Carvedilol Ameliorates Experimental Atherosclerosis by Regulating Cholesterol Efflux and Exosome Functions

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
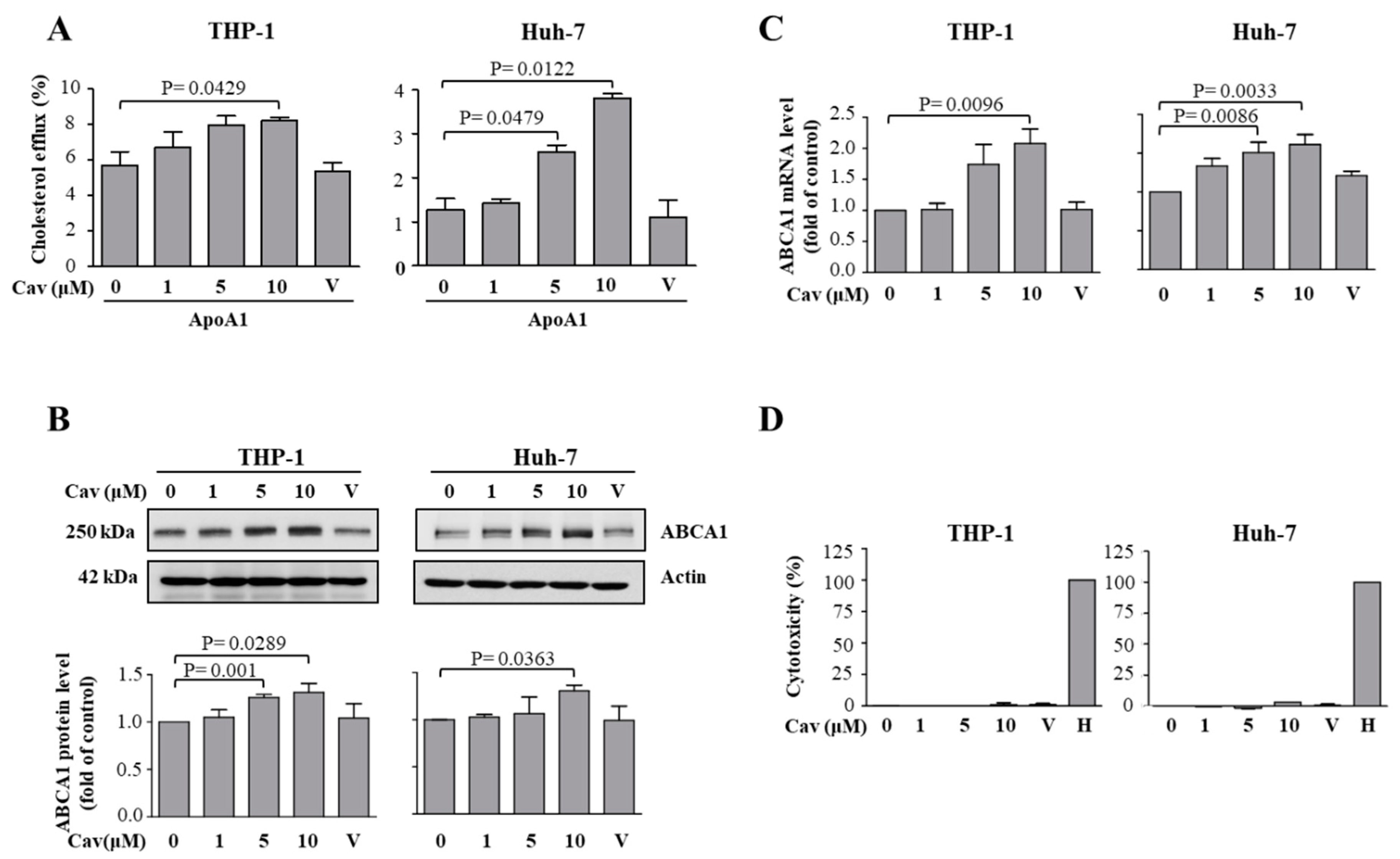
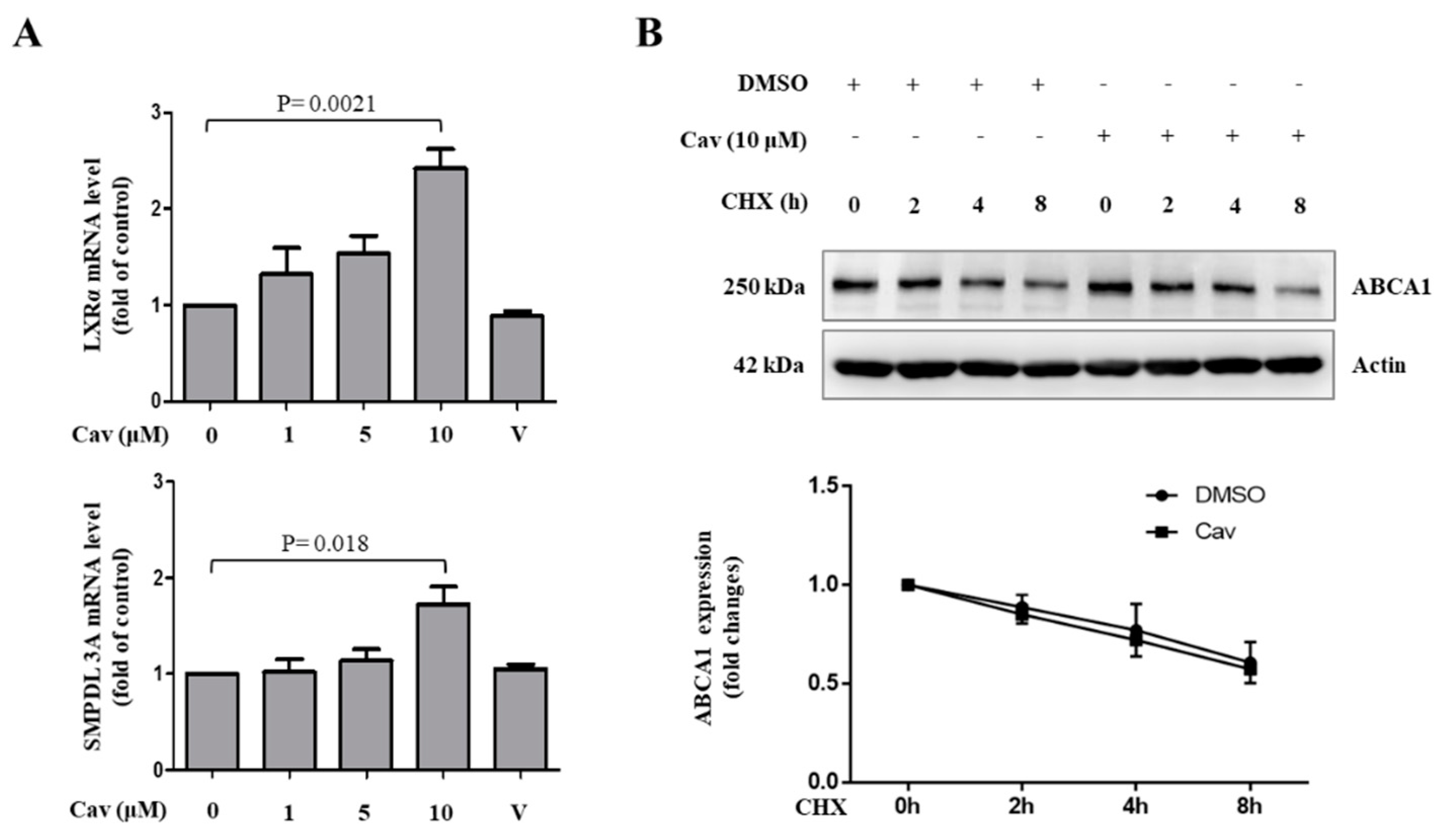
2.1. Treatment with Carvedilol Upregulated ABCA1 Expression and Cholesterol Efflux in Both Human Monocytic (THP-1) and Human Hepatic (Huh-7) Cells
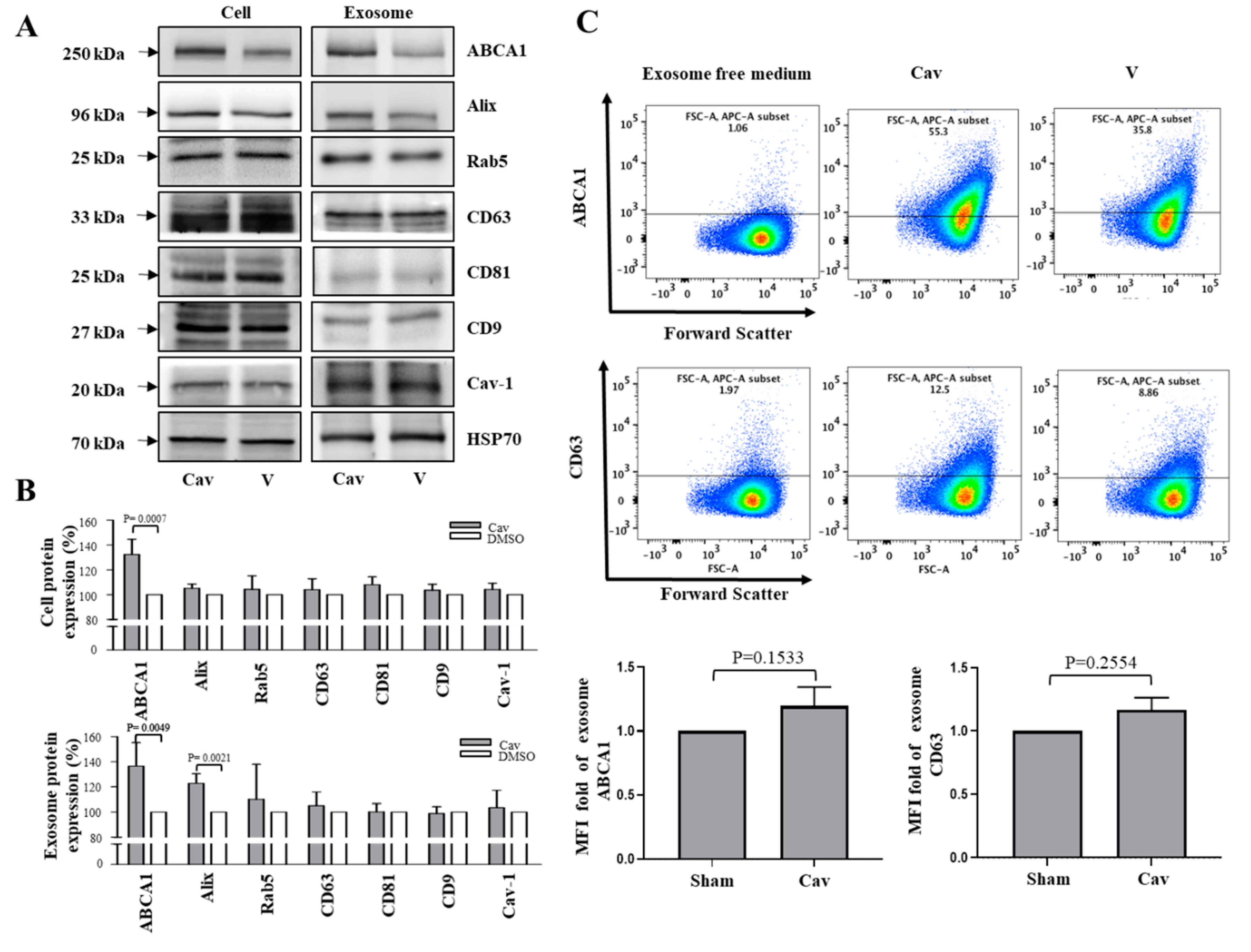
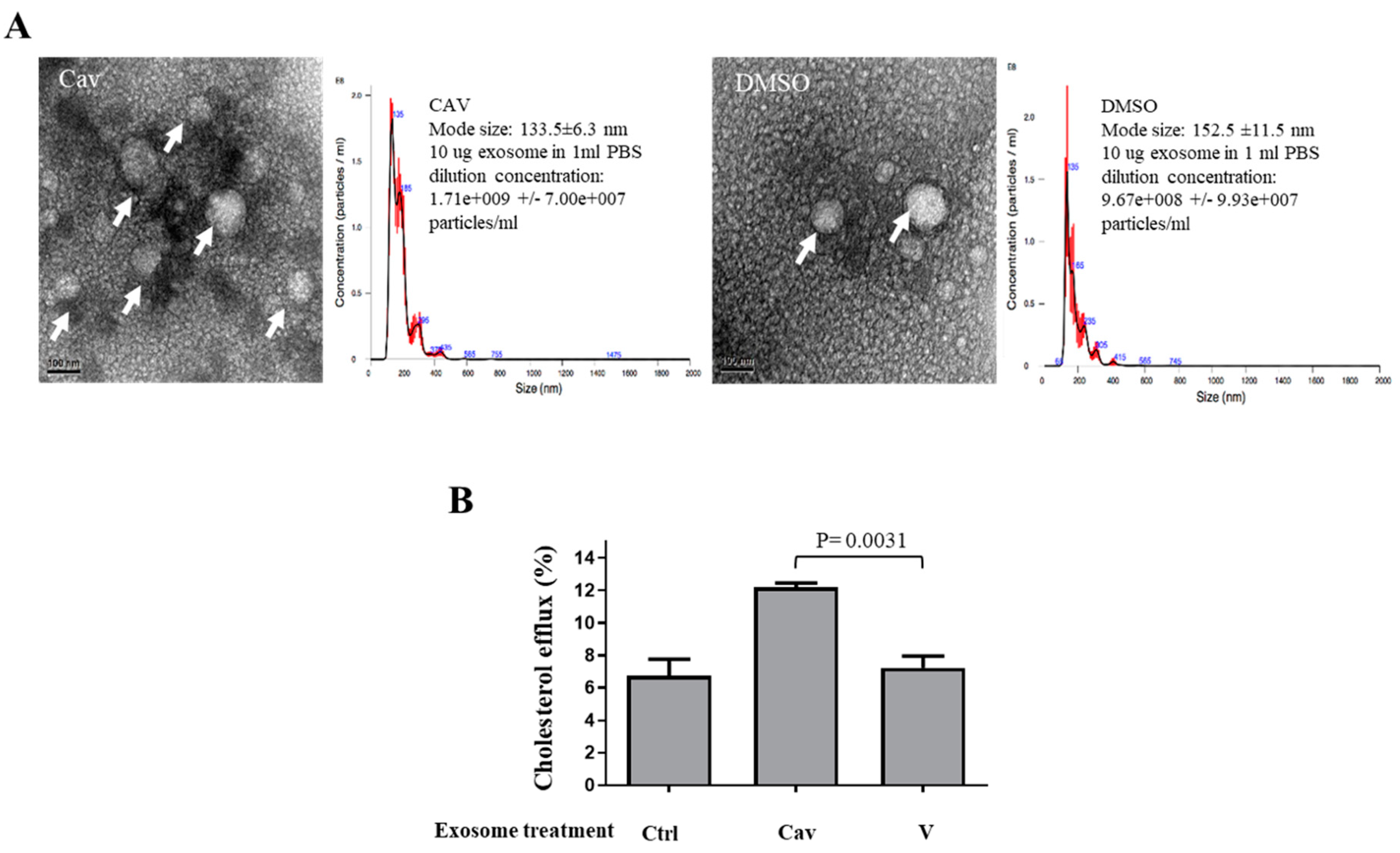
2.2. Increased ABCA1 Expression and Function in Exosomes of Carvedilol-Treated THP-1 Macrophages
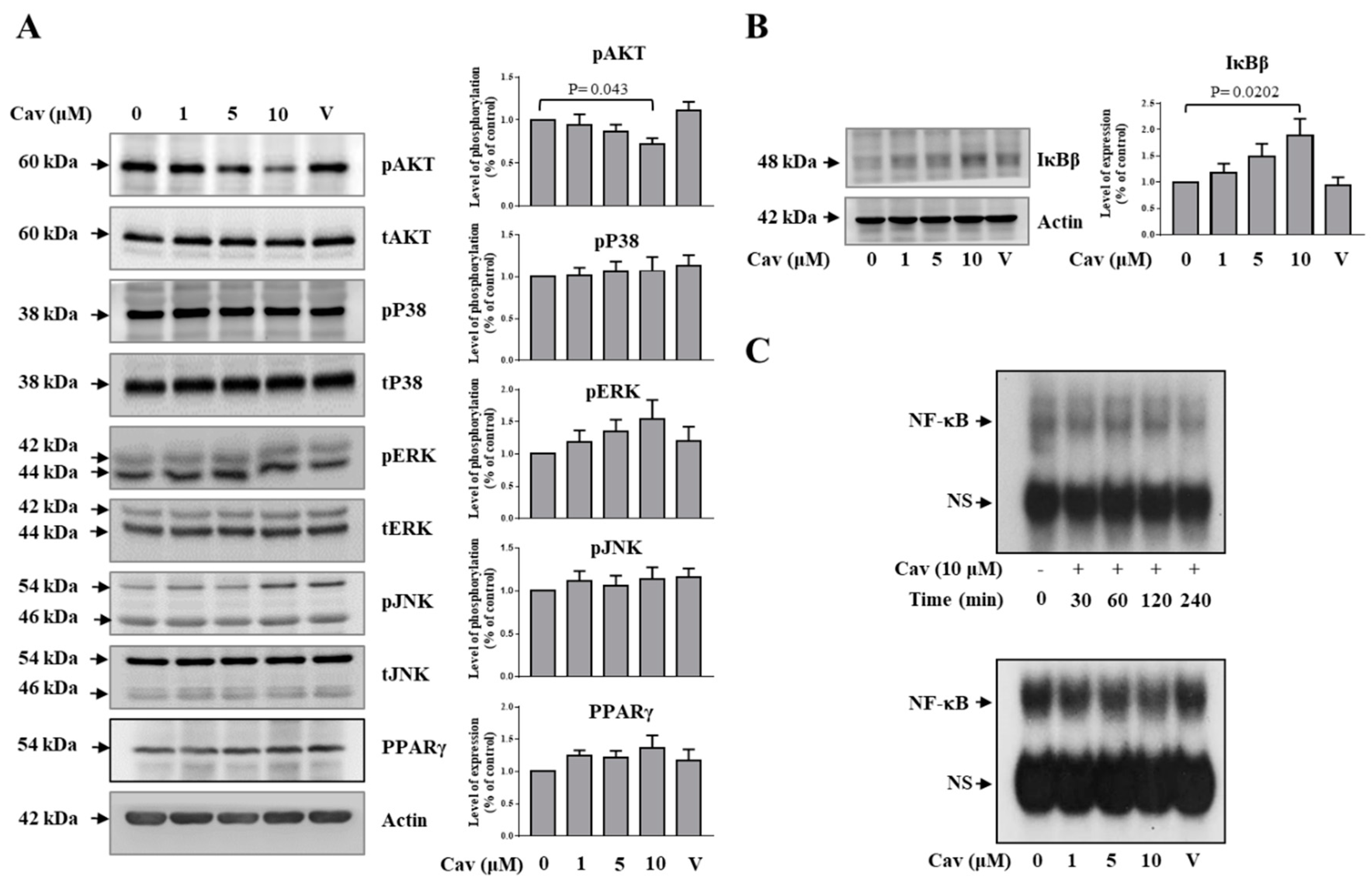
2.3. Carvedilol Inhibited Akt and NF-κB Signaling in THP-1 Macrophages
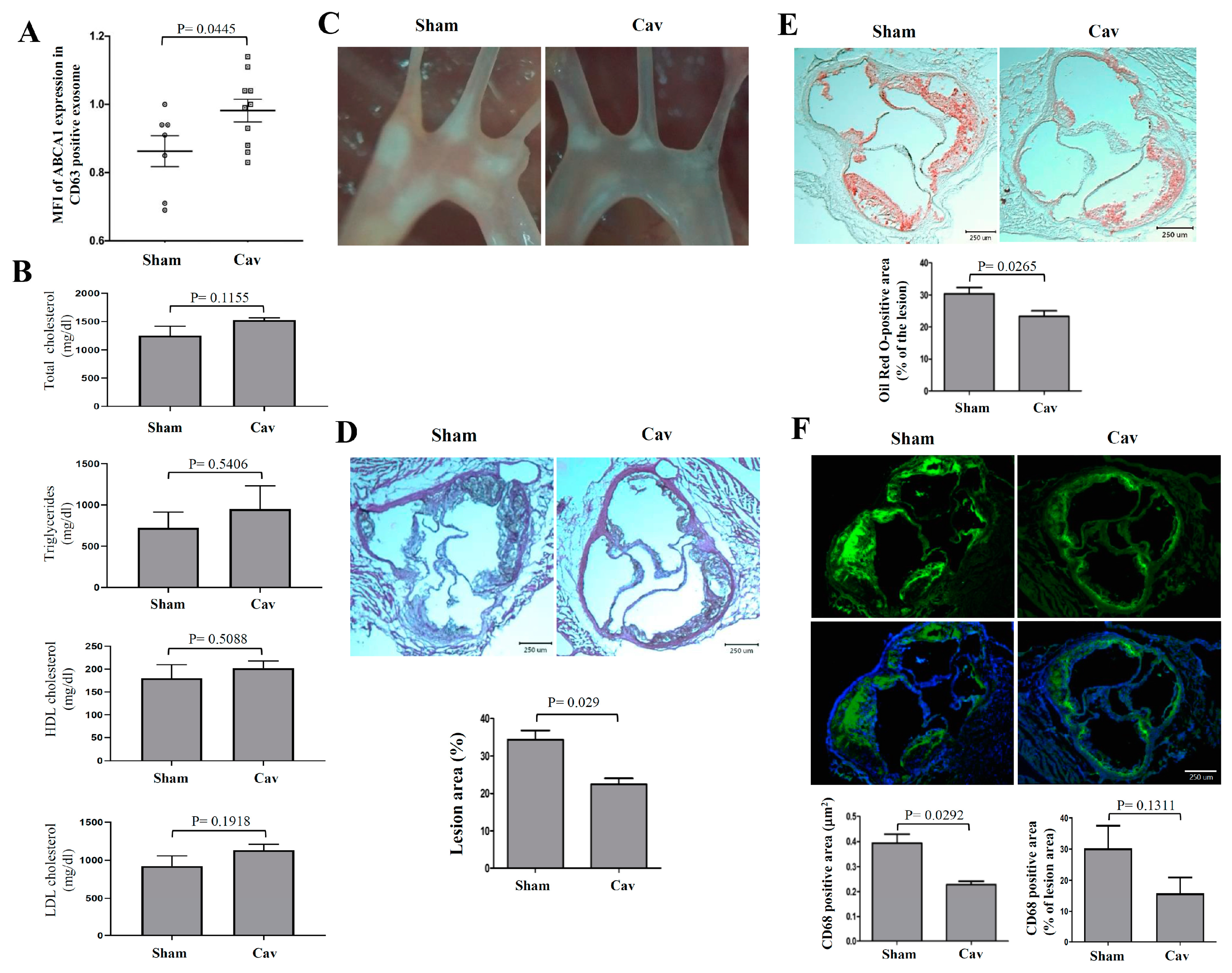
2.4. Treatment with Carvedilol Inhibited Atherosclerosis Progression in Atherosclerosis-Prone ldlr−/− Mice
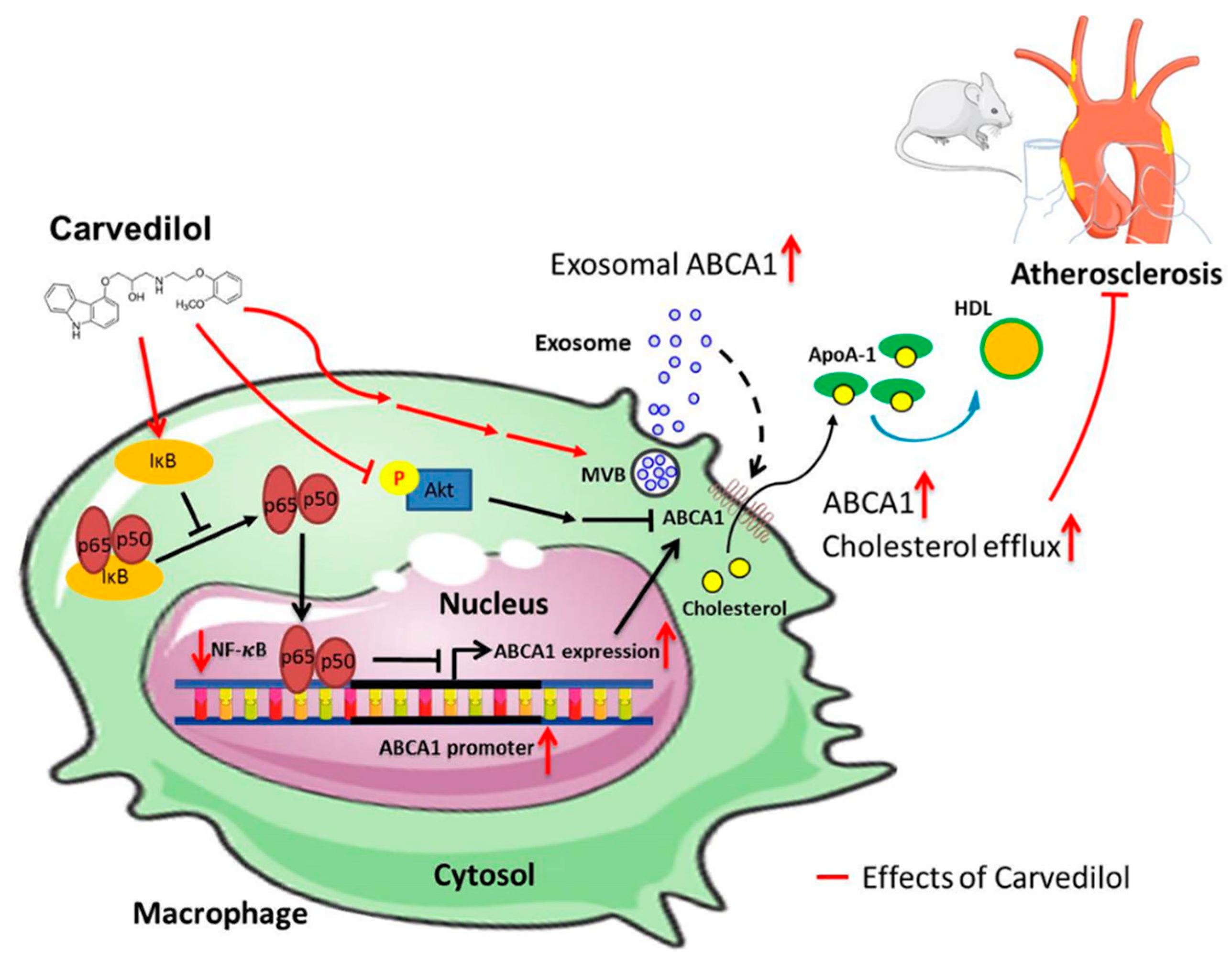
3. Discussion
4. Materials and Methods
4.1. Chemicals
4.2. Cell Culture
4.3. Measurement of Nonspecific Cytotoxicity of Cav
4.4. Cholesterol Efflux
4.5. Exosome Isolation by Polyethylene Glycol (PEG)
4.6. Semiquantification Analysis of Cell Culture Medium (CCM) Exosomes or Mice Serum Exosomes by Glycan-Recognition Bead, EXÖBead®
4.7. High-Resolution Liquid-Cell Transmission Electron Microscopy (TEM)
4.8. Nanoparticle Tracking Analysis
4.9. Western Blotting
4.10. Quantitative Reverse Transcription Polymerase Chain Reaction (qRT-PCR)
4.11. Nuclear Extract Preparation
4.12. Electrophoresis Mobility Shift Assay (EMSA)
4.13. Animals
4.14. Histology, Immunohistochemistry, and Plaque Analyses
4.15. Oil-Red O Staining (ORO)
4.16. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| ABCA1 | ATP-binding cassette transporter A1 |
| Akt | Protein kinase B |
| AP-1 | Activating protein-1 |
| CAD | Coronary artery disease |
| Cav | Carvedilol |
| CCM | Cell culture medium |
| ECs | Endothelial cells |
| EMSA | Electrophoresis mobility shift assay |
| ERK | Extracellular signal-regulated kinase |
| HDL | High-density lipoprotein |
| Huh-7 | Human hepatic cell line |
| IFN | Interferon |
| IκB | Inhibitor of kappa B |
| JNK | c-Jun N-terminal kinase |
| LDH | Lactate dehydrogenase |
| LDL | Low-density lipoprotein |
| Ldlr−/− | Low-density lipoprotein receptors knockout |
| LXR | Liver X receptor |
| MAPK | Mitogen-activated protein kinase |
| MTT | 3-[4,5-Dimethylthiazol-2-yl]-2,5-diphenyl tetrazolium bromide |
| NF-κB | Nuclear factor-kappa B |
| ORO | Oil-red O staining |
| PPAR | Peroxisome proliferator-activated receptor |
