Insights into the Interaction Mechanisms of the Proviral Integration Site of Moloney Murine Leukemia Virus (Pim) Kinases with Pan-Pim Inhibitors PIM447 and AZD1208: A Molecular Dynamics Simulation and MM/GBSA Calculation Study


Abstract
:
1. Introduction
2. Results and Discussion
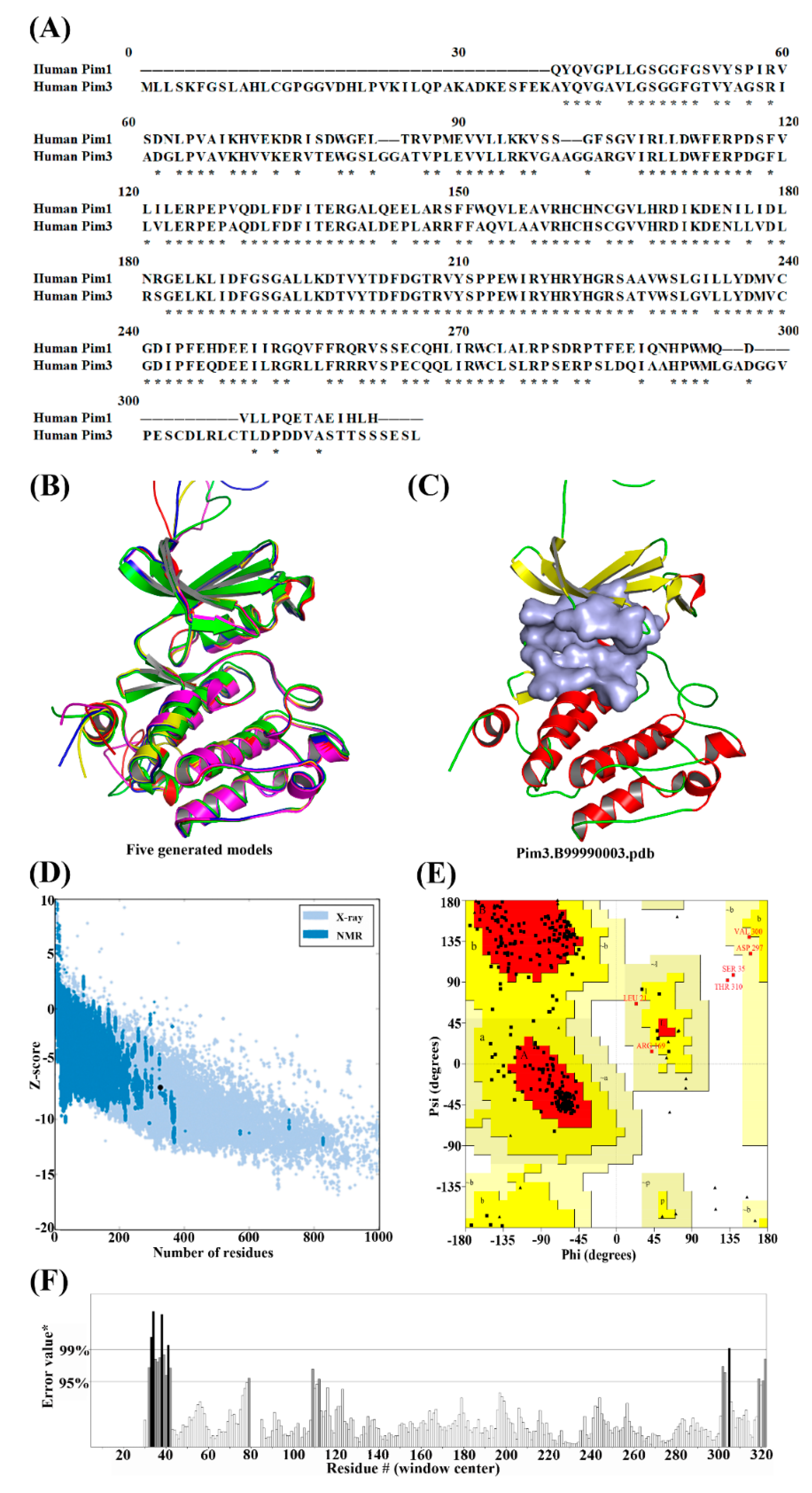
2.1. Molecular Modeling of Pim3
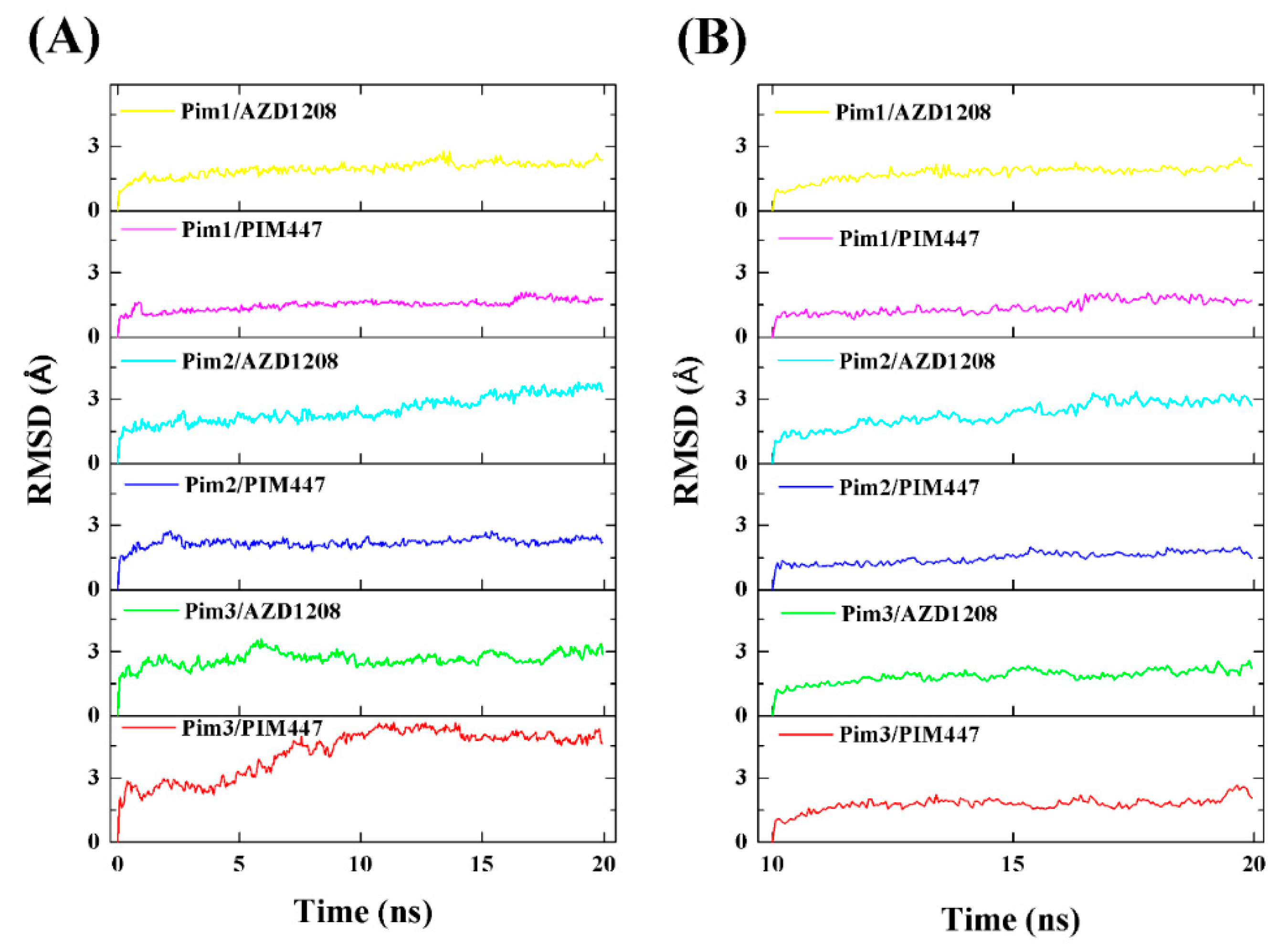
2.2. Overall Structure and Dynamics
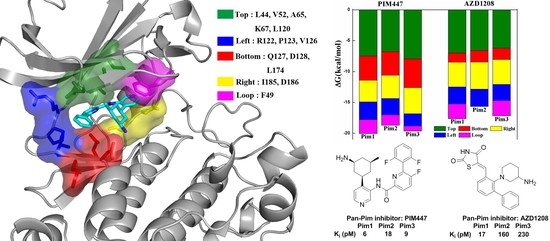
2.3. Binding Free Energies Estimated by MM/GBSA
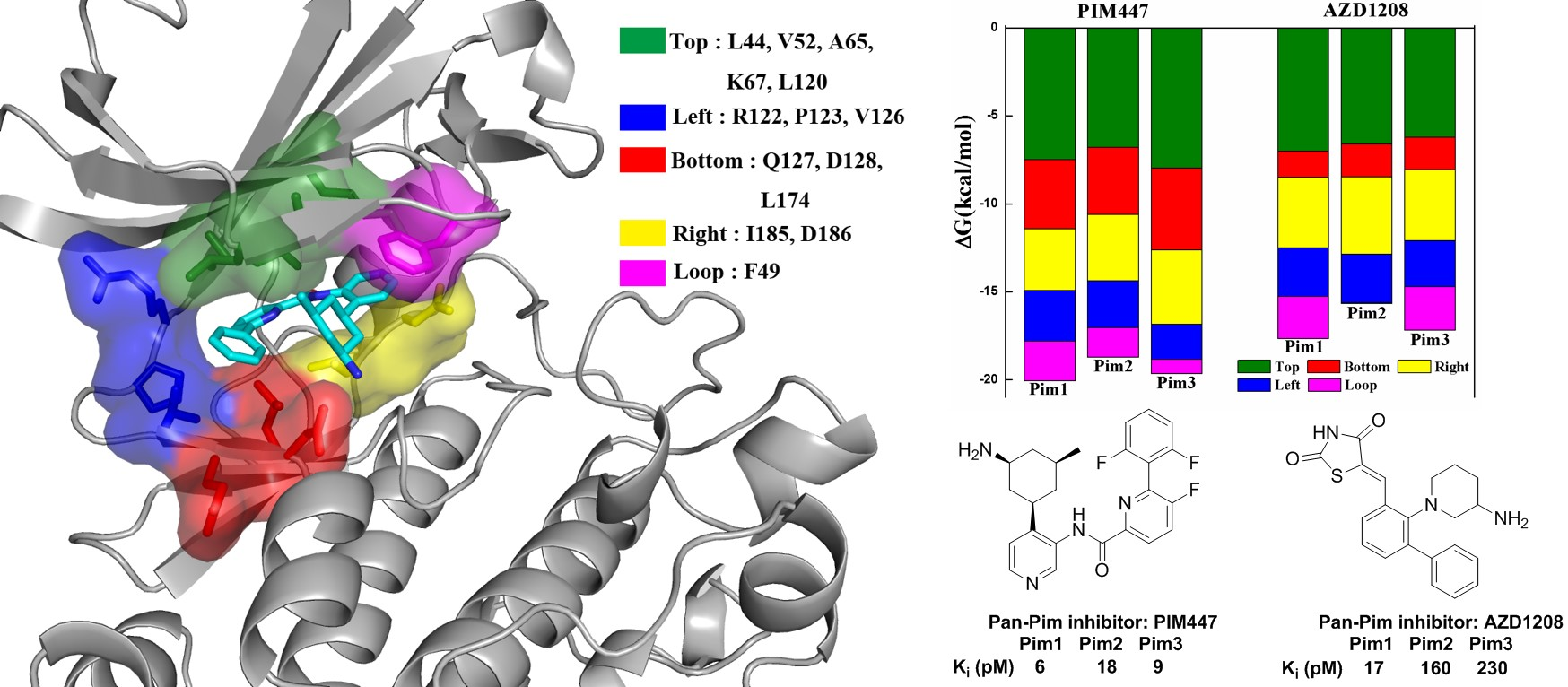
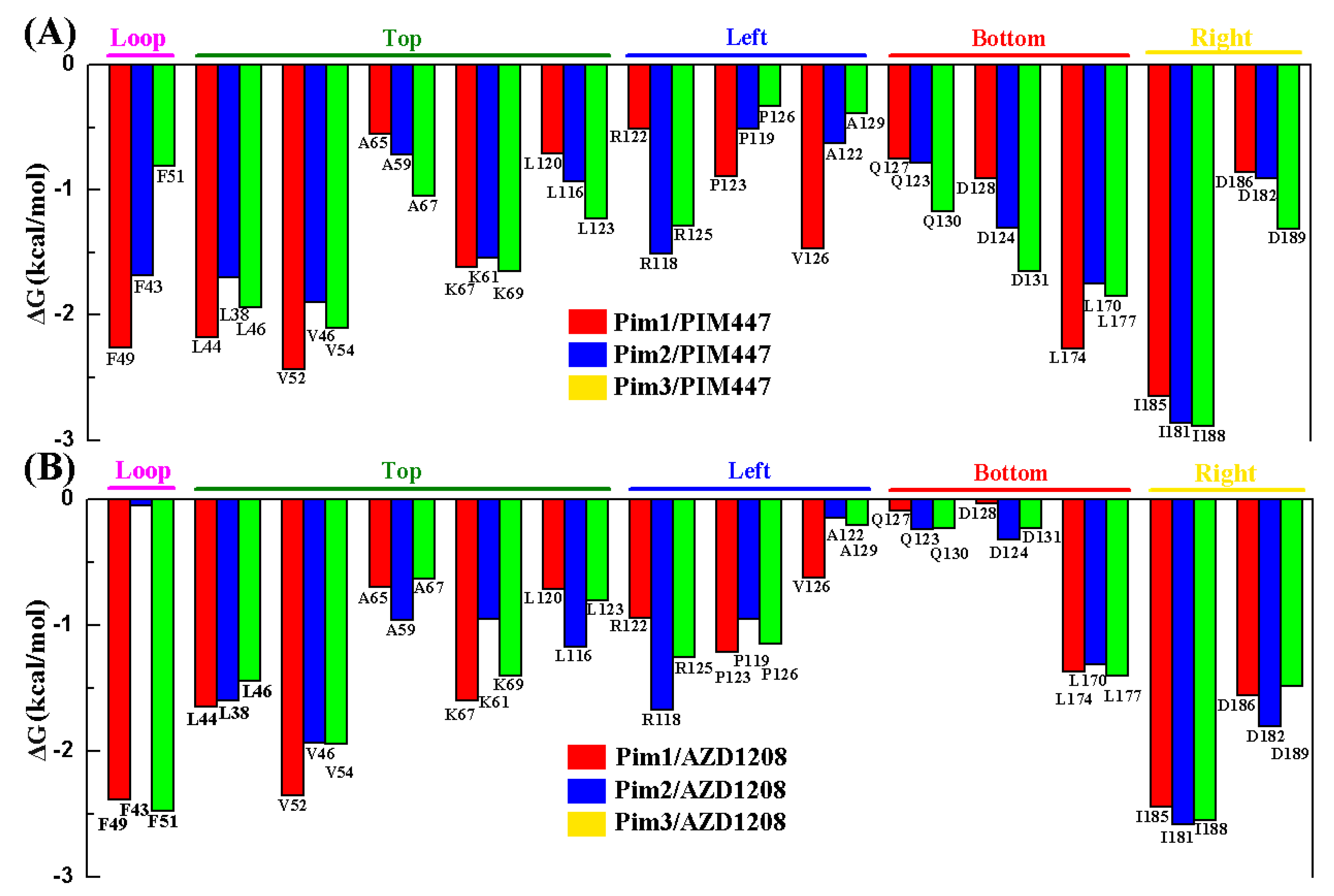
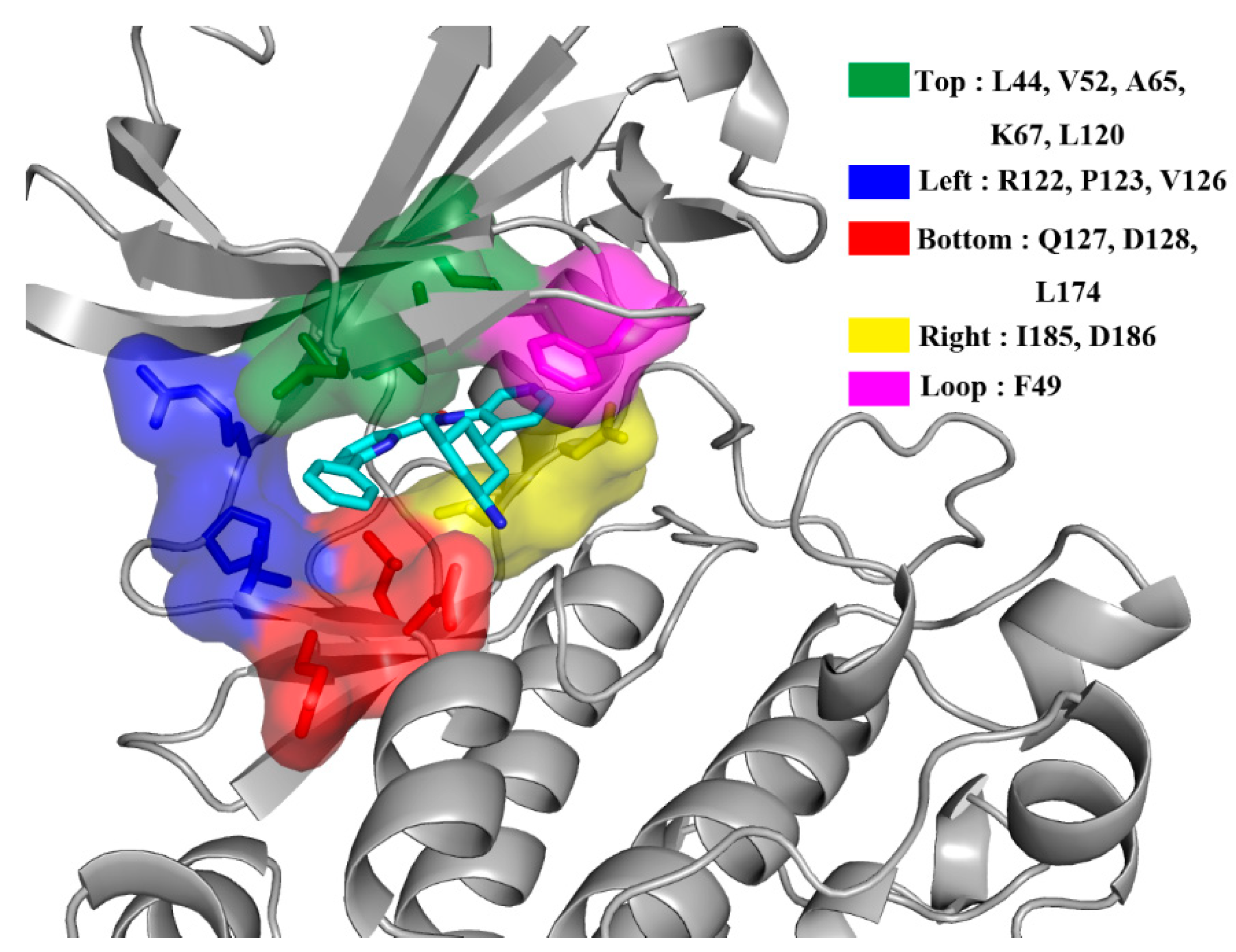
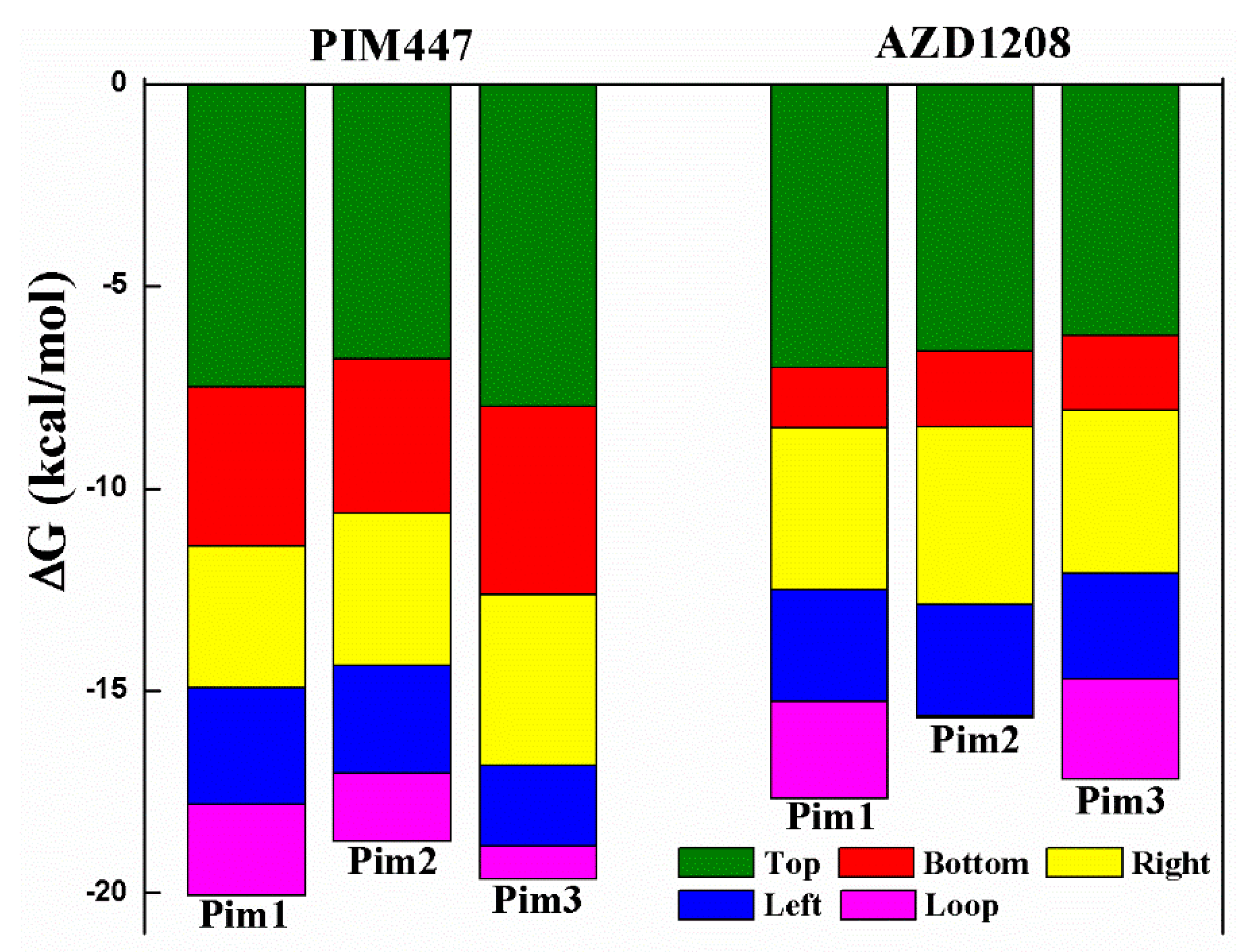
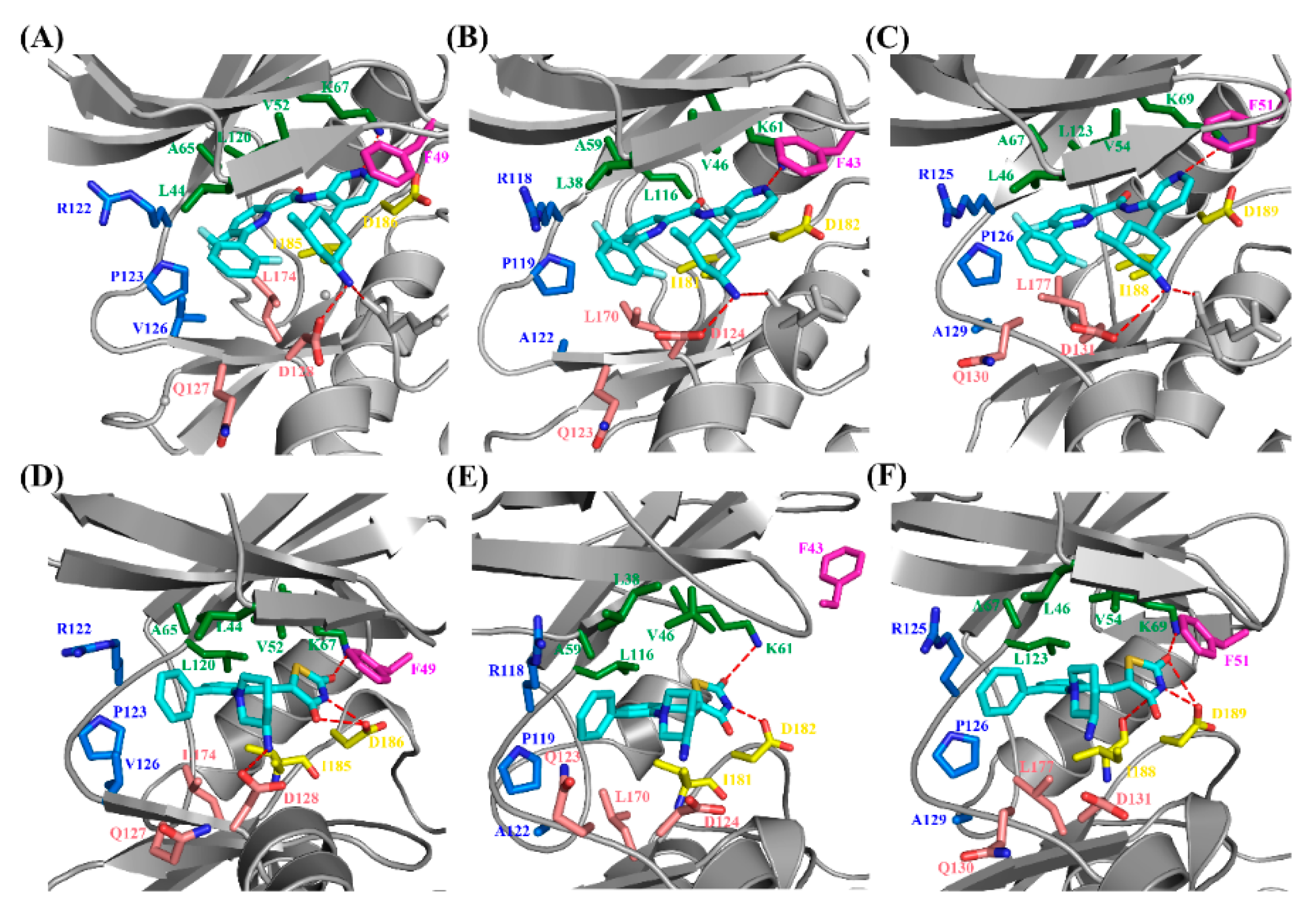
2.4. Decomposition Analysis of the Binding Free Energies
3. Materials and Methods
3.1. Homology Model of Pim3
3.2. Optimization of Homology Model of Pim3
3.3. Construction of Six Pim/Ligand Systems
3.4. Molecular Dynamics Simulations
3.5. MM/GBSA Calculation and MM/GBSA Free Energy Decomposition Analysis
= ΔEMM + ΔGGB + ΔGSA − TΔS
= ΔEvdw + ΔEele + ΔGGB + ΔGSA − TΔS
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| Pim | proviral integration site of the Moloney murine leukemia virus |
| MD | molecular dynamics |
| MM/GBSA | molecular mechanics generalized Born/surface area |
References
