APOBEC3-Mediated RNA Editing in Breast Cancer is Associated with Heightened Immune Activity and Improved Survival



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
2.1. Compilation of a Set of APOBEC3-Mediated RNA Editing Sites
2.2. Estimation of RNA Editing Levels in TCGA-BRCA Tissues
2.3. APOBEC3-Mediated C-to-U RNA Editing is Prevalent in Breast Cancer Tumors
2.4. APOBEC3 Gene Expression Correlates with RNA Editing Better Than APOBEC-Mediated Mutations
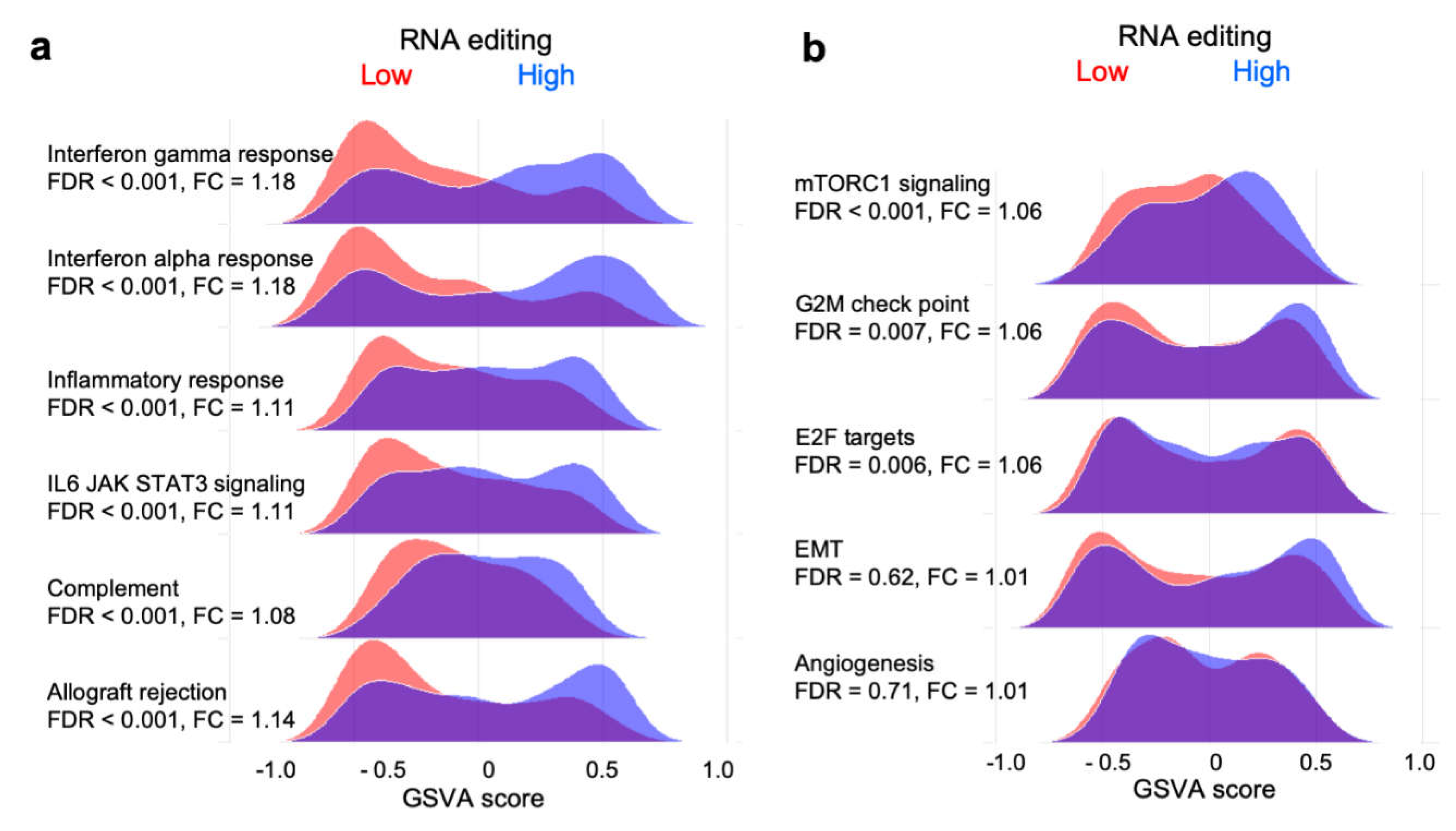
2.5. RNA Editing is Associated with Enriched Expression of Immune- but not Cancer Development-Related Gene Sets
2.6. Immune Activity is Elevated in Tumors with High RNA Editing
2.7. Patients with Tumors with High RNA Editing in Tumors have Better Survival
3. Discussion
4. Materials and Methods
4.1. Collation of APOBEC3-Mediated C-to-U RNA Editing Sites
4.2. Access to Controlled Sequence Alignment Data of TCGA Project
4.3. Estimation of RNA Editing Levels in TCGA Tissues
4.4. Validation of RNA Editing Level Estimates Using a Different Sequencing Read Alignment
4.5. Collection of Clinical and Pathologic Information of TCGA Subjects
4.6. Quantification of TCGA Tumor Genome Features
4.7. Generation of TCGA Tumor Gene Expression Dataset
4.8. Analyses of Gene Expression Data
4.9. Measurements of TCGA Tumor Immune Microenvironment
4.10. Other
Supplementary Materials
Author Contributions
Acknowledgments
Conflicts of Interest
References
- Bransteitter, R.; Prochnow, C.; Chen, X.S. The current structural and functional understanding of APOBEC deaminases. Cell Mol. Life Sci. 2009, 66, 3137–3147. [Google Scholar] [CrossRef] [PubMed]
- Fritz, E.L.; Rosenberg, B.R.; Lay, K.; Mihailovic, A.; Tuschl, T.; Papavasiliou, F.N. A comprehensive analysis of the effects of the deaminase AID on the transcriptome and methylome of activated B cells. Nat. Immunol. 2013, 14, 749–755. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smith, H.C.; Bennett, R.P.; Kizilyer, A.; McDougall, W.M.; Prohaska, K.M. Functions and regulation of the APOBEC family of proteins. Semin Cell Dev. Biol. 2012, 23, 258–268. [Google Scholar] [CrossRef] [PubMed]
- Gu, T.; Buaas, F.W.; Simons, A.K.; Ackert-Bicknell, C.L.; Braun, R.E.; Hibbs, M.A. Canonical A-to-I and C-to-U RNA editing is enriched at 3’UTRs and microRNA target sites in multiple mouse tissues. Plos ONE 2012, 7, e33720. [Google Scholar] [CrossRef] [PubMed]
- Peng, Z.; Cheng, Y.; Tan, B.C.; Kang, L.; Tian, Z.; Zhu, Y.; Zhang, W.; Liang, Y.; Hu, X.; Tan, X.; et al. Comprehensive analysis of RNA-Seq data reveals extensive RNA editing in a human transcriptome. Nat. Biotechnol. 2012, 30, 253–260. [Google Scholar] [CrossRef] [PubMed]