| RCT | Reverse cholesterol transport |
| SMPDL3A | Sphingomyelin Phosphodiesterase Acid Like 3A |
| TEM | Transmission electron microscopy |
| THP-1 | Human monocytic cell line |
References
- Antman, E.M.; Anbe, D.T.; Armstrong, P.W.; Bates, E.R.; Green, L.A.; Hand, M.; Hochman, J.S.; Krumholz, H.M.; Kushner, F.G.; Lamas, G.A.; et al. ACC/AHA Guidelines for the Management of Patients With ST-Elevation Myocardial Infarction. J. Am. Coll. Cardiol. 2004, 44, E1–E211. [Google Scholar] [CrossRef] [PubMed]
- Lopez-Sendon, J.; Swedberg, K.; McMurray, J.; Tamargo, J.; Maggioni, A.P.; Dargie, H.; Tendera, M.; Waagstein, F.; Kjekshus, J.; Lechat, P.; et al. Expert consensus document on beta-adrenergic receptor blockers. Eur. Heart J. 2004, 25, 1341–1362. [Google Scholar] [PubMed]
- Feuerstein, G.Z.; Ruffolo, R.R. Carvedilol, a novel vasodilating beta-blocker with the potential for cardiovascular organ protection. Eur. Heart J. 1996, 17, 24–29. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feuerstein, G.Z.; Ruffolo, R.R. Carvedilol, a novel multiple action antihypertensive agent with antioxidant activity and the potential for myocardial and vascular protection. Eur. Heart J. 1995, 16, 38–42. [Google Scholar] [CrossRef] [PubMed]
- Yue, T.-L.; McKenna, P.J.; Lysko, P.G.; Ruffolo, R.R.; Feuerstein, G.Z. Carvedilol, a new antihypertensive, prevents oxidation of human low density lipoprotein by macrophages and copper. Atherosclerosis 1992, 97, 209–216. [Google Scholar] [CrossRef]
- Li, B.; Liao, Y.-H.; Cheng, X.; Ge, H.; Guo, H.; Wang, M. Effects of carvedilol on cardiac cytokines expression and remodeling in rat with acute myocardial infarction. Int. J. Cardiol. 2006, 111, 247–255. [Google Scholar] [CrossRef]
- Wu, T.C.; Chen, Y.H.; Leu, H.B.; Chen, Y.L.; Lin, F.Y.; Lin, S.J.; Chen, J.W. Carvedilol, a pharmacological antioxidant, inhibits neointimal matrix metalloproteinase-2 and -9 in experimental atherosclerosis. Free Radic. Biol. Med. 2007, 43, 1508–1522. [Google Scholar] [CrossRef]
- Chen, J.W.; Lin, F.Y.; Chen, Y.H.; Wu, T.C.; Chen, Y.L.; Lin, S.J. Carvedilol inhibits tumor necrosis factor-alpha-induced endothelial transcription factor activation, adhesion molecule expression, and adhesiveness to human mononuclear cells. Arterioscler. Thromb. Vasc. Biol. 2004, 24, 2075–2081. [Google Scholar] [CrossRef]
- Donetti, E.; Soma, M.; Barberi, L.; Paoletti, R.; Fumagalli, R.; Roma, P.; Catapano, A.L. Dual effects of the antioxidant agents probucol and carvedilol on proliferative and fatty lesions in hypercholesterolemic rabbits. Atherosclerosis 1998, 141, 45–51. [Google Scholar] [CrossRef]