- Wang, H.L.; Cee, V.J.; Chavez, F.F., Jr.; Lanman, B.A.; Reed, A.B.; Wu, B.; Guerrero, N.; Lipford, J.R.; Sastri, C.; Winston, J.; et al. The discovery of novel 3-(pyrazin-2-yl)-1H-indazoles as potent pan-Pim kinase inhibitors. Bioorganic Med. Chem. Lett. 2015, 25, 834–840. [Google Scholar] [CrossRef] [PubMed]
- Koblish, H.; Li, Y.L.; Shin, N.; Hall, L.; Wang, Q.; Wang, K.; Covington, M.; Marando, C.; Bowman, K.; Boer, J.; et al. Preclinical characterization of INCB053914, a novel pan-PIM kinase inhibitor, alone and in combination with anticancer agents, in models of hematologic malignancies. PLoS ONE 2018, 13, e0199108. [Google Scholar] [CrossRef] [PubMed]
- Debreczeni, J.E.; Bullock, A.N.; Atilla, G.E.; Williams, D.S.; Bregman, H.; Knapp, S.; Meggers, E. Ruthenium half-sandwich complexes bound to protein kinase Pim-1. Angew. Chem. (Int. Ed. Engl.) 2006, 45, 1580–1585. [Google Scholar] [CrossRef] [PubMed]
- Bregman, H.; Meggers, E. Ruthenium half-sandwich complexes as protein kinase inhibitors: An N-succinimidyl ester for rapid derivatizations of the cyclopentadienyl moiety. Org. Lett. 2006, 8, 5465–5468. [Google Scholar] [CrossRef]
- Pogacic, V.; Bullock, A.N.; Fedorov, O.; Filippakopoulos, P.; Gasser, C.; Biondi, A.; Meyer-Monard, S.; Knapp, S.; Schwaller, J. Structural analysis identifies imidazo[1–b]pyridazines as PIM kinase inhibitors with in vitro antileukemic activity. Cancer Res. 2007, 67, 6916–6924. [Google Scholar] [CrossRef]
