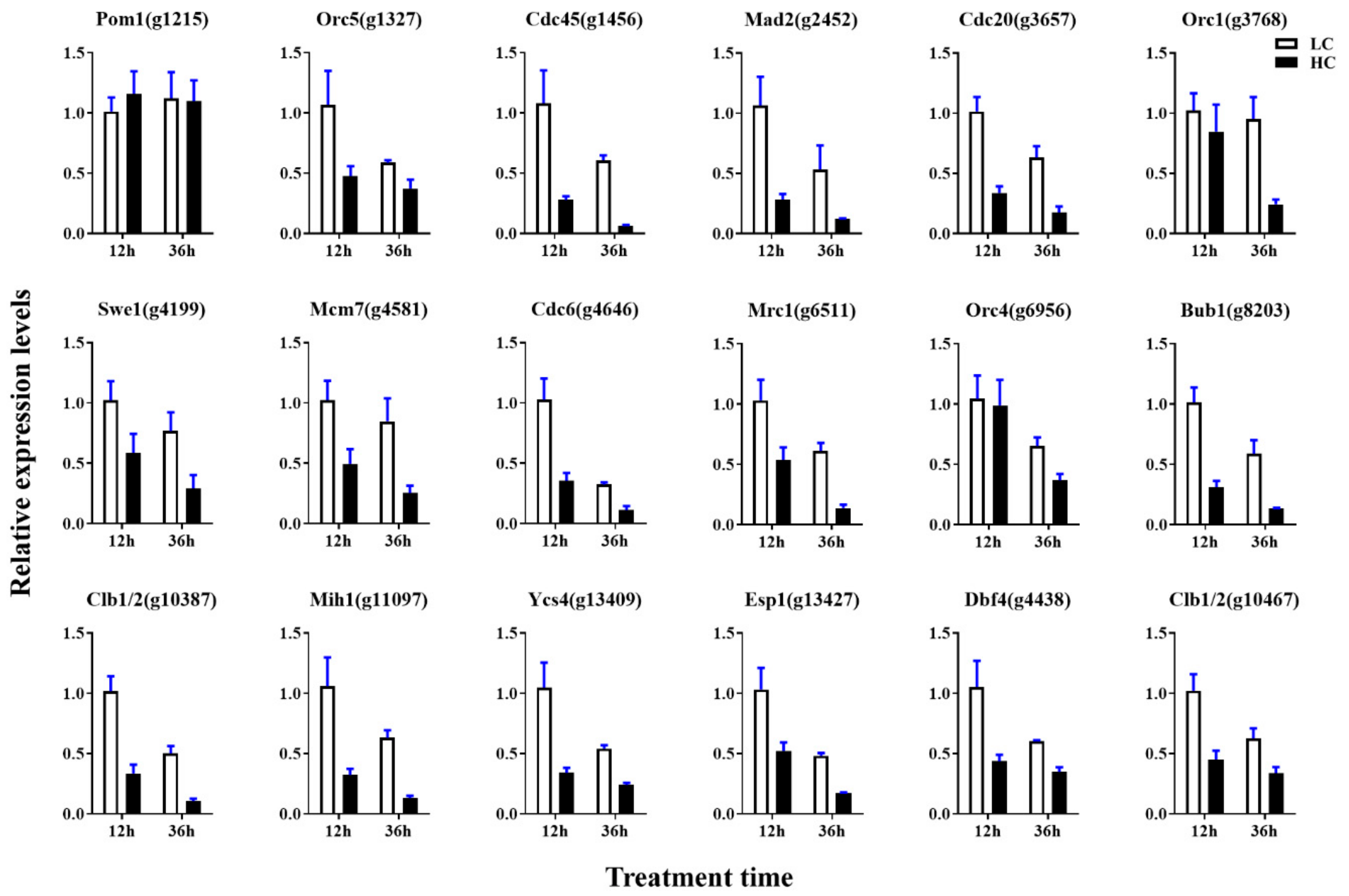
4.8. Validation of the DEGs by qRT-PCR
4.9. Preparation of Paraffin-Embedded Context Tissue for Morphology
4.10. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| cDNA | complementary DNA |
| CO2 | Carbon dioxide |
| DEGs | Differentially expressed genes |
| FC | Fold change |
| FDR | False discovery rate |
| FPKM | Fragments per Kilobase Million |
| GO | Gene ontology |
| HC | High concentration |
| KEGG | Kyoto Encyclopedia of Genes and Genomes |
| LC | Low concentration |
| qRT-PCR | Quantitative Real-time PCR |
References
- Kuees, U.; Navarro-Gonzalez, M. How do Agaricomycetes shape their fruiting bodies? 1. Morphological aspects of development. Fungal Biol. Rev. 2015, 29, 63–97. [Google Scholar] [CrossRef]
- Sakamoto, Y. Influences of environmental factors on fruiting body induction, development and maturation in mushroom-forming fungi. Fungal Biol. Rev. 2018, 32, 236–248. [Google Scholar] [CrossRef]
- Li, C.; Gong, W.; Zhang, L.; Yang, Z.; Nong, W.; Bian, Y.; Kwan, H.-S.; Cheung, M.-K.; Xiao, Y. Association mapping reveals genetic loci associated with important agronomic traits in Lentinula edodes, shiitake mushroom. Front. Microbiol. 2017, 8, 237. [Google Scholar] [CrossRef] [PubMed]
- Kinugawa, K.; Tanesaka, E. Changes in the rate of CO2 release from cultures of three basidiomycetes during cultivation. Trans. Mycol. Soc. Jpn. 1990, 31, 489–500. [Google Scholar]
- Kinugawa, K.; Suzuki, A.; Takamatsu, Y.; Kato, M.; Tanaka, K. Effects of concentrated carbon dioxide on the fruiting of several cultivated basidiomycetes (II). Mycoscience 1994, 35, 345–352. [Google Scholar] [CrossRef]
- Jang, K.-Y.; Jhune, C.-S.; Park, J.-S.; Cho, S.-M.; Weon, H.-Y.; Cheong, J.-C.; Choi, S.-G.; Sung, J.-M. Characterization of Fruitbody Morphology on Various Environmental Conditions in Pleurotus ostreatus. Mycobiology 2003, 31, 145–150. [Google Scholar] [CrossRef]
- Scrase, R.J.; Elliott, T.J. Biology and technology of mushroom culture. In Microbiology of Fermented Foods; Springer: Berlin, Germany, 1998; pp. 543–584. [Google Scholar]
- Morgan, J.A.; Milchunas, D.G.; LeCain, D.R.; West, M.; Mosier, A.R. Carbon dioxide enrichment alters plant community structure and accelerates shrub growth in the shortgrass steppe. Proc. Natl. Acad. Sci. USA 2007, 104, 14724–14729. [Google Scholar] [CrossRef]
- Talmage, S.C.; Gobler, C.J. Effects of past, present, and future ocean carbon dioxide concentrations on the growth and survival of larval shellfish. Proc. Natl. Acad. Sci. USA 2010, 107, 17246–17251. [Google Scholar] [CrossRef]
- Baumann, H.; Talmage, S.C.; Gobler, C.J. Reduced early life growth and survival in a fish in direct response to increased carbon dioxide. Nat. Clim. Change 2012, 2, 38–41. [Google Scholar] [CrossRef]