- Singaraja, R.R.; Fievet, C.; Castro, G.; James, E.R.; Hennuyer, N.; Clee, S.M.; Bissada, N.; Choy, J.C.; Fruchart, J.-C.; McManus, B.M.; et al. Increased ABCA1 activity protects against atherosclerosis. J. Clin. Investig. 2002, 110, 35–42. [Google Scholar] [CrossRef]
- Oram, J.F.; Lawn, R.M. ABCA1: The gatekeeper for eliminating excess tissue cholesterol. J. Lipid Res. 2001, 42, 1173–1179. [Google Scholar] [PubMed]
- Khera, A.V.; Cuchel, M.; De La Llera-Moya, M.; Rodrigues, A.; Burke, M.F.; Jafri, K.; French, B.C.; Phillips, J.A.; Mucksavage, M.L.; Wilensky, R.L.; et al. Cholesterol efflux capacity, high-density lipoprotein function, and atherosclerosis. N. Engl. J. Med. 2011, 364, 127–135. [Google Scholar] [CrossRef] [PubMed]
- Rohatgi, A.; Khera, A.; Berry, J.D.; Givens, E.G.; Ayers, C.R.; Wedin, K.E.; Neeland, I.J.; Yuhanna, I.S.; Rader, D.R.; De Lemos, J.A.; et al. HDL cholesterol efflux capacity and incident cardiovascular events. N. Engl. J. Med. 2014, 371, 2383–2393. [Google Scholar] [CrossRef] [PubMed]
- Sluijter, J.P.; Verhage, V.; Deddens, J.C.; Akker, F.V.D.; Doevendans, P.A. Microvesicles and exosomes for intracardiac communication. Cardiovasc. Res. 2014, 102, 302–311. [Google Scholar] [CrossRef] [Green Version]
- Chistiakov, D.A.; Orekhov, A.N.; Bobryshev, Y.V. Extracellular vesicles and atherosclerotic disease. Cell. Mol. Life Sci. 2015, 72, 2697–2708. [Google Scholar] [CrossRef]
- Hergenreider, E.; Heydt, S.; Treguer, K.; Boettger, T.; Horrevoets, A.J.G.; Zeiher, A.M.; Scheffer, M.P.; Frangakis, A.S.; Yin, X.; Mayr, M.; et al. Atheroprotective communication between endothelial cells and smooth muscle cells through miRNAs. Nat. Cell Biol. 2012, 14, 249–256. [Google Scholar] [CrossRef]
- Sheldon, H.; Heikamp, E.; Turley, H.; Dragovic, R.; Thomas, P.; Oon, C.E.; Leek, R.; Edelmann, M.; Kessler, B.; Sainson, R.C.; et al. New mechanism for Notch signaling to endothelium at a distance by Delta-like 4 incorporation into exosomes. Blood 2010, 116, 2385–2394. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Liu, D.; Chen, X.; Li, J.; Li, L.; Bian, Z.; Sun, F.; Lu, J.; Yin, Y.; Cai, X.; et al. Secreted Monocytic miR-150 Enhances Targeted Endothelial Cell Migration. Mol. Cell 2010, 39, 133–144. [Google Scholar] [CrossRef] [Green Version]