- Pierce, A.C.; Jacobs, M.; Stuver-Moody, C. Docking study yields four novel inhibitors of the protooncogene Pim-1 kinase. J. Med. Chem. 2008, 51, 1972–1975. [Google Scholar] [CrossRef]
- Qian, K.; Wang, L.; Cywin, C.L.; Farmer, B.T., 2nd; Hickey, E.; Homon, C.; Jakes, S.; Kashem, M.A.; Lee, G.; Leonard, S.; et al. Hit to lead account of the discovery of a new class of inhibitors of Pim kinases and crystallographic studies revealing an unusual kinase binding mode. J. Med. Chem. 2009, 52, 1814–1827. [Google Scholar] [CrossRef]
- Wang, X.; Blackaby, W.; Allen, V.; Chan, G.K.Y.; Chang, J.H.; Chiang, P.C.; Diene, C.; Drummond, J.; Do, S.; Fan, E.; et al. Optimization of Pan-Pim Kinase Activity and Oral Bioavailability Leading to Diaminopyrazole (GDC-0339) for the Treatment of Multiple Myeloma. J. Med. Chem. 2019, 62, 2140–2153. [Google Scholar] [CrossRef]
- Wang, X.; Kolesnikov, A.; Tay, S.; Chan, G.; Chao, Q.; Do, S.; Drummond, J.; Ebens, A.J.; Liu, N.; Ly, J.; et al. Discovery of 5-Azaindazole (GNE-955) as a Potent Pan-Pim Inhibitor with Optimized Bioavailability. J. Med. Chem. 2017, 60, 4458–4473. [Google Scholar] [CrossRef]
- Keeton, E.K.; McEachern, K.; Dillman, K.S.; Palakurthi, S.; Cao, Y.; Grondine, M.R.; Kaur, S.; Wang, S.; Chen, Y.; Wu, A.; et al. AZD1208, a potent and selective pan-Pim kinase inhibitor, demonstrates efficacy in preclinical models of acute myeloid leukemia. Blood 2014, 123, 905–913. [Google Scholar] [CrossRef] [Green Version]