- Bahn, J.H.; Lee, J.H.; Li, G.; Greer, C.; Peng, G.; Xiao, X. Accurate identification of A-to-I RNA editing in human by transcriptome sequencing. Genome Res. 2012, 22, 142–150. [Google Scholar] [CrossRef] [PubMed]
- Harris, R.S.; Dudley, J.P. APOBECs and virus restriction. Virology 2015, 479–480, 131–145. [Google Scholar] [CrossRef]
- Refsland, E.W.; Harris, R.S. The APOBEC3 family of retroelement restriction factors. Curr Top. Microbiol. Immunol. 2013, 371, 1–27. [Google Scholar]
- Burns, M.B.; Temiz, N.A.; Harris, R.S. Evidence for APOBEC3B mutagenesis in multiple human cancers. Nat. Genet. 2013, 45, 977–983. [Google Scholar] [CrossRef] [Green Version]
- Alexandrov, L.B.; Nik-Zainal, S.; Wedge, D.C.; Aparicio, S.A.; Behjati, S.; Biankin, A.V.; Bignell, G.R.; Bolli, N.; Borg, A.; Borresen-Dale, A.L.; et al. Signatures of mutational processes in human cancer. Nature 2013, 500, 415–421. [Google Scholar] [CrossRef] [Green Version]
- Leonard, B.; Hart, S.N.; Burns, M.B.; Carpenter, M.A.; Temiz, N.A.; Rathore, A.; Vogel, R.I.; Nikas, J.B.; Law, E.K.; Brown, W.L.; et al. APOBEC3B upregulation and genomic mutation patterns in serous ovarian carcinoma. Cancer Res. 2013, 73, 7222–7231. [Google Scholar] [CrossRef] [PubMed]
- Roberts, S.A.; Lawrence, M.S.; Klimczak, L.J.; Grimm, S.A.; Fargo, D.; Stojanov, P.; Kiezun, A.; Kryukov, G.V.; Carter, S.L.; Saksena, G.; et al. An APOBEC cytidine deaminase mutagenesis pattern is widespread in human cancers. Nat. Genet. 2013, 45, 970–976. [Google Scholar] [CrossRef]
- Nik-Zainal, S.; Wedge, D.C.; Alexandrov, L.B.; Petljak, M.; Butler, A.P.; Bolli, N.; Davies, H.R.; Knappskog, S.; Martin, S.; Papaemmanuil, E.; et al. Association of a germline copy number polymorphism of APOBEC3A and APOBEC3B with burden of putative APOBEC-dependent mutations in breast cancer. Nat. Genet. 2014, 46, 487–491. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Caval, V.; Suspene, R.; Shapira, M.; Vartanian, J.P.; Wain-Hobson, S. A prevalent cancer susceptibility APOBEC3A hybrid allele bearing APOBEC3B 3’UTR enhances chromosomal DNA damage. Nat. Commun. 2014, 5, 5129. [Google Scholar] [CrossRef] [PubMed]
- Prohaska, K.M.; Bennett, R.P.; Salter, J.D.; Smith, H.C. The multifaceted roles of RNA binding in APOBEC cytidine deaminase functions. Wiley Interdiscip Rev. RNA 2014. [Google Scholar] [CrossRef]
- Sharma, S.; Patnaik, S.K.; Kemer, Z.; Baysal, B.E. Transient overexpression of exogenous APOBEC3A causes C-to-U RNA editing of thousands of genes. RNA Biol. 2017, 14, 603–610. [Google Scholar] [CrossRef]