- Gao, W.; Liu, H.; Yuan, J.; Wu, C.; Huang, D.; Ma, Y.; Zhu, J.; Ma, L.; Guo, J.; Shi, H.; et al. Exosomes derived from mature dendritic cells increase endothelial inflammation and atherosclerosis via membrane TNF-alpha mediated NF-kappaB pathway. J. Cell. Mol. Med. 2016, 20, 2318–2327. [Google Scholar] [CrossRef]
- Martínez, M.C.; Andriantsitohaina, R. Extracellular Vesicles in Metabolic Syndrome. Circ. Res. 2017, 120, 1674–1686. [Google Scholar] [CrossRef]
- Oram, J.F.; Heinecke, J.W. ATP-Binding Cassette Transporter A1: A Cell Cholesterol Exporter That Protects Against Cardiovascular Disease. Physiol. Rev. 2005, 85, 1343–1372. [Google Scholar] [CrossRef] [PubMed]
- Bashore, A.C.; Liu, M.; Key, C.C.; Boudyguina, E.; Wang, X.; Carroll, C.M.; Sawyer, J.K.; Mullick, A.E.; Lee, R.G.; Macauley, S.L.; et al. Targeted Deletion of Hepatocyte Abca1 Increases Plasma HDL (High-Density Lipoprotein) Reverse Cholesterol Transport via the LDL (Low-Density Lipoprotein) Receptor. Arterioscler. Thromb. Vasc. Biol. 2019, 39, 1747–1761. [Google Scholar] [CrossRef] [PubMed]
- Bodzioch, M.; Orsó, E.; Klucken, J.; Langmann, T.; Böttcher, A.; Diederich, W.; Drobnik, W.; Barlage, S.; Büchler, C.; Porsch-Özcürümez, M.; et al. The gene encoding ATP-binding cassette transporter 1 is mutated in Tangier disease. Nat. Genet. 1999, 22, 347–351. [Google Scholar] [CrossRef] [PubMed]
- Basso, F.; Freeman, L.; Knapper, C.L.; Remaley, A.; Stonik, J.; Neufeld, E.B.; Tansey, T.; Amar, M.J.; Fruchart-Najib, J.; Duverger, N.; et al. Role of the hepatic ABCA1 transporter in modulating intrahepatic cholesterol and plasma HDL cholesterol concentrations. J. Lipid Res. 2003, 44, 296–302. [Google Scholar] [CrossRef] [Green Version]
- Chung, S.; Timmins, J.M.; Duong, M.; Degirolamo, C.; Rong, S.; Sawyer, J.K.; Singaraja, R.R.; Hayden, M.R.; Maeda, N.; Rudel, L.L.; et al. Targeted Deletion of Hepatocyte ABCA1 Leads to Very Low Density Lipoprotein Triglyceride Overproduction and Low Density Lipoprotein Hypercatabolism. J. Boil. Chem. 2010, 285, 12197–12209. [Google Scholar] [CrossRef] [Green Version]
- Bi, X.; Zhu, X.; Duong, M.; Boudyguina, E.Y.; Wilson, M.D.; Gebre, A.K.; Parks, J.S. Liver ABCA1 deletion in LDLrKO mice does not impair macrophage reverse cholesterol transport or exacerbate atherogenesis. Arter. Thromb. Vasc. Boil. 2013, 33, 2288–2296. [Google Scholar] [CrossRef]
- Colombo, M.; Moita, C.F.; Van Niel, G.; Kowal, J.; Vigneron, J.; Benaroch, P.; Manel, N.; Moita, L.F.; Théry, C.; Raposo, G. Analysis of ESCRT functions in exosome biogenesis, composition and secretion highlights the heterogeneity of extracellular vesicles. J. Cell Sci. 2013, 126, 5553–5565. [Google Scholar] [CrossRef]