- Nie, M.; Bell, C.; Wallenstein, M.D.; Pendall, E. Increased plant productivity and decreased microbial respiratory C loss by plant growth-promoting rhizobacteria under elevated CO 2. Sci. Rep. 2015, 5, 9212. [Google Scholar] [CrossRef]
- Bahn, Y.-S.; Cox, G.M.; Perfect, J.R.; Heitman, J. Carbonic anhydrase and CO2 sensing during Cryptococcus neoformans growth, differentiation, and virulence. Curr. Biol. 2005, 15, 2013–2020. [Google Scholar] [CrossRef] [PubMed]
- Klengel, T.; Liang, W.-J.; Chaloupka, J.; Ruoff, C.; Schröppel, K.; Naglik, J.R.; Eckert, S.E.; Mogensen, E.G.; Haynes, K.; Tuite, M.F. Fungal adenylyl cyclase integrates CO2 sensing with cAMP signaling and virulence. Curr. Biol. 2005, 15, 2021–2026. [Google Scholar] [CrossRef] [PubMed]
- Sharabi, K.; Lecuona, E.; Helenius, I.T.; Beitel, G.J.; Sznajder, J.I.; Gruenbaum, Y. Sensing, physiological effects and molecular response to elevated CO2 levels in eukaryotes. J. Cell. Mol. Med. 2009, 13, 4304–4318. [Google Scholar] [CrossRef] [PubMed]
- Azzam, Z.S.; Sharabi, K.; Guetta, J.; Bank, E.M.; Gruenbaum, Y. The physiological and molecular effects of elevated CO2 levels. Cell Cycle 2010, 9, 1528–1532. [Google Scholar] [CrossRef]
- Hall, R.A.; Sordi, L.D.; MacCallum, D.M.; Topal, H.; Eaton, R.; Bloor, J.W.; Robinson, G.K.; Levin, L.R.; Buck, J.; Wang, Y.; et al. CO2 Acts as a Signalling Molecule in Populations of the Fungal Pathogen Candida albicans. PLoS Pathog. 2010, 6, e1001193. [Google Scholar] [CrossRef] [PubMed]
- Kazan, K. Plant-biotic interactions under elevated CO2: A molecular perspective. Environ. Exp. Bot. 2018, 153, 249–261. [Google Scholar] [CrossRef]
- Bahn, Y.-S.; Mühlschlegel, F.A. CO2 sensing in fungi and beyond. Curr. Opin. Microbiol. 2006, 9, 572–578. [Google Scholar] [CrossRef]
- Martin, R.; Pohlers, S.; Mühlschlegel, F.A.; Kurzai, O. CO2 sensing in fungi: At the heart of metabolic signaling. Curr. Genet. 2017, 63, 965–972. [Google Scholar] [CrossRef]
- Lu, Y.; Su, C.; Ray, S.; Yuan, Y.; Liu, H. CO2 Signaling through the Ptc2-Ssn3 Axis Governs Sustained Hyphal Development of Candida albicans by Reducing Ume6 Phosphorylation and Degradation. mBio 2019, 10, e02320-18. [Google Scholar] [CrossRef]
- Royse, D.J.; Baars, J.; Tan, Q. Current overview of mushroom production in the world. Edible Med. Mushrooms Technol. Appl. 2017, 5–13. [Google Scholar] [CrossRef]
- Wang, P.M.; Liu, X.B.; Dai, Y.C.; Horak, E.; Steffen, K.; Yang, Z.L. Phylogeny and species delimitation of Flammulina: Taxonomic status of winter mushroom in East Asia and a new European species identified using an integrated approach. Mycol. Prog. 2018, 17, 1013–1030. [Google Scholar] [CrossRef]
- Attaran Dowom, S.; Rezaeian, S.; Pourianfar, H.R. Agronomic and environmental factors affecting cultivation of the winter mushroom or Enokitake: Achievements and prospects. Appl. Microbiol. Biotechnol. 2019, 103, 2469–2481. [Google Scholar] [CrossRef] [PubMed]