- Burger, M.T.; Nishiguchi, G.; Han, W.; Lan, J.; Simmons, R.; Atallah, G.; Ding, Y.; Tamez, V.; Zhang, Y.; Mathur, M.; et al. Identification of N-(4-((1R,3S,5S)-3-Amino-5-methylcyclohexyl)pyridin-3-yl)-6-(2,6-difluorophenyl)- 5-fluoropicolinamide (PIM447), a Potent and Selective Proviral Insertion Site of Moloney Murine Leukemia (PIM) 1, 2, and 3 Kinase Inhibitor in Clinical Trials for Hematological Malignancies. J. Med. Chem. 2015, 58, 8373–8386. [Google Scholar] [CrossRef] [PubMed]
- Garba, L.; Mohamad Yussoff, M.A.; Abd Halim, K.B.; Ishak, S.N.H.; Mohamad Ali, M.S.; Oslan, S.N.; Raja Abd Rahman, R.N.Z. Homology modeling and docking studies of a Delta9-fatty acid desaturase from a Cold-tolerant Pseudomonas sp. AMS8. PeerJ 2018, 6, e4347. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Liang, Y.; Zhang, Y. Atomic-level protein structure refinement using fragment-guided molecular dynamics conformation sampling. Structure 2011, 19, 1784–1795. [Google Scholar] [CrossRef] [PubMed]
- Marti-Renom, M.A.; Stuart, A.C.; Fiser, A.; Sanchez, R.; Melo, F.; Sali, A. Comparative protein structure modeling of genes and genomes. Annu. Rev. Biophys. Biomol. Struct. 2000, 29, 291–325. [Google Scholar] [CrossRef] [PubMed]