- Jeppesen, D.K.; Fenix, A.M.; Franklin, J.L.; Higginbotham, J.N.; Zhang, Q.; Zimmerman, L.J.; Liebler, D.C.; Ping, J.; Liu, Q.; Evans, R.; et al. Reassessment of Exosome Composition. Cell 2019, 177, 428–445. [Google Scholar] [CrossRef]
- Nandi, S.; Ma, L.; Denis, M.; Karwatsky, J.; Li, Z.; Jiang, X.C.; Zha, X. ABCA1-mediated cholesterol efflux generates microparticles in addition to HDL through processes governed by membrane rigidity. J. Lipid Res. 2009, 50, 456–466. [Google Scholar] [CrossRef] [Green Version]
- Hafiane, A.; Genest, J. ATP binding cassette A1 (ABCA1) mediates microparticle formation during high-density lipoprotein (HDL) biogenesis. Atherosclerosis 2017, 257, 90–99. [Google Scholar] [CrossRef]
- Gaceb, A.; Martinez, M.C.; Andriantsitohaina, R. Extracellular vesicles: New players in cardiovascular diseases. Int. J. Biochem. Cell Biol. 2014, 50, 24–28. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nguyen, M.A.; Karunakaran, D.; Geoffrion, M.; Cheng, H.S.; Tandoc, K.; Perisic Matic, L.; Hedin, U.; Maegdefessel, L.; Fish, J.E.; Rayner, K.J. Extracellular Vesicles Secreted by Atherogenic Macrophages Transfer MicroRNA to Inhibit Cell Migration. Arterioscler. Thromb. Vasc. Biol. 2018, 38, 49–63. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xie, Z.; Wang, X.; Liu, X.; Du, H.; Sun, C.; Shao, X.; Tian, J.; Gu, X.; Wang, H.; Tian, J.; et al. Adipose-Derived Exosomes Exert Proatherogenic Effects by Regulating Macrophage Foam Cell Formation and Polarization. J. Am. Heart Assoc. 2018, 7, e007442. [Google Scholar] [CrossRef] [PubMed]
- Niu, C.; Wang, X.; Zhao, M.; Cai, T.; Liu, P.; Li, J.; Willard, B.; Zu, L.; Zhou, E.; Li, Y.; et al. Macrophage Foam Cell-Derived Extracellular Vesicles Promote Vascular Smooth Muscle Cell Migration and Adhesion. J. Am. Heart Assoc. 2016, 5, e004099. [Google Scholar] [CrossRef] [PubMed]
- Vion, A.-C.; Ramkhelawon, B.; Loyer, X.; Chironi, G.; Devue, C.; Loirand, G.; Tedgui, A.; Lehoux, S.; Boulanger, C.M. Shear Stress Regulates Endothelial Microparticle Release. Circ. Res. 2013, 112, 1323–1333. [Google Scholar] [CrossRef] [PubMed]
- Baker, R.G.; Hayden, M.S.; Ghosh, S. NF-kappaB, inflammation, and metabolic disease. Cell Metab. 2011, 13, 11–22. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, V.; van Dijk, K.W.; Groen, A.K.; Vos, R.M.; van der Kaa, J.; Gijbels, M.J.; Havekes, L.M.; Pannekoek, H. Macrophage-specific inhibition of NF-kappaB activation reduces foam-cell formation. Atherosclerosis 2007, 192, 283–290. [Google Scholar] [CrossRef]