- Nowrousian, M. Genomics and transcriptomics to study fruiting body development: An update. Fungal Biol. Rev. 2018, 32, 231–235. [Google Scholar] [CrossRef]
- Park, Y.-J.; Baek, J.H.; Lee, S.; Kim, C.; Rhee, H.; Kim, H.; Seo, J.-S.; Park, H.-R.; Yoon, D.-E.; Nam, J.-Y. Whole genome and global gene expression analyses of the model mushroom Flammulina velutipes reveal a high capacity for lignocellulose degradation. PLoS ONE 2014, 9, e93560. [Google Scholar] [CrossRef]
- Wang, W.; Liu, F.; Jiang, Y.; Wu, G.; Guo, L.; Chen, R.; Chen, B.; Lu, Y.; Dai, Y.; Xie, B. The multigene family of fungal laccases and their expression in the white rot basidiomycete Flammulina velutipes. Gene 2015, 563, 142–149. [Google Scholar] [CrossRef]
- Liu, F.; Wang, W.; Chen, B.-Z.; Xie, B.-G. Homocitrate synthase expression and lysine content in fruiting body of different developmental stages in Flammulina velutipes. Curr. Microbiol. 2015, 70, 821–828. [Google Scholar] [CrossRef]
- Wang, W.; Lian, L.; Xu, P.; Chou, T.; Mukhtar, I.; Osakina, A.; Waqas, M.; Chen, B.; Liu, X.; Liu, F. Advances in understanding mating type gene organization in the mushroom-forming fungus Flammulina velutipes. G3 Genes Genomes Genet. 2016, 6, 3635–3645. [Google Scholar]
- Liu, J.; Chang, M.; Meng, J.; Feng, C.; Zhao, H.; Zhang, M. Comparative proteome reveals metabolic changes during the fruiting process in Flammulina velutipes. J. Agric. Food Chem. 2017, 65, 5091–5100. [Google Scholar] [CrossRef]
- Liu, J.-Y.; Men, J.; Chang, M.; Feng, C.; Yuan, L.-G. iTRAQ-based quantitative proteome revealed metabolic changes of Flammulina velutipes mycelia in response to cold stress. J. Proteom. 2017, 156, 75–84. [Google Scholar] [CrossRef]
- Wang, W.; Mukhtar, I.; Chou, T.; Jiang, S.; Liu, X.; Van Peer, A.F.; Xie, B. A second HD mating type sublocus of Flammulina velutipes is at least di-allelic and active: New primers for identification of HD-a and HD-b subloci. PeerJ 2019, 7, e6292. [Google Scholar] [CrossRef]
- Ciechanover, A. The ubiquitin-proteasome pathway: On protein death and cell life. EMBO J. 1998, 17, 7151–7160. [Google Scholar] [CrossRef] [PubMed]
- Salazar-Roa, M.; Malumbres, M. Fueling the cell division cycle. Trends Cell Biol. 2017, 27, 69–81. [Google Scholar] [CrossRef] [PubMed]
- Mocciaro, A.; Rape, M. Emerging regulatory mechanisms in ubiquitin-dependent cell cycle control. J. Cell Sci. 2012, 125, 255–263. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, T.; Liu, Z.; Wang, Y.; Cheng, H.; Yang, Q.; Guo, A.; Ren, J.; Xue, Y. UUCD: A family-based database of ubiquitin and ubiquitin-like conjugation. Nucleic Acids Res. 2013, 41, D445–D451. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, F.; Zheng, J.; Tian, X.; Wang, J.; Niu, L.; Feng, W. An automatic sorting system for fresh white button mushrooms based on image processing. Comput. Electron. Agric. 2018, 151, 416–425. [Google Scholar] [CrossRef]