- Sharma, S.; Patnaik, S.K.; Taggart, R.T.; Baysal, B.E. The double-domain cytidine deaminase APOBEC3G is a cellular site-specific RNA editing enzyme. Sci. Rep. 2016, 6, 39100. [Google Scholar] [CrossRef]
- Sharma, S.; Patnaik, S.K.; Taggart, R.T.; Kannisto, E.D.; Enriquez, S.M.; Gollnick, P.; Baysal, B.E. APOBEC3A cytidine deaminase induces RNA editing in monocytes and macrophages. Nat. Commun. 2015, 6, 6881. [Google Scholar] [CrossRef]
- Patnaik, S.K.; Kannisto, E.D. APOBEC3B is a new RNA editing enzyme. In Proceedings of the RNA 2016, Annual Meeting of RNA Society, Kyoto, Japan, 28 June–2 July 2016. [Google Scholar]
- Cescon, D.W.; Haibe-Kains, B. DNA replication stress: A source of APOBEC3B expression in breast cancer. Genome Biol. 2016, 17, 202. [Google Scholar] [CrossRef]
- Trevino, V. Integrative genomic analysis identifies associations of molecular alterations to APOBEC and BRCA1/2 mutational signatures in breast cancer. Mol. Genet. Genom. Med. 2019, 7, e810. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harris, R.S. Molecular mechanism and clinical impact of APOBEC3B-catalyzed mutagenesis in breast cancer. Breast Cancer Res. 2015, 17, 8. [Google Scholar] [CrossRef] [PubMed]
- Sieuwerts, A.M.; Doebar, S.C.; de Weerd, V.; Verhoef, E.I.; Beauford, C.M.; Agahozo, M.C.; Martens, J.W.M.; van Deurzen, C.H.M. APOBEC3B Gene Expression in Ductal Carcinoma In Situ and Synchronous Invasive Breast Cancer. Cancers (Basel) 2019, 11, 1062. [Google Scholar] [CrossRef] [PubMed]
- Liao, Y.; Smyth, G.K.; Shi, W. The Subread aligner: Fast, accurate and scalable read mapping by seed-and-vote. Nucleic Acids Res. 2013, 41, e108. [Google Scholar] [CrossRef]
- Hanzelmann, S.; Castelo, R.; Guinney, J. GSVA: Gene set variation analysis for microarray and RNA-seq data. Bmc Bioinform. 2013, 14, 7. [Google Scholar] [CrossRef]
- Chen, B.; Khodadoust, M.S.; Liu, C.L.; Newman, A.M.; Alizadeh, A.A. Profiling Tumor Infiltrating Immune Cells with CIBERSORT. Methods Mol. Biol. 2018, 1711, 243–259. [Google Scholar]
- Liu, J.; Lichtenberg, T.; Hoadley, K.A.; Poisson, L.M.; Lazar, A.J.; Cherniack, A.D.; Kovatich, A.J.; Benz, C.C.; Levine, D.A.; Lee, A.V.; et al. An Integrated TCGA Pan-Cancer Clinical Data Resource to Drive High-Quality Survival Outcome Analytics. Cell 2018, 173, 400–416.e11. [Google Scholar] [CrossRef] [Green Version]
- Hong, H.; An, O.; Chan, T.H.M.; Ng, V.H.E.; Kwok, H.S.; Lin, J.S.; Qi, L.; Han, J.; Tay, D.J.T.; Tang, S.J.; et al. Bidirectional regulation of adenosine-to-inosine (A-to-I) RNA editing by DEAH box helicase 9 (DHX9) in cancer. Nucleic Acids Res. 2018, 46, 7953–7969. [Google Scholar] [CrossRef]