- Bie, J.; Zhao, B.; Song, J.; Ghosh, S. Improved insulin sensitivity in high fat- and high cholesterol-fed Ldlr−/− mice with macrophage-specific transgenic expression of cholesteryl ester hydrolase: Role of macrophage inflammation and infiltration into adipose tissue. J. Biol. Chem. 2010, 285, 13630–13637. [Google Scholar] [CrossRef]
- Gerbod-Giannone, M.C.; Li, Y.; Holleboom, A.; Han, S.; Hsu, L.C.; Tabas, I.; Tall, A.R. TNFalpha induces ABCA1 through NF-kappaB in macrophages and in phagocytes ingesting apoptotic cells. Proc. Natl. Acad. Sci. USA 2006, 103, 3112–3117. [Google Scholar] [CrossRef]
- Dong, F.; Mo, Z.; Eid, W.; Courtney, K.C.; Zha, X. Akt Inhibition Promotes ABCA1-Mediated Cholesterol Efflux to ApoA-I through Suppressing mTORC1. PLoS ONE 2014, 9, e113789. [Google Scholar] [CrossRef]
- Fernández-Hernando, C.; Ackah, E.; Yu, J.; Suárez, Y.; Murata, T.; Iwakiri, Y.; Prendergast, J.; Miao, R.Q.; Birnbaum, M.J.; Sessa, W.C. Loss of Akt1 leads to severe atherosclerosis and occlusive coronary artery disease. Cell Metab. 2007, 6, 446–457. [Google Scholar] [CrossRef] [PubMed]
- Bensinger, S.J.; Bradley, M.N.; Joseph, S.B.; Zelcer, N.; Janssen, E.M.; Hausner, M.A.; Shih, R.; Parks, J.S.; Edwards, P.A.; Jamieson, B.D.; et al. LXR signaling couples sterol metabolism to proliferation in the acquired immune response. Cell 2008, 134, 97–111. [Google Scholar] [CrossRef] [PubMed]
- Kappus, M.S.; Murphy, A.J.; Abramowicz, S.; Ntonga, V.; Welch, C.L.; Tall, A.R.; Westerterp, M. Activation of Liver X Receptor Decreases Atherosclerosis in Ldlr−/− Mice in the Absence of ATP-Binding Cassette Transporters A1 and G1 in Myeloid Cells. Arterioscler. Thromb. Vasc. Boil. 2014, 34, 279–284. [Google Scholar] [CrossRef] [PubMed]
- Joseph, S.B.; Castrillo, A.; Laffitte, B.A.; Mangelsdorf, D.J.; Tontonoz, P. Reciprocal regulation of inflammation and lipid metabolism by liver X receptors. Nat. Med. 2003, 9, 213–219. [Google Scholar] [CrossRef] [PubMed]
- Venkateswaran, A.; Laffitte, B.A.; Joseph, S.B.; Mak, P.A.; Wilpitz, D.C.; Edwards, P.A.; Tontonoz, P. Control of cellular cholesterol efflux by the nuclear oxysterol receptor LXR alpha. Proc. Natl. Acad. Sci. USA 2000, 97, 12097–12102. [Google Scholar] [CrossRef] [PubMed]