- Tamura, K.; Peterson, D.; Peterson, N.; Stecher, G.; Nei, M.; Kumar, S. MEGA5: Molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol. Biol. Evol. 2011, 28, 2731–2739. [Google Scholar] [CrossRef] [PubMed]
- Webb, B.; Sali, A. Protein Structure Modeling with MODELLER. In Functional Genomics: Methods and Protocols; Kaufmann, M., Klinger, C., Savelsbergh, A., Eds.; Springer New York: New York, NY, USA, 2017; pp. 39–54. [Google Scholar]
- Melo, F.; Sali, A. Fold assessment for comparative protein structure modeling. Protein Sci. 2007, 16, 2412–2426. [Google Scholar] [CrossRef] [Green Version]
- Eramian, D.; Shen, M.-y.; Devos, D.; Melo, F.; Sali, A.; Marti-Renom, M.A. A composite score for predicting errors in protein structure models. Protein Sci. 2006, 15, 1653–1666. [Google Scholar] [CrossRef] [Green Version]
- Case, D.A.; Cheatham, T.E., 3rd; Darden, T.; Gohlke, H.; Luo, R.; Merz, K.M.; Onufriev, A.; Simmerling, C.; Wang, B.; Woods, R.J. The Amber biomolecular simulation programs. J. Comput. Chem. 2005, 26, 1668–1688. [Google Scholar] [CrossRef] [Green Version]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 09 Rev. A.02; Gaussian Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Gao, J.; Zhang, Y.; Chen, H.; Chen, Q.; Feng, D.; Zhang, L.; Li, C. Computational insights into the interaction mechanism of transcription cofactor vestigial-like protein 4 binding to TEA domain transcription factor 4 by molecular dynamics simulation and molecular mechanics generalized Born/surface area) calculation. J. Biomol. Struct. Dyn. 2019, 37, 2538–2545. [Google Scholar] [CrossRef]