- Kung, C.P.; Maggi, L.B., Jr.; Weber, J.D. The Role of RNA Editing in Cancer Development and Metabolic Disorders. Front. Endocrinol. (Lausanne) 2018, 9, 762. [Google Scholar] [CrossRef] [Green Version]
- Ruan, H.; Han, L. Regulatory RNA Editing Sites in Cancer: Prediction and Beyond. EBioMedicine 2018, 27, 7–8. [Google Scholar] [CrossRef]
- Chigaev, M.; Yu, H.; Samuels, D.C.; Sheng, Q.; Oyebamiji, O.; Ness, S.; Yue, W.; Zhao, Y.Y.; Guo, Y. Genomic Positional Dissection of RNA Editomes in Tumor and Normal Samples. Front. Genet. 2019, 10, 211. [Google Scholar] [CrossRef] [PubMed]
- Peng, X.; Xu, X.; Wang, Y.; Hawke, D.H.; Yu, S.; Han, L.; Zhou, Z.; Mojumdar, K.; Jeong, K.J.; Labrie, M.; et al. A-to-I RNA Editing Contributes to Proteomic Diversity in Cancer. Cancer Cell 2018, 33, 817–828.e7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sagredo, E.A.; Blanco, A.; Sagredo, A.I.; Perez, P.; Sepulveda-Hermosilla, G.; Morales, F.; Muller, B.; Verdugo, R.; Marcelain, K.; Harismendy, O.; et al. ADAR1-mediated RNA-editing of 3’UTRs in breast cancer. Biol. Res. 2018, 51, 36. [Google Scholar] [CrossRef] [PubMed]
- Saraconi, G.; Severi, F.; Sala, C.; Mattiuz, G.; Conticello, S.G. The RNA editing enzyme APOBEC1 induces somatic mutations and a compatible mutational signature is present in esophageal adenocarcinomas. Genome Biol. 2014, 15, 417. [Google Scholar] [CrossRef]
- Swanton, C.; McGranahan, N.; Starrett, G.J.; Harris, R.S. APOBEC Enzymes: Mutagenic Fuel for Cancer Evolution and Heterogeneity. Cancer Discov 2015, 5, 704–712. [Google Scholar] [CrossRef] [Green Version]
- Gao, G.F.; Parker, J.S.; Reynolds, S.M.; Silva, T.C.; Wang, L.B.; Zhou, W.; Akbani, R.; Bailey, M.; Balu, S.; Berman, B.P.; et al. Before and After: Comparison of Legacy and Harmonized TCGA Genomic Data Commons’ Data. Cell Syst. 2019, 9, 24–34.e10. [Google Scholar] [CrossRef]
- Wang, T.; Liu, J.; Shen, L.; Tonti-Filippini, J.; Zhu, Y.; Jia, H.; Lister, R.; Whitaker, J.W.; Ecker, J.R.; Millar, A.H.; et al. STAR: An integrated solution to management and visualization of sequencing data. Bioinformatics 2013, 29, 3204–3210. [Google Scholar] [CrossRef]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef] [Green Version]
- Jacobson, R.S.; Becich, M.J.; Bollag, R.J.; Chavan, G.; Corrigan, J.; Dhir, R.; Feldman, M.D.; Gaudioso, C.; Legowski, E.; Maihle, N.J.; et al. A Federated Network for Translational Cancer Research Using Clinical Data and Biospecimens. Cancer Res. 2015, 75, 5194–5201. [Google Scholar] [CrossRef]
- Cerami, E.; Gao, J.; Dogrusoz, U.; Gross, B.E.; Sumer, S.O.; Aksoy, B.A.; Jacobsen, A.; Byrne, C.J.; Heuer, M.L.; Larsson, E.; et al. The cBio cancer genomics portal: An open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012, 2, 401–404. [Google Scholar] [CrossRef]