- Fonarow, G.C.; Deedwania, P.; Fonseca, V.; Nesto, R.W.; Watson, K.; Tarka, E.; Lukas, M.A.; Madan, A.; Shabbout, M. Differential effects of extended-release carvedilol and extended-release metoprolol on lipid profiles in patients with hypertension: Results of the Extended-Release Carvedilol Lipid Trial. J. Am. Soc. Hypertens. 2009, 3, 210–220. [Google Scholar] [CrossRef]
- Seguchi, H.; Nakamura, H.; Aosaki, N.; Homma, Y.; Mikami, Y.; Takahashi, S. Effects of carvedilol on serum lipids in hypertensive and normotensive subjects. Eur. J. Clin. Pharmacol. 1990, 38, S139–S142. [Google Scholar] [CrossRef]
- Shimada, K.; Hirano, E.; Kimura, T.; Fujita, M.; Kishimoto, C. Carvedilol reduces the severity of atherosclerosis in apolipoprotein E-deficient mice via reducing superoxide production. Exp. Biol. Med. 2012, 237, 1039–1044. [Google Scholar] [CrossRef] [Green Version]
- Dandona, P.; Ghanim, H.; Brooks, D.P. Antioxidant activity of carvedilol in cardiovascular disease. J. Hypertens. 2007, 25, 731–741. [Google Scholar] [CrossRef]
- Yang, S.-P.; Ho, L.-J.; Lin, Y.-L.; Cheng, S.-M.; Tsao, T.-P.; Chang, D.-M.; Hsu, Y.-L.; Shih, C.-Y.; Juan, T.-Y.; Lai, J.-H. Carvedilol, a new antioxidative beta-blocker, blocks in vitro human peripheral blood T cell activation by downregulating NF-kappaB activity. Cardiovasc. Res. 2003, 59, 776–787. [Google Scholar] [CrossRef]
- Chen, S.-J.; Kao, Y.-H.; Jing, L.; Chuang, Y.-P.; Wu, W.-L.; Liu, S.-T.; Huang, S.-M.; Lai, J.-H.; Ho, L.-J.; Tsai, M.-C.; et al. Epigallocatechin-3-gallate Reduces Scavenger Receptor A Expression and Foam Cell Formation in Human Macrophages. J. Agric. Food Chem. 2017, 65, 3141–3150. [Google Scholar] [CrossRef] [PubMed]
- Thery, C.; Amigorena, S.; Raposo, G.; Clayton, A. Isolation and characterization of exosomes from cell culture supernatants and biological fluids. Curr. Protoc. Cell Biol. 2006, 30, 3–22. [Google Scholar] [CrossRef] [PubMed]
- Rider, M.A.; Hurwitz, S.N.; Meckes, D.G. ExtraPEG: A Polyethylene Glycol-Based Method for Enrichment of Extracellular Vesicles. Sci. Rep. 2016, 6, 23978. [Google Scholar] [CrossRef] [PubMed]
- Nawaz, M. Abstract Book: ISEV2017. J. Extracell. Vesicles 2017, 6, 1310414. [Google Scholar]
- Hida, K.; Zheng, L.; Wauben, M.; Ikuo, M.; Shen, L.; Gho, Y.S.; Takakura, Y.; Thery, C. ISEV2019 Abstract Book. J. Extracell. Vesicles 2019, 8, 1593587. [Google Scholar]
- Liao, H.-G.; Zheng, H. Liquid Cell Transmission Electron Microscopy. Annu. Rev. Phys. Chem. 2016, 67, 719–747. [Google Scholar] [CrossRef]