- Sakamoto, Y.; Ando, A.; Tamai, Y.; Yajima, T. Pileus differentiation and pileus-specific protein expression in Flammulina velutipes. Fungal Genet. Biol. 2007, 44, 14–24. [Google Scholar] [CrossRef]
- Muraguchi, H.; Fujita, T.; Kishibe, Y.; Konno, K.; Ueda, N.; Nakahori, K.; Yanagi, S.O.; Kamada, T. The exp1 gene essential for pileus expansion and autolysis of the inky cap mushroom Coprinopsis cinerea (Coprinus cinereus) encodes an HMG protein. Fungal Genet. Biol. 2008, 45, 890–896. [Google Scholar] [CrossRef]
- Niederpruem, D.J. Role of carbon dioxide in the control of fruiting of Schizophyllum commune. J. Bacteriol. 1963, 85, 1300–1308. [Google Scholar]
- Turner, E.M. Development of excised sporocarps of Agaricus bisporus and its control by CO2. Trans. Br. Mycol. Soc. 1977, 69, 183–186. [Google Scholar] [CrossRef]
- Zhao, J.; Tenev, T.; Martins, L.M.; Downward, J.; Lemoine, N.R. The ubiquitin-proteasome pathway regulates survivin degradation in a cell cycle-dependent manner. J. Cell Sci. 2000, 113, 4363–4371. [Google Scholar]
- Teixeira, L.K.; Reed, S.I. Ubiquitin ligases and cell cycle control. Annu. Rev. Biochem. 2013, 82, 387–414. [Google Scholar] [CrossRef] [PubMed]
- Tao, Y.; Chen, R.; Yan, J.; Long, Y.; Tong, Z.; Song, H.; Xie, B. A hydrophobin gene, Hyd9, plays an important role in the formation of aerial hyphae and primordia in Flammulina filiformis. Gene 2019, 706, 84–90. [Google Scholar] [CrossRef] [PubMed]
- Kües, U. Life history and developmental processes in the basidiomycete Coprinus cinereus. Microbiol. Mol. Biol. Rev. 2000, 64, 316–353. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zeng, X.; Liu, F.; Chen, J.; Wang, W.; Xie, B.G.; Guo, S.X. Genomic sequencing and analysis of genes related to terpenoid compound biosynthesis of Flammulina velutipes. Mycosystema 2015, 34, 670–682. [Google Scholar]
- Kim, D.; Langmead, B.; Salzberg, S.L. HISAT: A fast spliced aligner with low memory requirements. Nat. Methods 2015, 12, 357–360. [Google Scholar] [CrossRef] [Green Version]
- Pertea, M.; Pertea, G.M.; Antonescu, C.M.; Chang, T.-C.; Mendell, J.T.; Salzberg, S.L. StringTie enables improved reconstruction of a transcriptome from RNA-seq reads. Nat. Biotechnol. 2015, 33, 290–295. [Google Scholar] [CrossRef] [Green Version]
- Frazee, A.C.; Pertea, G.; Jaffe, A.E.; Langmead, B.; Salzberg, S.L.; Leek, J.T. Ballgown bridges the gap between transcriptome assembly and expression analysis. Nat. Biotechnol. 2015, 33, 243–246. [Google Scholar] [CrossRef] [Green Version]
- Pertea, M.; Kim, D.; Pertea, G.M.; Leek, J.T.; Salzberg, S.L. Transcript-level expression analysis of RNA-seq experiments with HISAT, StringTie and Ballgown. Nat. Protoc. 2016, 11, 1650–1667. [Google Scholar] [CrossRef]