- Chen, H.; Wang, Y.; Gao, Z.; Yang, W.; Gao, J. Assessing the performance of three resveratrol in binding with SIRT1 by molecular dynamics simulation and MM/GBSA methods: The weakest binding of resveratrol 3 to SIRT1 triggers a possibility of dissociation from its binding site. J. Comput.-Aided Mol. Des. 2019, 33, 437–446. [Google Scholar] [CrossRef]
- Genheden, S.; Ryde, U. The MM/PBSA and MM/GBSA methods to estimate ligand-binding affinities. Expert Opin. Drug Discov. 2015, 10, 449–461. [Google Scholar] [CrossRef] [PubMed]
- Onufriev, A.; Bashford, D.; Case, D.A. Exploring protein native states and large-scale conformational changes with a modified generalized born model. Proteins 2004, 55, 383–394. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weiser, J.; Shenkin, P.S.; Still, W.C. Approximate atomic surfaces from linear combinations of pairwise overlaps (LCPO). J. Comput. Chem. 1999, 20, 217–230. [Google Scholar] [CrossRef]
- Chao, S.-W.; Su, M.-Y.; Chiou, L.-C.; Chen, L.-C.; Chang, C.-I.; Huang, W.-J. Total Synthesis of Hispidulin and the Structural Basis for Its Inhibition of Proto-oncogene Kinase Pim-1. J. Nat. Prod. 2015, 78, 1969–1976. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Models | molpdf a | DOPE Score b | GA341 Score c |