- Sokolova, V.; Ludwig, A.-K.; Hornung, S.; Rotan, O.; Horn, P.A.; Epple, M.; Giebel, B. Characterisation of exosomes derived from human cells by nanoparticle tracking analysis and scanning electron microscopy. Colloids Surf. B Biointerfaces 2011, 87, 146–150. [Google Scholar] [CrossRef]
- Schmittgen, T.D.; Livak, K.J. Analyzing real-time PCR data by the comparative C(T) method. Nat. Protoc. 2008, 3, 1101–1108. [Google Scholar] [CrossRef]
- Cheng, S.-M.; Lai, J.-H.; Yang, S.-P.; Tsao, T.-P.; Ho, L.-J.; Liou, J.-T.; Cheng, C.-C. Modulation of human T cells signaling transduction by lovastatin. Int. J. Cardiol. 2010, 140, 24–33. [Google Scholar] [CrossRef]
- Kumar, S.; Chen, M.; Li, Y.; Wong, F.H.S.; Thiam, C.W.; Hossain, M.Z.; Poh, K.K.; Hirohata, S.; Ogawa, H.; Angeli, V.; et al. Loss of ADAMTS4 reduces high fat diet-induced atherosclerosis and enhances plaque stability in ApoE−/− mice. Sci. Rep. 2016, 6, 31130. [Google Scholar] [CrossRef]
- Seimon, T.A.; Wang, Y.; Han, S.; Senokuchi, T.; Schrijvers, D.M.; Kuriakose, G.; Tall, A.R.; Tabas, I.A. Macrophage deficiency of p38alpha MAPK promotes apoptosis and plaque necrosis in advanced atherosclerotic lesions in mice. J. Clin. Investig. 2009, 119, 886–898. [Google Scholar] [PubMed]
- Lin, C.S.; Lin, F.Y.; Ho, L.J.; Tsai, C.S.; Cheng, S.M.; Wu, W.L.; Huang, C.Y.; Lian, C.H.; Yang, S.P.; Lai, J.H. PKCdelta signalling regulates SR-A and CD36 expression and foam cell formation. Cardiovasc. Res. 2012, 95, 346–355. [Google Scholar] [CrossRef] [PubMed]







© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chen, S.-J.; Tsui, P.-F.; Chuang, Y.-P.; Chiang, D.M.-L.; Chen, L.W.; Liu, S.-T.; Lin, F.-Y.; Huang, S.-M.; Lin, S.-H.; Wu, W.-L.; et al. Carvedilol Ameliorates Experimental Atherosclerosis by Regulating Cholesterol Efflux and Exosome Functions. Int. J. Mol. Sci. 2019, 20, 5202. https://doi.org/10.3390/ijms20205202
Chen S-J, Tsui P-F, Chuang Y-P, Chiang DM-L, Chen LW, Liu S-T, Lin F-Y, Huang S-M, Lin S-H, Wu W-L, et al. Carvedilol Ameliorates Experimental Atherosclerosis by Regulating Cholesterol Efflux and Exosome Functions. International Journal of Molecular Sciences. 2019; 20(20):5202. https://doi.org/10.3390/ijms20205202
Chicago/Turabian StyleChen, Sy-Jou, Pi-Fen Tsui, Yi-Ping Chuang, Dapi Meng-Lin Chiang, Liv Weichien Chen, Shu-Ting Liu, Feng-Yen Lin, Shih-Ming Huang, Shih-Hua Lin, Wan-Lin Wu, and et al. 2019. "Carvedilol Ameliorates Experimental Atherosclerosis by Regulating Cholesterol Efflux and Exosome Functions" International Journal of Molecular Sciences 20, no. 20: 5202. https://doi.org/10.3390/ijms20205202