- Conesa, A.; Götz, S.; García-Gómez, J.M.; Terol, J.; Talón, M.; Robles, M. Blast2GO: A universal tool for annotation, visualization and analysis in functional genomics research. Bioinformatics 2005, 21, 3674–3676. [Google Scholar] [CrossRef] [Green Version]
- Moriya, Y.; Itoh, M.; Okuda, S.; Yoshizawa, A.C.; Kanehisa, M. KAAS: An automatic genome annotation and pathway reconstruction server. Nucleic Acids Res. 2007, 35, W182–W185. [Google Scholar] [CrossRef] [Green Version]
- Yan, J.-J.; Zhang, L.; Wang, R.-Q.; Xie, B.; Li, X.; Chen, R.-L.; Guo, L.-X.; Xie, B.-G. The sequence characteristics and expression models reveal superoxide dismutase involved in cold response and fruiting body development in Volvariella volvacea. Int. J. Mol. Sci. 2016, 17, 34. [Google Scholar] [CrossRef] [Green Version]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔCT method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef]
- Tao, Y.; Van Peer, A.F.; Huang, Q.; Shao, Y.; Zhang, L.; Xie, B.; Jiang, Y.; Zhu, J.; Xie, B. Identification of novel and robust internal control genes from Volvariella volvacea that are suitable for RT-qPCR in filamentous fungi. Sci. Rep. 2016, 6, 29236. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Canene-Adams, K. Preparation of formalin-fixed paraffin-embedded tissue for immunohistochemistry. Meth. Enzymol. 2013, 533, 225–233. [Google Scholar] [PubMed]
- Swift, M.L. GraphPad prism, data analysis, and scientific graphing. J. Chem. Inf. Comput. Sci. 1997, 37, 411–412. [Google Scholar] [CrossRef]














© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yan, J.-J.; Tong, Z.-J.; Liu, Y.-Y.; Li, Y.-N.; Zhao, C.; Mukhtar, I.; Tao, Y.-X.; Chen, B.-Z.; Deng, Y.-J.; Xie, B.-G. Comparative Transcriptomics of Flammulina filiformis Suggests a High CO2 Concentration Inhibits Early Pileus Expansion by Decreasing Cell Division Control Pathways. Int. J. Mol. Sci. 2019, 20, 5923. https://doi.org/10.3390/ijms20235923
Yan J-J, Tong Z-J, Liu Y-Y, Li Y-N, Zhao C, Mukhtar I, Tao Y-X, Chen B-Z, Deng Y-J, Xie B-G. Comparative Transcriptomics of Flammulina filiformis Suggests a High CO2 Concentration Inhibits Early Pileus Expansion by Decreasing Cell Division Control Pathways. International Journal of Molecular Sciences. 2019; 20(23):5923. https://doi.org/10.3390/ijms20235923
Chicago/Turabian StyleYan, Jun-Jie, Zong-Jun Tong, Yuan-Yuan Liu, Yi-Ning Li, Chen Zhao, Irum Mukhtar, Yong-Xin Tao, Bing-Zhi Chen, You-Jin Deng, and Bao-Gui Xie. 2019. "Comparative Transcriptomics of Flammulina filiformis Suggests a High CO2 Concentration Inhibits Early Pileus Expansion by Decreasing Cell Division Control Pathways" International Journal of Molecular Sciences 20, no. 23: 5923. https://doi.org/10.3390/ijms20235923