|---|---|---|---|
| Pim3.B99990001 | 1709.42212 | −33,078.14063 | 1.00000 |
| Pim3.B99990002 | 1679.96997 | −33,142.85938 | 1.00000 |
| Pim3.B99990003 | 1775.07397 | −33,461.44922 | 1.00000 |
| Pim3.B99990004 | 1816.04749 | −33,243.78516 | 1.00000 |
| Pim3.B99990005 | 1728.24146 | −33,196.65234 | 1.00000 |
| System | ΔEvdw | ΔEele | ΔGGB | ΔGSA | TΔS | ΔGbind | Ki (pM) |
|---|---|---|---|---|---|---|---|
| Pim1/AZD1208 | −39.65 ± 3.16 | −5.45 ± 3.33 | 9.39 ± 1.99 | −5.90 ± 0.18 | −10.44 ± 2.28 | −31.17 ± 2.64 | 17 |
| Pim1/PIM447 | −47.98 ± 2.68 | −9.99 ± 2.56 | 15.94 ± 2.16 | −6.36 ± 0.17 | −11.24 ± 2.71 | −37.15 ± 2.46 | 6 |
| Pim2/AZD1208 | −37.27 ± 2.98 | −6.16 ± 3.02 | 10.83 ± 2.38 | −5.92 ± 0.19 | −13.33 ± 2.14 | −25.19 ± 2.67 | 160 |
| Pim2/PIM447 | −46.81 ± 3.00 | −10.69 ± 2.27 | 15.86 ± 1.78 | −6.38 ± 0.12 | −12.10 ± 2.45 | −35.92 ± 2.88 | 18 |
| Pim3/AZD1208 | −39.24 ± 3.21 | −9.55 ± 2.50 | 12.61 ± 1.87 | −5.90 ± 0.13 | −13.47 ± 2.63 | −28.61 ± 2.95 | 230 |
| Pim3/PIM447 | −49.55 ± 2.33 | −9.41 ± 1.59 | 15.76 ± 1.17 | −6.63 ± 0.11 | −11.26 ± 2.36 | −38.58 ± 2.19 | 9 |
| System | Top | Bottom | Right | Left | Loop | Sum |
|---|---|---|---|---|---|---|
| Pim1/PIM447 | −7.49 | −3.93 | −3.51 | −2.87 | −2.26 | −20.06 |
| Pim2/PIM447 | −6.79 | −3.83 | −3.77 | −2.65 | −1.68 | −18.72 |
| Pim3/PIM447 | −7.97 | −4.67 | −4.20 | −2.01 | −0.81 | −19.66 |