- Thorsson, V.; Gibbs, D.L.; Brown, S.D.; Wolf, D.; Bortone, D.S.; Ou Yang, T.H.; Porta-Pardo, E.; Gao, G.F.; Plaisier, C.L.; Eddy, J.A.; et al. The Immune Landscape of Cancer. Immunity 2018, 48, 812–830.e14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mayakonda, A.; Lin, D.C.; Assenov, Y.; Plass, C.; Koeffler, H.P. Maftools: Efficient and comprehensive analysis of somatic variants in cancer. Genome Res. 2018, 28, 1747–1756. [Google Scholar] [CrossRef] [PubMed]
- Colaprico, A.; Silva, T.C.; Olsen, C.; Garofano, L.; Cava, C.; Garolini, D.; Sabedot, T.S.; Malta, T.M.; Pagnotta, S.M.; Castiglioni, I.; et al. TCGAbiolinks: An R/Bioconductor package for integrative analysis of TCGA data. Nucleic Acids Res. 2016, 44, e71. [Google Scholar] [CrossRef] [PubMed]
- Durinck, S.; Moreau, Y.; Kasprzyk, A.; Davis, S.; De Moor, B.; Brazma, A.; Huber, W. BioMart and Bioconductor: A powerful link between biological databases and microarray data analysis. Bioinformatics 2005, 21, 3439–3440. [Google Scholar] [CrossRef]
- Smyth, G. Limma: Linear models for microarray data. In Bioinformatics and Computational Biology Solutions using R and Bioconductor; Gentleman, R., Carey, V.J., Huber, W., Dudoit, S., Irizarry, R.A., Eds.; Springer: New York, NY, USA, 2005; pp. 397–420. [Google Scholar]
- Rooney, M.S.; Shukla, S.A.; Wu, C.J.; Getz, G.; Hacohen, N. Molecular and genetic properties of tumors associated with local immune cytolytic activity. Cell 2015, 160, 48–61. [Google Scholar] [CrossRef]
- Mi, H.; Thomas, P. PANTHER pathway: An ontology-based pathway database coupled with data analysis tools. Methods Mol. Biol. 2009, 563, 123–140. [Google Scholar]






© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Asaoka, M.; Ishikawa, T.; Takabe, K.; Patnaik, S.K. APOBEC3-Mediated RNA Editing in Breast Cancer is Associated with Heightened Immune Activity and Improved Survival. Int. J. Mol. Sci. 2019, 20, 5621. https://doi.org/10.3390/ijms20225621
Asaoka M, Ishikawa T, Takabe K, Patnaik SK. APOBEC3-Mediated RNA Editing in Breast Cancer is Associated with Heightened Immune Activity and Improved Survival. International Journal of Molecular Sciences. 2019; 20(22):5621. https://doi.org/10.3390/ijms20225621
Chicago/Turabian StyleAsaoka, Mariko, Takashi Ishikawa, Kazuaki Takabe, and Santosh K. Patnaik. 2019. "APOBEC3-Mediated RNA Editing in Breast Cancer is Associated with Heightened Immune Activity and Improved Survival" International Journal of Molecular Sciences 20, no. 22: 5621. https://doi.org/10.3390/ijms20225621