| Pim1/AZD1208 | −7.01 | −1.50 | −4.00 | −2.77 | −2.38 | −17.66 |
| Pim2/AZD1208 | −6.61 | −1.87 | −4.38 | −2.77 | −0.05 | −15.68 |
| Pim3/AZD1208 | −6.21 | −1.86 | −4.03 | −2.61 | −2.47 | −17.18 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chen, Q.; Wang, Y.; Shi, S.; Li, K.; Zhang, L.; Gao, J. Insights into the Interaction Mechanisms of the Proviral Integration Site of Moloney Murine Leukemia Virus (Pim) Kinases with Pan-Pim Inhibitors PIM447 and AZD1208: A Molecular Dynamics Simulation and MM/GBSA Calculation Study. Int. J. Mol. Sci. 2019, 20, 5410. https://doi.org/10.3390/ijms20215410
Chen Q, Wang Y, Shi S, Li K, Zhang L, Gao J. Insights into the Interaction Mechanisms of the Proviral Integration Site of Moloney Murine Leukemia Virus (Pim) Kinases with Pan-Pim Inhibitors PIM447 and AZD1208: A Molecular Dynamics Simulation and MM/GBSA Calculation Study. International Journal of Molecular Sciences. 2019; 20(21):5410. https://doi.org/10.3390/ijms20215410
Chicago/Turabian StyleChen, Qingqing, Yan Wang, Shanshan Shi, Kaihang Li, Ling Zhang, and Jian Gao. 2019. "Insights into the Interaction Mechanisms of the Proviral Integration Site of Moloney Murine Leukemia Virus (Pim) Kinases with Pan-Pim Inhibitors PIM447 and AZD1208: A Molecular Dynamics Simulation and MM/GBSA Calculation Study" International Journal of Molecular Sciences 20, no. 21: 5410. https://doi.org/10.3390/ijms20215410